

Abstract
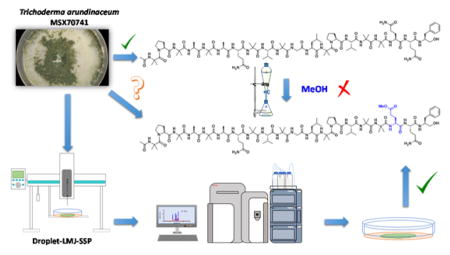
In the field of natural products chemistry, a common question pertains to the authenticity of an isolated compound, i.e. are the interesting side chains biosynthesized naturally or an artefact of the isolation/purification processes? The droplet-liquid microjunction-surface sampling probe (droplet-LMJ-SSP) coupled to a hyphenated system (UPLC-UV-HRESIMS) empowers the analysis of natural product sources in situ, providing data on the biosynthetic timing and spatial distribution of secondary metabolites. In this study the droplet-LMJ-SSP was utilized to validate the authenticity of two new peptaibols (2 and 3) as biosynthesized secondary metabolites, even though both them had structural features that could be perceived as artefacts. Compounds 2 and 3 were isolated from the scaled up fermentation of Trichoderma arundinaceum (strain MSX70741), along with a new member of the trichobrevin BIII complex (1), and four known compounds (4–7). The structures of the isolates were established using a set of spectroscopic and spectrometric methods, and their absolute configurations were determined by Marfey’s analysis. The cytotoxic activity of compounds 1, 3, 4 and 6 was evaluated against a panel of cancer cell lines, where cytotoxic activity in the single digit μM range was observed.
Graphical Abstract
A new technique was used to monitor the chemistry of fungal cultures in situ, thereby confirming authenticity of secondary metabolites.
Introduction
Fungi are a rich source of bioactive secondary metabolites with therapeutic activities.1–3 The recent estimates for fungal biodiversity range from 2.2 to 3.8 million species;4 however, only a fraction of these fungi (~135,000 species) have been taxonomically characterized and an even smaller percentage examined chemically.5 Thus, our understanding of the chemical diversity and pharmacological applications of most fungi is still quite limited. Amongst the well-studied fungi, Trichoderma species are known for their ability to produce bioactive secondary metabolites, including polyketides, alkaloids, terpenoids, non-ribosomally biosynthesized peptides (NRPs), and metabolites of mixed biogenesis.6–8 Between the NRPs, peptaibols represent the largest group, with more than 1000 compounds reported to date.9 These molecules, typically composed of 5–20 amino acid residues, are characterized by a high content of α-aminoisobutyric acid (Aib), an acylated N-terminus, and a C-terminus that may consist of a free or methoxy substituted 2-amino alcohol, amine, amide, free amino acid or sugar alcohol.10–14
When working on a well-studied structural class, it is relatively easy to pose questions about the biosynthetic authenticity of a new analogue. Artefacts of the isolation process are a well accepted problem.15–17 In addition to adding some confusion to the natural products literature, the biosynthetic authenticity of a compound can be important when considering how certain compounds either support or refute a biosynthetic pathway. In this context, there is value to the development of strategies to either authenticate or invalidate isolated compounds as true secondary metabolites.
Herein, we communicate the application of the recently described droplet-liquid microjunction-surface sampling probe (droplet-LMJ-SSP), which was coupled to a UPLC chromatographic system and paired with both UV and HRESIMS detectors,18 to validate the biosynthetic authenticity of fungal secondary metabolites. The droplet-LMJ-SSP is a manually controlled surface sampling tool that performs a microextraction on the culture surface. The droplet-LMJ-SSP has been used in various types of analysis of fungal cultures, including the in situ scouting and identification of peptaibols,19 dereplication18 and mapping of the spatial and temporal distribution of fungal metabolites,20, 21 and to monitor the biosynthesis of targeted non-natural natural products.22
As part of our ongoing research to discover new anticancer leads from filamentous fungi,23–27 Trichoderma arundinaceum strain MSX70741,10 a well-known peptaibols producer from the Mycosynthetix library (with more than 55,000 fungal accessions28) was reinvestigated. Consequently, the scaled up culture of strain MSX70741 on rice led to the isolation of three new peptaibols (1–3, Fig. 1), together with the four known compounds alamethicin F50 (4), alamethicin II (5), atroviridin J (6), and trichobranchin D-I (7).10 The structures of the isolates were established using a set of spectroscopic (1D and 2D NMR) and spectrometric (HRESIMS/MSn) techniques. The absolute configurations of these isolates were determined by Marfey’s analysis.10, 29 The cytotoxic activities of compounds 1, 3, 4 and 6 were evaluated in an MTT assay against a panel of human cancer cell lines: HCT 116 (colorectal carcinoma), DLD-1, HT-29, and SW948 (colorectal adenocarcinomas), Hep-G2, and Huh-7 (hepatocellular carcinomas), and HeLa (adenocarcinoma).30 The evaluated compounds displayed cytotoxic activities with IC50 values in the single digit μM range. Finally, in situ analysis of the cultures of MSX70741 cultured on potato-dextrose-agar (PDA) confirmed the authenticity of the compounds as natural products, discarding the possibility of artefacts of the purification process.
Fig. 1.
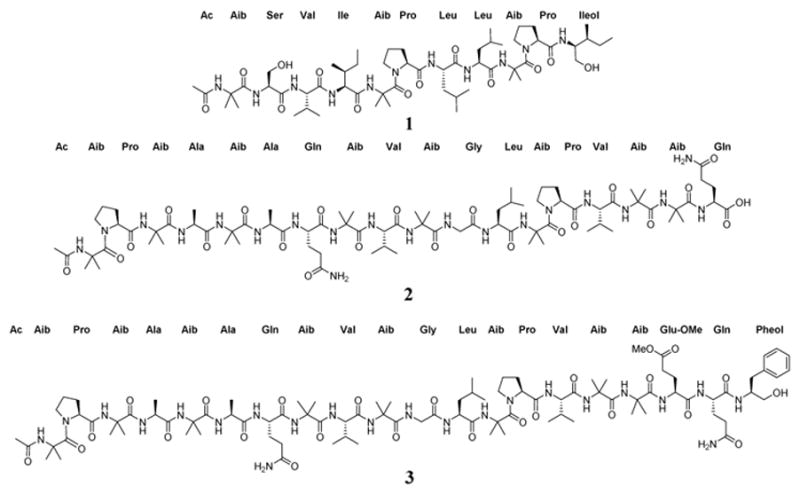
Structures of compound 1–3.
Results and Discussion
Structure elucidation
Previous studies on Trichoderma arundinaceum strain MSX70741 stimulated this project,10 largely to amplify the supply of key peptaibols for pharmacological assays, as will be reported in the future. Thus, to increase the supply of those peptaibols, four large scale cultures were set up, extracted with 1:1 CHCl3-MeOH and partitioned with 4:1:5 CHCl3/MeOH/H2O.25, 26, 31 The pooled organic soluble extract was then fractionated using normal phase flash chromatography to yield five fractions. Further purification of the fraction that eluted with 100% MeOH employing reverse phase flash chromatography, followed by preparative and semi-preparative HPLC, led to the isolation of three new metabolites (trichobrevin BIII-D, prealamethicin F50, and Glu(OMe)18-alamethicin F50, 1–3, respectively) (Fig. 1), along with multi-mg to hundred-mg samples of the known peptaibols, alamethicin F50 (4), alamethicin II (5), atroviridin J (6), and trichobranchin D-I (7).
Compounds 1 and 7 were isolated as white amorphous powders, and their molecular formulae were determined to be C56H99N11O13 and C55H97N11O13 on the basis of HRESIMS data (m/z 1134.7496 [M+H]+, and 1120.7343 [M+H]+, respectively (calc. for C56H100N11O13 and C55H98N11O13). In combination with NMR studies, these data indicated 13 degrees of unsaturation for both compounds. Literature searches based on molecular formulae and exact masses in the Dictionary of Natural Products and Peptaibiotics databases indicated that compound 7 was the known peptaibol trichobranchin D-I, previously isolated by Ayers et al.,10 while 1 was closely related to the trichobrevin subclass of peptaibols. The amino acid sequence of compound 1 was established on the basis of HRESIMS/MS data. For example, the full scan spectrum showed characteristic ions at m/z 1134.7506 [M+H]+, 920.5819 (b9+, N-terminal fragment), 512.3082 (b5+), 623.4493 (y6+, C-terminal fragment) and 215.1754 (y2+), all fragments generated by cleavage of the labile bonds between Aib5-Pro6 and Aib9-Pro10 (Fig. 2).28 Further fragmentation of the ion b5+ generated a series of b+ ions (Ac-b1+-b4+; m/z = 128.0709, 215.1014, 314.1720, and 427.2575, respectively), providing information about the successive losses of Aib5, Lxx4 (Leu or Ile), Vxx3 (Val or Iva), and Ser2 (Fig. 2). HRESIMS/MS of the C-terminal fragment y6+ yielded a series of ions corresponding to the loss of the fragment y2+ (m/z = 409.2804), Aib9 (m/z = 324.2305), and Lxx8 (m/z = 211.1445), the last accounting for the molecular formula C11H19N2O2 and corresponding with a b fragment constituted by Pro and Lxx (Fig. 2). Finally, MS3 of fragment y2+ confirmed the presence of Lxxol (Leuol or Ileol) as the C-terminal amino acid by generation of the y1+ fragment at m/z = 118.1228, consistent with the formula C6H16NO. In view of the data obtained from mass spectrometry, the putative amino acid sequence of compound 1 was Ac-Aib1-Ser2-Vxx3-Lxx4-Aib5-Pro6-Lxx7-Lxx8-Aib9-Pro10-Lxxol11.
Fig. 2.
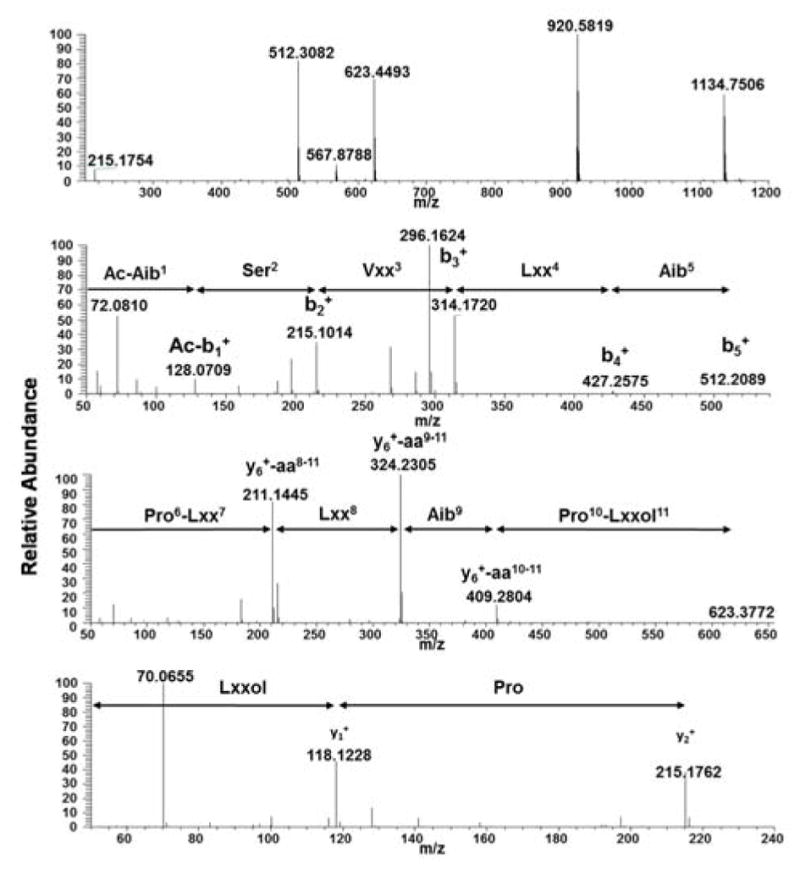
Positive HRESIMS of compound 1. A) Full scan showing in source fragmentation, B–D) HRESIMS/MSn of fragments b5+, y6+ and y2+, respectively.
Based upon a detailed search in the Peptaibiotics Database (https://peptaibiotics-database.boku.ac.at), which contains 1350 entries, including over 980 peptaibols, the putative amino acid arrangement of compound 1 resembled the structure of the trichobrevin BIII complex, a family of three compounds identified by HPLC-Ion-Trap-ESIMS in the extract of some strains of Trichoderma brevicompactum.32 Members of the trichobrevin BIII complex share the amino acid sequence Ac-Aib1-Ser2-Vxx3-Lxx4-Aib5-Pro6-Lxx7-Lxx8-Aib9-Pro10-Lxxol11; however, in the study published by Degenkolb in 2006, the unambiguous identity of Vxx3, Lxx4, Lxx7, Lxx8, and Lxxol11 was not established.32 In this communication we report the isolation and structural elucidation of a member of this family, which was assigned the trivial name trichobrevin BIII-D (1). The planar structure of compound 1 was established based on exhaustive interpretation of 1D and 2D NMR data, and the absolute configuration of the individual amino acids was established from the complete acid hydrolysis and chemical derivatization with Marfey’s reagent of 1, followed by UPLC-UV analysis.
In brief, the 1H-NMR of 1 recorded in DMSO-d6 exhibited resonances for nine exchangeable protons between δH 7.02–8.77 ppm (showed by recording the spectra in MeOH-d3, signals among δH 3.22–4.29 ppm belonging to the α-H’s, six singlet signals among δH 1.32–1.45 ppm, attributed to Aib1, Aib5 and Aib9, several doublets ascribed to Val3, Leu7, Leu8, and two triplets at δH 0.76 (J = 7.4 Hz), and δH 0.83 (J = 7.5 Hz), which were unambiguously assigned to Ile4 and Ileol11, respectively. In addition, the presence of a sharp singlet at δH 1.91 ppm in the 1H NMR spectrum of 1 confirmed the presence of an acetylated N-terminal residue (Table 1). The 13C-NMR spectrum displayed
Table 1.
NMR spectroscopic data for compound 1 (700 and 175 MHz, 1H and 13C, respectively) in DMSO-d6
| position | δC | type | δH, mult. (J in Hz) | position | δC | type | δH, mult. (J in Hz) |
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Ac | δ 2 | 3.65a | |||||
| C═O | 171.0 | C | Leu7 | ||||
| CH3 | 23.0 | CH3 | 1.91, s | C═O | 172.6 | C | |
| Aib1 | α | 53.1 | CH | 3.96a | |||
| C═O | 176.1 | C | β 1 | 38.7 | CH2 | 1.47a | |
| α | 55.7 | C | β 2 | 1.85a | |||
| β1 | 23.8 | CH3 | 1.34, s | γ | 24.4 | CH | 1.72a |
| β2 | 26.3 | CH3 | 1.36, s | δ1 | 20.8 | CH3 | 0.83, d (6.6) |
| NH | 8.77, s | δ2 | 23.0 | CH3 | 0.92, d (6.6) | ||
| Ser2 | NH | 7.53, d (7.9) | |||||
| C═O | 171.2 | C | Leu8 | ||||
| α | 58.3 | CH | 3.991 | C═O | 172.2 | C | |
| β | 60.5 | CH2 | 3.66–3.75, m | α | 50.6 | CH | 4.29a |
| NH | 8.42, d (4.3) | β | 39.6a | CH2 | 1.55a | ||
| OH | 5.34, brs | γ | 24.1 | CH | 1.59a | ||
| Val3 | δ1 | 20.3 | CH3 | 0.74, d (6.4) | |||
| C═O | 171.5 | C | δ2 | 22.8 | CH3 | 0.79, d (6.4) | |
| α | 60.4 | CH | 3.86, dd (7.5, 7.4) | NH | 7.16, d (9.1) | ||
| β | 29.0 | CH | 2.13, dq-like (13.7, 6.9) | Aib9 | |||
| γ1 | 19.1 | CH3 | 0.89, d (6.8) | C═O | 171.5 | C | |
| γ1 | 18.9 | CH3 | 0.93, d (6.8) | α | 55.7 | C | |
| NH | 7.68, d (7.4) | β1 | 25.5 | CH3 | 1.32, s | ||
| Ile4 | β2 | 23.8 | CH3 | 1.36, s | |||
| C═O | 172.5 | C | NH | 7.52, s | |||
| α | 57.6 | CH | 4.12, t (7.8) | Pro10 | |||
| β | 35.5 | CH | 1.89, m | C═O | 171.1 | C | |
| γ1a | 24.6 | CH2 | 1.24, m | α | 62.2 | CH | 4.24, dd (8.3, 6.4) |
| γ1b | 1.41, m | β1 | 28.9 | CH2 | 1.64, m | ||
| δ | 10.5 | CH3 | 0.76, t (7.4) | β2 | 2.08, m | ||
| γ2 | 15.5 | CH3 | 0.84, d (6.9) | γ | 25.4 | CH2 | 1.76, m |
| NH | 7.30, d (8.5) | δ1 | 48.0 | CH2 | 3.22, dt (11.3, 7.2) | ||
| Aib5 | δ2 | 3.65, m | |||||
| C═O | 173.0 | C | Ileol11 | ||||
| α | 56.1 | C | α | 54.5 | CH | 3.66a | |
| β1 | 25.5 | CH3 | 1.38, s | β | 34.9 | CH | 1.59a |
| β2 | 23.0 | CH3 | 1.45, s | γ1a | 23.9 | CH2 | 0.99, ddd (13.5, 9.2, 7.0) |
| NH | 7.97, s | γ1b | 1.46a | ||||
| Pro6 | δ | 11.6 | CH3 | 0.83, t (7.5) | |||
| C═O | 173.3 | C | γ2 | 16.0 | CH3 | 0.81, d (7.0) | |
| α | 63.3 | CH | 4.20, dd (8.1, 7.6) | β1′ | 61.2 | CH2 | 3.36, dd (11.3, 6.7) |
| β1 | 28.6 | CH2 | 1.59a | β2′ | 3.45a | ||
| β2 | 2.24, m | NH | 7.02, d (9.4) | ||||
| γ | 25.7 | CH2 | 1.86a | OH | 3.98a | ||
| δ1 | 48.5 | CH2 | 3.39, dt (11.7, 7.9) | ||||
Overlapping signals; chemical shifts were determined from 1H-13C HSQC correlations
11 signals between δC 171.0–176.1 ppm assigned to amide carbonyl groups, 15 signals ranging δC 48–70 ppm, and several resonances in the alkyl region.28, 33, 34 Interpretation of the 2D-NMR data (1H-1H TOCSY, 1H-1H COSY, 1H-13C HSQC and 1H-13C HMBC), in particular TOCSY, permitted the assembly of each amino acid through the total spin correlations observed. NOESY correlations of the amide NH’s protons with their corresponding neighbouring amino acids, along with HMBC cross peaks of the amide protons through 2JCH with their i-1 carbonyl group, and αC, and 3JCH of the αH’s with the i carbonyl group (Fig. 3), confirmed the sequence of 1 as shown.34
Fig. 3.
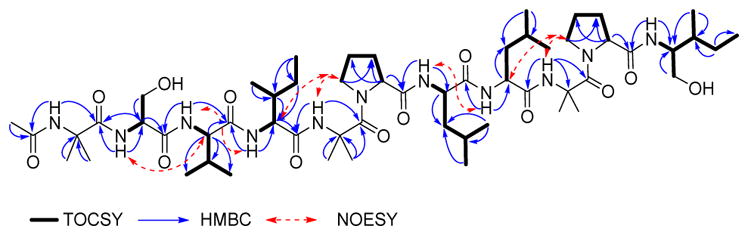
Selected TOCSY, HMBC and NOESY correlations observed for compound 1.
The absolute configuration of the individual amino acids in compound 1 was established using a 10 min Marfey’s-UPLC-UV method.29 The general procedure started with the acid hydrolysis of 1, followed by derivatization of the hydrolysate under alkaline conditions with Nα-(2,4-dinitro-5-fluorophenyl)-L-alaninamide (L-FDAA, Marfey’s reagent), followed by UPLC-UV analysis at 340 nm. Finally, the UPLC retention times of the derivatized amino acids in 1 were compared with those of derivatized L and D standards for each amino acid (Fig. S7).10 Thus the unambiguous structure of compound 1 was assigned as Ac-Aib1-L-Ser2-L-Val3-L-Ile4-Aib5-L-Pro6-L-Leu7-L-Leu8-Aib9-L-Pro10-L-Ileol11 and given the trivial name trichobrevin BIII-D. In all of our previous studies on peptaibols, the amino acids were L,10, 28 as is typical for this particular class of compounds.
Compound 2 was isolated as a white powder with a molecular formula of C78H132N20O22 as evidenced by HRESIMS data and analysis of the 1H, 13C, and edited-HSQC NMR data (Table S1, Figs. S10, S11 and S14), revealing an index of hydrogen deficiency of 23. The positive full scan MS spectrum of 2 displayed four intense peaks at m/z 1701.9910 ([M+H]+, calc. for C78H133N20O22, 1701.9897), 1189.6946 (b13+, fragment), 851.4989 ([M+2H]2+), and 513.3032 (y5+, fragment) (Fig. S8). The composition and absolute configuration of the amino acids was confirmed on the basis of MS2 and Marfey’s analysis (Figs. 4 and S9). Thus, the residues were confirmed to be Aib (8), L-Ala (2), L-Pro (2), L-Val (2), L-Gln (2), and L-Leu (1). By acid hydrolysis, the Gln residues in the molecule were converted into Glu,35 however, losses of 146.069 and 128.056 a.m.u. in the MS2 spectrum of fragments y5+ (m/z 513.3032) and b7+ (m/z 665.3597), respectively, supported the presence of L-Gln (Fig. S9).11 Analysis of 1D and 2D NMR data revealed that all the amino acids and connections were the same as those in alamethicin F50 (4). The main differences in 2 compared to 4 were the absence of two signals around at δH 6.63 and 7.34, assigned to the NH2 group of Gln19 in 4, as well as the lack of resonances attributed to the aromatic ring of the Pheol20 moiety (Table S1). Therefore, the structure of compound 2 was established as Ac-Aib1-Pro2-Aib3-Ala4-Aib5-Ala6-Gln7-Aib8-Val9-Aib10-Gly11-Leu12-Aib13-Pro14-Val15-Aib16-Aib17-Gln18, and assigned the trivial name of prealamethicin F50 (2), since it had the same amino acid sequence minus two C-terminal residues, indicating its close relationship to 4.
Fig. 4.
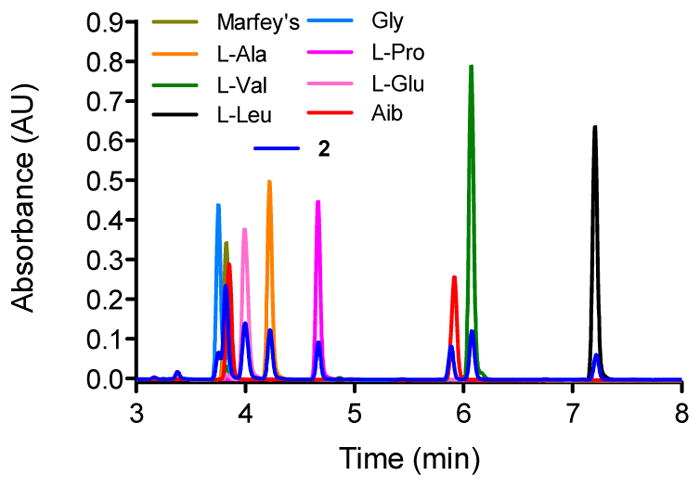
Marfey’s analysis of compound 2.
Compound 3 was also isolated as a white powder, and its molecular formula was established as C93H152N22O25 on the basis of HRESIMS data, which displayed a protonated molecular ion at m/z 1978.1385 [M+H]+ (calc. for C93H153N22O25, m/z 1978.1371). In source fragmentation of compound 3 generated peaks corresponding to the fragments b13+ and y7+ at m/z 1189.6943 and 789.4504, respectively, along with the [M+2H]2+ ion at m/z 989.5727. MS2 of the fragment b13+ supported the sequence Ac-Aib1-Pro2-Aib3-Ala4-Aib5-Ala6-Gln7-Aib8-Val9-Aib10-Gly11-Leu12-Aib13. MS2 of fragment y7+ showed the loss of m/z 279.15987 (C14H21N3O3), accounting for Gln19 and Pheol20.
Further fragmentation of the ion at m/z 510.2917 gave origin to a peak at m/z 367.2338, indicating the loss of m/z 143.0581 (C6H9NO3+), matching accurately for glutamic acid δ-methyl ester. Thus, MS2 analysis allowed the elucidation of the sequence Pro14-Val15-Aib16-Aib17-Glu-OMe18-Gln19-Pheol20 (Figs. S16 and S17). 1H, 13C, and 2D-NMR data of compound 3 were similar to those recorded for alamethicin F50 (4) (Table S1), the main differences being the absence of protons attributed to the
NH2 group of Gln18 (Table S1), which were replaced by a methoxy group at δH/δC (3.57/51.2), showing strong HMBC correlations with the δ carbon of glutamic acid at δ(C) 172.8, supporting the presence of glutamic acid δ-methyl ester. As expected, Marfey’s analysis of compound 3 was the same as that for alamethicin F50 (4), allowing the establishment of the absolute configuration of the individual amino acids in compound 3 as L-Pro, L-Ala, L-Gln, L-Val, L-Leu, L-Glu, and L-Pheol (Fig. S24). Thus, compound 3 was characterized as the δ-methyl ester of Glu18 in alamethicin F50, and assigned the name Glu(OMe)18-alamethicin F50 (3). This compound was previously obtained via semisynthesis in 1977 by Pandey et al, in a study focused on the structure elucidation of alamethicin I and II, however it is the first report as natural product.36
The occurrence of methyl esters of glutamic acid in peptaibols is rare. For example, of the over 1350 peptaibiotics reported in the Peptaibiotics Database, there are just four examples of this subclass of analogues, TA1896, TA1924, TA1910 and TA1924a, all of which are peptaibols with 19 amino acid residues isolated from Trichoderma atroviride by Panizel et al. in 2013.37 Thus, the isolation and characterization of compound 3 represents the second report of glutamic acid methyl ester containing peptaibols isolated from nature, and the first belonging to the alamethicin class, the most extensively studied peptaibol.38
In situ chemical analysis
A question that may arise in natural products research pertains to the authenticity of a compound. Is the isolated compound actually biosynthesized by the organism or is it an artefact of the extraction, fractionation and/or the purification processes? Indeed, this question is of particular relevance when isolating compounds of extremely low yield, for instance those in the baseline of a chromatographic run after scaling up an extraction process, and/or when isolating compounds that represent minor derivatives of known compounds, for example methyl esters or close related biosynthetic precursors. This project encompasses all of those aspects. We isolated compounds 2 and 3 as minor side fractions when targeting the large-scale development of compound 4. Moreover, the analogues, while structurally interesting, particularly from a biosynthetic standpoint, represent minor deviations of well-known compounds, in this case the lack of two amino acid residues in 2 or the presence of a glutamic acid methyl ester in 3.
Thus, the possibility of isolation of compound 3 as an artefact was discarded based on the outcomes obtained from two experiments. First, alamethicin F50 (4), the compound isolated in high yield from the extract of MSX70741, was stored in MeOH for one month at room temperature. Afterwards, the 1H-NMR spectrum was recorded, and the presence of the –OMe singlet at δH 3.27 was not observed (data not shown). In addition, in situ UPLC-PDA-HRMS-MS/MS analysis of the culture of strain MSX70741 grown in potato-dextrose-agar (PDA) for 21 days, using a droplet-LMJ-SSP,18, 20 was used to validate the authenticity of the 3 as true secondary metabolites. The results obtained from this analysis showed a peak matching the retention time and characteristic in-source ion peaks for compound 3, especially the fragment b13+ and y7+ at m/z 1189.69 and 789.45, respectively, along with the [M+2H]2+ ion at m/z 989.57 (Fig. 5). Similar results were obtained for compounds 1 and 2, validating the authenticity of all the new compounds (1–3) as true fungal metabolites.
Fig. 5.

In situ analysis of Trichoderma arundinaceum strain MSX70741 grown in PDA using a droplet-liquid microjunction-surface sampling probe (droplet-LMJ-SSP) coupled to a hyphenated system (UPLC-PDA-HRMS-MS/MS). A) Base peak extracted chromatograms for compounds 1 (maroon), 2 (green) and 3 (blue). B) full-scan HRESIMS for compounds 1 (top), 2 (middle), and 3 (bottom). C) Base peak chromatogram of the In situ analysis of Trichoderma arundinaceum strain MSX70741 (grey); extracted ion chromatograms (XIC) of m/z 920.57 (1; maroon), m/z 513.30 (2, green) and m/z 789.44 (3, blue) matching retention times for compounds 1–3. D) Full-scan MS at 5.78 (top) and 6.95 min (bottom). Extracted HRESIMS at 5.78 and 6.95 min matched the retention times and in source fragmentation for compounds 2 (top; in source fragments boxed in green), 1 (bottom, in source fragments boxed in maroon), and 3 (bottom, in source fragments boxed in blue) within ± 5.0 ppm. Application of this methodology allowed the identification of compounds 1–3 as natural products, discarding the possibility of artifacts of the isolation process.
Cytotoxic activity
The activity of compounds 1, 3, 4 and 6, were evaluated against a panel of cancer cell lines, including strains HCT 116 (colorectal carcinoma), DLD-1, HT-29, and SW948 (colorectal adenocarcinomas), Hep-G2, and Huh-7 (hepatocellular carcinomas), and HeLa (adenocarcinoma)30, using an MTT assay (Table 2). Compound 1 exhibited moderate activity against strains HCT 116 and HT-29 with IC50 values of 6.8 and 6.7 μM, respectively, and no activity against hepatocellular carcinomas and adenocarcinoma cell lines. Glu(OMe)18-alamethicin F50 (3) was the most active compound, with IC50 values ranging from 2.5 through 6.5 μM, and no selectivity against different cell lines (Table 2). The bioactivity results of these compounds correlates with their hydrophobicity and are in harmony with the findings previously reported for peptaibols biosynthesized from 11 amino acids39 and alamethicin F50 derivatives.10
Table 2.
Cytotoxic activity of compounds 1, 3, 4 and 6 against a panel of cancer cell lines
| Compounds | IC50 Values (μM) | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| HCT-116 | DLD-1 | HT-29 | SW948 | HepG2 | Huh-7 | HeLa | |
| 1 | 6.8 | >8 | 6.7 | >8 | >8 | >8 | >8 |
| 3 | 2.6 | 6.1 | 3.2 | 4.0 | 6.5 | 3.0 | 2.5 |
| 4 | 3.2 | NT | NT | NT | NT | 6.5 | 5.7 |
| 6 | 3.5 | 4.7 | 3.7 | 4.0 | 6.3 | 4.0 | 3.0 |
NT: Not tested.
Experimental Section
General experimental procedures
NMR experiments were conducted in DMSO-d6 or MeOH-d3 with presaturation of the OH peak at δH 4.92 ppm (wet experiment). NMR instrumentation was an Agilent 700 MHz NMR spectrometer (Agilent Technologies, Inc., Santa Clara, CA, USA) equipped with a cryoprobe, operating at 700 MHz for 1H and 175 MHz for 13C. HRESIMS data were obtained using a Thermo QExactive Plus mass spectrometer (ThermoFisher, San Jose, CA, USA) combined with an electrospray ionization source. Dereplication of the fungal culture by in situ sampling was performed using the droplet-LMJ-SSP coupled with a Waters Acquity ultraperformance liquid chromatography (UPLC) system (Waters Corp.) to a Thermo QExactive Plus. The droplet-LMJ-SSP experiments were carried out using procedures described previously by Sica et al.18 Briefly, extractions were performed using Fisher Optima LC/MS grade solvents consisting of 25:25:50 MeOH-Dioxane-H2O. An initial 5 μL of solvent was drawn into the syringe. Droplets of 4 μL were dispensed onto the surface of the sample at a rate of 2 μL/s, held on the surface for 2 s, and withdrawn back into the syringe at the same rate. This extraction process was repeated in triplicate for a single spot prior to injection into the UPLC-MS system. The HCD fragmentation used a normalized collision energy of 35 for all the compounds to obtain MS/MS data. The UPLC separations were performed using an Acquity BEH C18 column (50 mm × 2.1 mm, internal diameter, 1.7 μm) equilibrated at 40 °C and a flow rate set at 0.3 mL/min. The mobile phase consisted of a linear MeCN-H2O (acidified with 0.1% formic acid) gradient starting at 15% MeCN to 100% MeCN over 8 min. The mobile phase was held for another 1.5 min at 100% MeCN before returning to the starting conditions. The HPLC separations were performed using a Varian ProStar HPLC system connected to a ProStar 335 photodiode array detector (PDA) with UV detection set at 195 nm and 210 nm. Preparative HPLC purification of isolated compounds was performed on a Phenomenex Synergi 4 μm particle size C12 column (21 × 250 mm) at a flow rate of 15.0 or 20.0 mL/min. Semipreparative HPLC was carried out on a Phenomenex Gemini-NX 5 μm particle size C18 column (10 × 250 mm) or Phenomenex Synergi 4 μm particle size C12 column (10 × 250 mm) at 4.6 mL/min. Flash column chromatography was carried out with a Teledyne ISCO Combiflash Rf connected to an ELSD, and with UV detection set at 200–400 nm according with established protocols.10, 20, 26, 28, 31, 40
Fungal strain identification
Fungal strain identification was carried out following the procedures outlined in detail previously.41 Also, the Supporting Information from Rivera-Chávez et al., 2017,22 has specific details on the identification of this strain.
Fermentation, extraction and isolation
The MSX70741 strain (Trichoderma arundinaceum) was isolated in April 1993 by Dr. Barry Katz from wood collected in a humid mountain forest (April 1993).10 A seed culture of the fungal strain MSX70741 was grown on a malt extract agar slant, and a small piece of agar with mycelium was transferred into YESD media (followed by incubation for 7 days at 22 °C with agitation at 125 rpm). The seed culture was subsequently transferred into a 2.8 L Fernbach flask containing 150 g of rice and 300 mL of H2O to which was added a vitamin solution. Each flask was incubated at 22 °C until the cultures showed good growth. To the large scale solid fermentation (×4) of MSX70741 was added 500 mL of 1:1 CHCl3-MeOH, and the mixtures were shaken for 16 h at 100 rpm in a reciprocating shaker separately. The solution was filtered, and equal volumes of H2O and CHCl3 were added to a final volume of 2 L, the mixture was stirred for 2 h and then transferred into a separatory funnel. The bottom layer was drawn off and evaporated to dryness. The extract was defatted by partitioning between 300 mL of a mixture of 1:1 MeOH-MeCN and 300 mL of hexane in a separatory funnel. The bottom layer was collected and evaporated to dryness. The defatted large scale extract (7.5 g) was adsorbed onto a minimal amount of Celite 545 (Acros Organics) and mixed using mortar and pestle. This material was fractionated via flash chromatography on a 120 g RediSep Rf Gold Si-gel column, using a gradient solvent system of hexane-CHCl3-MeOH at 85 mL/min flow rate and 30 column volumes (CV) over 67.7 min, to afford five fractions (F1–F5). Fraction F5 (2.8 g), eluted with 100% MeOH, was subjected to reverse phase flash chromatography on a 130 g C18 RediSep column using a gradient solvent system of MeOH-H2O 20:80 → 100:0 over 20 CV and hold at 100:0 for 5 CV at 85 mL/min flow rate (a total of 25.0 CV over 38.2 min), to afford seven fractions (F5I-F5VII). Part of fraction F5V (~250 mg) was subjected to preparative HPLC using a gradient system initiated with 40:60 MeCN-H2O (0.1% formic acid) to 100 % MeCN over 30 min at a flow rate of 15.0 mL/min to generate 16 fractions (F5V1-16). Resolution of fraction F5V-4 by semipreparative HPLC (Gemini-NX) using an isocratic system of 60:40 MeCN-H2O (0.1% formic acid) at 4.6 mL/min afforded compound 7 (3.1 mg, tR 14.5 min) and compound 4 (2.0 mg, tR 17.0 min). Fraction F5V-7 was resolved by semipreparative HPLC (Synergi) using an isocratic method of 60:40 MeCN-H2O (0.1% formic acid) to afford compound 2 (4.3 mg, tR 14.0 min). Resolution of fraction F5V-8 using an isocratic method of 60:40 MeCN-H2O (0.1% formic acid) led to the isolation of 3 (4.0 mg, tR 17.5–20.0 min). Fractions F5I, and F5II were combined and fractionated by preparative HPLC using a gradient system initiated with 40:60 MeCN-H2O (0.1% formic acid) to 100 % MeCN over 30 min at a flow rate of 15.0 mL/min to generate 10 fractions. Resolution of fraction F5I-II-3 using a gradient method starting at 40:60 MeCN-H2O (0.1% formic acid) to 100 % MeCN over 30 min at a flow rate of 4.6 mL/min in a Synergi column led to the isolation of 1 (1.2 mg, tR 17.5–20.0 min). Compounds 5 and 6 were obtained from fractions F5V-6 and F5I-II, respectively (5, 50.4 mg; 6, 13.5 mg), approximately 130 mg more of compound 4 were isolated readily from different fractions.
Trichobrenin BIII-D (1)
White powder; (c = 0.20, MeoH); UV (MeOH) λmax (log ε) 211 (3.75) nm; 1H NMR ((D6)DMSO-d6, 700 MHz) and 13C NMR (DMSO-d6, 175 MHz), see Table 1; HRESIMS m/z 1134.7506 [M + H]+; calc. for C56H100N11O13 1134.7496.
Prealamethicin F50 (2)
White powder; (c = 0.10, MeoH); UV (MeOH) λmax (log ε) 212 (4.25) nm; 1H NMR ((D3)MeOH, 700 MHz) and 13C NMR (MeOH-d3, 175 MHz), see Table S1; HRESIMS m/z 1701.9910 [M + H]+; calc. for C78H132N20O22 1701.9897.
Glu(OMe)18-alamethicin F50 (3)
White powder; (c = 0.27, MeoH); UV (MeOH) λmax (log ε) 212 (3.89) nm; 1H NMR ((D6)DMSO, 700 MHz) and 13C NMR (DMSO-d6, 175 MHz), see Table S1; HRESIMS m/z 1978.1337 [M + H]+; calc. for C93H153N22O25, m/z 1978.1371.
Marfey’s analysis
This was carried out as described in detail previously.10, 19, 22 Briefly, approximately 0.2 mg of each amino acid standard was weighed into 2 mL reaction vials. To each standard was added 50 μl of H2O, 20 μl of 1 M NaHCO3, and 100 μl 1% Marfey’s reagent (Nα-(2,4-dinitro-5-fluorophenyl)-L-alaninamide, Acros Organics) in acetone. The reaction mixtures were agitated at 40 °C for 1 h. The reactions were stopped by adding 10 μl of 2 N HCl. The product of the reactions was dried under a stream of N2 and dissolved in ~1.7 mL of MeOH. Each derivatized standard was injected individually (1.0 μl) onto the UPLC. The UPLC conditions were 10–70% MeOH in 0.1 % of formic acid in water over 10 min on a BEH C18 column, and the eluent was monitored at 340 nm.
The digested and derivatized peptaibols were generated using the following procedure: approximately 0.2–0.3 mg of compounds 1–3 were weighed separately into 2 mL reaction vials, to which was added 0.5 mL of 6 N HCl. The compounds were hydrolyzed at 90 °C for 24 h, and then evaporated under a stream of N2. To each hydrolysis product was then added 25 μL H2O, 10 μl 1 M NaHCO3, and 50 μl of 1% Marfey’s reagent in acetone. The reaction mixtures were agitated at 40 °C for 1 h. The reactions were halted by the addition of 5 μl of 2 N HCl. The mixtures were dried under a stream of N2 and brought up in ~200 μL of MeOH and injected onto the UPLC using the same conditions as for the standards.
Cytotoxicity assay
Cell culture
Cell-Culture reagents, antibiotics and other supplements were purchased from Sigma, USA. Fetal Bovine Serum (FBS, US certified) was from Invitrogen Life Sciences, USA. HCT-116, DLD-1, HT29, SW948, HepG2, Huh-7, HeLa cells were grown in Dulbeco’s Modified Eagle’s Medium (Sigma-Aldrich, USA) or RPMI-1640 (with 10% FBS, 100 units/mL pencillin and 100 μg/mL streptomycin) (Invitrogen Life Sciences, USA). The cells were maintained at 37 °C in a humidified atmosphere with 5% CO2. All cell-culture work was performed under aseptic conditions inside a laminar airflow chamber.
Assay
Toxicity of the compounds in different cell lines in the presence of 0.2% FBS was determined using 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide reduction assay (MTT assay). Compounds were dissolved in DMSO at 10 mg/mL concentration and stored at –20 °C. The dilutions were made in culture medium before treatment. 5×103 cells/well was plated in a 96-well plate. After 24 h of plating, the cells were treated with different concentrations of compounds in triplicates. MTT (20 μl of 5mg/mL) was added to the cells after 44 h. The medium was removed from the wells 4 h after MTT addition. 200 μl of DMSO were added to dissolve the formazan crystals, and the absorbance was then measured at 570 nm in an enzyme-linked immunosorbent assay reader. Alamethicin F50 (4) was used as a positive control, as it has been tested extensively in the literature.10, 22
Conclusions
In conclusion, three new peptaibols (1–3) were isolated and characterized from the organic extract of the fungus Trichoderma arundinaceum (strain MSX70741). The structures of the isolates were established using high field 1D and 2D NMR (700 MHz) and spectrometric (HRESIMS/MSn) techniques, and their absolute configuration determined by Marfey’s analysis of the individual amino acids using a 10 min UPLC method. Compound 3 represents the second report of peptaibols containing a δ-methyl ester of glutamic acid. Compounds 1, 3, 4 and 6 were evaluated for their cytotoxicity against a panel of cancer cell lines. Products 3 and 6 displayed promising activity with IC50 values in the lower micromolar range. Importantly, in situ UPLC-PDA-HRMS-MS/MS analysis of the culture of strain MSX70741 grown in potato-dextrose-agar allowed the identification of compounds 2–3 as natural products, eliminating the possibility of artefacts of the isolation process. This study added another application of the droplet-LMJ-SSP, confirming it as a powerful and effective tool to address some of the common problems and questions in natural products research and drug discovery, such as those regarding the biosynthesis of targeted compounds, chemical ecology (spatial distribution of metabolites) and improving the efficacy of protocols focused on the discovery of new drug leads from nature (dereplication protocols). Finally, this study highlights the benefit of scaling up the fermentation conditions of a targeted microorganism, as doing so often present opportunities to uncover new chemical diversity.
Supplementary Material
Acknowledgments
This research was supported in part by P01 CA125066 from the National Cancer Institute and R01 GM088241 from the National Institute of General Medical Sciences, both part of the National Institutes of Health, Bethesda, MD, USA. Mycology technical support was provided by B. Darveaux (Mycosynthetix). The authors acknowledge the technical assistance of Dr. Franklin Moy (the University of North Carolina at Greensboro) for the 700 MHz NMR data. The high-resolution mass spectrometry data were acquired in the Triad Mass Spectrometry Laboratory at the University of North Carolina at Greensboro. We thank Drs. Vilmos Kertesz and Gary J. Van Berkel (Mass Spectrometry and Laser Spectroscopy Group, Chemical Sciences Division, Oak Ridge National Laboratory) for inspiration and guidance with the droplet-LMJ-SSP.
Footnotes
Electronic Supplementary Information (ESI) available: 1D and 2D NMR data for compounds 1–3. Mass spectrometry analysis for compounds 1–3. Marfey’s analysis for compounds 1 and 3. Tabulated NMR data for compounds 2–4. See DOI: 10.1039/x0xx00000x
References
- 1.Barkal LJ, Theberge AB, Guo CJ, Spraker J, Rappert L, Berthier J, Brakke KA, Wang CCC, Beebe DJ, Keller NP, Berthier E. Nat Commun. 2016;7 doi: 10.1038/ncomms10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evidente A, Kornienko A, Cimmino A, Andolfi A, Lefranc F, Mathieu V, Kiss R. Nat Prod Rep. 2014;31:617–627. doi: 10.1039/c3np70078j. [DOI] [PubMed] [Google Scholar]
- 3.Schueffler A, Anke T. Nat Prod Rep. 2014;31:1425–1448. doi: 10.1039/c4np00060a. [DOI] [PubMed] [Google Scholar]
- 4.Hawksworth DL, Lücking R. Microbiology Spectrum. 2017:5. doi: 10.1128/microbiolspec.funk-0052-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hibbett D, Abarenkov K, Kõljalg U, Öpik M, Chai B, Cole J, Wang Q, Crous P, Robert V, Helgason T, Herr JR, Kirk P, Lueschow S, O’Donnell K, Nilsson RH, Oono R, Schoch C, Smyth C, Walker DM, Porras-Alfaro A, Taylor JW, Geiser DM. Mycologia. 2016;108:1049–1068. doi: 10.3852/16-130. [DOI] [PubMed] [Google Scholar]
- 6.Almassi F, Ghisalberti EL, Narbey MJ, Sivasithamparam K. J Nat Prod. 1991;54:396–402. [Google Scholar]
- 7.Mukherjee PK, Horwitz BA, Kenerley CM. Microbiology. 2012;158:35–45. doi: 10.1099/mic.0.053629-0. [DOI] [PubMed] [Google Scholar]
- 8.Reino JL, Guerrero RF, Hernández-Galán R, Collado IG. Phytochem Rev. 2008;7:89–123. [Google Scholar]
- 9.Neumann NKN, Stoppacher N, Zeilinger S, Degenkolb T, Brückner H, Schuhmacher R. Chem Biodiver. 2015;12:743–751. doi: 10.1002/cbdv.201400393. [DOI] [PubMed] [Google Scholar]
- 10.Ayers S, Ehrmann BM, Adcock AF, Kroll DJ, Carcache de Blanco EJ, Shen Q, Swanson SM, Falkinham JO, Wani MC, Mitchell SM, Pearce CJ, Oberlies NH. J Pept Sci. 2012;18:500–510. doi: 10.1002/psc.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Degenkolb T, Berg A, Gams W, Schlegel B, Gräfe U. J Pept Sci. 2003;9:666–678. doi: 10.1002/psc.497. [DOI] [PubMed] [Google Scholar]
- 12.Shi M, Wang HN, Xie ST, Luo Y, Sun CY, Chen XL, Zhang YZ. Mol Cancer. 2010;9:1–16. doi: 10.1186/1476-4598-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toniolo C, Brückner H. Chem Biodiver. 2007;4:1021–1022. [Google Scholar]
- 14.Whitmore L, Chugh JK, Snook CF, Wallace BA. J Pept Sci. 2003;9:663–665. doi: 10.1002/psc.533. [DOI] [PubMed] [Google Scholar]
- 15.Cao CM, Zhang H, Gallagher RJ, Timmermann BN. J Nat Prod. 2013;76:2040–2046. doi: 10.1021/np400296s. [DOI] [PubMed] [Google Scholar]
- 16.Maltese F, van der Kooy F, Verpoorte R. Nat Prod Commun. 2009;4:447–454. [PubMed] [Google Scholar]
- 17.Reimer D, Hughes CC. J Nat Prod. 2017;80:126–133. doi: 10.1021/acs.jnatprod.6b00773. [DOI] [PubMed] [Google Scholar]
- 18.Sica VP, Raja HA, El-Elimat T, Kertesz V, Van Berkel GJ, Pearce CJ, Oberlies NH. J Nat Prod. 2015;78:1926–1936. doi: 10.1021/acs.jnatprod.5b00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sica VP, Rees ER, Raja HA, Rivera-Chávez J, Burdette JE, Pearce CJ, Oberlies NH. Phytochemistry. 2017;143:45–53. doi: 10.1016/j.phytochem.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paguigan ND, Raja HA, Day CS, Oberlies NH. Phytochemistry. 2016;126:59–65. doi: 10.1016/j.phytochem.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 21.Sica VP, Rees ER, Tchegnon E, Bardsley RH, Raja HA, Oberlies NH. Frontiers in Microbiology. 2016:7. doi: 10.3389/fmicb.2016.00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivera-Chávez J, Raja HA, Graf TN, Burdette JE, Pearce CJ, Oberlies NH. J Nat Prod. 2017;80:1883–1892. doi: 10.1021/acs.jnatprod.7b00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ayers S, Ehrmann BM, Adcock AF, Kroll DJ, Wani MC, Pearce CJ, Oberlies NH. Tetrahedron Lett. 2011;52:5733–5735. doi: 10.1016/j.tetlet.2011.08.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ayers S, Graf TN, Adcock AF, Kroll DJ, Shen Q, Swanson SM, Matthew S, Carcache de Blanco EJ, Wani MC, Darveaux BA, Pearce CJ, Oberlies NH. J Antibiot (Tokyo) 2012;65:3–8. doi: 10.1038/ja.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Elimat T, Figueroa M, Raja HA, Graf TN, Swanson SM, Falkinham JO, 3rd, Wani MC, Pearce CJ, Oberlies NH. Eur J Org Chem. 2015;2015:109–121. doi: 10.1002/ejoc.201402984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Figueroa M, Graf TN, Ayers S, Adcock AF, Kroll DJ, Yang J, Swanson SM, Munoz-Acuna U, Carcache de Blanco EJ, Agrawal R, Wani MC, Darveaux BA, Pearce CJ, Oberlies NH. J Antibiot (Tokio) 2012;65:559–564. doi: 10.1038/ja.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sy-Cordero AA, Figueroa M, Raja HA, Meza Aviña ME, Croatt MP, Adcock AF, Kroll DJ, Wani MC, Pearce CJ, Oberlies NH. Tetrahedron. 2015;71:8899–8904. doi: 10.1016/j.tet.2015.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Figueroa M, Raja H, Falkinham JO, Adcock AF, Kroll DJ, Wani MC, Pearce CJ, Oberlies NH. J Nat Prod. 2013;76:1007–1015. doi: 10.1021/np3008842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vijayasarathy S, Prasad P, Fremlin LJ, Ratnayake R, Salim AA, Khalil Z, Capon RJ. J Nat Prod. 2016;79:421–427. doi: 10.1021/acs.jnatprod.5b01125. [DOI] [PubMed] [Google Scholar]
- 30.Van Meerloo J, Kaspers GJL, Cloos J. In: Cancer Cell Culture: Methods and Protocols. Cree AI, editor. Humana Press; Totowa, NJ: 2011. pp. 237–245. [DOI] [Google Scholar]
- 31.Ayers S, Graf TN, Adcock AF, Kroll DJ, Shen Q, Swanson SM, Wani MC, Darveaux BA, Pearce CJ, Oberlies NH. Tetrahedron Lett. 2011;52:5128–5130. doi: 10.1016/j.tetlet.2011.07.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Degenkolb T, Gräfenhan T, Nirenberg HI, Gams W, Brückner H. J Agric Food Chem. 2006;54:7047–7061. doi: 10.1021/jf060788q. [DOI] [PubMed] [Google Scholar]
- 33.Liu D, Lin H, Proksch P, Tang X, Shao Z, Lin W. Org Lett. 2015;17:1220–1223. doi: 10.1021/acs.orglett.5b00172. [DOI] [PubMed] [Google Scholar]
- 34.Otto A, Laub A, Wendt L, Porzel A, Schmidt J, Palfner G, Becerra J, Krüger D, Stadler M, Wessjohann L, Westermann B, Arnold N. J Nat Prod. 2016;79:929–938. doi: 10.1021/acs.jnatprod.5b01018. [DOI] [PubMed] [Google Scholar]
- 35.Isaka M, Palasarn S, Komwijit S, Somrithipol S, Sommai S. Tetrahedron Lett. 2014;55:469–471. [Google Scholar]
- 36.Pandey RC, Cook JC, Rinehart KL. J Ame Chem Soc. 1977;99:8469–8483. doi: 10.1021/ja00457a064. [DOI] [PubMed] [Google Scholar]
- 37.Panizel I, Yarden O, Ilan M, Carmeli S. Mar Drugs. 2013;11:4937. doi: 10.3390/md11124937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leitgeb B, Szekeres A, Manczinger L, Vágvölgyi C, Kredics L. Chem Biodiver. 2007;4:1027–1051. doi: 10.1002/cbdv.200790095. [DOI] [PubMed] [Google Scholar]
- 39.Ruiz N, Wielgosz-Collin G, Poirier L, Grovel O, Petit KE, Mohamed-Benkada M, du Pont TR, Bissett J, Vérité P, Barnathan G, Pouchus YF. Peptides. 2007;28:1351–1358. doi: 10.1016/j.peptides.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 40.El-Elimat T, Figueroa M, Raja HA, Graf TN, Adcock AF, Kroll DJ, Day CS, Wani MC, Pearce CJ, Oberlies NH. J Nat Prod. 2013;76:382–387. doi: 10.1021/np300749w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raja HA, Miller AN, Pearce CJ, Oberlies NH. J Nat Prod. 2017;80:756–770. doi: 10.1021/acs.jnatprod.6b01085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.