

ABSTRACT
Voltage-gated sodium channels (VGSCs) are the basic ion channels for neuronal excitability, which are crucial for the resting potential and the generation and propagation of action potentials in neurons. To date, at least nine distinct sodium channel isoforms have been detected in the nervous system. Recent studies have identified that voltage-gated sodium channels not only play an essential role in the normal electrophysiological activities of neurons but also have a close relationship with neurological diseases. In this study, the latest research findings regarding the structure, type, distribution, and function of VGSCs in the nervous system and their relationship to neurological diseases, such as epilepsy, neuropathic pain, brain tumors, neural trauma, and multiple sclerosis, are reviewed in detail.
KEYWORDS: function, nervous system, neurological diseases, voltage-gated sodium channel
Introduction
The voltage-gated Na+ channel (VGSC) is a type of microporous transmembrane protein that is widely distributed on the membranes of excitable cells such as neurons, and it is mainly responsible for the transmembrane transport of Na+. The VGSC is the most important ion channel required for neuronal cells to generate excitability and execute normal physiological functions.1–5 Structurally, the VGSC consists of a highly glycosylated pore-forming α subunit (240–260 kDa), which can function independently together with 1–4 auxiliary β subunits (30.4–45 kDa).3–6 To date, 10 VGSC subtypes have been successfully identified in mammals. They were named Nav1.1-Nav1.9 and NavX according to differences in the α subunit.4,5,7,8 The coding gene, structure, electrophysiological function, tetrodotoxin (TTX) resistance, and tissue distribution of different VGSC subtypes are relatively specific. Currently, except for the Nav1.4 Na+ channel, the expression of the other nine subtypes of VGSCs can be detected in the nervous system,9–11 but their specific expression patterns, electrophysiological characteristics and relationship with nervous system diseases are distinct from each other. In this study, based on the latest research findings and the results from our group, we reviewed the structure, type, distribution, and function of VGSCs in the nervous system and their relationship to nervous system diseases such as epilepsy, pain, brain tumors, neural trauma, and multiple sclerosis.
Structure, type, distribution, and function of the VGSC α subunit in the nervous system
Structure of the VGSC α subunit
The α subunit is the core subunit of the VGSC, with a molecular weight as large as 240–260 kDa.4,5,7,9 It is composed of the following three parts: four highly homologous transmembrane domains (DI-IV, with amino acid homology >75%); three intracellular loops (2 long loops, L1 and L2, and 1 short loop, L3); and the N-terminus and C-terminus (NT and CT)12,13 (Fig. 1). Each transmembrane domain contains six hydrophobic α-helical transmembrane segments (S1-S6). Transmembrane segment S4 contains 5–6 positively charged arginine residues, which are extremely sensitive to changes in the membrane potential. Therefore, segment S4 is called the “voltage sensor” of the VGSC and is supposed to be closely related to the activation of the VGSC. The fragment between transmembrane segments S5 and S6 contains the short fragments SS1 and SS2, which serve as a “re-entrant” across the cell membrane (i.e., the P domain). The intracellular loop L3 between the transmembrane domains DIII and DIV has three hydrophobic amino acid residues (isoleucine (I), phenylalanine (F), and methionine (M)). This region is referred to as the IFM structure, which is involved in the rapid inactivation of the VGSC by facilitating intracellular signaling.5,8,12 The CT and NT of the α subunit mainly regulate the function of the VGSC. For example, the CT is not only involved in the regulation of the VGSC inactivation process but also contains many domains with interactions between proteins and intracellular regulators such as the calmodulin (CaM)-binding IQ motif (I1908-R1918),14 Nedd4-like ubiquitin protein-binding PY motif (P1974-Y1977),15 PDZ-binding PY motif, and FHF1B-binding 1773–1832 motif.16 The above proteins or intracellular regulators participate in the regulation of the electrophysiological activity of VGSCs through interactions with the CT.
Figure 1.
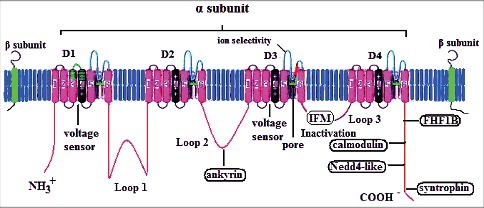
Schematic representation and modulation sites of the voltage-gated sodium channel (modified according to12). The predicted membrane topology of the α-subunit of voltage-gated sodium channels is illustrated together with two β subunits. DI∼Dχ indicate the four homologous domains of the α-subunit. Segments 5 and 6 are the pore-lining segments, and the S4 helices serve as the voltage sensor. The isoleucine-phenylalanine-methionine (IFM) residues are key amino acids for fast inactivation gating. Schematic representation of five proteins that have been reported to interact with voltage-gated sodium channels.
Until now, the three-dimensional or crystal structures of voltage-gated sodium channels in the human nervous system have remained unknown. However, the crystal structures of a voltage-gated sodium channel from bacteria (Arcobacter butzleri or marine alphaproteobacterium HIMB114) have been reported by two independent research groups.17–19 These authors showed the basic structure of sodium channels (inactivated states) at Å-level resolution and analyzed its biophysical properties.17–19 In those studies, the authors determined how one segment of the voltage-gated sodium channel interacted with other molecular structures.17 These studies are prominent notable landmarks in the research field of voltage-gated sodium channels, providing new insights into the molecular basis of the voltage-sensing, ion conductance, and voltage-gated dependence of sodium channels at the atomic level. Furthermore, for clinical investigations, these studies investigating inactivated-state structures of voltage-gated sodium channels provide novel avenues of drug development and therapy for a range of debilitating Nav channelopathies. Very recently, Shen H et al.20 reported the cryogenic electron microscopy structure of a putative Nav channel from American cockroach (designated NavPaS) at a resolution of 3.8 Å. The results of their study demonstrated that the voltage-sensing domains (VSDs) of the four repeats exhibit distinct conformations, and the entrance to the asymmetric selectivity filter vestibule is guarded by heavily glycosylated and disulfide bond-stabilized extracellular loops.20 Their results also indicated that a conserved amino-terminal domain is placed below VSDI, and a carboxy-terminal domain is bound to the III-IV linker on the cytoplasmic side.20 The significance of this study is that the structure of NavPaS established an important foundation for understanding the function of Nav channels as well as disease-related mechanisms.
Types of VGSC α subunits
To date, at least nine distinct sodium channel isoforms (Nav1.1-Nav1.3, Nav1.5 to Nav1.9, and NavX) have been detected in the nervous system.5,9-11,21,22 According to their sensitivity to tetrodotoxin (TTX), the specific inhibitor of sodium channels, these voltage-gated sodium channels are classified as TTX-sensitive (Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.6, and Nav1.7 channels) and TTX-resistant or insensitive types (Nav1.5, Nav1.8, and Nav1.9 channels). The pharmacological properties of the NavX Na+ channel remain unclear. Different types of Na+ channels have different coding genes, and their gene localization on human chromosomes is different (the specific coding genes and chromosome locations are described in detail by Goldin AL. et al.).8 In addition, the Na+ channels of each subtype may have various isoforms due to the selective splicing of their coding genes or different modifications during the transcription process. Alternative splicing of the sodium channel gene is known to alter the pharmacological sensitivities, kinetics, and channel distribution under physiological or pathological conditions. This phenomenon of gene expression and regulation is present in all sodium channels. For example, the inactivation kinetics of Nav1.1 channels containing the alternate exon 5N are more sensitive to intracellular fluoride ions and to changing temperature than channels containing exon 5A, although only three amino acids differ between exons 5A and 5N.23 Moreover, Nav1.1 channels containing exon 5N exhibit a more rapid recovery from inactivation at physiological temperatures.23 Similar results were also obtained for Nav1.5Na+ channels. To date, nine distinct sodium channel isoforms of Nav1.5 have been discovered due to alternative splicing of the SCN5A gene, including Nav1.5a-f and Nav1.5 E28B-D, and four of these isoforms can generate different functional variants.24,25 Therefore, not only does the voltage-gated Na+ channel itself have many subtypes, but its subtypes may also be divided into various isoforms. It is believed that the specific isoforms, distinct distribution and function of different subtypes of voltage-gated Na+ channels will be clarified with more extensive and detailed studies.
Distribution and function of the VGSC α subunit in the nervous system
The nervous system consists of the central nervous system (CNS) and peripheral nervous system (PNS). The distributions of the above 9 VGSC α subunits in the CNS and PNS are different. In general, the Nav1.1, Nav1.2, Nav1.3, Nav1.5, and Nav1.6 Na+ channels are more widely distributed in the CNS, while the Nav1.7, Nav1.8, and Nav1.9 Na+ channels are mainly distributed in the PNS (especially in the spinal nerve system).9-11,21,22,25-30 Normal VGSCs have three main features: (1) voltage-dependent activation; (2) rapid inactivation; and (3) Na+ selectivity.3,5,7,8 The α subunit is the dominant subunit of VGSCs and is responsible for normal electrophysiological function. It is also the main factor that determines the normal physiological activities of VGSCs, such as voltage-gating activity, ion-selectivity, and tetrodotoxin resistance. The α subunit itself can independently play a role, but only when it is co-expressed with the β subunits will the electrophysiological characteristics of an intact Na+ channel be exhibited.6,31 The specific electrophysiological and pharmacological characteristics of different α subunits are different, and different subtypes of the same α subunit also have different electrophysiological characteristics.
VGSC α subunits in the CNS
The distribution and function of the VGSC α subunit (from the mRNA to the protein level) in the CNS has been studied for decades (Table 1). Initially, those studies were performed based mostly on animal experiments. In the 1980s, Noda et al.32,33 elucidated the primary structure of the electrophorus sodium channel by cloning and sequencing the DNA complementary to its messenger RNA and isolating complementary DNA clones derived from two distinct rat brain mRNAs encoding sodium channel large polypeptides. Auld et al.34 cloned a full-length rat brain Na+ channel alpha subunit cDNA and analyzed its functional properties by transcribing the cDNA in vitro and injecting it into Xenopus oocytes to synthesize functional Na+ channels. Westenbroek et al.35 first analyzed the differential localization of the sodium channel subtypes Nav1.1 and Nav1.2 in rat brain neurons by immunocytochemical methods. The results of that study indicated that Nav1.1 is expressed in cell bodies and axon initial segments, while Nav1.2 is expressed in axons and dendrites.35 Gong B et al.36 further investigated the differences in Nav1.1 and Nav1.2 sodium channel expression in rat brain and showed that although the expression of these two Na(+) channel alpha subunits in heterologous systems yielded currents with very similar electrophysiological and pharmacological properties, their distinct spatial and temporal patterning, as well as their association with auxiliary subunits in brain, suggested that they performed distinct, nonoverlapping functions in situ.
Table 1.
Distribution of voltage-gated sodium channel α subunits in neurons of CNS.
| Nav isoforms | Cell body | Proximal process | Proximal AIS | Distal AIS | Nodes of Ranvier |
|---|---|---|---|---|---|
| Nav1.1 | + | + | + | + | |
| Nav1.2 | + | ||||
| Nav1.3 | + | + | |||
| Nav1.5 | + | + | |||
| Nav1.6 | + | + | + | + |
Note. AIS: axon initial segment. + : Major distribution of voltage-gated sodium channel α subunits in neurons of CNS.
Subsequently, in 1992, Ahmed et al.37 isolated and cloned a complete amino acid sequence of the α subunit of a human brain sodium channel from a cDNA library derived from human cerebral cortex. Since then, based on the foundational studies in this field, scholars have begun examining the cloning, distribution, and functional analysis of sodium channels in the human nervous system.38 In 2000, Whitaker et al.27 systematically investigated the specific distribution of four brain sodium channel subtypes in selected human CNS regions by a comparative immunohistochemical method. Their results showed that the Nav1.1, Nav1.3, and Nav1.6 Na+ channels were mainly distributed in the neuronal cell bodies and proximal processes, while the Nav1.2 Na+ channel was mainly distributed in the neuronal axons and nerve fiber bundles.27 Whitaker et al. also systematically analyzed the specific expression of the above four types of VGSC α subunits in the cerebral cortex (middle frontal gyrus (MFG), middle temporal gyrus (MTG), sensory and motor cortex (SMC), and visual cortex (VC)), deep brain nuclei (globus pallidus (Gp), putamen (Put), substantia nigra, and thalamic nuclei (Tha)), hippocampal structures (hippocampus (Hip), CA3, CA2, and dentate gyrus (DG)), insular cortex, and cerebellum (Purkinje cell body).27 The results demonstrated that the Nav1.1, Nav1.2, Nav1.3, and Nav1.6 Na+ channels were expressed in all the above tissues but that the expression levels differed.
The following studies further confirmed the subcellular distribution and function of these four brain sodium subtypes in neurons, indicating that Nav1.1 and Nav1.3 are predominantly localized to the neuronal soma and proximal dendrites, where neuronal excitability is controlled through the integration of synaptic impulses to establish the threshold for action potential initiation and propagation to the dendritic and axonal compartments.10 Using affinity-purified isoform-specific antibodies, Caldwell JH et al.39 found that Na(v)1.6 is highly concentrated at the nodes of Ranvier of both sensory and motor axons in the peripheral nervous system and at the nodes in the central nervous system. The following study further confirmed that Nav1.6 is prominently expressed at axon initial segments, where action potentials are initiated, and at the nodes of Ranvier, the gaps in the myelin sheaths of myelinated axons, where it propagates action potentials.10 Kaplan MR et al.40,41 showed that Nav1.2, but not Nav1.6, is clustered in developing central nervous system nodes and that clustering of Nav1.2 and Nav1.6 is differentially controlled. Their studies also indicated that oligodendrocyte-conditioned medium is sufficient to induce clustering of the Nav1.2 alpha subunit along central nervous system axons in vitro and that this clustering is regulated by electrical activity and requires an intact actin cytoskeleton and synthesis of a non-sodium channel protein.40,41 Their results revealed that the sequential clustering of Nav1.2 and Nav1.6 channels is differentially controlled and suggested that myelination induces Nav1.6 clustering.41 Gasser A et al.42 further confirmed that an ankyrin G-binding motif is necessary and sufficient for targeting Nav1.6 sodium channels to axon initial segments and nodes of Ranvier. In addition to Nav1.6, localization of the Nav1.8 sodium channel isoform at nodes of Ranvier in normal human radicular tooth pulp was also identified.43 In the following studies, Van Wart A et al.44 used quantitative and real-time PCR to determine whether the developmental appearance of Nav1.6 channels is accompanied by an increase in the steady-state level of Nav1.6 mRNA in the retina. They further demonstrated that Nav1.6 levels did not change between postnatal day 2 (P2) and P10, but there was an approximately 3-fold increase in Nav1.6 transcript levels between P10 and P1944. The results indicated a developmental switch from sodium channel isoform Nav1.2 to isoform Nav1.6 at initial segments and nodes of Ranvier in rat retinal ganglion cells during the second and third postnatal weeks. The above findings indicated that the Nav1.1, Nav1.3, and Nav1.6 Na+ channels may be closely related to the generation of neuronal action potentials, while the Nav1.2 Na+ channel may play an important role in transmission of these action potentials. It is well known that the distal end of the axon initial segment (AIS) is the site for action potential initiation in cortical pyramidal neurons. However, it is not clear which VGSC subtype plays a key role in the generation of action potentials. A study exploring the subcellular distribution of Nav1.1 in the nervous system indicated that Nav1.1 is expressed in nodes of Ranvier throughout the mouse spinal cord and in many brain regions.45 The authors identified three populations of nodes expressing Nav1.1, Nav1.6, or both. They also observed Nav1.1 expression in a proximal AIS subcompartment in spinal cord neurons, including 80% of motor neurons, and in multiple brain areas, suggesting that Nav1.1 is involved in the control of action potential generation and propagation45. Subsequently, Hu W, et al.46 found that low-threshold Na(v)1.6 and high-threshold Na(v)1.2 channels preferentially accumulate at the distal and proximal AIS, respectively, and have distinct functions in action potential initiation and back-propagation. Patch-clamp recordings from the cut end of pyramidal neuron axons in the rat prefrontal cortex revealed a high density of Na(+) currents and a progressive reduction in the half-activation voltage (up to 14 mV) with increasing distance from the soma at the AIS.46 Their further modeling studies and simultaneous somatic and axonal recordings showed that distal Na(v)1.6 promotes action potential initiation, whereas proximal Na(v)1.2 promotes its back-propagation to the soma.46 Recently, Lorincz et al.47 revealed the presence of the Nav1.6 channel subunit in proximal and distal dendrites of hippocampal CA1 cortical pyramidal cells by using a highly sensitive electron microscopic immunogold technique. The results demonstrated a lower subunit density by a factor of 35 to 80 than that found in axon initial segments. In addition, a gradual decrease in Nav1.6 density was also detected along the proximodistal axis of the dendritic tree without any labeling of the dendritic spines.47 These results revealed the characteristic subcellular distributions of VGSCs in neurons, which serve different functions in action potential initiation and back-propagation.
The Nav1.5 Na+ channel α subunit, the so-called cardiac sodium channel, is also widely expressed in the CNS. The Nav1.5 Na+ channel is a tetrodotoxin-resistant (TTX-R) Na+ channel. It was first successfully cloned in myocardium and has been considered to be a myocardial tissue-specific Na+ channel.9,16,24–26,48 However, the results of previous studies and our preliminary research have shown that Nav1.5 Na+ channel mRNA and protein are also expressed in neurons of the human brain, and this TTX-R Na+ current was also detected.11,24,25,49,50 In 2005, the function of the Nav1.5 Na+ channel α subunit was analyzed in the human neuroblastoma cell line NB-1.51 The results demonstrated that the Nav1.5 Na+ channel in NB-1 is a novel Nav1.5 Na+ channel subtype that differs from that of myocardial tissue and has different electrophysiological characteristics from those of the myocardial Nav1.5 Na+ channel.51 This result confirmed the functional expression of Nav1.5 channels in neural precursor cells. Recently, another study demonstrated that Nav1.5 sodium channel window currents contributed to spontaneous firing in olfactory sensory neurons.52 It is well known that olfactory sensory neurons (OSNs) fire spontaneously as well as in response to odor, and both forms of firing are physiologically important. The study revealed that whole cell patch-clamp recordings from OSNs demonstrated both tetrodotoxin-sensitive and tetrodotoxin-resistant components of the Na(+) current.52 However, RT-PCR showed that only one tetrodotoxin-resistant mRNA, the so-called cardiac subtype Nav1.5, was expressed in olfactory tissue.52 Additionally, the immunohistochemical analysis indicated that Nav1.5 is present in the apical knob of OSN dendrites but not in the axon. Further studies using the patch-clamp recording method indicated that Nav1.5 channels are required for spontaneous activity despite resting inactivation.52 In summary, this is the first study to confirm that Nav1.5 channels are not only expressed in olfactory neurons but also play a key role in the generation of action potentials. These results indicate the significance of Nav1.5 in odor sensitivities and may provide novel insights into the treatment of olfactory disorders.
Our previous studies have indicated that Nav1.5 mRNAs are expressed in the cortical neurons of human or rat brain tissues.25,53–56 Recently, we systematically investigated the expression of various Nav1.5 splice variants and their associated electrophysiological properties in rat brain tissue via biochemical analyses and whole-cell patch-clamp recording methods.57 The results demonstrated that several Nav1.5 splice variants are expressed in the rat brain at different expression ratios. Electrophysiological analysis of Nav1.5 in rat brain slices revealed that the TTX-resistant Na current is activated at −40 mV and reaches a maximum amplitude at 0 Mv.57 This study with electrophysiological analysis further confirmed the functional expression of Nav1.5 in brain neurons. Thus, the significance of specific Nav1.5 channel variants in the CNS and their relationship with the normal and abnormal electrophysiological activities of neurons warrants further investigation.
VGSC α subunits in the PNS (especially the spinal nerves)
With the exception of the Nav1.4 Na+ channel, expression of the other 9 types of VGSC α subunits was detected in the PNS (especially the spinal nerve system), but the expression levels varied between the different types of Na+ channel α subunits. The expression levels of the Nav1.7-Nav1.9 Na+ channels were relatively high, while the expression levels of the Nav1.1-Nav1.3, Nav1.6, and NavX Na+ channels were lower. The Nav1.5 Na+ channel was also expressed and considered to be the main Na+ channel to generate the third TTX-R current.9,28,31,58–60 In the PNS, VGSCs in the dorsal root ganglion (DRG) neurons of the spinal nerve have been studied most extensively. DRG neurons can be classified into several subtypes according to different classification criteria, such as myelinated A-fiber neurons and unmyelinated C-fiber neurons. The N52− (negative)-labeled C-fiber neurons can be divided into two subtypes: peptidergic neurons and nonpeptidergic neurons, such as TrkA+ (positive) neurons (peptidergic) and IB4+ neurons (nonpeptidergic).28,60 Studies have found that the expression levels of different types of VGSCs in different subtypes of DRG neurons are different (Table 2). The expression of the Nav1.1 and Nav1.6 Na+ channels are limited to A-fiber neurons, especially TrkC+ neurons, and these channels are highly expressed in the axon origin of spinal cord neurons and near the nodes of Ranvier.28,60 These results suggest that these Na+ channels are expressed in proprioceptive neurons and that the expression levels of Nav1.7-Nav1.9 Na+ channels in C-fiber neurons are higher than those in A-fiber neurons, whereas NavX Na+ can be expressed in both neurons, exhibiting basically the same expression level. According to another set of classification criteria, the mRNAs of the Nav1.7-Nav1.9 Na+ channels, but not those of the Nav1.1 and Nav1.6 Na+ channels, are highly expressed in N52−/TrkA± DRG neurons. The expression levels of the Nav1.7 and Nav1.8 Na+ channels in N52+/TrkA+ DRG neurons are similar to those in N52− DRG neurons, but the expression level of the Nav1.9 Na+ channel is low. The mRNA expression levels of the Nav1.1 and Nav1.6 Na+ channels in N52+/TrkA− DRG neurons are high, with no expression of the Nav1.8 and Nav1.9 Na+ channels. The expression levels of the Nav1.8 and Nav1.9 Na+ channels in the IB4+ neurons are high, while the expression levels of the Nav1.1 and Nav1.6 Na+ channels are low. Additionally, neurons that do not express the Nav1.7 Na+ channel are mostly N52+/TrkA− neurons.28,60 To summarize, VGSC α subunits are widely distributed in the neurons of the PNS, which may be closely related to certain diseases such as neuropathic pain.
Table 2.
Distribution of voltage-gated sodium channel α subunits in DRG neurons.
| Nav isoforms | N52-/TrkA+ | N52-/TrkA− | N52+/TrkA+ | N52+/TrkA− | IB4+ |
|---|---|---|---|---|---|
| Nav1.1 | Rare | Minor | ++ | ++ | + |
| Nav1.6 | Rare | Rare | ++ | ++ | + |
| Nav1.7 | ++ | ++ | ++ | + | |
| Nav1.8 | ++ | ++ | ++ | ++ | |
| Nav1.9 | ++ | ++ | + | Rare | ++ |
Note. ++Higher expression of sodium channels in DRG neurons of PNS (positive cells: more than 60%). + : High expression of sodium channels in DRG neurons of PNS (positive cells: more than 30% but less than 60%). A detailed expression pattern of voltage-gated sodium α subunits in different DRG neuronal populations is described by Fukuoka T et al (reference 28). Note to mention, voltage-gated sodium channels Nav1.2, Nav1.3 and Nav1.5 are also expressed in DRG neurons, however, their expressions are not detected by this classification.
Structure, type, distribution, and function of the VGSC β subunits in the nervous system
Structure of the VGSC β subunits
In the VGSC structure, β subunits have been considered auxiliary subunits of the α subunit5,7–9,31,61–63 and mainly consist of three parts: an independent transmembrane domain; a small intracellular C-terminus; and a large extracellular N-terminus.61
The crystal structure of β subunits has been investigated for several years. For example, Gilchrist et al.64 explored the crystal structure of the extracellular β4 domain and identified (58)Cys as an exposed residue that eliminates the influence of β4 on toxin pharmacology. Their results also suggested the presence of a docking site that is maintained by a cysteine bridge buried within the hydrophobic core of β4. Furthermore, Namadurai et al.65 reported the crystal structure of the human β3 subunit immunoglobulin (Ig) domain and found that the β3 subunit Ig assembles as a trimer in the crystal asymmetric unit. The results also indicate a more complex picture of the Nav α/β subunit interaction. Recently, Yan Z et al.66 presented the cryo-EM structure of EeNav1.4, the Nav channel from electric eel, in complex with the β1 subunit at 4.0 Å resolution. The study results indicated that the immunoglobulin domain of β1 docks onto the extracellular L5I and L6IV loops of EeNav1.4 via extensive polar interactions, and the single transmembrane helix interacts with the third voltage-sensing domain (VSDIII).66 The authors also performed a structural comparison with closed NavPaS, and the results showed that the outward transfer of gating charges is coupled to the iris-like pore domain dilation through intricate force transmissions involving multiple channel segments.66 To summarize, the above studies provide new insights into the structure and function of the β subunits as well as some pathological Nav channel mutations.
Types of VGSC β subunits
There are four subtypes of β subunits, β1-β4 (30.4–45 kDa), of which the β1 subunit has two variants, β1A and β1B. The coding genes are SCN1B-SCN4B. Subunits β1, β1A, and β3 have similar amino acid sequences, in which the β1 and β3 subunits have an amino acid identity of 45%. In the case of β1B, normal splicing of the exon 3/intron 3 boundary does not occur, leading to in-frame retention of intron 3 and generation of an alternate C-terminal sequence that does not include a transmembrane domain.67 Thus, β1B is called a developmentally regulated secreted protein, and its amino acid identities with β1 and β1A are 17% and 33%, respectively. These subunits are linked to the α subunit by non-covalent bonds. The amino acid identity of the β2 and β4 subunits is approximately 35%, but they are linked to the α subunit by covalent bonds (disulfide bonds).61–63,68–71 In vivo, the α subunit is generally associated with two β subunits to form a large heteromultimeric complex, of which one is linked by a non-covalent bond (β1 or β3), and the other subunit is linked by a covalent bond (β2 or β4).61,63,72,73
Distribution of VGSC β subunits
The four known types of β subunits are widely distributed in the CNS and PNS, in which the β1 subunit is widely distributed in large or medium (>30 μm) DRG neurons, while they are distributed to a reduced degree in smaller neurons (<25 μm).9,61,62 The β2 subunit is widely distributed in CNS and PNS neurons, including the cerebral cortex, cerebellum, hippocampus, brainstem, spinal cord, and DRG neurons. The β3 subunit is highly expressed in small (<25 μm) and medium (25–45 μm) neurons, with a reduced distribution in large (>45 μm) neurons. The β4 subunit is highly expressed in the PNS, especially in DRG neurons. At the cellular level, the β4 subunit is mainly expressed in large neurons, with relatively lower expression in medium and small neurons (Table 3).8,61–63,68–71
Table 3.
Distribution of voltage-gated sodium channel β subunits in neurons.
| Navβ isoforms | Distribution in neurons |
|---|---|
| β1 | mainly in large or medium (>30 μm) DRG neurons, less in smaller neurons (<25 μm) |
| β2 | widely distributed in CNS and PNS neurons, including cerebral and DRG neurons |
| β3 | mainly in small (<25 μm) and medium (25–45 μm) neurons, less in large (>45 μm) neurons |
| β4 | mainly in large neurons, less in medium and small neurons |
Function of VGSC β subunits
Although β subunits are the auxiliary subunits of the α subunit, their co-expression with the α subunit is considered to be very important in the regulation of the α subunit and even the function of that Na+ channel. Their specific roles include the following: (1) regulating the gated kinetics of the whole Na+ channel; (2) regulating the voltage-dependence of the Na+ channels; (3) regulating the expression of the Na+ channel on the surface of the cell membrane; and (4) playing a similar role as adhesion molecules, which interact with cytoskeleton proteins, extracellular motifs, and other cytokines, thus regulating cell migration and aggregation.5,8,31,61,62 The adhesive functions of β subunits are critical for brain development and thus excitability, including the processes of neurite outgrowth, axon pathfinding, and fasciculation, and they are postulated to play similar roles in the heart and peripheral nerves.67 In summary, a growing body of studies has identified the importance of β subunit proteins not only in normal physiology but also in numerous pathophysiologies, including channelopathies.
The β subunits are localized to a number of important subcellular compartments, enabling them to engage in specific functions. In the brain and peripheral nerves, β subunits are clustered in the AIS and nodes of Ranvier, where VGSC α subunits are preferentially accumulated.67 At the AIS and nodes, a C-terminal association with the adaptor protein ankyrin G is postulated to anchor β subunits to the cytoskeletal protein βχ spectrin.67,74 At the nodes of Ranvier, β subunits can associate with α subunits tightly clustered within the nodal gap and thus positioned for channel modulation during rapid saltatory conduction.67,75 β subunits also modulate the function of the α subunit, with effects including alterations in persistent and resurgent sodium currents, peak sodium current density, and voltage-dependence activation and inactivation. For instance, previous studies have found that the co-expression of the β1 subunit and Nav1.7 and Nav1.8 in Xenopus oocytes can enhance the Na+ current and cause a hyperpolarized shift of the Na+ channel during the process of steady-state inactivation. Furthermore, the β1 subunit could specifically enhance the expression of the Nav1.8 Na+ channel without affecting the expression of the Nav1.7 Na+ channel.61,63 Recent studies on mammalians indicated that the co-expression of the β1 subunit with Nav1.8 could increase the Na+ current by 2-fold, and a hyperpolarization shift was detected regardless of the activation or inactivation of the Na+ channel.68 In DRG neurons, knocking down the β1 subunit can reduce the current of Na +, slightly changing the amplitude of the current curve and the gating characteristics of the Na+ channel. In vivo, mice with SCN1B gene silencing show many neurological disorders, including hyper-excitability of DRG neurons, epilepsy and ataxia, among others.61,69 The expression of the β1A subunit is highest in the embryonic period, and it begins to decrease after birth. Co-expression of β1A with Nav1.2 in Chinese hamster eggs was found to increase the Na+ current by 2.5 times and to generate a slight hyperpolarization shift, although this shift did not affect its steady-state inactivation potential or channel kinetics. Furthermore, the β1A subunit also increased the expression level of Nav1.2Na+ channels on the cell membrane, similar to its parental β1 subunit.61,70
The regulatory role of the β2 subunit is more apparent when it is co-expressed with the α subunit of sensory neuron Na+ channels. Its co-expression with Nav1.8 in Xenopus oocytes can cause a relatively moderate depolarizing shift (4 mV) during the inactivation of the Nav1.8 Na+ channel, while the activation process, current dynamics, and Na+ current peak are unaffected. Moreover, when the β2 subunit was co-expressed with Nav1.3, Nav1.6, Nav1.7, little to no change was detected in the voltage-dependence, channel dynamics and current intensities of these sodium channels.61,68 The above in vitro studies have shown that the β2 subunit has a minor effect on the overall function of the Na+ channel. However, in contrast, the amplitude of the Na+ current and the expression level of mRNA and protein all decreased in mice carrying the β2 subunit knockout,61 indicating that expression of the β2 subunit in neurons can enhance the amplitude of the Na+ current and channel dynamics, among others. The specific reasons for the inconsistency of in vitro and in vivo experiments are unclear. Additionally, studies have found that after the SCN2B gene was knocked out, the expression level of TTX-S Na+ decreased in DRG neurons. More importantly, the mechanical pain caused by peripheral nerve injury in mice with SCN2B gene silencing was decreased.61 The above findings suggest that the β2 subunit may play an important role in the generation of neuropathic pain.
The β3 subunit is widely expressed in the neurons of the CNS and PNS, but it is not expressed in glial cells or in most non-neuronal cells. The β3 subunit as well as the Nav1.8 and Nav1.9 of TTX-R are widely expressed in peripheral nerve receptors, suggesting that the β3 subunit may play an important role in the generation of neuropathic pain. When the peripheral nerve is injured by chronic compression or avulsion, the mRNA expression level of the β3 subunit in C-fiber neuronal receptors and DRG neurons was increased.61,63,68 The β3 subunit may also play an important role in regulating the entire function of Na+ channels. Early studies have found that the current intensity of Nav1.8Na+ was increased when the β3 subunit was co-expressed with Nav1.8 in Xenopus oocytes. However, follow-up studies showed that when β3 subunits were co-expressed with Nav1.8 in mammalian cells, the Nav1.8Na+ current peak could be reduced by 31%, but there was no effect on its activation or steady inactivation.61,68 The above results indicate that the β3 subunit and Nav1.8 co-expression may affect the electrophysiological function of Nav1.8Na+ channels, but this hypothesis requires further confirmation in subsequent studies. As mentioned above, Namadurai et al.65 detected full-length β3 subunit trimers on the plasma membrane of transfected HEK293 cells by using a fluorescence photoactivated localization microscopy method. They further showed that β3 subunits can bind to more than one site on the Nav1.5 α subunit and induce the formation of α subunit oligomers, including trimers.65 Their results suggest a novel and unexpected role for the β3 subunits in Nav channel cross-linking and provide new structural insights into some pathological sodium channel mutations.
The β4 subunit is a newly discovered auxiliary subunit, the function of which has recently been systematically studied. This subunit is widely expressed in the nervous system, and its expression level in DRG neurons of the PNS is higher than that in the brain and spinal cord. The β4 subunit regulates the function of the entire Na+ channel by containing many positively charged lysines and hydrophobic residues at the terminus. Recent studies have shown that the β4 subunit plays a specific role in the generation of resurgent currents in Na+ channels, as the co-expression of Nav1.6 and the β4 subunit can generate a resurgent current31,61,76; resurgent currents are the electrophysiological basis for the occurrence of pain and neurological disorders such as epilepsy, suggesting that the β4 subunit may play an important role in the occurrence and development of certain diseases in the nervous system.
In conclusion, the four types of β-subunits are widely expressed in the nervous system, particularly in the peripheral nervous system. They play a role in regulating the voltage-dependence, gated properties of sodium channels by co-expression with the α subunit and thus affect the normal electrophysiological activities of neurons.
VGSCs and neurological diseases
The activity of voltage-gated sodium channels has long been linked to neurological disorders of neuronal excitability, such as epilepsy and pain. Therefore, these types of diseases are also known as “channelopathies”. In addition to epilepsy and pain, mutations in or dysfunctions of α subunits or β-subunits both can lead to other neurological diseases. In the following section, the relationship between α subunits of voltage-gated sodium channel and these neurological diseases are mainly discussed (β-subunits related channelopathies are described in detail by O'Malley et al.67).
VGSCs and epilepsy
VGSCs are the most important initiating ion channels for neurons to generate action potentials and are involved primarily in the formation of the rising phase of the action potential.46 They also play an important role in the normal and abnormal electrophysiological activities of neurons. Epilepsy is a brain dysfunction syndrome caused by excessive synchronization of brain neuron discharge. Since mutations in the genes encoding many types of ion channels have been found to be the main etiology causing abnormal neuronal discharge and leading to epilepsy, epilepsy is also considered an ion channel disease (channelopathy).77–82 The relationship between VGSCs and epilepsy, especially the relationship between the gene encoding the α subunit of the Na+ channels and primary epilepsy, has long been a popular topic in the field of epilepsy research. A variety of epilepsy syndromes are caused by mutations of the gene encoding the VGSC α subunit or β subunits.83–89 Currently, the most commonly used clinical antiepileptic drugs are essentially Na+ channel inhibitors, further indicating that VGSCs are closely related to epilepsy.
To date, the mutation of the SCN1A gene encoding the Nav1.1 Na+ channel α subunit has been demonstrated to be the etiology of many primary epilepsies.85–87,90–94 In addition, mutations of the Nav1.2, Nav1.3, Nav1.5, and Nav1.6 Na+ channel α subunit coding genes SCN2A, SCN3A, SCN5A, and SCN8A may also be related to some types of epilepsy. Among them, the relationship between mutations of the gene encoding the Nav1.1 Na+ channel α subunit and epilepsy has been extensively studied.86,95–98 It is now known that Dravet syndrome (SMEI, severe myoclonic epilepsy in infancy), GEFS (+) (generalized epilepsy with febrile seizures, plus), and FS (familial febrile seizure) can be caused by mutations of the SCN1A gene encoding the VGSC Nav1.1 Na+ channel α subunit.85–87,90–94 There are up to one hundred known gene mutation sites, as described by Catterall WA. et al..85 Currently, the specific mechanism of epilepsy caused by SCN1A gene mutation is not yet fully understood, but it is thought to be closely related to the impaired generation of the action potential and current in GABA-ergic inhibitory neuronal Na+ channels.85,91 Catterall WA et al.85 summarized the relationship between SCN1A gene mutation and epilepsy severity. Studies have also found that a missense mutation of the SCN2A gene encoding the Nav1.2 Na+ channel α subunit could also trigger GEFS (+),86 and the results of an animal study showed that the Navl.2/SCN2A gene mutation (SCN2AGAL879-88QQQ) in Q54 transgenic mice could inhibit the inactivation of VGSCs, causing the progressive epilepsy associated with hippocampal sclerosis. This study also found that the epilepsy of these Q54 transgenic mice was very similar to the temporal lobe epilepsy observed in humans,80 which is important for future studies of human epilepsy and for the establishment of corresponding animal models.79,80,82,84
The expression level of the Nav1.3 Na+ channel is relatively high in embryonic brain tissue. With increasing age, the expression level decreases, showing a low expression level in adult brain tissue. However, studies have found that this protein may play a role in the pathogenesis of epilepsy.95–97 The results of animal studies showed that the expression levels of Nav1.1, Nav1.3, and the β1 subunit in hippocampal neurons of mice with spontaneous epilepsy were significantly higher than those in the control group,96 indicating that the Nav1.3 Na+ channel may also play an important role in the pathophysiology of the abnormal neuronal discharge and thus be closely related to the occurrence and development of epilepsy. However, the exact relationship between Nav1.3 and human epilepsy is still unknown and merits further investigation..
The expression level of the Nav1.5 Na+ channel in the brain is similar to that of the Nav1.3 Na+ channel, which is also high in the embryonic period and low in the adult brain tissue of both humans and mice. Previous studies have shown that the Nav1.5 Na+ channel is widely distributed in the limbic system of the brains of both humans and mice, such as in the structure of the hippocampus.24,25,49,50,53–56 As the limbic system is a predilection site of epilepsy, previous studies have speculated that the Nav1.5 Na+ channel may be closely related to the occurrence and development of epilepsy. Until recently, the above speculation was only preliminarily confirmed. A study experimentally examined the brain tissue of a patient who died of sudden unexpected death in epilepsy (SUDEP) and found a missense mutation (R523C in exon 12) in the SCN5A gene encoding the Nav1.5 Na+ channel α subunit in human brain tissue.98 Although the changes in electrophysiological function due to the mutation are not yet clear, this is the first time that a mutation in the Nav1.5 Na+ channel α subunit in the human brain tissue from a patient with epilepsy has been reported in the literature. Subsequently, another study identified, for the first time, a family showing an association between Brugada syndrome and epilepsy with a known mutation in the SCN5A gene (p.W1095X, c.3284G>A).99 The authors suggested that this mutation could be responsible for cardiac and brain involvement and confirmed the possibility that SCN5A mutations might confer susceptibility to recurrent seizure activity, supporting the emerging concept of a genetically determined cardiocerebral channelopathy.99 Although these observations indicate that SCN5A mutations may play a role in the pathogenesis of epilepsy, this role might not be a major one due to the low expression level of these proteins in human brain neurons.
The sodium channel Nav1.6, encoded by the SCN8A gene, is one of the major voltage-gated channels in brain neurons and plays an important role in regulating the initiation and propagation of action potentials in the nervous system.46 Loss of Nav1.6 activity results in reduced neuronal excitability, while gain-of-function mutations can increase neuronal excitability.100 Previous studies have demonstrated that de novo mutations of human SCN8A detected by exome sequencing reveal a role for Nav1.6 in epilepsy.100 Future studies should provide insight into the specific mechanisms of Nav1.6 pathogenesis in epilepsy.
In addition to mutations of the genes encoding the VGSC α subunit that is known to trigger epilepsy, mutations of the genes encoding the β subunits could also lead to epilepsy. Studies have found that a mutation in the SCN1B gene encoding the β1 subunit could induce GEFS+ because the missense mutation of the Navβ1/SCN1B gene located on chromosome 19q13 changed the Cys at position 121 to Trp, leading to a change in the normal electrophysiological function of the VGSC and resulting in the abnormal discharge of neurons and thus causing epilepsy.101 It has also been reported that mutations in sodium channel β subunits can cause Dravet syndrome.102 In conclusion, studies examining the relationship between mutation of the β subunit-encoding gene and epilepsy are being reported (see detailed review by O'Malley et al.67), and it is believed that more attention will be paid to β subunits to further understand their functions.
Currently, the specific mechanism of epilepsy induced by VGSC coding gene mutations is unclear, but it is closely related to the normal electrophysiological characteristics of VGSCs. For example, a delay in Na+ channel inactivation will generate a persistent sodium current, which reduces the discharge threshold of a persistent action potential. The rapid recovery of the Na+ channel from the inactivated state will result in an increased discharge frequency of the action potential, and the expression of VGSCs in the inhibitory neuronal cell membrane is decreased.78,83–85,87 However, the entire process from mutation of the VGSC-encoding gene to epileptogenesis is particularly complex, as reflected by not only the changes in the normal electrophysiological function of neurons but also the signal transduction between neurons and neurons and between neurons and glial cells at the cellular level, receptor-ligand regulation and abnormal signal transduction pathways at the molecular level, and mutations in the VGSC coding genes as well as changes in the genes encoding other related ion channels and cytokines at the genetic level. Thus, further studies are still needed to elucidate the specific mechanisms of epileptic seizures.
VGSCs and pain
Pain is an acute or chronic disease caused by a variety of harmful factors that stimulate the peripheral or central nervous system and lead to its structural and functional disorder. Studies have found that VGSCs play an important role in the generation and conduction of pain, especially in the generation of peripheral neuropathic pain.103–105 The somatic sensory afferent neurons are aggregated in DRG neurons of the peripheral nerves. A variety of harmful stimuli may cause changes in the type, quantity, distribution and electrophysiological activity of the VGSCs in the DRG neurons, resulting in increased excitability of the DRG neurons and increased discharge frequency or ectopic discharge, thereby causing pain. The types of VGSC that are known to be closely related to pain include the Nav1.7, Nav1.8, Nav1.9, and Nav1.3 Na+ channels. Electrophysiological studies have shown that the occurrence of VGSC-mediated pain is the result of the total effect of the entire sodium channel, which means the effect is a combined action of the α subunit and β subunits (especially β1 and β3).68,70,103–108
The Nav1.7 Na+ channel is expressed in peripheral neurons, including DRG sensory neurons, sympathetic ganglion neurons, myenteric neurons, and trigeminal ganglion neurons.109 Three known pain syndromes are caused by mutations in the gene SCN9A encoding the Nav1.7 Na+ channel α subunit: inherited erythromelalgia (IEM), paroxysmal extreme pain disorder (PEPD), and congenital inability to experience pain (CIP).107,109–115 The mutation sites and types of the SCN9A gene encoding the Nav1.7 Na+ channel α subunit corresponding to these three pain syndromes have been described by Dib-Hajj SD. et al..109 Although all three pain syndromes are caused by mutations of the SCN9A gene, their mechanisms are different. The pain of IEM caused by SCN9A mutations occurs because the mutated Nav1.7 Na+ channel is more easily activated during hyperpolarization. In PEPD, the SCN9A mutation causes incomplete rapid inactivation of the Nav1.7 Na+ channel, leading to a persistent Na+ current. The CIP is caused by the functional loss of the mutated Nav1.7 Na+ channel.109,111,114,115
The Nav1.8 Na+ channel is a sensory neuron-specific Na+ channel that is widely expressed in DRG neurons and trigeminal ganglion neurons.116 It is characterized by slow inactivation and mainly involves the formation of the rising phase of the action potential in DRG neurons, playing an important role in the regulation of neuronal excitability. The relationship of the Nav1.8 Na+ channel and pain has mainly been tested in animal experiments. Studies have shown that the Nav1.8 Na+ channel plays an important role in peripheral neuropathic pain, such as inflammatory, neuropathic, and neurofibromatologic pain.109,116–119 Furthermore, the Nav1.8 Na+ channel is closely associated with temperature-related pain, such as cold stimulation, and it is considered to be the most important Na+ channel for pain induction at a low temperature.120 Animal studies have found that animals with Nav1.8 Na+ channel gene silencing are almost insensitive to nociceptive cold stimulation. Additionally, neurons with Nav1.8/SCN10A gene silencing could be passively depolarized only by current stimulation at 10°C. If expression of the Nav1.8 Na+ channel was not altered in the previously designed low temperature conditions, then generation of the neuronal action potential would be normal.109,117,120 This result suggests that the Nav1.8 Na+ channel is the most important Na+ channel for generation of the action potential in peripheral neurons at a low temperature. More importantly, the Nav1.8 Na+ channel-specific blockers uO-conotoxin, A-803467, and Compound 2 could significantly inhibit or reduce neuropathological and inflammatory pain in experimental animals.109 Furthermore, A-803467 could significantly reduce chronic compression pain in the sciatic nerve at a low temperature,109 further demonstrating that the Nav1.8 Na+ channel plays an important role in inflammatory and neuropathic pain.
The Nav1.9 Na+ channel is mainly expressed in the primary sensory neurons and enteric neurons of peripheral nerves. Its activation and inactivation are slower than those in other types of Na+ channels, and this feature is mainly involved in the regulation of the neuronal action potential threshold. Recent animal studies have shown that the Nav1.9 Na+ channel mediates neuropathological, inflammatory and visceral pain.113,116,121 For example, after gene knockout mice (Nav1.9 Na+ channel α subunit) received a foot injection of a variety of inflammatory mediators, none or only mild symptoms of inflammatory pain was observed. A similar result was obtained for the intraperitoneal injection of complete Freund's adjuvant (CFA).116 These findings demonstrate that the Nav1.9 Na+ channel plays an important role in the pathophysiology of pain generation. However, the specific role of the Nav1.9 Na+ channel in human pain is still under investigation.
The relationship between the Nav1.3 Na+ channel and pain was found by studying nerve injury. The Nav1.3 Na+ channel is mainly expressed at the embryonic stage and at a young age, with low expression in mature DRG neurons; however, when the nerve is injured, expression of the Nav1.3 Na+ channel in DRG neurons is significantly increased.122,123 Similar to the Nav1.8 Na+ channel, the Nav1.3 Na+ channel has the characteristic of slow deactivation, and the Nav1.3 Na+ channel can produce a ramp current and short-term persistent current near the resting membrane potential, which allows the Nav1.3 Na+ channel to generate the action potential at subthreshold stimulation.123 In an animal model of chronic compression injury of the nerve root, the expression level of the Nav1.3 Na+ channel was significantly increased, accompanied by rapid repolarization of the excitatory potential, leading to persistent abnormal discharge and resulting in the manifestation of neuropathic pain. In contrast, inhibition of Nav1.3 Na+ channel expression (such as by glial cell line-derived neurotrophic factor, GDNF) or the use of its inhibitors could reduce the excitability of DRG neurons and reduce the hyperalgesia phenomenon.122,123 These results suggest that the Nav1.3 Na+ channel plays an important role in the pathophysiology of pain.
The VGSC β subunits also play an important role in the pathophysiology of pain, and these subunits are co-expressed and function with the α subunit of the Na+ channel. Studies using an animal model of pain caused by sciatic nerve transection have found that expression of the β3 subunit in the ipsilateral DRG neurons of the nerve injury was significantly increased compared to that of the contralateral DRG neurons, consistent with the up-regulation of Nav1.3.122,124 This result indicates that the β3 subunit may regulate the excitability of neurons by regulating the function of the co-expressed Nav1.3 and thus mediating the generation of pain.
Presently, the drugs for clinical treatment of peripheral neuropathic pain are almost all Na+ channel blockers, such as carbamazepine, the preferred drug therapy for trigeminal neuralgia, and the peripheral nerve blocker lidocaine. However, since these drugs can inhibit all types of Na+ channels, side effects are inevitable. Therefore, if the Na+ channel responsible for different types of pain can be clinically identified, and a blocking drug specific to this type of Na+ channel can be developed, the side effects of these common Na+ channel blockers can be avoided. Such studies are currently ongoing. For example, McCormack K et al.125 have described two unique structurally related nanomolar potent small molecule Nav channel inhibitors that exhibit up to 1,000-fold selectivity for human Nav1.3/Nav1.1 (ICA-121431, IC50, 19 nM) or Nav1.7 (PF-04856264, IC50, 28 nM) vs. other TTX-sensitive or resistant (i.e., Nav1.5) sodium channels. Alexandrou et al.126 reported two potent and selective arylsulfonamide Nav1.7 inhibitors (PF-05198007 and PF-05089771), which they have used to directly interrogate the role of Nav1.7 in nociceptor physiology. They have demonstrated that Nav1.7 is the predominant functional TTX-sensitive Nav in mouse and human nociceptors and contributes to the initiation and the upstroke phase of the nociceptor action potential.126 Cao L et al.127 reported that five patients with IEM were treated with a new potent and selective compound that blocked the Nav1.7 sodium channel, resulting in a decrease in heat-induced pain in most patients. In summary, since the present studies provide a potential framework for identifying subtype selective small molecule sodium channel inhibitors targeting the specific voltage-gated sodium channels, it would be possible to achieve a real targeted therapy in the future.
VGSCs and glioma
Glioma is the most common primary tumor of the nervous system, accounting for 40–60% of primary intracranial tumors.128 According to their pathological differences, gliomas can be divided into pilocytic astrocytoma, diffuse (hypertrophic) astrocytoma, oligodendroglioma, and glioblastoma. According to their level of malignancy, gliomas can be divided into WHO grades I-IV, of which grade I is benign, while grades II-IV refer to malignant tumors.128 Previous studies have found that VGSCs on the cell membrane of malignant gliomas play an important role in the cell proliferation and migration of glioma.129–131 Moreover, the types of VGSCs that can be detected in glioma cells include Nav1.1-Nav1.3, Nav1.5-Nav1.7, and NavX (Nav2.1) Na+ channels.129,130,132 Among them, Nav1.1-Nav1.3, Nav1.6, and NavX (Nav2.1) Na+ channels can be detected in pilocytic astrocytomas (grade I); Nav1.1-Nav1.3, Nav1.5, Nav1.6, and NavX (Nav2.1) Na+ channels can detected in diffuse astrocytomas (hypertrophic, grade II-III); Nav1.1-Nav1.3 and Nav1.6 Na+ channels can be detected in oligodendroglioma (grade II-III); and Nav1.1-Nav1.3 and Nav1.5-Nav1.7 Na+ channels can be detected in glioblastoma (grade IV).129,130,132–134 The Nav1.1-Nav1.3 and Nav1.6 Na+ channels were detected in all types of the above gliomas, but the expression type and expression level of the VGSCs in the gliomas of different pathological types or different grades are different.
Analysis of the electrophysiological characteristics of different types of VGSCs in glioma cells is a prerequisite to elucidate their specific functions. As mentioned above, the specific electrophysiological function of the voltage-gated Nav1.5 Na+ channels in the human neuroblastoma cell line NB-1 were analyzed in 2005.51 Our subsequent studies showed that the mRNA and protein expression levels of the Nav1.5 Na+ channel in gliomas of all malignancy grades were significantly higher than those in normal brain tissue.133 This result is consistent with a previous study.135 In addition, reduced Nav1.5 expression significantly suppressed the proliferation and invasive ability of astrocytoma cells, indicating that Nav1.5 might play a role in promoting the growth and metastasis of astrocytoma cells.133 Furthermore, a recent study also found that patients with SCN5A mutations in GBM (glioblastoma multiforme, a malignant glioma) had a significantly shorter survival.134 This result further suggests that the voltage-gated Nav1.5 Na+ channel may play an important role in the proliferation and migration of glioma cells.
Very recently, a study analyzed the effects of antiepileptic drugs on the growth of glioblastoma cell lines and found that temozolomide and oxcarbazepine, supposed Na+ channel blockers, significantly inhibited glioblastoma cell growth and reached IC50 at the therapeutic concentrations.136 These results suggested the possibility of using antiepileptic drugs as a future treatment method for gliomas. However, since the antiepileptic drugs are unable to selectively block the subtypes of sodium channels in glioma tissue, a high dose of these compounds may also block sodium channels in other functional tissues, such as the heart, causing serious side effects. Therefore, future studies should clarify which subtype of sodium channel plays a key role in the proliferation and migration of gliomas and which type of drug is specific for blocking this subtype of sodium channel. If all these issues are clarified, to the best of our knowledge, local treatment (surgical resection with local anti-sodium channel drug therapy) of glioma using these compounds should be a practical therapeutic strategy in the future.
VGSCs and neural injury
The pathophysiological changes and neurochemical reaction after injury in the brain, spinal cord, and nerve root depend not only the primary injury but also the subsequent secondary injury, and these secondary injuries will result in the loss or apoptosis of neurons that were not directly injured. The expression of VGSCs and the electrophysiological activity of neurons after injury were found to be altered, which would affect the excitability and stability of the whole cell.122,137–143 The study of lateral fluid percussion injury in rats revealed that the mRNA and protein expression levels of the Nav1.6 Na+ channel α subunit in bilateral hippocampal neurons began to increase 2 hours after brain injury, and the expression level was 7 times that of the control group at approximately 12 hours, which was still significantly higher than that of the control group at 72 hours.137 These results suggest that the Nav1.6 Na+ channel may be involved in the secondary reaction after nerve injury.
Cerebral ischemia-reperfusion injury is similar to traumatic brain injury. A study found that in the cerebral hemisphere of the injured side of rats with reperfusion 2 hours after arterial embolization, the expression levels of the Nav1.1-Nav1.3 and Nav1.7 Na+ channels were significantly down-regulated, indicating that VGSCs may play an important role in cerebral ischemia-reperfusion injury.138 Similarly, the expression level of VGSCs was also altered when the spinal nerve root was injured, but the changes in Na+ channel in different injury types varied. Generally, in the injured DRG neurons, the expression level of the Nav1.3 Na+ channel was up-regulated, while the expression levels of the Nav1.8 and Nav1.9 Na+ channels showed a significant downward trend.122,139 The difference is that although the expression levels of the Nav1.1, Nav1.6, and Nav1.7 Na+ channels also showed a downward trend, their decline was reduced.140 Some studies also showed that the expression levels of the Nav1.8 and Nav1.9 Na+ channels were down-regulated in patients with brachial plexus avulsion injury.141,142 The reason for the different expression patterns of sodium channels between animal experiments and actual clinical injury is not yet clear. Additionally, in a model of chronic nerve root compression injury, although the expression level of the Nav1.8 Na+ channel was not decreased, and the expression level of the Nav1.3 Na+ channel was not increased, the total electrophysiological activity of the Na+ channel had been changed. For example, the action potential of the TTX-S Na+ channel shifted to hyperpolarization, whereas the peak of the slowly inactivated TTX-R current was increased.143,144 These studies have shown that VGSCs also play an important role in the pathophysiology after nerve root injury.
Overall, alterations in VGSC subunit expressions were detected in multiple traumatic neural injury models, demonstrating the importance of these proteins in the regulations of normal or abnormal excitability in sensory neurons. Future studies should focus on the functional alterations of different subtypes of sodium channels expressed in the neurons of traumatic neural injury models and reveal the specific role played by each subtype of sodium channel in the generation and propagation of pain.
VGSCs and multiple sclerosis
Multiple sclerosis (MS) is a chronic inflammatory disease of the CNS that is mainly characterized by white matter demyelination, and its pathogenesis remains unclear.145 Previous studies have focused less on the changes in VGSCs in patients with MS lesions and their possible role in the occurrence and development of MS.145–147 However, a recent study found that the expression level of Nav1.5 was significantly increased in MS lesions (reactive astrocytes), while the expression levels of the Nav1.1-Nav1.3 and Nav1.6 Na+ channels exhibited only slight changes compared to astrocytes in normal white matter.135 The results of this study indicated that VGSCs may play a role in the functional response of reactive glial cells in MS lesions, and the researchers speculated that the increased expression level of the voltage-gated Nav1.5 Na+ channel is a compensatory mechanism. This compensatory mechanism may play an important role in the maintenance of intracellular and extracellular ion homeostasis for Na+-K+-ATPase.135 This is the first study to report changes of the voltage-gated Nav1.5 Na+ channel in MS, thus providing an experimental basis for further clarification of the pathophysiology of MS.
Similar to MS, the pathological changes of experimental autoimmune encephalomyelitis (EAE) are very complex, including inflammation, demyelination, and glial cell reaction.148–150 Studies have found that VGSC blockers can reduce the expression of microglia and phagocytes in EAE lesions,148 while several studies have shown that VGSC blockers can reduce the degree of injury in cell axons and improve the clinical status of the patients.148–150 It is worth noting that the drugs flecainide (has a strong inhibitory effect on the voltage-gated Nav1.5 Na+ channel), carbamazepine, and phenytoin showed a protective effect in EAE injury.148–150 This protective effect is reflected by the inhibition of microglial activity, including the reduction in phagocytosis as well as apoptosis and inhibition of the release of IL-1 and TNF-α preinflammatory mediators,148–150 suggesting that VGSCs may play an important role in the pathophysiology of EAE. Clinical studies of applications of Na+ channel blockers in patients with MS and other neurological disorders are currently underway.151 Such studies will elucidate the significance of voltage-gated Na+ channel blockers in the treatment of MS and other lesions caused by the glial cell response.
In conclusion, the relationship between sodium channels and nervous system diseases has been studied extensively and has been provided many important discoveries and developments. Future studies will enhance our understanding of the structures and functions of sodium channels and their relationship with human diseases.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by the National Natural Science Foundation of China (No. 31100770) and the Liaoning Provincial Natural Science Foundation of China (No. 2014021097).
References
- 1.Corry B, Thomas M. Mechanism of ion permeation and selectivity in a voltage gated sodium channel. J Am Chem Soc. 2012;134:1840–6. doi: 10.1021/ja210020h. PMID:22191670 [DOI] [PubMed] [Google Scholar]
- 2.Leterrier C, Brachet A, Fache MP, Dargent B. Voltage-gated sodium channel organization in neurons: protein interactions and trafficking pathways. Neurosci Letters. 2010;486:92–100. doi: 10.1016/j.neulet.2010.08.079. [DOI] [PubMed] [Google Scholar]
- 3.Wood JN, Baker M. Voltage-gated sodium channels. Curr Opin Pharmacol. 2001;1:17–21. doi: 10.1016/S1471-4892(01)00007-8. PMID:11712529 [DOI] [PubMed] [Google Scholar]
- 4.Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacological Rev. 2005;57:387–95. doi: 10.1124/pr.57.4.13. [DOI] [PubMed] [Google Scholar]
- 5.Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol. 2003;4:207. doi: 10.1186/gb-2003-4-3-207. PMID:12620097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/S0896-6273(00)81133-2. PMID:10798388 [DOI] [PubMed] [Google Scholar]
- 7.Diss JK, Fraser SP, Djamgoz MB. Voltage-gated Na+ channels: multiplicity of expression, plasticity, functional implications and pathophysiological aspects. Eur Biophys J. 2004;33:180–93. doi: 10.1007/s00249-004-0389-0. PMID:14963621 [DOI] [PubMed] [Google Scholar]
- 8.Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, et al.. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–8. doi: 10.1016/S0896-6273(00)00116-1. PMID:11144347 [DOI] [PubMed] [Google Scholar]
- 9.Candenas L, Seda M, Noheda P, Buschmann H, Cintado CG, Martin JD, Pinto FM. Molecular diversity of voltage-gated sodium channel alpha and beta subunit mRNAs in human tissues. Eur J Pharmacol. 2006;541:9–16. doi: 10.1016/j.ejphar.2006.04.025. PMID:16750188 [DOI] [PubMed] [Google Scholar]
- 10.Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev. 2008;88:1407–47. doi: 10.1152/physrev.00002.2008. PMID:18923186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pappalardo LW, Black JA, Waxman SG. Sodium channels in astroglia and microglia. Glia. 2016;64:1628–45. doi: 10.1002/glia.22967. PMID:26919466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovascular Med. 2005;15:35–40. doi: 10.1016/j.tcm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Motoike HK, Liu H, Glaaser IW, Yang AS, Tateyama M, Kass RS. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J General Physiol. 2004;123:155–65. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herzog RI, Liu C, Waxman SG, Cummins TR. Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J Neurosci. 2003;23:8261–70. PMID:12967988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rougier JS, van Bemmelen MX, Bruce MC, Jespersen T, Gavillet B, Apotheloz F, Cordonier S, Staub O, Rotin D, Abriel H. Molecular determinants of voltage-gated sodium channel regulation by the Nedd4/Nedd4-like proteins. Am J Physiol Cell Physiol. 2005;288:C692–701. doi: 10.1152/ajpcell.00460.2004. PMID:15548568 [DOI] [PubMed] [Google Scholar]
- 16.Ou Y, Strege P, Miller SM, Makielski J, Ackerman M, Gibbons SJ, Farrugia G. Syntrophin gamma 2 regulates SCN5A gating by a PDZ domain-mediated interaction. J Biol Chem. 2003;278:1915–23. doi: 10.1074/jbc.M209938200. PMID:12429735 [DOI] [PubMed] [Google Scholar]
- 17.Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012;486:135–9. PMID:22678296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Ren W, DeCaen P, Yan C, Tao X, Tang L, Wang J, Hasegawa K, Kumasaka T, He J, et al.. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature. 2012;486:130–4. PMID:22678295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–8. doi: 10.1038/nature10238. PMID:21743477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen H, Zhou Q, Pan X, Li Z, Wu J, Yan N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science. 2017;355(6328). pii: eaal4326. doi: 10.1126/science.aal4326. [DOI] [PubMed] [Google Scholar]
- 21.Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7:548–62. doi: 10.1038/nrn1938. PMID:16791144 [DOI] [PubMed] [Google Scholar]
- 22.Whitaker WR, Clare JJ, Powell AJ, Chen YH, Faull RL, Emson PC. Distribution of voltage-gated sodium channel alpha-subunit and beta-subunit mRNAs in human hippocampal formation, cortex, and cerebellum. J Comparative Neurol. 2000;422:123–39. doi: 10.1002/(SICI)1096-9861(20000619)422:1%3c123::AID-CNE8%3e3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 23.Fletcher EV, Kullmann DM, Schorge S. Alternative splicing modulates inactivation of type 1 voltage-gated sodium channels by toggling an amino acid in the first S3-S4 linker. J Biol Chem. 2011;286:36700–8. doi: 10.1074/jbc.M111.250225. PMID:21890636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schroeter A, Walzik S, Blechschmidt S, Haufe V, Benndorf K, Zimmer T. Structure and function of splice variants of the cardiac voltage-gated sodium channel Na(v)1.5. J Mol Cell Cardiol. 2010;49:16–24. doi: 10.1016/j.yjmcc.2010.04.004. PMID:20398673 [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Ou SW, Wang YJ, Kameyama M, Kameyama A, Zong ZH. Analysis of four novel variants of Nav1.5/SCN5A cloned from the brain. Neurosci Res.2009;64:339–47. doi: 10.1016/j.neures.2009.04.003. PMID:19376164 [DOI] [PubMed] [Google Scholar]
- 26.Onkal R, Mattis JH, Fraser SP, Diss JK, Shao D, Okuse K, Djamgoz MB. Alternative splicing of Nav1.5: an electrophysiological comparison of ‘neonatal’ and ‘adult’ isoforms and critical involvement of a lysine residue. J Cell Physiol. 2008;216:716–26. doi: 10.1002/jcp.21451. PMID:18393272 [DOI] [PubMed] [Google Scholar]
- 27.Whitaker WR, Faull RL, Waldvogel HJ, Plumpton CJ, Emson PC, Clare JJ. Comparative distribution of voltage-gated sodium channel proteins in human brain. Brain Res Mol Brain Res. 2001;88:37–53. doi: 10.1016/S0169-328X(00)00289-8. PMID:11295230 [DOI] [PubMed] [Google Scholar]
- 28.Fukuoka T, Noguchi K. Comparative study of voltage-gated sodium channel alpha-subunits in non-overlapping four neuronal populations in the rat dorsal root ganglion. Neurosci Res. 2011;70:164–71. doi: 10.1016/j.neures.2011.01.020. PMID:21303679 [DOI] [PubMed] [Google Scholar]
- 29.Schaller KL, Caldwell JH. Expression and distribution of voltage-gated sodium channels in the cerebellum. Cerebellum. 2003;2:2–9. doi: 10.1080/14734220309424. PMID:12882229 [DOI] [PubMed] [Google Scholar]
- 30.Schaller KL, Caldwell JH. Developmental and regional expression of sodium channel isoform NaCh6 in the rat central nervous system. J Comparative Neurol. 2000;420:84–97. doi: 10.1002/(SICI)1096-9861(20000424)420:1%3c84::AID-CNE6%3e3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 31.Aman TK, Grieco-Calub TM, Chen C, Rusconi R, Slat EA, Isom LL, Raman IM. Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J Neurosci. 2009;29:2027–42. doi: 10.1523/JNEUROSCI.4531-08.2009. PMID:19228957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, et al.. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 1984; 312:121–7. doi: 10.1038/312121a0. PMID:6209577 [DOI] [PubMed] [Google Scholar]
- 33.Noda M, Ikeda T, Kayano T, Suzuki H, Takeshima H, Kurasaki M, Takahashi H, Numa S. Existence of distinct sodium channel messenger RNAs in rat brain. Nature 1986; 320:188–92. doi: 10.1038/320188a0. PMID:3754035 [DOI] [PubMed] [Google Scholar]
- 34.Auld VJ, Goldin AL, Krafte DS, Marshall J, Dunn JM, Catterall WA, Lester HA, Davidson N, Dunn RJ. A rat brain Na+ channel alpha subunit with novel gating properties. Neuron. 1988;1:449–61. doi: 10.1016/0896-6273(88)90176-6. PMID:2856097 [DOI] [PubMed] [Google Scholar]
- 35.Westenbroek RE, Merrick DK, Catterall WA. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron. 1989;3:695–704. doi: 10.1016/0896-6273(89)90238-9. PMID:2561976 [DOI] [PubMed] [Google Scholar]
- 36.Gong B, Rhodes KJ, Bekele-Arcuri Z, Trimmer JS. Type I and type II Na(+) channel alpha-subunit polypeptides exhibit distinct spatial and temporal patterning, and association with auxiliary subunits in rat brain. J Comparative Neurol. 1999;412:342–52. doi: 10.1002/(SICI)1096-9861(19990920)412:2%3c342::AID-CNE11%3e3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Ahmed CM, Ware DH, Lee SC, Patten CD, Ferrer-Montiel AV, Schinder AF, McPherson JD, Wagner-McPherson CB, Wasmuth JJ, Evans GA, et al.. Primary structure, chromosomal localization, and functional expression of a voltage-gated sodium channel from human brain. Proc Natl Acad Sci U S A. 1992;89:8220–4. doi: 10.1073/pnas.89.17.8220. PMID:1325650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen YH, Dale TJ, Romanos MA, Whitaker WR, Xie XM, Clare JJ. Cloning, distribution and functional analysis of the type III sodium channel from human brain. Eur J Neurosci. 2000;12:4281–9. PMID:11122339 [PubMed] [Google Scholar]
- 39.Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A. 2000;97:5616–20. doi: 10.1073/pnas.090034797. PMID:10779552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaplan MR, Meyer-Franke A, Lambert S, Bennett V, Duncan ID, Levinson SR, Barres BA. Induction of sodium channel clustering by oligodendrocytes. Nature. 1997;386:724–8. doi: 10.1038/386724a0. PMID:9109490 [DOI] [PubMed] [Google Scholar]
- 41.Kaplan MR, Cho MH, Ullian EM, Isom LL, Levinson SR, Barres BA. Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier. Neuron. 2001;30:105–19. doi: 10.1016/S0896-6273(01)00266-5. PMID:11343648 [DOI] [PubMed] [Google Scholar]
- 42.Gasser A, Ho TS, Cheng X, Chang KJ, Waxman SG, Rasband MN, Dib-Hajj SD. An ankyrinG-binding motif is necessary and sufficient for targeting Nav1.6 sodium channels to axon initial segments and nodes of Ranvier. J Neurosci. 2012;32:7232–43. doi: 10.1523/JNEUROSCI.5434-11.2012. PMID:22623668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henry MA, Sorensen HJ, Johnson LR, Levinson SR. Localization of the Nav1.8 sodium channel isoform at nodes of Ranvier in normal human radicular tooth pulp. Neurosci Letters. 2005;380:32–6. doi: 10.1016/j.neulet.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 44.Van Wart A, Matthews G. Expression of sodium channels Nav1.2 and Nav1.6 during postnatal development of the retina. Neurosci Letters. 2006;403:315–7. doi: 10.1016/j.neulet.2006.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duflocq A, Le Bras B, Bullier E, Couraud F, Davenne M. Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments. Mol Cell Neurosci. 2008;39:180–92. doi: 10.1016/j.mcn.2008.06.008. PMID:18621130 [DOI] [PubMed] [Google Scholar]
- 46.Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12:996–1002. doi: 10.1038/nn.2359. PMID:19633666 [DOI] [PubMed] [Google Scholar]
- 47.Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010;328:906–9. doi: 10.1126/science.1187958. PMID:20466935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rook MB, Evers MM, Vos MA, Bierhuizen MF. Biology of cardiac sodium channel Nav1.5 expression. Cardiovascular Res. 2012;93:12–23. doi: 10.1093/cvr/cvr252. [DOI] [PubMed] [Google Scholar]
- 49.Hartmann HA, Colom LV, Sutherland ML, Noebels JL. Selective localization of cardiac SCN5A sodium channels in limbic regions of rat brain. Nat Neurosci. 1999;2:593–5. doi: 10.1038/10147. PMID:10404176 [DOI] [PubMed] [Google Scholar]
- 50.Donahue LM, Coates PW, Lee VH, Ippensen DC, Arze SE, Poduslo SE. The cardiac sodium channel mRNA is expressed in the developing and adult rat and human brain. Brain Res. 2000;887:335–43. doi: 10.1016/S0006-8993(00)03033-X. PMID:11134623 [DOI] [PubMed] [Google Scholar]
- 51.Ou SW, Kameyama A, Hao LY, Horiuchi M, Minobe E, Wang WY, Makita N, Kameyama M. Tetrodotoxin-resistant Na+ channels in human neuroblastoma cells are encoded by new variants of Nav1.5/SCN5A. Eur J Neurosci. 2005;22:793–801. doi: 10.1111/j.1460-9568.2005.04280.x. PMID:16115203 [DOI] [PubMed] [Google Scholar]
- 52.Frenz CT, Hansen A, Dupuis ND, Shultz N, Levinson SR, Finger TE, Dionne VE. NaV1.5 sodium channel window currents contribute to spontaneous firing in olfactory sensory neurons. J Neurophysiol. 2014;112:1091–104. doi: 10.1152/jn.00154.2014. PMID:24872539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, Ou SW, Wang YJ, Zong ZH, Lin L, Kameyama M, Kameyama A. New variants of Nav1.5/SCN5A encode Na+ channels in the brain. J Neurogenetics. 2008;22:57–75. doi: 10.1080/01677060701672077. [DOI] [PubMed] [Google Scholar]
- 54.Ou SW, Zong ZH, Wang J, Wang YJ. Nav1.5 Na+ Channels in human brain are encoded by new variants of Nav1.5/SCN5A. PIBB. 2007;34:935–44. [Google Scholar]
- 55.Wang J, Zong ZH, OU SW, Wang YJ, Ren CT, Lin Y. Cloning, distribution and analysis of the new exon encoding Nav1.5 channel in brain tissues. PIBB. 2007;34:255–9. [Google Scholar]
- 56.Ren CT, Li DM, Ou SW, Wang YJ, Lin Y, Zong ZH, Kameyama M, Kameyama A. Cloning and expression of the two new variants of Nav1.5/SCN5A in rat brain. Mol Cell Biochem. 2012;365:139–48. doi: 10.1007/s11010-012-1253-7. PMID:22331407 [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Ou SW, Bai YF, Wang YJ, Xu ZD, Luan GM. Multiple Nav1.5 isoforms are functionally expressed in the brain and present distinct expression patterns compared with cardiac Nav1.5. Mol Med Reports. 2017;16:719–29. doi: 10.3892/mmr.2017.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raymond CK, Castle J, Garrett-Engele P, Armour CD, Kan Z, Tsinoremas N, Johnson JM. Expression of alternatively spliced sodium channel alpha-subunit genes. Unique splicing patterns are observed in dorsal root ganglia. J Biol Chem. 2004;279:46234–41. doi: 10.1074/jbc.M406387200. PMID:15302875 [DOI] [PubMed] [Google Scholar]
- 59.Kerr NC, Gao Z, Holmes FE, Hobson SA, Hancox JC, Wynick D, James AF. The sodium channel Nav1.5a is the predominant isoform expressed in adult mouse dorsal root ganglia and exhibits distinct inactivation properties from the full-length Nav1.5 channel. Mol Cell Neurosci. 2007;35:283–91. doi: 10.1016/j.mcn.2007.03.002. PMID:17433712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fukuoka T, Kobayashi K, Yamanaka H, Obata K, Dai Y, Noguchi K. Comparative study of the distribution of the alpha-subunits of voltage-gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J Comparative Neurol. 2008;510:188–206. doi: 10.1002/cne.21786. [DOI] [PubMed] [Google Scholar]
- 61.Chahine M, O'Leary ME. Regulatory role of Voltage-Gated Na channel beta Subunits in sensory neurons. Frontiers Pharmacol. 2011;2:70. doi: 10.3389/fphar.2011.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Isom LL. Sodium channel beta subunits: anything but auxiliary. Neuroscientist. 2001;7:42–54. doi: 10.1177/107385840100700108. PMID:11486343 [DOI] [PubMed] [Google Scholar]
- 63.Vijayaragavan K, Powell AJ, Kinghorn IJ, Chahine M. Role of auxiliary beta1-, beta2-, and beta3-subunits and their interaction with Na(v)1.8 voltage-gated sodium channel. Biochem Biophys Res Communications. 2004;319:531–40. doi: 10.1016/j.bbrc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 64.Gilchrist J, Das S, Van Petegem F, Bosmans F. Crystallographic insights into sodium-channel modulation by the beta4 subunit. Proc Natl Acad Sci U S A. 2013;110:E5016–24. doi: 10.1073/pnas.1314557110. PMID:24297919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Namadurai S, Balasuriya D, Rajappa R, Wiemhofer M, Stott K, Klingauf J, Edwardson JM, Chirgadze DY, Jackson AP. Crystal structure and molecular imaging of the Nav channel beta3 subunit indicates a trimeric assembly. J Biol Chem. 2014;289:10797–811. doi: 10.1074/jbc.M113.527994. PMID:24567321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yan Z, Zhou Q, Wang L, Wu J, Zhao Y, Huang G, Peng W, Shen H, Lei J, Yan N. Structure of the Nav1.4-beta1 complex from Electric Eel. Cell. 2017;170:470–82 e11. doi: 10.1016/j.cell.2017.06.039. [DOI] [PubMed] [Google Scholar]
- 67.O'Malley HA, Isom LL. Sodium channel beta subunits: emerging targets in channelopathies. Annual Rev Physiol. 2015;77:481–504. doi: 10.1146/annurev-physiol-021014-071846. PMID:25668026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao J, O'Leary ME, Chahine M. Regulation of Nav1.6 and Nav1.8 peripheral nerve Na+ channels by auxiliary beta-subunits. J Neurophysiol. 2011;106:608–19. doi: 10.1152/jn.00107.2011. PMID:21562192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lopez-Santiago LF, Brackenbury WJ, Chen C, Isom LL. Na+ channel Scn1b gene regulates dorsal root ganglion nociceptor excitability in vivo. J Biol Chem. 2011;286:22913–23. doi: 10.1074/jbc.M111.242370. PMID:21555511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qin N, D'Andrea MR, Lubin ML, Shafaee N, Codd EE, Correa AM. Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit. Eur J Biochem. 2003;270:4762–70. doi: 10.1046/j.1432-1033.2003.03878.x. PMID:14622265 [DOI] [PubMed] [Google Scholar]
- 71.Casula MA, Facer P, Powell AJ, Kinghorn IJ, Plumpton C, Tate SN, Bountra C, Birch R, Anand P. Expression of the sodium channel beta3 subunit in injured human sensory neurons. Neuroreport 2004; 15:1629–32. doi: 10.1097/01.wnr.0000134927.02776.ae. PMID:15232296 [DOI] [PubMed] [Google Scholar]
- 72.Catterall WA. The molecular basis of neuronal excitability. Science. 1984;223:653–61. doi: 10.1126/science.6320365. PMID:6320365 [DOI] [PubMed] [Google Scholar]
- 73.Catterall WA. Molecular properties of voltage-sensitive sodium channels. Annual Rev Biochem. 1986;55:953–85. doi: 10.1146/annurev.bi.55.070186.004513. [DOI] [PubMed] [Google Scholar]
- 74.Malhotra JD, Koopmann MC, Kazen-Gillespie KA, Fettman N, Hortsch M, Isom LL. Structural requirements for interaction of sodium channel beta 1 subunits with ankyrin. J Biol Chem. 2002;277:26681–8. doi: 10.1074/jbc.M202354200. PMID:11997395 [DOI] [PubMed] [Google Scholar]
- 75.Buffington SA, Rasband MN. Na+ channel-dependent recruitment of Navbeta4 to axon initial segments and nodes of Ranvier. J Neurosci. 2013;33:6191–202. doi: 10.1523/JNEUROSCI.4051-12.2013. PMID:23554500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, et al.. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci. 2003;23:7577–85. PMID:12930796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chahine M, Chatelier A, Babich O, Krupp JJ. Voltage-gated sodium channels in neurological disorders. CNS Neurol Disord Drug Targets. 2008;7:144–58. doi: 10.2174/187152708784083830. PMID:18537643 [DOI] [PubMed] [Google Scholar]
- 78.Jarecki BW, Piekarz AD, Jackson JO 2nd, Cummins TR. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Invest. 2010;120:369–78. doi: 10.1172/JCI40801. PMID:20038812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jacobs MP, Fischbach GD, Davis MR, Dichter MA, Dingledine R, Lowenstein DH, Morrell MJ, Noebels JL, Rogawski MA, Spencer SS, et al.. Future directions for epilepsy research. Neurology. 2001;57:1536–42. doi: 10.1212/WNL.57.9.1536. PMID:11706087 [DOI] [PubMed] [Google Scholar]
- 80.Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115:2010–7. doi: 10.1172/JCI25466. PMID:16075041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.George AL., Jr Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–9. doi: 10.1172/JCI25505. PMID:16075039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berkovic SF, Mulley JC, Scheffer IE, Petrou S. Human epilepsies: interaction of genetic and acquired factors. Trends Neurosci. 2006;29:391–7. doi: 10.1016/j.tins.2006.05.009. PMID:16769131 [DOI] [PubMed] [Google Scholar]
- 83.Waxman SG. Channel, neuronal and clinical function in sodium channelopathies: from genotype to phenotype. Nat neurosci. 2007;10:405–9. doi: 10.1038/nn1857. PMID:17387329 [DOI] [PubMed] [Google Scholar]
- 84.Stafstrom CE. Persistent sodium current and its role in epilepsy. Epilepsy Currents. 2007;7:15–22. doi: 10.1111/j.1535-7511.2007.00156.x. PMID:17304346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. J Physiol. 2010;588:1849–59. doi: 10.1113/jphysiol.2010.187484. PMID:20194124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kwan P, Poon WS, Ng HK, Kang DE, Wong V, Ng PW, Lui CH, Sin NC, Wong KS, Baum L. Multidrug resistance in epilepsy and polymorphisms in the voltage-gated sodium channel genes SCN1A, SCN2A, and SCN3A: correlation among phenotype, genotype, and mRNA expression. Pharmacogenetics Genomics. 2008;18:989–98. doi: 10.1097/FPC.0b013e3283117d67. PMID:18784617 [DOI] [PubMed] [Google Scholar]
- 87.Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia. 2010;51:1650–8. doi: 10.1111/j.1528-1167.2010.02640.x. PMID:20831750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hahn A, Neubauer BA. Sodium and potassium channel dysfunctions in rare and common idiopathic epilepsy syndromes. Brain devel. 2009;31:515–20. doi: 10.1016/j.braindev.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 89.Catterall WA, Dib-Hajj S, Meisler MH, Pietrobon D. Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J Neurosci. 2008;28:11768–77. doi: 10.1523/JNEUROSCI.3901-08.2008. PMID:19005038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Depienne C, Trouillard O, Saint-Martin C, Gourfinkel-An I, Bouteiller D, Carpentier W, Keren B, Abert B, Gautier A, Baulac S, et al.. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. 2009;46:183–91. doi: 10.1136/jmg.2008.062323. PMID:18930999 [DOI] [PubMed] [Google Scholar]
- 91.Martin MS, Dutt K, Papale LA, Dube CM, Dutton SB, de Haan G, Shankar A, Tufik S, Meisler MH, Baram TZ, et al.. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J Biol Chem. 2010;285:9823–34. doi: 10.1074/jbc.M109.078568. PMID:20100831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010;9:413–24. doi: 10.1016/S1474-4422(10)70059-4. PMID:20298965 [DOI] [PubMed] [Google Scholar]
- 93.Dravet C. Dravet syndrome history. Dev Med Child Neurol. 2011;53 Suppl 2:1–6. doi: 10.1111/j.1469-8749.2011.03964.x. PMID:21504424 [DOI] [PubMed] [Google Scholar]
- 94.Schutte SS, Schutte RJ, Barragan EV, O'Dowd DK. Model systems for studying cellular mechanisms of SCN1A-related epilepsy. J Neurophysiol. 2016;115:1755–66. doi: 10.1152/jn.00824.2015. PMID:26843603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Estacion M, Gasser A, Dib-Hajj SD, Waxman SG. A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons. Exp Neurol. 2010;224:362–8. doi: 10.1016/j.expneurol.2010.04.012. PMID:20420834 [DOI] [PubMed] [Google Scholar]
- 96.Guo F, Yu N, Cai JQ, Quinn T, Zong ZH, Zeng YJ, Hao LY. Voltage-gated sodium channel Nav1.1, Nav1.3 and beta1 subunit were up-regulated in the hippocampus of spontaneously epileptic rat. Brain Res Bulletin. 2008;75:179–87. doi: 10.1016/j.brainresbull.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 97.Holland KD, Kearney JA, Glauser TA, Buck G, Keddache M, Blankston JR, Glaaser IW, Kass RS, Meisler MH. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci Letters. 2008;433:65–70. doi: 10.1016/j.neulet.2007.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aurlien D, Leren TP, Tauboll E, Gjerstad L. New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure. 2009;18:158–60. doi: 10.1016/j.seizure.2008.07.008. PMID:18752973 [DOI] [PubMed] [Google Scholar]
- 99.Parisi P, Oliva A, Coll Vidal M, Partemi S, Campuzano O, Iglesias A, Pisani D, Pascali VL, Paolino MC, Villa MP, et al.. Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. 2013;105:415–8. doi: 10.1016/j.eplepsyres.2013.02.024. PMID:23538271 [DOI] [PubMed] [Google Scholar]
- 100.O'Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Frontiers Genetics. 2013;4:213. doi: 10.3389/fgene.2013.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Patino GA, Brackenbury WJ, Bao Y, Lopez-Santiago LF, O'Malley HA, Chen C, Calhoun JD, Lafrenière RG, Cossette P, Rouleau GA, et al.. Voltage-gated Na+ channel beta1B: a secreted cell adhesion molecule involved in human epilepsy. J Neurosci. 2011;31:14577–91. doi: 10.1523/JNEUROSCI.0361-11.2011. PMID:21994374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Patino GA, Claes LR, Lopez-Santiago LF, Slat EA, Dondeti RS, Chen C, O'Malley HA, Gray CB, Miyazaki H, Nukina N, et al.. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci. 2009;29:10764–78. doi: 10.1523/JNEUROSCI.2475-09.2009. PMID:19710327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dib-Hajj SD, Black JA, Waxman SG. Voltage-gated sodium channels: therapeutic targets for pain. Pain Med. 2009;10:1260–9. doi: 10.1111/j.1526-4637.2009.00719.x. PMID:19818036 [DOI] [PubMed] [Google Scholar]
- 104.Lampert A, O'Reilly AO, Reeh P, Leffler A. Sodium channelopathies and pain. Pflugers Archiv: European J Physiol. 2010;460:249–63. doi: 10.1007/s00424-009-0779-3. [DOI] [PubMed] [Google Scholar]
- 105.Cregg R, Momin A, Rugiero F, Wood JN, Zhao J. Pain channelopathies. J Physiol. 2010;588:1897–904. doi: 10.1113/jphysiol.2010.187807. PMID:20142270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Andavan GS, Lemmens-Gruber R. Voltage-gated sodium channels: mutations, channelopathies and targets. Curr Med Chem. 2011;18:377–97. doi: 10.2174/092986711794839133. PMID:21143119 [DOI] [PubMed] [Google Scholar]
- 107.Devor M. Sodium channels and mechanisms of neuropathic pain. J Pain. 2006;7:S3–S12. doi: 10.1016/j.jpain.2005.09.006. PMID:16426998 [DOI] [PubMed] [Google Scholar]
- 108.Waxman SG, Dib-Hajj S. Erythermalgia: molecular basis for an inherited pain syndrome. Trends Mol Med. 2005;11:555–62. doi: 10.1016/j.molmed.2005.10.004. PMID:16278094 [DOI] [PubMed] [Google Scholar]
- 109.Dib-Hajj SD, Binshtok AM, Cummins TR, Jarvis MF, Samad T, Zimmermann K. Voltage-gated sodium channels in pain states: role in pathophysiology and targets for treatment. Brain Res Rev. 2009;60:65–83. doi: 10.1016/j.brainresrev.2008.12.005. PMID:19150627 [DOI] [PubMed] [Google Scholar]
- 110.Benarroch EE. Sodium channels and pain. Neurology. 2007;68:233–6. doi: 10.1212/01.wnl.0000252951.48745.a1. PMID:17224580 [DOI] [PubMed] [Google Scholar]
- 111.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, et al.. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–8. doi: 10.1038/nature05413. PMID:17167479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cummins TR, Sheets PL, Waxman SG. The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain. 2007;131:243–57. doi: 10.1016/j.pain.2007.07.026. PMID:17766042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu M, Wood JN. The roles of sodium channels in nociception: implications for mechanisms of neuropathic pain. Pain Med. 2011;12 Suppl 3:S93–9. doi: 10.1111/j.1526-4637.2011.01158.x. PMID:21752183 [DOI] [PubMed] [Google Scholar]
- 114.Ahmad S, Dahllund L, Eriksson AB, Hellgren D, Karlsson U, Lund PE, Meijer IA, Meury L, Mills T, Moody A, et al.. A stop codon mutation in SCN9A causes lack of pain sensation. Hum Mol Genetics. 2007;16:2114–21. doi: 10.1093/hmg/ddm160. [DOI] [PubMed] [Google Scholar]
- 115.Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, et al.. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–74. doi: 10.1016/j.neuron.2006.10.006. PMID:17145499 [DOI] [PubMed] [Google Scholar]
- 116.Leo S, D'Hooge R, Meert T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behavioural Brain Res. 2010;208:149–57. doi: 10.1016/j.bbr.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 117.Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–5. doi: 10.1126/science.1156916. PMID:18669863 [DOI] [PubMed] [Google Scholar]
- 118.Kerr BJ, Souslova V, McMahon SB, Wood JN. A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport. 2001;12:3077–80. doi: 10.1097/00001756-200110080-00019. PMID:11568640 [DOI] [PubMed] [Google Scholar]
- 119.Laird JM, Souslova V, Wood JN, Cervero F. Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null mice. J Neurosci. 2002;22:8352–6. PMID:12351708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JN, Reeh PW. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature. 2007;447:855–8. doi: 10.1038/nature05880. PMID:17568746 [DOI] [PubMed] [Google Scholar]
- 121.Maingret F, Coste B, Padilla F, Clerc N, Crest M, Korogod SM, Delmas P. Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J General Physiol. 2008;131:211–25. doi: 10.1085/jgp.200709935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003;23:8881–92. PMID:14523090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lampert A, Hains BC, Waxman SG. Upregulation of persistent and ramp sodium current in dorsal horn neurons after spinal cord injury. Exp Brain Res. 2006;174:660–6. doi: 10.1007/s00221-006-0511-x. PMID:16718433 [DOI] [PubMed] [Google Scholar]
- 124.Takahashi N, Kikuchi S, Dai Y, Kobayashi K, Fukuoka T, Noguchi K. Expression of auxiliary beta subunits of sodium channels in primary afferent neurons and the effect of nerve injury. Neuroscience. 2003;121:441–50. doi: 10.1016/S0306-4522(03)00432-9. PMID:14522002 [DOI] [PubMed] [Google Scholar]
- 125.McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, Krambis MJ, Antonio BM, Zellmer SG, Printzenhoff D, et al.. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci U S A. 2013;110:E2724–32. doi: 10.1073/pnas.1220844110. PMID:23818614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Alexandrou AJ, Brown AR, Chapman ML, Estacion M, Turner J, Mis MA, Wilbrey A, Payne EC, Gutteridge A, Cox PJ, et al.. Subtype-Selective small molecule inhibitors reveal a fundamental role for Nav1.7 in Nociceptor electrogenesis, axonal conduction and presynaptic release. PloS One. 2016;11:e0152405. doi: 10.1371/journal.pone.0152405. PMID:27050761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cao L, McDonnell A, Nitzsche A, Alexandrou A, Saintot PP, Loucif AJ, Brown AR, Young G, Mis M, Randall A, et al.. Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci Transl Med. 2016;8:335ra56. doi: 10.1126/scitranslmed.aad7653. PMID:27099175 [DOI] [PubMed] [Google Scholar]
- 128.Binello E, Germano IM. Targeting glioma stem cells: a novel framework for brain tumors. Cancer Sci. 2011;102:1958–66. doi: 10.1111/j.1349-7006.2011.02064.x. PMID:21848914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schrey M, Codina C, Kraft R, Beetz C, Kalff R, Wolfl S, Patt S. Molecular characterization of voltage-gated sodium channels in human gliomas. Neuroreport. 2002;13:2493–8. doi: 10.1097/00001756-200212200-00023. PMID:12499855 [DOI] [PubMed] [Google Scholar]
- 130.Kawaguchi A, Asano H, Matsushima K, Wada T, Yoshida S, Ichida S. Enhancement of sodium current in NG108-15 cells during neural differentiation is mainly due to an increase in NaV1.7 expression. Neurochem Res. 2007;32:1469–75. doi: 10.1007/s11064-007-9334-9. PMID:17404832 [DOI] [PubMed] [Google Scholar]
- 131.Fiske JL, Fomin VP, Brown ML, Duncan RL, Sikes RA. Voltage-sensitive ion channels and cancer. Cancer Metastasis Rev. 2006;25:493–500. doi: 10.1007/s10555-006-9017-z. PMID:17111226 [DOI] [PubMed] [Google Scholar]
- 132.Lefranc F, Mijatovic T, Kondo Y, Sauvage S, Roland I, Debeir O, Krstic D, Vasic V, Gailly P, Kondo S, et al.. Targeting the alpha 1 subunit of the sodium pump to combat glioblastoma cells. Neurosurgery. 2008;62:211–21; discussion 21–2. doi: 10.1227/01.NEU.0000311080.43024.0E. PMID:18300910 [DOI] [PubMed] [Google Scholar]
- 133.Xing D, Wang J, Ou S, Wang Y, Qiu B, Ding D, Guo F, Gao Q. Expression of neonatal Nav1.5 in human brain astrocytoma and its effect on proliferation, invasion and apoptosis of astrocytoma cells. Oncol Reports. 2014;31:2692–700. doi: 10.3892/or.2014.3143. [DOI] [PubMed] [Google Scholar]
- 134.Joshi AD, Parsons DW, Velculescu VE, Riggins GJ. Sodium ion channel mutations in glioblastoma patients correlate with shorter survival. Mol Cancer. 2011;10:17. doi: 10.1186/1476-4598-10-17. PMID:21314958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Black JA, Newcombe J, Waxman SG. Astrocytes within multiple sclerosis lesions upregulate sodium channel Nav1.5. Brain: a J Neurol. 2010;133:835–46. doi: 10.1093/brain/awq003. [DOI] [PubMed] [Google Scholar]
- 136.Lee CY, Lai HY, Chiu A, Chan SH, Hsiao LP, Lee ST. The effects of antiepileptic drugs on the growth of glioblastoma cell lines. J Neuro-Oncol. 2016;127:445–53. doi: 10.1007/s11060-016-2056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mao Q, Jia F, Zhang XH, Qiu YM, Ge JW, Bao WJ, Luo QZ, Jiang JY. The up-regulation of voltage-gated sodium channel Nav1.6 expression following fluid percussion traumatic brain injury in rats. Neurosurgery. 2010;66:1134–9; discussion 9. doi: 10.1227/01.NEU.0000369612.31946.A2. PMID:20421839 [DOI] [PubMed] [Google Scholar]
- 138.Yao C, Williams AJ, Cui P, Berti R, Hunter JC, Tortella FC, Dave JR. Differential pattern of expression of voltage-gated sodium channel genes following ischemic brain injury in rats. Neurotoxicity Res. 2002;4:67–75. doi: 10.1080/10298420290007646. [DOI] [PubMed] [Google Scholar]
- 139.Yin R, Liu D, Chhoa M, Li CM, Luo Y, Zhang M, Lehto SG, Immke DC, Moyer BD. Voltage-gated sodium channel function and expression in injured and uninjured rat dorsal root ganglia neurons. Int J Neurosci. 2016;126:182–92. doi: 10.3109/00207454.2015.1004172. PMID:25562420 [DOI] [PubMed] [Google Scholar]
- 140.Yao C, Williams AJ, Hartings JA, Lu XC, Tortella FC, Dave JR. Down-regulation of the sodium channel Na(v)1.1 alpha-subunit following focal ischemic brain injury in rats: in situ hybridization and immunohistochemical analysis. Life Sci. 2005;77:1116–29. doi: 10.1016/j.lfs.2005.02.008. PMID:15878599 [DOI] [PubMed] [Google Scholar]
- 141.Sleeper AA, Cummins TR, Dib-Hajj SD, Hormuzdiar W, Tyrrell L, Waxman SG, Black JA. Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J Neurosci. 2000;20:7279–89. PMID:11007885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Coward K, Plumpton C, Facer P, Birch R, Carlstedt T, Tate S, Bountra C, Anand P. Immunolocalization of SNS/PN3 and NaN/SNS2 sodium channels in human pain states. Pain. 2000;85:41–50. doi: 10.1016/S0304-3959(99)00251-1. PMID:10692601 [DOI] [PubMed] [Google Scholar]
- 143.Abe M, Kurihara T, Han W, Shinomiya K, Tanabe T. Changes in expression of voltage-dependent ion channel subunits in dorsal root ganglia of rats with radicular injury and pain. Spine. 2002;27:1517–24; discussion 25. doi: 10.1097/00007632-200207150-00007. PMID:12131710 [DOI] [PubMed] [Google Scholar]
- 144.Tan ZY, Donnelly DF, LaMotte RH. Effects of a chronic compression of the dorsal root ganglion on voltage-gated Na+ and K+ currents in cutaneous afferent neurons. J Neurophysiol. 2006;95:1115–23. doi: 10.1152/jn.00830.2005. PMID:16424456 [DOI] [PubMed] [Google Scholar]
- 145.Waxman SG. Mechanisms of disease: sodium channels and neuroprotection in multiple sclerosis-current status. Nat Clin Practice Neurol. 2008;4:159–69. doi: 10.1038/ncpneuro0735. [DOI] [PubMed] [Google Scholar]
- 146.O'Malley HA, Shreiner AB, Chen GH, Huffnagle GB, Isom LL. Loss of Na+ channel beta2 subunits is neuroprotective in a mouse model of multiple sclerosis. Mol Cell Neurosci. 2009;40:143–55. doi: 10.1016/j.mcn.2008.10.001. PMID:19013247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Craner MJ, Damarjian TG, Liu S, Hains BC, Lo AC, Black JA, Newcombe J, Cuzner ML, Waxman SG. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia. 2005;49:220–9. doi: 10.1002/glia.20112. PMID:15390090 [DOI] [PubMed] [Google Scholar]
- 148.Black JA, Waxman SG. Phenytoin protects central axons in experimental autoimmune encephalomyelitis. J Neurological Sci. 2008;274:57–63. doi: 10.1016/j.jns.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 149.Black JA, Liu S, Carrithers M, Carrithers LM, Waxman SG. Exacerbation of experimental autoimmune encephalomyelitis after withdrawal of phenytoin and carbamazepine. Annals Neurol. 2007;62:21–33. doi: 10.1002/ana.21172. [DOI] [PubMed] [Google Scholar]
- 150.Bechtold DA, Kapoor R, Smith KJ. Axonal protection using flecainide in experimental autoimmune encephalomyelitis. Annals Neurol. 2004;55:607–16. doi: 10.1002/ana.20045. [DOI] [PubMed] [Google Scholar]
- 151.Bechtold DA, Miller SJ, Dawson AC, Sun Y, Kapoor R, Berry D, Smith KJ. Axonal protection achieved in a model of multiple sclerosis using lamotrigine. J Neurol. 2006;253:1542–51. doi: 10.1007/s00415-006-0204-1. PMID:17219031 [DOI] [PubMed] [Google Scholar]