

Abstract
Trafficking of memory CD8+ T cells out of the circulation is essential to provide protective immunity against intracellular pathogens in non-lymphoid tissues. However, the molecular mechanisms that dictate the trafficking potential of diverse memory CD8+ T cell populations are not completely defined. Here, we show that following infection or inflammatory challenge, central memory (TCM) CD8+ T cells rapidly traffic into non-lymphoid tissues, whereas most effector memory (TEM) cells remain in the circulation. Furthermore, we demonstrate that cellular migration of memory CD8+ T cells into non-lymphoid tissues is driven by IL-15-stimulated enzymatic synthesis of core 2 O-glycans, which generates functional ligands for E- and P-selectin. Given that IL-15 stimulated expression of glycosyltransferase enzymes is largely a feature of TCM CD8+ T cells, this allows TCM to selectively migrate out of the circulation and into non-lymphoid tissues. Thus, these data show that the capacity to synthesize core 2 O-glycans identifies the memory CD8+ T cells with tissue-trafficking potential and that TCM, and not TEM, is the major subset that enters non-lymphoid tissues following infection or tissue injury.
INTRODUCTION
Trafficking of leukocytes out of the circulation and into lymphoid and non-lymphoid tissues requires the collective action of a variety of receptor - ligand interactions (1). Following maturation in the thymus, naïve CD8+ T cells enter the periphery, but are confined to the circulation and lymphoid organs, as their gene expression profile limits their trafficking to these compartments. In contrast, antigen-experienced, long-lived memory CD8+ T cells can directly infiltrate non-lymphoid tissues during episodes of local inflammation (2–4). Furthermore, the capacity for memory CD8+ T cells to traffic directly into inflamed tissues occurs independent of antigen re-stimulation and prior to the re-expansion of memory CD8+ T cells in lymphoid organs during an infection (5). Indeed, this feature of memory CD8+ T cells contributes significantly to antigen-specific protective immunity in non-lymphoid tissues (6, 7). Although understanding the factors that govern the tissue-trafficking potential of memory CD8+ T cells is highly relevant for vaccine design and host defense, the molecular and biochemical mechanisms that contribute to trafficking of memory CD8+ T cells are largely undefined.
Memory T cells are often classified based on the expression of receptors required for lymph node homing (8, 9). Naïve and central memory (TCM) CD8+ T cells express the chemokine receptor CCR7 and L-selectin (CD62L), which are required for T cells to extravasate across high endothelial venules and into lymph nodes. Effector memory (TEM) CD8+ T cells do not express these receptors, which excludes them from entering lymph nodes directly from the circulation. Because TEM cannot enter lymph nodes, it has been predicted that these cells actively patrol non-lymphoid tissues and also be the first T cells to arrive following infection or tissue injury. In fact, TEM CD8+ T cells express a variety of inflammatory chemokine receptors such as CCR5 and CX3CR1 (8, 10, 11), which could cause them to infiltrate non-lymphoid tissues during episodes of inflammation. TEM CD8+ T cells are also rich in granzymes, highly cytolytic, and provide robust protective immunity against some infections (12–14). Nevertheless, studies directly comparing the trafficking potential of TCM and TEM CD8+ T cells subsets or identifying the mechanisms that dictate their trafficking into non-lymphoid tissues during inflammatory challenges have not been rigorously performed.
Following infection, tissue damage, or other inflammatory event within non-lymphoid tissues, the associated vascular endothelium becomes activated and expresses adhesion molecules and chemokines that function to recruit circulating leukocytes. E- and P-selectin are C-type lectin, oligosaccharide-binding proteins that function to capture circulating leukocytes as the first step of the extravasation process (15). The capacity for CD8+ T cells to generate functional ligands for E- and P-selectin relies on post-translational O-linked glycosylation of cell surface proteins such as PSGL-1, ESL-1, CD44, and CD43 (16). Specifically, core 2 O-glycans decorated with sialyl Lewis x (sLex) provide the ligand binding site for the C-type lectin domains of E- and P-selectin (17). Enzymatic synthesis of core 2 O-glycans in CD8+ T cells requires core 2 β1,6 N-acetylglucosaminyltransferase-I (C2GlcNAcT-I; Gcnt1) to synthesize the complex core 2 O-glycans that function as E- and P-selectin ligands (18). Thus, expression of Gcnt1 and the mechanisms that regulate core 2 O-glycan synthesis in memory CD8+ T cells may ultimately control their tissue-trafficking potential.
Previously, we have demonstrated that core 2 O-glycan synthesis is dynamically regulated in memory CD8+ T cells in an IL-15-dependent, antigen-independent manner (7). Because core 2 O-glycan synthesis is critical for memory CD8+ T cells to traffic into inflamed non-lymphoid tissues, we investigated whether the capacity to synthesize core 2 O-glycans was differentially regulated within the memory CD8+ T cell compartment. Unexpectedly, we found that in disagreement with the TCM/TEM trafficking paradigm, core 2 O-glycan synthesis was highly active in TCM, but was less active or even absent in TEM subsets. Furthermore, we show that in response to repetitive rounds of antigen-stimulated differentiation, memory CD8+ T cells become KLRG1+ TEM that lose the capacity to synthesize core 2 O-glycans and are therefore unable to effectively generate functional ligands for E- and P-selectin and trafficking into inflamed, non-lymphoid tissues is significantly diminished. Overall, these results identify core 2 O-glycan synthesis as a critical regulator of memory CD8+ T cell trafficking and demonstrate that TCM CD8+ T cells, and not TEM, are the major subset that infiltrates non-lymphoid tissues in response to inflammation.
RESULTS
Core 2 O-glycan expressing memory CD8+ T cells traffic into inflamed skin in an antigen-independent manner
It has recently been demonstrated that naïve, pathogen-free mice may not accurately model typical inflammatory responses during infections because these animals have never acquired a mature, functional memory T cell compartment (19). Thus, to define the extent of memory CD8+ T cell trafficking that occurs in non-lymphoid tissues during a viral infection, we compared the quantity of CD8+ T cells in the skin of naïve, pathogen-free B6 mice infected with Vaccinia virus (VacV) versus mice that had been previously infected 60 days earlier with lymphocytic choriomeningitis virus (LCMV) (Fig S1A). Because LCMV and VacV do not share any T cell epitopes, this strategy allowed us to specifically quantify the trafficking of LCMV-specific memory CD8+ T cells into an inflamed tissue. On day 3 post-infection, there were 5-10 times more CD8+ T cells in the VacV-infected skin of mice that were LCMV-immune compared to naïve controls (Fig 1A, Fig S1B). LCMV-specific memory CD8+ T cells (H2-Db-GP33 and H2-Db-GP276) trafficked into the VacV-infected skin and were reactive to the monoclonal antibody 1B11 (Fig 1B,C), which binds to the core 2 O-linked glycosylated isoform of CD43 and identifies CD8+ T cells that can bind to E- and P-selectin (7, 20). This demonstrates that during a local inflammatory event, core 2 O-glycan expressing memory CD8+ T cells, regardless of specificity, traffic into the skin in an antigen-independent manner.
Figure 1. Memory CD8+ T cells traffic into VacV-infected skin independent of antigen specificity.
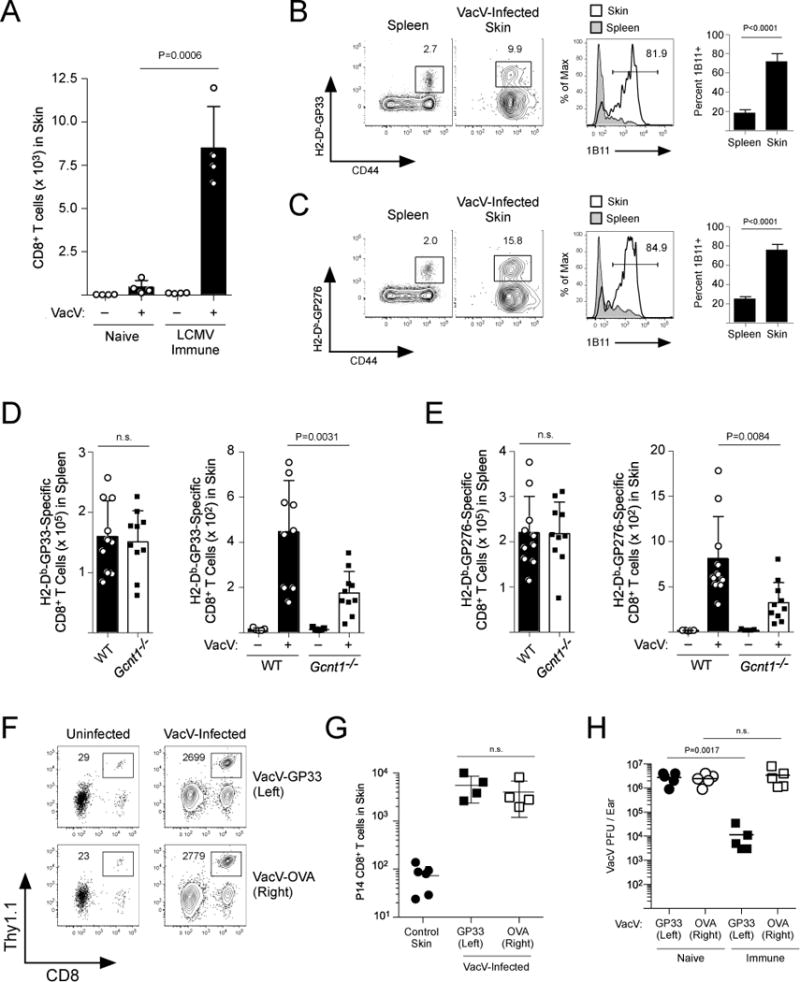
(A) Naïve B6 mice were infected with LCMV. On day 60 post-LCMV infection, LCMV-immune or naïve age matched controls were infected with VacV on the left ear skin by scarification. On day 3 post-infection with VacV, trafficking of CD8+ T cells into the skin was quantified. (B,C) LCMV-specific CD8+ T cells (H2-Db-GP33-41 and -GP276-284) were identified in the VacV-infected skin and spleen. Core 2 O-glycosylated CD43 was measured with the 1B11 antibody. (D,E) WT or Gcnt1−/− B6 mice were infected with LCMV. On day 60 post-LCMV infection, mice were infected with VacV on the left ear skin. Trafficking of (D) GP33-specific and (E) GP276-specific CD8+ T cells was quantified on day 3 post-infection. (F) Naïve P14 CD8+ T cells were transferred into naïve B6 mice and infected with LCMV-Armstrong. On day 75 post-infection, LCMV-immune mice were challenged with VacV-GP33 on the left ear skin and VacV-OVA on the right. Trafficking of P14 CD8+ T cells into the skin was quantified on day 3 post-VacV-infection. (G) Quantification of (F). (H) Same as (F) except VacV was quantified from naïve or LCMV immune mice on day 4 post-infection of the skin.
Previous studies have demonstrated that activated CD8+ T cells require Gcnt1 to initiate synthesis of core 2 O-glycans (17, 18). In fact, following acute LCMV infection, antigen-specific Gcnt1−/− CD8+ T cells expanded the same as WT controls, but did not express the 1B11 epitope, and binding to both E- and P-selectin was significantly reduced (Fig S2A–F). Because Gcnt1−/− CD8+ T cells were deficient in generating functional ligands for E- and P-selectin, we next tested whether Gcnt1 was required for trafficking of memory CD8+ T cells into the skin. LCMV-immune WT and Gcnt1−/− mice were infected with VacV and trafficking of GP33- and GP276-specific CD8+ T cells into the skin was analyzed. Similar numbers of GP33- and GP276-specific memory CD8+ T cells were present in the spleens of WT and Gcnt1−/− mice, but trafficking of Gcnt1−/− memory CD8+ T cells into the skin was significantly reduced compared to WT controls (Fig 1D,E). Thus, the capacity to synthesize core 2 O-glycans is critical for memory CD8+ T cells to traffic into the skin in response to local inflammation.
To determine whether the antigen-independent trafficking of memory CD8+ T cells into VacV-infected skin was critical for these cells to provide protective immunity, naïve TCR-tg P14 CD8+ T cells (specific for the LCMV epitope H2-Db-GP33) were transferred into naïve B6 mice and infected with LCMV. At >60 days post-infection, LCMV-immune mice were challenged with VacV expressing the GP33-41 epitope of LCMV (VacV-GP33) on the skin of the left ear and VacV expressing ovalbumin (VacV-OVA) on the right ear. GP33-specific memory P14 CD8+ T cells trafficked into the skin of both VacV infections in an E- and P-selectin-dependent manner, further demonstrating that local inflammation is responsible for recruiting memory CD8+ T cells into a site of infection (Fig 1D,E and Fig S3). However, memory P14 CD8+ T cells reduced the viral burden of the VacV-GP33 infection, but provided no protective immunity against VacV-OVA (Fig 1F). Therefore, these data demonstrate that memory CD8+ T cells of diverse specificities, when present, are a major component of early inflammation following a viral skin infection and antigen-independent trafficking is required for these cells to provide antigen-specific protective immunity.
IL-15 stimulates core 2 O-glycan synthesis in memory CD8+ T cells
Our previous work revealed that memory CD8+ T cells, but not naïve CD8+ T cells, synthesize core 2 O-glycans and generate functional ligands for E- and P-selectin in response to stimulation with IL-15 (7). Indeed, benzyl 2-acetamido-2-deoxy-α-D-galactopyranoside (BADG), which blocks incorporation of N-acetylglucosamine (GlcNAc) into O-glycans, prevented IL-15-stimulated binding to recombinant E- and P-selectin proteins (Fig 2A). Furthermore, removing sialic acids with neuraminidase also eliminated the IL-15-stimulated E- and P-selectin binding (Fig 2B). Synthesis of mature, core 2 O-glycans bearing sLex requires the collective action of a number of enzymes (Fig 2C). To determine whether a specific set of glycosyltransferase genes that facilitates O-glycan synthesis was transcriptionally regulated by IL-15, we purified memory P14 CD8+ T cells, stimulated them in vitro, and analyzed the expression of candidate genes that could potentially contribute to mature O-glycan synthesis. Expression of the core 2 O-glycan initiation enzyme Gcnt1 was significantly increased (Fig 2D). However, we also detected expression of a number of other glycosyltransferases that have been previously implicated in contributing to formation of E- and P-selectin ligands. IL-15 caused memory CD8+ T cells to increase expression of B3gnt3 and B4galt5, which could function cooperatively to extend lactosamine repeats on core 2 (and/or core 1) O-glycans (21, 22). α1,3 fucosyltransferase-7 (Fut7) and ST3 β-galactoside α-2,3-sialyltransferase-4 (St3gal4) were also found to be expressed following IL-15 stimulation. Notably, there was no change in expression of ST3 β-galactoside α-2,3-sialyltransferase 1 (St3gal1), the enzyme responsible for “capping” core 1 O-glycans with sialic acid and preventing core 2 O-glycan synthesis (23). Expression of Fut4 and St3gal6, which have been shown to contribute to sLex formation in other leukocyte populations, were also not increased by IL-15 (Fig S4A). Collectively, these data demonstrate that IL-15 stimulates expression of glycosyltransferase enzymes in memory CD8+ T cells that facilitate synthesis of sLex containing O-linked glycans.
Figure 2. Core 2 O-glycan synthesis is stimulated by IL-15 signaling.
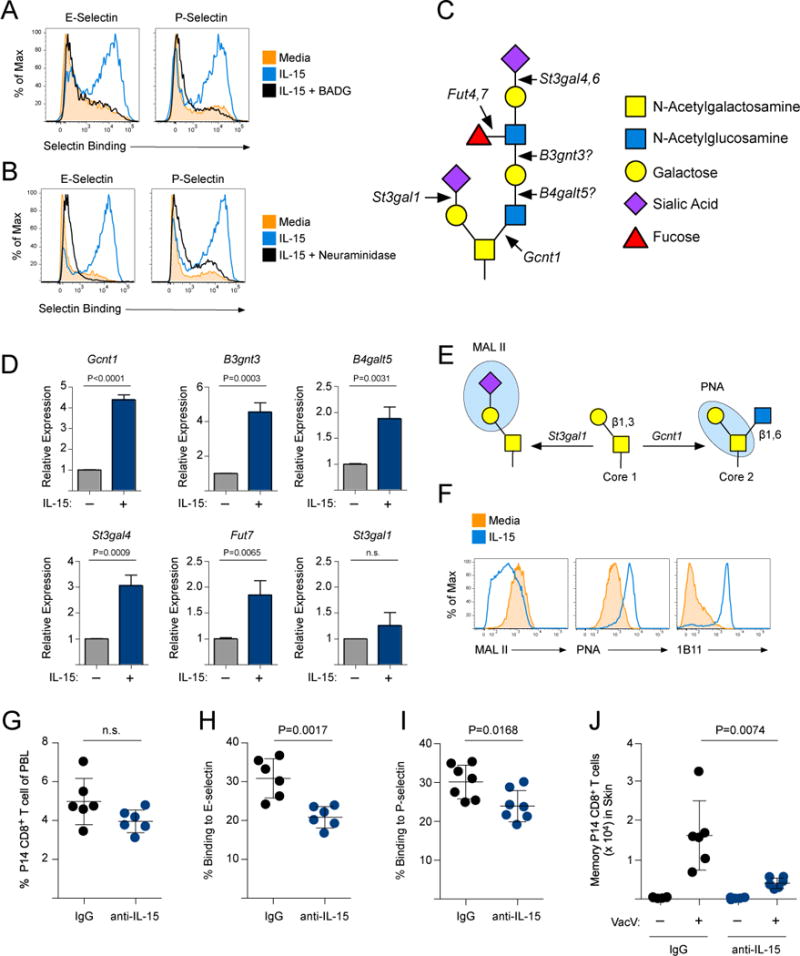
(A) Memory P14 CD8+ T cells were purified and stimulated with 250 ng/ml IL-15 or IL-15 + 5 mM BADG for 3 days. Binding of E- and P-selectin chimeric proteins was quantified. (B) Same as (A) except after IL-15 stimulation, memory P14 CD8+ T cells were treated with neuraminidase prior to analyzing E- and P-selectin binding. (C) The synthesis of complex core 2 O-glycans requires the collective action of a number of glycosyltransferases. (D) Memory P14 CD8+ T cells were purified and stimulated in vitro with 500 ng/ml of IL-15 for 3 days. Changes in gene expression of Gcnt1, B3gnt3, B4galt5, St3gal4, Fut7, and St3gal1 were determined by qPCR. (E) St3gal1 caps the core 1 O-glycan substrate with α2,3-linked sialic acid, which prevents core 2 O-glycan synthesis (by Gcnt1) and can be detected with the lectin MAL II. PNA binds to core 1 O-glycans only if they do not contain an α2,3-linked sialic acid. (F) Memory P14 CD8+ T cells were stimulated as in (A) and binding of MAL II, PNA, or the 1B11 antibody was analyzed. (G) LCMV-immune mice (90 days post-infection) were given control IgG or IL-15 neutralizing antibody for 10 days. The frequency, (H) E-selectin binding and (I) P-selectin binding of the memory P14 CD8+ T cell population in the circulation were quantified. Mice from (G-I) were infected with VacV and memory P14 CD8+ T cells were quantified in the skin on day 3 post-VacV infection.
Addition of the α2,3-linked sialic acid to the core 1 O-glycan substrate inhibits core 2 O-glycan synthesis by Gcnt1. The lectin MAL II binds directly to this sialic acid (24) and PNA binds only to unsialylated core 1 O-glycans (Fig 2E). IL-15-stimulated memory CD8+ T cells decreased MAL II binding and increased PNA binding compared to those cultured in media alone, demonstrating a loss of sialylated core 1 O-glycans (Fig 2F). Because St3gal1 expression was not decreased by IL-15 (Fig 2D), this suggests that Gcnt1 is able to “outcompete” St3gal1 for the core 1 O-glycan substrate. In fact, IL-15 also increased expression of the 1B11 epitope (Fig 2F), which requires Gcnt1 activity. This analysis demonstrates that IL-15 alters the core 1 status of O-glycans and stimulates core 2 O-glycan synthesis in memory CD8+ T cells.
IL-15 is often described as a homeostatic cytokine that is important for the long-term maintenance of memory CD8+ T cell populations, but also functions to stimulate effector functions of T cells (25, 26). To investigate whether steady-state exposure to IL-15 maintained the synthesis of selectin ligands on memory CD8+ T cells, we utilized an IL-15 neutralizing antibody (27). Indeed, neutralizing IL-15 caused circulating memory CD8+ T cells to reduce binding to E- and P-selectin, but did not affect the circulating frequency (Fig 2G–I). When IL-15 was neutralized, memory CD8+ T cells did not traffic into VacV-infected skin as well as cells in control treated animals (Fig 2J). Notably, VacV infection caused memory CD8+ T cells in the circulation to increase binding to E- and P-selectin, which was also dependent on IL-15 (Fig S4B–D). Collectively, these data demonstrate that IL-15 is a critical regulator of core 2 O-glycan synthesis during both homeostasis and following infection, and that interfering with this signaling axis alters the trafficking potential of memory CD8+ T cells.
Core 2 O-glycan synthesis is active primarily in TCM CD8+ T cells
It has been demonstrated using both functional readouts and gene expression profiles coupled with principle component analysis that memory CD8+ T cells lie within a differentiation continuum. This includes central memory cells, cells of intermediate differentiation, and finally, terminally differentiated effector memory cells (28, 29). Using the differentiation markers CD62L and KLRG1, these three types of memory CD8+ T cell populations can be readily identified after an acute LCMV infection (Fig 3A). Because our studies revealed a critical role for core 2 O-glycan synthesis in controlling the trafficking of memory CD8+ T cells into inflamed tissues, we next investigated whether core 2 O-glycan synthesis was restricted to specific subsets. Interestingly, the TCM CD8+ T cell subset (CD62L+/KLRG1−) expressed more core 2 O-glycans compared to effector memory (CD62L−) populations (Fig 3A,B). Memory CD8+ T cells that did not express CD62L (i.e. TEM) could be further classified based on expression of KLRG1. Notably, essentially no KLRG1+ TEM CD8+ T cells expressed core 2 O-glycans (Fig 3B). Expression of the IL-2/15Rβ (CD122) within each of the subsets correlated with the percentage of memory CD8+ T cells that expressed core 2 O-glycans (Fig 3C), suggesting that sensitivity to IL-15 is an important regulator of core 2 O-glycan synthesis within the memory CD8+ T cell compartment. In fact, TCM CD8+ T cells from the circulation could bind to both E- and P-selectin, whereas most TEM do not (Fig 3D,E).
Figure 3. Core 2 O-glycan synthesis in memory CD8+ T cell subsets.
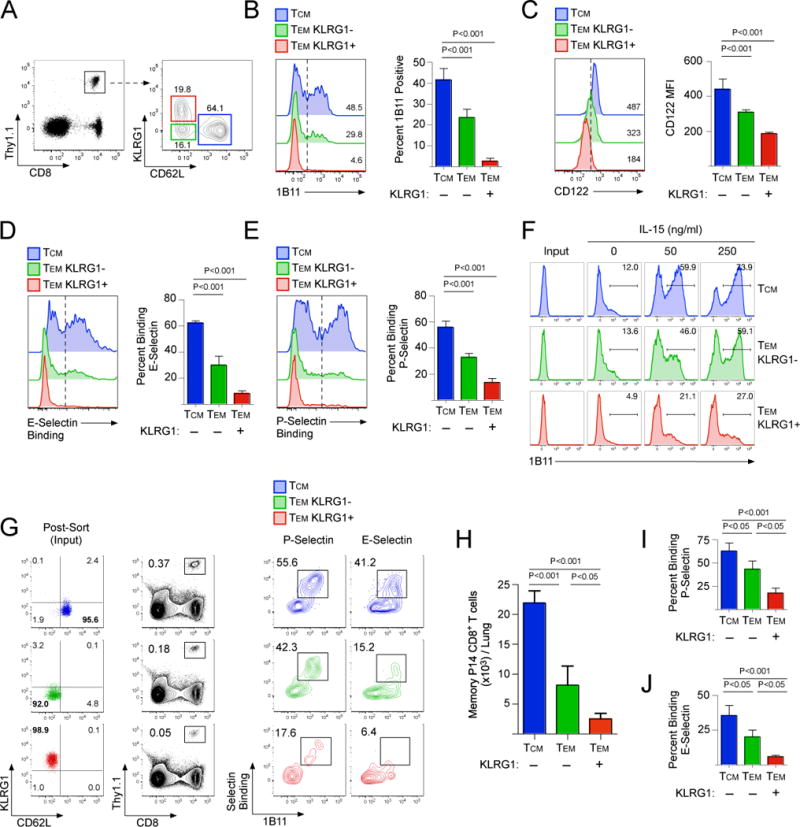
(A) Memory P14 CD8+ T cells were analyzed 75 days post-LCMV infection for expression of CD62L and KLRG1. (B-E) Memory P14 CD8+ T cells subsets from (A) were analyzed for expression of (B) core 2 O-glycans (1B11), (C) CD122, (D) E-selectin binding, and (E) P-selectin binding. (F) 1B11- cells from each subset as shown in (A) were sorted by FACS and cultured in vitro for 3 days in media alone or supplemented with 50 or 250 ng/ml of IL-15. (G) Memory CD8+ T cell subsets were sorted and 5 × 105 were transferred into B6 mice and challenged with CpG intranasally. Recruitment into the challenged lung and binding of E- and P-selectin was analyzed at 60 hours post-challenge. (H-J) Quantification of data shown in (G)
To determine whether core 2 O-glycan synthesis was fixed or could be stimulated on either TCM or TEM, we sorted memory CD8+ T cells that were not expressing core 2 O-glycans (1B11-) from each of the three subsets and cultured them in vitro with IL-15. TCM strongly increased core 2 O-glycan synthesis as measured by a de novo increase in 1B11 reactivity, but most KLRG1+ TEM CD8+ T cells could not (Fig 3F). Consistent with previous findings (30), stimulation of TCM with IL-15 did not alter expression of CD62L or KLRG1, but did reduce expression of CCR7 (Fig S5A). Finally, TCM isolated from lymph nodes also synthesized core 2 O-glycans and bound to E- and P-selectin following stimulation with IL-15 (Fig S5B). Therefore, these data demonstrate that core 2 O-glycan synthesis is largely restricted to the TCM subset and can be stimulated by IL-15.
It has generally been accepted that TEM are the memory CD8+ T cells that are recruited into non-lymphoid tissues in response to inflammation. However, because we found that expression of core 2 O-glycans was primarily a feature of TCM CD8+ T cells and not TEM, we next tested whether these subsets would traffic differently into non-lymphoid tissue following a sterile inflammatory challenge. Each population (TCM, KLRG1- TEM, and KLRG1+ TEM) was sorted and transferred into naïve B6 mice (Fig 3G) and then challenged intranasally with the TLR9 agonist CpG. TCM CD8+ T cells trafficked into the inflamed lung mucosa better than either of the TEM subsets (KLRG1+/−) (Fig 3G,H). Furthermore, TCM that trafficked into the lung expressed core 2 O-glycans and bound to both P- and E-selectin better than either of the TEM subsets (Fig 3I,J). Collectively, these data demonstrate that within the memory CD8+ T cell compartment, core 2 O-glycan expressing TCM are the major subset that infiltrates non-lymphoid tissue in response to inflammatory challenge.
TCM CD8+ T cells possess more core 2 O-glycan synthesis activity than terminally differentiated TEM cells
The proportions of TCM and TEM CD8+ T cells that form following infection or vaccination can be influenced by many variables (31). For example, high levels of inflammatory cytokines (e.g. IL-12, IFNα/β) and multiple rounds of antigen encounter are known to cause memory CD8+ T cells to differentiate largely into KLRG1+ TEM. Using a previously described method of generating primary or tertiary memory CD8+ T cells with a single or three consecutive LCMV infections (10, 32) (Fig S6A), we generated memory P14 CD8+ T cell populations that became predominantly TCM (primary) or KLRG1+ TEM (tertiary) (Fig 4A, Fig S6B–D). In agreement with Fig 3D,E, terminally differentiated tertiary TEM CD8+ T cells exhibited limited capacity to bind to E- and P-selectin compared to primary memory CD8+ T cells (Fig 4B–D), but expressed similar levels of proteins that function as selectin ligands (e.g. PSGL-1, CD44, and CD43) (Fig S6E) suggesting that the difference in the generation of E- and P-selectin ligands occurs at the level of post-translational glycosylation. Indeed, tertiary memory CD8+ T cells expressed less CD122 and the common γ chain, CD132 (Fig 4E–G). Jacalin, a lectin that binds core 1 O-glycans regardless of sialylation status, bound to both primary and tertiary memory CD8+ T cells; demonstrating that similar levels of total core 1 O-glycans were present. In contrast, PNA reacted more strongly on primary memory CD8+ T cells, consistent with our finding that sensitivity to IL-15 can alter the sialylation state of the core 1 O-glycan substrate (Fig 4H–J).
Figure 4. Memory CD8+ T cells that become terminally differentiated by multiple antigen encounters lose core 2 O-glycan synthesis activity.

(A) Expression of CD62L and KLRG1 on primary and tertiary memory P14 CD8+ T cells 90 days post-LCMV infection and (B) binding to E- and P-selectin. (C,D) Quantification of (B). (E) Expression of CD122 and CD132. (F,G) Quantification of (E). (H) Expression of total core 1 O-glycans (Jacalin) and unsialylated core 1 O-glycans (PNA). (I,J) Quantification of (H). (K) Primary and tertiary memory P14 CD8+ T cells were purified and stimulated with the indicated concentration of IL-15 for 15 minutes and phosphorylation of STAT5 (Y694) was analyzed by immunoblot. (L) Primary and tertiary memory CD8+ T cells were purified and cultured with IL-15 for 3 days. Binding of MAL II, PNA and the 1B11 antibody was analyzed. (M) Same as (L) except the glycosylation of PSGL-1 was analyzed by immunoblot. (N) Same as (L) except binding to E-selectin was analyzed by flow cytometry. (O) Quantification of (N) from 4 independent experiments. (P,Q) Same as (N,O) except for P-selectin.
Stimulation through the IL-15 receptor activates a canonical Jak/STAT signaling pathway, resulting in the activation of the STAT5 transcription factor by tyrosine phosphorylation. Primary memory CD8+ T cells activated STAT5 better than tertiary memory CD8+ T cells when stimulated with IL-15 (Fig 4K). Furthermore, as we had shown previously (Fig 2F), IL-15 caused primary memory CD8+ T cells to decrease MAL II and increase PNA and 1B11 reactivity, but had no affect on the O-linked glycosylation status of tertiary memory CD8+ T cells (Fig 4L). PSGL-1 functions as both a P- and E-selectin ligand and stimulation of primary memory CD8+ T cells with IL-15 caused PSGL-1 to become glycosylated, indicated by slower migration of the glycosylated version of PSGL-1 following separation by SDS-PAGE (33). PSGL-1 was mostly in a non-glycosylated form in tertiary memory CD8+ T cells and stimulation with IL-15 did not change its glycosylation state (Fig 4M). Finally, when stimulated with IL-15, primary memory CD8+ T cells significantly increased binding to both E- and P-selectin, but tertiary memory CD8+ T cells did not (Fig 4N–Q). Collectively, these findings demonstrate that core 2 O-glycan synthesis is highly active in TCM CD8+ T cells that form following a primary infection, but not in terminally differentiated KLRG1+ TEM CD8+ T cells.
Primary TCM CD8+ T cells traffic into inflamed, non-lymphoid tissues better than terminally differentiated tertiary KLRG1+ TEM cells
Our analysis of IL-15 stimulated core 2 O-glycan synthesis of memory CD8+ T cell subsets suggested that tertiary memory CD8+ T cells generated through multiple antigen-specific infections would be impaired in trafficking, similar to the small percentage of KLRG1+ TEM that can be detected following a single, acute LCMV infection (Fig 3A). To directly determine in a competitive manner whether primary or tertiary memory CD8+ T cells trafficked better to the lung mucosa during an inflammatory event, we transferred equal number of primary (Thy1.1/1.2) and tertiary (Thy1.1/1.1) memory P14 CD8+ T cells into naïve B6 mice (Thy1.2/1.2), which were then challenged intranasally with CpG (Fig 5A). Primary memory CD8+ T cells trafficked to the inflamed lung mucosa better than tertiary memory CD8+ T cells (Fig 5B–D). In addition, more primary memory CD8+ T cells that trafficked to the CpG-treated lungs expressed core 2 O-glycans and bound to both E- and P-selectin than tertiary memory CD8+ T cells (Fig 5E–H). Indeed, these data agree with Fig 3G–J, demonstrating that TCM CD8+ T cells that are formed following a primary infection are recruited to the inflamed lung mucosa better than tertiary KLRG1+ TEM cells.
Figure 5. Primary memory CD8+ T cells traffic into the inflamed lung mucosa better than tertiary memory.
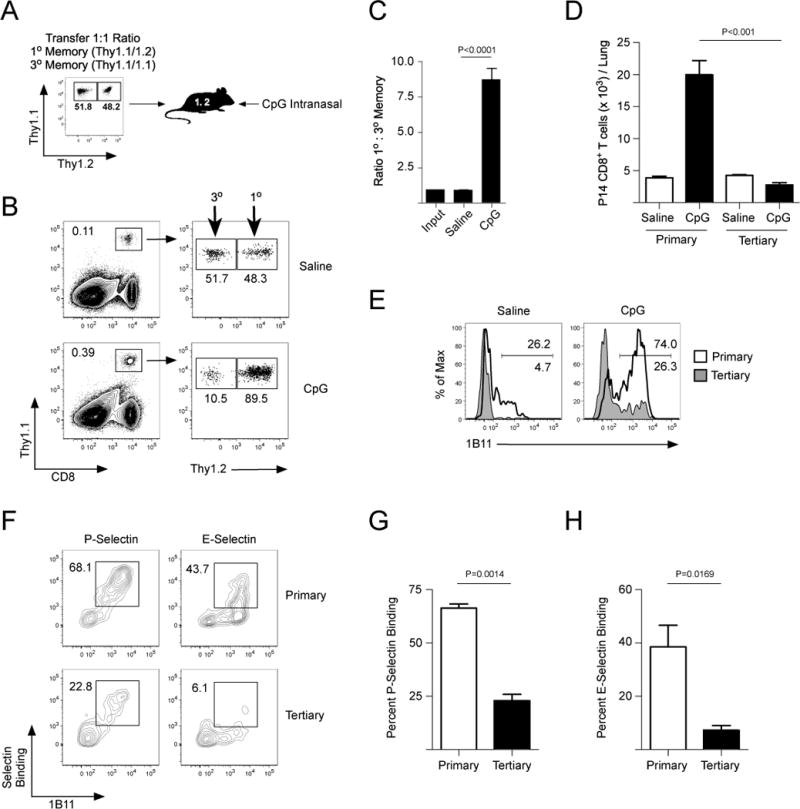
(A) Primary (Thy1.1/1.2) and tertiary (Thy1.1/1.1) memory P14 CD8+ T cells were purified and equal numbers were transferred into naïve B6 (Thy1.2/1.2) mice. (B) Mice from (A) were challenged with saline or CpG and recruitment to the lung was analyzed 60 hours post-challenge. (C,D) Quantification of (B). (E) Expression of core 2 O-glycans (1B11) from (B). (F) Cells from (B) were analyzed for binding to P- and E-selectin. (G,H) Quantification of (F).
Trafficking of memory CD8+ T cells into VacV-infected skin is also dependent on core 2 O-glycan synthesis and E- and P-selectin (7). To analyze memory CD8+ T cell trafficking during this infection without antigen influence, mice containing equal frequencies of either primary or tertiary memory P14 CD8+ T cells (Fig 6A) were infected on the ear skin with VacV-OVA. Primary memory GP33-specific P14 CD8+ T cells rapidly trafficked into the VacV-OVA infected skin, but significantly fewer tertiary memory CD8+ T cells reached the site of infection (Fig 6B,C). Expression of core 2 O-glycans was highly enriched on primary memory CD8+ T cells that trafficked into the VacV-OVA infected skin compared to the same antigen-specific cells in the circulation (Fig 6D). Fewer tertiary memory CD8+ T cells expressed core 2 O-glycans in the circulation and in the infected ear skin (Fig 6D), resulting in less binding to P- and E-selectin (Fig 6E–G). Next, to determine whether differential trafficking affected protective immunity, we purified primary and tertiary memory P14 CD8+ T cells and transferred them into naïve B6 mice, which were then infected on the skin with VacV-GP33. Primary memory CD8+ T cells rapidly trafficked into the skin and provided protection against the infection (Fig 6H,I). In contrast, limited numbers of tertiary memory P14 CD8+ T cells trafficked into the infected skin and were unable to provide significant protective immunity (Fig 6H,I). Therefore, these data demonstrate that the capacity to synthesize core 2 O-glycans that function as P- and E-selectin ligands is essential for memory CD8+ T cells to leave the circulation and traffic into skin to provide protective immunity.
Figure 6. Primary memory CD8+ T cells traffic into VacV-infected skin and provide protective immunity against viral infection.
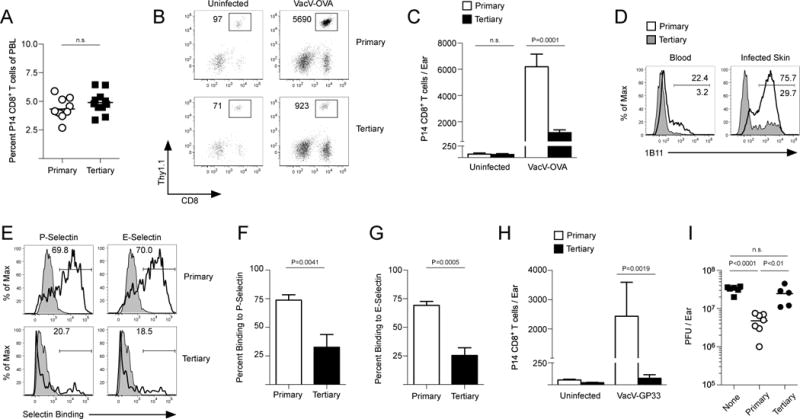
(A) Frequencies of primary and tertiary memory P14 CD8+ T cells in individual B6 mice. (B) Mice containing equal frequencies of either primary or tertiary memory CD8+ T cells were infected with VacV-OVA on the left ear skin and recruitment of memory GP33-specific P14 CD8+ T cells was analyzed on day 3 post-infection. (C) Quantification of (B). (D) Expression of core 2 O-glycans (1B11) on primary and tertiary memory P14 CD8+ T cells in the blood and VacV-infected skin. (E) Binding to P- and E-selectin on P14 CD8+ T cells isolated from the skin. (F,G) Quantification of (E). (H) Primary and tertiary memory P14 CD8+ T cells were purified from spleens and 2 × 106 were transferred into naïve B6 mice and the left ear skin was infected with VacV-GP33. Recruitment of memory P14 CD8+ T cells into the infected and control skin was analyzed on day 3 post-infection. (I) Same as (H) except viral load was measured by standard plaque assay on day 4 post-infection.
Finally, we tested whether these findings could be generalized to another form of skin inflammation. Sensitization and subsequent local challenge with the chemical hapten DNFB causes contact hypersensitivity, a type of inflammation mediated by activated T cells. In fact, more memory CD8+ T cells are recruited to DNFB challenged skin in sensitized mice compared to mice that were not sensitized (Fig S7A,B). We initiated contact hypersensitivity on LCMV-immune mice containing equal frequencies of circulating primary or tertiary memory P14 CD8+ T cells. Primary memory CD8+ T cells trafficked to the DNFB-challenged skin better than tertiary memory CD8+ T cells and expressed more core 2 O-glycans (Fig S7C–F). Thus, in three different models of inflammation, primary memory CD8+ T cells traffic better than tertiary memory cells to inflamed non-lymphoid tissue.
Core 2 O-glycan synthesis is active in conventional, but not inflationary MCMV-specific memory CD8+ T cells
Murine cytomegalovirus (MCMV) is a β-herpes virus that, following a robust acute infection, establishes life-long latency in the host. Because our studies using acute LCMV infections demonstrated that TCM CD8+ T cells actively synthesize core 2 O-glycans, whereas KLRG1+ TEM CD8+ T cells did not, we next assessed core 2 O-glycan synthesis and tissue-trafficking of endogenous MCMV-specific memory CD8+ T cells. In contrast to most acute infections, two major types of antigen-specific CD8+ T cell responses occur in mice infected with MCMV(34). ‘Conventional’ CD8+ T cells (M57- and M45-specific) undergo typical expansion and contraction, similar to what is found following most acute infections, and give rise to a memory population that is primarily TCM and KLRG1- TEM (Fig 7A–C). In contrast, a separate group of antigen-specific CD8+ T cells (M38- and m139-specific) become ‘inflationary’ and continue to expand after MCMV has entered the latent stage of the infection (Fig 7A) and become predominantly KLRG1+ TEM (Fig 7B,C) (35). A larger percentage of conventional memory CD8+ T cells in the circulation expressed core 2 O-glycans compared to the inflationary CD8+ T cells (Fig 7D). Furthermore, IL-15 stimulation caused the conventional memory CD8+ T cells to synthesize core 2 O-glycans, but not the inflationary MCMV-specific CD8+ T cells (Fig 7E). These data are consistent with our findings using LCMV infections, demonstrating that terminally differentiated KLRG1+ TEM CD8+ T cells lose the capacity to synthesize core 2 O-glycans.
Figure 7. Inflationary KLRG1+ TEM MCMV-specific CD8+ T cells lose core 2 O-glycan synthesis.
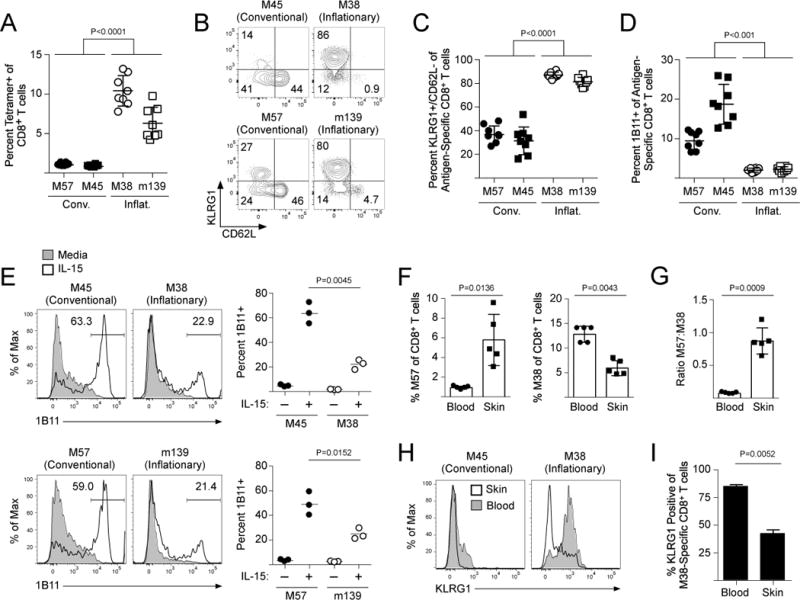
(A) Naïve B6 mice were infected with MCMV. On day 120 post-infection, the frequency of MCMV-specific CD8+ T cells was quantified by MCMV-specific MHC class I tetramers. (B,C) Expression of CD62L and KRLG1 on MCMV-specific CD8+ T cells from (A). (D) Core 2 O-glycan expression (1B11) from (A). (E) CD8+ T cells of MCMV-infected mice were purified and stimulated in vitro with 250 ng/mL IL-15 for 3 days. Expression of core 2 O-glycans (1B11) was analyzed. (F) MCMV-infected mice were infected with VacV on the left ear skin. Frequency of M57-specific and M38-specific CD8+ T in the blood and skin on day 5 post-VacV infection. (G) Quantification of trafficking efficiency from (F). (H) Same as (F). Expression of KLRG1 on conventional (M45-specific) and inflationary (M38-specific) CD8+ T cells from the blood and the skin following VacV infection. (I) Quantification of (H).
To test whether there were differences in the trafficking of these two types of memory CD8+ T cells, we challenged mice latently infected with MCMV with VacV on the skin. The frequency of conventional, M57-specific memory CD8+ T cell frequency in the skin increased compared to the circulation, whereas the frequency of inflationary M38-specific CD8+ T cells in the skin was lower than in the blood (Fig 7F). Because M57- and M38-specific CD8+ T cells were analyzed in the same animal, we could quantify the relative frequencies of these two antigen-specific cell types in the circulation compared to those that trafficked into the skin. This revealed that M57-specific CD8+ T cells infiltrated the VacV-infected skin nearly 10 times better than inflationary M38-specific CD8+ T cells (Fig 7G). Notably, most of the inflationary M38-specific CD8+ T cells that trafficked into the VacV-infected skin were actually KLRG1-, even though most of these antigen-specific cells in the circulation were KLRG1+ (Fig 7H,I). Collectively, these data demonstrate that inflationary MCMV-specific CD8+ T cells that become KLRG1+ TEM exhibit limited core 2 O-glycan synthesis activity, thereby excluding most of them from entering non-lymphoid tissues during a local inflammatory event.
DISCUSSION
For nearly two decades, the central and effector memory T cell paradigm has been the foundation for describing memory T cell trafficking potentials, even though studies directly testing the trafficking of defined memory T cell subsets have not been rigorously performed. To further complicate matters, the recent identification of tissue-resident memory (TRM) CD8+ T cells (36) presents the possibility that previous, classic studies that identified effector memory CD8+ T cells in non-lymphoid tissues (9, 37) could have actually been observing this newly described subset, not TEM actively patrolling non-lymphoid tissues. In fact, it has recently been reported that circulating T cells exhibiting a TCM phenotype can be identified in human skin (38), demonstrating that TCM are not confined to the circulation and lymph nodes, but likely traffic into non-lymphoid tissues. A recent study by Gerlach et al found that the TCM/TEM classification also does not accurately predict the homeostatic trafficking patterns of memory CD8+ T cells (39). Notably, the authors found that CX3CR1Hi/CD62LLo/CD27Lo TEM CD8+ T cells were not present in lymph collected from the thoracic duct, suggesting that these cells were, in fact, confined to the circulation. Here, using models of infection and inflammation, we show the importance of core 2 O-glycan synthesis activity in regulating the tissue trafficking of memory CD8+ T cells, which we found is predominantly a feature of the TCM subset.
We have recently shown that core 2 O-glycan synthesis is highly dynamic and occurs independent of antigen re-stimulation in memory CD8+ T cells (7). The capacity for memory, but not naïve CD8+ T cells to synthesize core 2 O-glycans was attributed to epigenetic changes of Gcnt1. Interestingly, essentially all memory CD8+ T cells are also reactive to the lectin PNA (40), which identifies unsialylated core 1 O-glycans. Because sialylation of core 1 O-glycans prevents core 2 O-glycan synthesis, expression or function of St3gal1 may also be actively suppressed in long-lived memory cells. These findings support the model that, following activation, antigen-experienced memory CD8+ T cells “unlock” core 2 O-glycan synthesis through epigenetic mechanisms, which is then ultimately controlled by external inflammatory factors. Although our experiments indicate a clear role for IL-15 in both the maintenance and the induction of core 2 O-glycans, other activators of STAT5 (e.g. IL-2, IL-7, IL-21) could certainly stimulate core 2 O-glycan synthesis during some infections or pathological conditions. Importantly, IL-15 controls the homeostatic proliferation of TCM, but also the persistence of TEM CD8+ T cells (10, 41). Thus, we cannot conclusively rule out that there may be reduced survival of KLRG1+ TEM compared to TCM after they traffic into non-lymphoid tissues. However, IL-15-driven homeostatic proliferation could be one of the mechanisms that maintains core 2 O-glycans on TCM CD8+ T cells in the circulation, and potentially, these are the memory T cells that infiltrate non-lymphoid tissues in response to local inflammation.
In summary, our studies here show that core 2 O-glycan synthesis is highly active in TCM, but diminishes as memory CD8+ T cells become terminally differentiated, which essentially excludes them from entering non-lymphoid tissues in response to inflammation. Of note, the term “tertiary memory” in this study refers to antigen-specific memory CD8+ T cells that have been activated using three consecutive LCMV infections, generating a nearly homogenous KLRG1+ TEM population of memory T cells. As mentioned previously, a number of factors influence the proportions of TCM and TEM CD8+ T cells that form following infection or vaccination. Although most studies have found that repeated, antigen-specific challenges give rise to predominantly KLRG1+ TEM CD8+ T cells (12, 32, 42), there are certainly exceptions and in some instances, can generate memory CD8+ T cells with a lower proportion of KLRG1+ TEM compared to a primary infection (43). Nevertheless, our findings demonstrate that memory CD8+ T cell differentiation into TCM and TEM ultimately dictates their trafficking potential and thus, understanding the mechanisms that shape the composition of memory T cell populations remain critical for improving host defense and vaccine design against specific pathogens.
MATERIALS AND METHODS
Study Design
The goal of this study was to identify which memory CD8+ T cells trafficked into non-lymphoid tissues in response to local inflammation. We used adoptive transfers of TCR-transgenic T cells and LCMV infections to generate diverse populations of memory CD8+ T cells in mice. Phenotyping and ex vivo cytokine stimulation assays were used to investigate the mechanisms regulating core 2 O-glycan synthesis and to identify which T cell subsets could actively synthesize core 2 O-glycans. To quantify trafficking of memory CD8+ T cells in vivo, LCMV-immune mice were 1) infected on the skin with VacV, 2) challenged intranasally with the TLR9 agonist CpG, or 3) subjected to contact hypersensitivity with DNFB. All experiments were performed two or more times.
Mice and Viral Infections
C57BL/6J mice were purchased from Jackson Laboratories and used for experiments at 6-10 weeks of age. Gcnt1−/− mice (44) and P14 TCR-transgenic mice (45) have been previously described. For adoptive transfers, 1.0 – 2.5 × 104 naïve Thy1.1+ P14 CD8+ T cells from either blood or spleen were injected intravenously in 200 μL of PBS. LCMV-Armstrong was injected i.p (2 × 105 PFU). Vaccinia virus (VacV) expressing GP33-41 (VacV-GP33) and ovalbumin257-264 (VacV-OVA) have been previously described (46, 47). Infections with VacV were performed on anesthetized mice by placing 5 × 106 PFU of virus in 10 μl of PBS on the ventral side of the ear pinna and then poking the virus coated skin 25 times with a 27G needle. Murine cytomegalovirus (MCMV) strain MW97.01 was injected i.p (2 × 105 PFU). All animal experiments and infectious agents were approved by the OHSU Institutional Animal Care and Use Committee and Institutional Biosafety Committee.
Inflammatory Challenges and IL-15 Neutralization
Intranasal challenge with CpG was performed by applying 50 nmoles of CpG ODN 1826 (Integrative DNA Technology) in saline onto the nares of anesthetized mice (7). To induce contact hypersensitivity, the shaved abdomens of LCMV-immune mice were sensitized with 0.5% DNFB (Sigma) in 4:1 acetone:olive oil. On day 5 post-sensitization, the ear skin was challenged with 20 μl of 0.1% DNFB in acetone:olive oil. IL-15 neutralizing antibody (clone M96) was kindly provided by Amgen. To neutralize IL-15, 200 μg of anti-IL-15 antibody or control rat IgG (Sigma) was administered every other day for 10 days by i.p injection in 200 μl of saline. Blocking antibodies against E-selectin (clone 9A9) and P-selectin (clone RB40.34) were purified from hybridomas and 250 μg was injected i.p.
Leukocyte Isolation from Skin or Lung
Ears from infected mice were removed and the dorsal and ventral sides of the ear pinna were separated and allowed to incubate for 30-60 minutes at 37°C with 1-2 ml HBSS (Gibco) containing CaCl2 and MgCl2 supplemented with 125 U/ml collagenase (Invitrogen) and 60 U/ml DNase-I (Sigma-Aldrich) at 37°C. To isolate leukocytes from the lung following CpG challenge, mice were anesthetized with ketamine/xylazine followed by whole body perfusion with 10 mls of PBS injected into the left ventricle of the heart. For skin and lung, whole tissue suspensions were generated by gently forcing the tissue through a wire mesh screen. Leukocytes were then purified from whole tissue suspensions by re-suspending the cells in 10 mL of 35% Percoll (GE Healthcare)/HBSS in 50 mL conical tubes followed by centrifugation (500 × g) for 10 minutes at room temp.
Lectin and Selectin Binding
Fluorescein or biotin-conjugated Jacalin, Peanut Agglutinin (PNA), and MAL II (VectorLabs) were incubated with cells for 30 minutes in 1% FBS/PBS at room temperature. Binding of biotin-conjugated lectins was detected with FITC-Streptavidin (BioLegend). E-selectin (5 μg/ml) and P-selectin (1.5 μg/ml) human IgG Fc chimeric proteins (R & D Systems) were incubated with cells for 30 minutes in 1% FBS/DPBS containing Ca2+ and Mg2+ (Gibco) at room temperature. Binding of selectins was detected using anti-human IgG-Fc PE (eBioscience). Cells were then stained with fluorescent antibodies as described in Flow Cytometry and Antibodies.
Statistical Analysis
Statistical analyses were performed with Prism software (version 6.0, GraphPad Software) using either the paired or unpaired Student’s t test or ANOVA with Tukey’s post-test for significance.
Supplementary Material
Figure S1: Trafficking of memory CD8+ T cells into VacV-infected skin.
Figure S2: CD8+ T cells require Gcnt1 to synthesize core 2 O-glycans for binding to E- and P-selectin.
Figure S3: Trafficking of memory CD8+ T cells into VacV-infected skin requires interactions with E- and P-selectin.
Figure S4: IL-15 regulates E- and P-selectin binding during VacV infection.
Figure S5: Central memory CD8+ T cells synthesize core 2 O-glycans and maintain expression of CD62L.
Figure S6: Phenotype of terminally differentiated tertiary memory CD8+ T cells using repetitive LCMV-Armstrong infection.
Figure S7: Primary memory CD8+ T cells are recruited into the skin better than tertiary memory CD8+ T cells during contact hypersensitivity.
Table S1: Primer pairs for gene expression analysis in Figure 2 and Supplemental Figure 4.
Table S2: Raw data sets and statistical analyses
One Sentence Summary.
Core 2 O-glycan expressing TCM CD8+ T cells are the major memory CD8+ T cell subset that traffics into inflamed, non-lymphoid tissues.
Acknowledgments
We thank David Parker for critical review of the manuscript, Hermann Ziltener and Doug Carlow (University of British Columbia) for providing Gcnt1−/− mice, and Amgen for IL-15 neutralizing antibody.
Funding: Supported by NIH grants K22-AI102981 to JCN, R01-AI47206 to ABH, and R01-AI42767 to JTH.
Footnotes
Author Contributions: JFO, JLM, SJH, and JCN designed and performed experiments, analyzed data and performed statistical analyses. CB performed experiments. MWM, ABH, and JTH provided consultation in experimental design and interpretation of the data and contributed intellectually to the project. JCN wrote the manuscript.
The authors declare that they have no competing interests.
References
- 1.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nature Reviews Immunology. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 2.Nolz JC, Starbeck-Miller GR, Harty JT. Naive, effector and memory CD8 T-cell trafficking: parallels and distinctions. Immunotherapy. 2011;3:1223–1233. doi: 10.2217/imt.11.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ely KH, et al. Nonspecific recruitment of memory CD8+ T cells to the lung airways during respiratory virus infections. Journal of Immunology. 2003;170:1423–1429. doi: 10.4049/jimmunol.170.3.1423. [DOI] [PubMed] [Google Scholar]
- 4.Gebhardt T, et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. 2011;477:216–219. doi: 10.1038/nature10339. [DOI] [PubMed] [Google Scholar]
- 5.Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nature Reviews Immunology. 2009;9:153–161. doi: 10.1038/nri2496. [DOI] [PubMed] [Google Scholar]
- 6.Kohlmeier JE, et al. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity. 2008;29:101–113. doi: 10.1016/j.immuni.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nolz JC, Harty JT. IL-15 regulates memory CD8+ T cell O-glycan synthesis and affects trafficking. The Journal of Clinical Investigation. 2014;124:1013–1026. doi: 10.1172/JCI72039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 9.Wherry EJ, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nature Immunology. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 10.Wirth TC, et al. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity. 2010;33:128–140. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bottcher JP, et al. Functional classification of memory CD8(+) T cells by CX3CR1 expression. Nat Commun. 2015;6:8306. doi: 10.1038/ncomms9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jabbari A, Harty JT. Secondary memory CD8+ T cells are more protective but slower to acquire a central-memory phenotype. The Journal of Experimental Medicine. 2006;203:919–932. doi: 10.1084/jem.20052237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olson JA, McDonald-Hyman C, Jameson SC, Hamilton SE. Effector-like CD8(+) T cells in the memory population mediate potent protective immunity. Immunity. 2013;38:1250–1260. doi: 10.1016/j.immuni.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bachmann MF, Wolint P, Schwarz K, Jager P, Oxenius A. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. Journal of Immunology. 2005;175:4686–4696. doi: 10.4049/jimmunol.175.7.4686. [DOI] [PubMed] [Google Scholar]
- 15.Ley K. The role of selectins in inflammation and disease. Trends Mol Med. 2003;9:263–268. doi: 10.1016/s1471-4914(03)00071-6. [DOI] [PubMed] [Google Scholar]
- 16.Sperandio M, Gleissner CA, Ley K. Glycosylation in immune cell trafficking. Immunological Reviews. 2009;230:97–113. doi: 10.1111/j.1600-065X.2009.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hobbs SJ, Nolz JC. Regulation of T Cell Trafficking by Enzymatic Synthesis of O-Glycans. Front Immunol. 2017;8:600. doi: 10.3389/fimmu.2017.00600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlow DA, Corbel SY, Williams MJ, Ziltener HJ. IL-2, -4, and -15 differentially regulate O-glycan branching and P-selectin ligand formation in activated CD8 T cells. Journal of Immunology. 2001;167:6841–6848. doi: 10.4049/jimmunol.167.12.6841. [DOI] [PubMed] [Google Scholar]
- 19.Beura LK, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532:512–516. doi: 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlow DA, Ardman B, Ziltener HJ. A novel CD8 T cell-restricted CD45RB epitope shared by CD43 is differentially affected by glycosylation. Journal of Immunology. 1999;163:1441–1448. [PubMed] [Google Scholar]
- 21.van Die I, et al. The acceptor substrate specificity of human beta4-galactosyltransferase V indicates its potential function in O-glycosylation. FEBS Lett. 1999;450:52–56. doi: 10.1016/s0014-5793(99)00462-7. [DOI] [PubMed] [Google Scholar]
- 22.Yeh JC, et al. Novel sulfated lymphocyte homing receptors and their control by a Core1 extension beta 1,3-N-acetylglucosaminyltransferase. Cell. 2001;105:957–969. doi: 10.1016/s0092-8674(01)00394-4. [DOI] [PubMed] [Google Scholar]
- 23.Priatel JJ, et al. The ST3Gal-I sialyltransferase controls CD8+ T lymphocyte homeostasis by modulating O-glycan biosynthesis. Immunity. 2000;12:273–283. doi: 10.1016/s1074-7613(00)80180-6. [DOI] [PubMed] [Google Scholar]
- 24.Geisler C, Jarvis DL. Effective glycoanalysis with Maackia amurensis lectins requires a clear understanding of their binding specificities. Glycobiology. 2011;21:988–993. doi: 10.1093/glycob/cwr080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richer MJ, et al. Inflammatory IL-15 is required for optimal memory T cell responses. The Journal of Clinical Investigation. 2015;125:3477–3490. doi: 10.1172/JCI81261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity. 2012;37:549–562. doi: 10.1016/j.immuni.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lebrec H, et al. Homeostasis of human NK cells is not IL-15 dependent. Journal of Immunology. 2013;191:5551–5558. doi: 10.4049/jimmunol.1301000. [DOI] [PubMed] [Google Scholar]
- 28.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nature Reviews Immunology. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36:142–152. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gattinoni L, et al. A human memory T cell subset with stem cell-like properties. Nature Medicine. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nature Reviews Immunology. 2008;8:107–119. doi: 10.1038/nri2251. [DOI] [PubMed] [Google Scholar]
- 32.Nolz JC, Harty JT. Protective capacity of memory CD8+ T cells is dictated by antigen exposure history and nature of the infection. Immunity. 2011;34:781–793. doi: 10.1016/j.immuni.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore KL, et al. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. The Journal of Cell Biology. 1995;128:661–671. doi: 10.1083/jcb.128.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munks MW, et al. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. Journal of Immunology. 2006;177:450–458. doi: 10.4049/jimmunol.177.1.450. [DOI] [PubMed] [Google Scholar]
- 35.Kuijpers TW, et al. Frequencies of circulating cytolytic, CD45RA+CD27-, CD8+ T lymphocytes depend on infection with CMV. Journal of Immunology. 2003;170:4342–4348. doi: 10.4049/jimmunol.170.8.4342. [DOI] [PubMed] [Google Scholar]
- 36.Carbone FR. Tissue-Resident Memory T Cells and Fixed Immune Surveillance in Nonlymphoid Organs. Journal of Immunology. 2015;195:17–22. doi: 10.4049/jimmunol.1500515. [DOI] [PubMed] [Google Scholar]
- 37.Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 38.Watanabe R, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Science Translational Medicine. 2015;7:279ra239. doi: 10.1126/scitranslmed.3010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerlach C, et al. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity. 2016;45:1270–1284. doi: 10.1016/j.immuni.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galvan M, Murali-Krishna K, Ming LL, Baum L, Ahmed R. Alterations in cell surface carbohydrates on T cells from virally infected mice can distinguish effector/memory CD8+ T cells from naive cells. Journal of Immunology. 1998;161:641–648. [PubMed] [Google Scholar]
- 41.Sandau MM, Kohlmeier JE, Woodland DL, Jameson SC. IL-15 regulates both quantitative and qualitative features of the memory CD8 T cell pool. Journal of Immunology. 2010;184:35–44. doi: 10.4049/jimmunol.0803355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masopust D, Ha SJ, Vezys V, Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. Journal of Immunology. 2006;177:831–839. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- 43.Davies B, et al. Cutting Edge: Tissue-Resident Memory T Cells Generated by Multiple Immunizations or Localized Deposition Provide Enhanced Immunity. Journal of Immunology. 2017;198:2233–2237. doi: 10.4049/jimmunol.1601367. [DOI] [PubMed] [Google Scholar]
- 44.Ellies LG, et al. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. 1998;9:881–890. doi: 10.1016/s1074-7613(00)80653-6. [DOI] [PubMed] [Google Scholar]
- 45.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 46.Restifo NP, et al. Antigen processing in vivo and the elicitation of primary CTL responses. Journal of Immunology. 1995;154:4414–4422. [PMC free article] [PubMed] [Google Scholar]
- 47.Oldstone MB, et al. Vaccination to prevent persistent viral infection. Journal of Virology. 1993;67:4372–4378. doi: 10.1128/jvi.67.7.4372-4378.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Trafficking of memory CD8+ T cells into VacV-infected skin.
Figure S2: CD8+ T cells require Gcnt1 to synthesize core 2 O-glycans for binding to E- and P-selectin.
Figure S3: Trafficking of memory CD8+ T cells into VacV-infected skin requires interactions with E- and P-selectin.
Figure S4: IL-15 regulates E- and P-selectin binding during VacV infection.
Figure S5: Central memory CD8+ T cells synthesize core 2 O-glycans and maintain expression of CD62L.
Figure S6: Phenotype of terminally differentiated tertiary memory CD8+ T cells using repetitive LCMV-Armstrong infection.
Figure S7: Primary memory CD8+ T cells are recruited into the skin better than tertiary memory CD8+ T cells during contact hypersensitivity.
Table S1: Primer pairs for gene expression analysis in Figure 2 and Supplemental Figure 4.
Table S2: Raw data sets and statistical analyses