

Mitochondria, traditionally known for their role in power (ATP) generation, are important signalling organelles. To support these vital functions, mitochondria need to undergo continuous remodelling- a process known as “mitochondrial dynamics.”1 Failure to do so compromises their function, leading to a spectrum of age-related human diseases, including diabetes, cardiovascular (ischaemia), neuronal (stroke, Parkinson's and Alzheimer's) and renal diseases, and cancer.2 It is generally acknowledged that a rise in oxidative stress, characterised by an increased production of reactive oxygen species (ROS), is responsible for abnormal mitochondrial dynamics. However, how ROS signal abnormal mitochondrial dynamics was unclear.
Mitochondrial dynamics involves continuous fission and fusion of the mitochondrial tubular network.1,2 Fission is initiated by the ER-assisted constriction of the mitochondrial tubules followed by their cleavage by Drp1 (dynamin-related protein1) and dynamin 2. Drp1 is normally localised to the cytoplasm where it needs to undergo modifications before it can be recruited to mitochondria. One such modification requires a Ca2+ signal from an undetermined source. By activating calcineurin, Ca2+ removes the inhibitory phosphate group from Drp1, thereby promoting its recruitment to mitochondria. Fission results in the generation of functional and dysfunctional fragments. The latter are removed by mitophagy. Functional mitochondrial fragments merge with the healthy mitochondrial network, with the help of mitofusin-1/2 (MFN-1/2) and OPA-1(Optic atrophy type 1), which catalyse the fusion of the outer and inner membranes of mitochondria respectively.
Previous studies have shown that free fatty acids (FFAs), whose levels increase in obesity and contribute to type 2 diabetes, cause extensive fragmentation of the mitochondrial network in pancreatic β-cells by stimulating cellular ROS production.3 In our recent publication,4 we tested the possibility that the rise in ROS would activate Ca2+ channels to provide the Ca2+ required for Drp1 recruitment to mitochondria. We focussed our attention on TRPM2 (transient receptor potential melastatin2) channels because these are activated by ROS and conduct cations including Ca2+.5
Consistent with our prediction, inhibition of TRPM2 channels by chemical, siRNA and gene knockout approaches prevented FFA-induced mitochondrial fragmentation as well as β-cell death.4 ROS required for mitochondrial fission are provided by the FFA-mediated activation of cytoplasmic NADPH-oxidase-2. At first, these findings seemed to support our initial hypothesis that TRPM2 activation provides the Ca2+ required for Drp1 recruitment and the subsequent mitochondrial fragmentation. However, we have decided to test the role of Zn2+ because our previous study demonstrated that Zn2+ chelation prevents TRPM2-mediated β-cell death,6 and other studies suggested that mitochondrial fragmentation precedes β-cell death.3 Surprisingly, chelating Zn2+ alone was sufficient to prevent mitochondrial fission. On probing further, we found that free Zn2+ found largely in the lysosomes was transferred to mitochondria. In a related study, we have demonstrated that TRPM2-mediated Ca2+ entry causes lysosomal membrane permeabilisation and Zn2+ release.7 Although we do not know how this Zn2+ is transferred to mitochondria, rise in the mitochondrial Zn2+ led to a marked loss of mitochondrial membrane potential, promoting Drp1 recruitment to mitochondria and β-cell apoptosis.
This study has thus led to the discovery of a novel signalling pathway where Ca2+ and Zn2+ collaborate to transduce the signal from FFA to the mitochondria of pancreatic β-cells to cause cell death (Fig. 1). Importantly, the two ions facilitate communication between different cellular compartments. Thus extracellular Ca2+ enters the cytoplasm via ROS-activated plasma membrane TRPM2 channels. The resultant rise in cytosolic Ca2+ triggers escape of lysosomal free Zn2+ to mitochondria. Zn2+, being an inhibitor of the electron transport chain, leads to the loss of mitochondrial membrane potential required for the recruitment cytoplasmic Drp1 to mitochondria. Drp1 then induces excessive mitochondrial fission, leading to increased apoptotic β-cell death.
Figure 1.
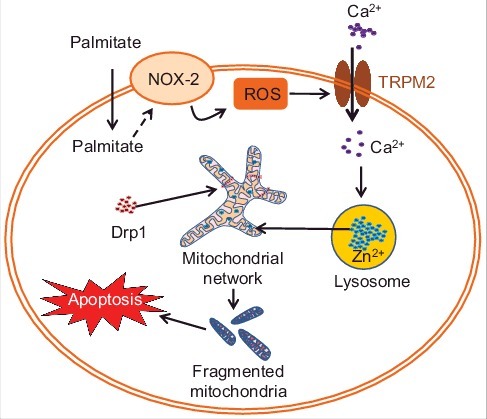
Schematic of how palmitate induces mitochondrial fragmentation in pancreatic β-cells. Palmitate activates NOX-2 (NADPH oxidase-2) to induce ROS (reactive oxygen species) production. ROS activates TRPM2 channel leading to extracellular Ca2+ (purple dots) entry, and Ca2+-induced lysosomal Zn2+ (blue dots) transfer to mitochondria. Rise in mitochondrial Zn2+ induces Drp1 (red dots) recruitment, breakdown of the mitochondrial network and eventually β-cell death.
These new findings raise a number of unanswered questions: (1) How does the rise in cytosolic Ca2+ induce lysosomal Zn2+ release? (2) How does Zn2+ enter mitochondria? Is it via the mitochondrial uniporter (MCU) or other transport molecules? (3) How does Zn2+ promote Drp1 recruitment? Which of the multiple posttranslational mechanisms required for mitochondrial Drp1 recruitment does Zn2+ affect? (4) Does Drp-1 recruitment to mitochondria involve Ca2+ signalling? If so, where does this Ca2+ come from? Interestingly, a recent study suggested Ca2+-calcineurin signalling occurs at the ER-mitochondria junction.8 Answers to these key questions are important if we are to understand how the oxidative stress signals are translated into mitochondrial fragmentation, which is a common feature of most age-related human illnesses.
In conclusion, our findings highlight a previously unappreciated role for ionic signalling, in particular a role for Zn2+ signalling, in mitochondrial dynamics. Furthermore, they suggest that ionic signalling is not limited to communication between just two organelles (for example, between the plasma membrane and the ER, and mitochondria and the ER), but could involve multiple organelles. Our findings emphasise the importance of an integrated approach to investigate how abnormal inter-organelle signalling can affect organelle homeostasis and contribute to human diseases. Such approaches may reveal hitherto unknown therapeutic targets that may be common to a wide range of human diseases.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Friedman JR, Nunnari J. Mitochondrial form and function. Nature 2014;505:335-43. doi: 10.1038/nature12985. PMID:24429632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236-5110. doi: 10.1056/NEJMra1215233. PMID:24304053 [DOI] [PubMed] [Google Scholar]
- [3].Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, Walzer G, Twig G, Katz S, Corkey BE, et al. . Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 2009;58:2303-15. doi: 10.2337/db07-1781. PMID:19581419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Li F, Munsey TS, Sivaprasadarao A. TRPM2-mediated rise in mitochondrial Zn2+ promotes palmitate-induced mitochondrial fission and pancreatic beta-cell death in rodents. Cell Death Differ. 2017. doi: 10.1038/cdd.2017.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sumoza-Toledo A, Penner R. TRPM2: a multifunctional ion channel for calcium signalling. J Physiol-London 2011;589:1515-2510. doi: 10.1113/jphysiol.2010.201855. PMID:21135052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Manna PT, Munsey TS, Abuarab N, Li F, Asipu A, Howell G, Sedo A, Yang W, Naylor J, Beech DJ, et al. . TRPM2 mediated intracellular Zn2+ release triggers pancreatic beta cell death. Biochem J. 2015;466:537-46. doi: 10.1042/BJ20140747. PMID:25562606 [DOI] [PubMed] [Google Scholar]
- [7].Abuarab N, Munsey TS, Jiang LH, Li J, Sivaprasadarao A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci Signal. 2017;10. doi: 10.1126/scisignal.aal4161. PMID:28765513 [DOI] [PubMed] [Google Scholar]
- [8].Mehta S, Aye-Han NN, Ganesan A, Oldach L, Gorshkov K, Zhang J. Calmodulin-controlled spatial decoding of oscillatory Ca2+ signals by calcineurin. Elife 2014;3:e03765. doi: 10.7554/eLife.03765. PMID:25056880 [DOI] [PMC free article] [PubMed] [Google Scholar]