

Abstract
Purpose
To characterize the mediators of 5-HT2A serotonin receptor–driven retinal neuroprotection.
Methods
Albino mice were treated intraperitoneally with saline or sarpogrelate, a 5-HT2A antagonist, immediately before light exposure (LE). Following LE, retinas were harvested for a high-throughput phosphorylation microarray to quantify activated phosphorylated proteins in G protein–coupled receptor (GPCR) signaling. To confirm microarray results and define temporal changes, Western blots of select GPCR signaling proteins were performed. Since both methodologies implicated MAPK/ERK activation, the functional significance of sarpogrelate-mediated ERK1/2 activation was examined by inhibition of ERK1/2 phosphorylation via pretreatment with the MEK inhibitor (MEKi) PD0325901. The degree of neuroprotection was evaluated with spectral-domain optical coherence tomography (SD-OCT) and electroretinography (ERG). To determine the effects of sarpogrelate on gene expression, a qPCR array measuring the expression of 84 genes involved in oxidative stress and cell death was performed 48 hours post LE.
Results
Sarpogrelate led to an activation of the MAPK/ERK pathway. Temporal analysis further demonstrated a transient activation of ERK1/2, starting with an early inhibition 20 minutes into LE, a maximum activation at 3 hours post LE, and a return to baseline at 7 hours post LE. Inhibition of ERK1/2 with MEKi pretreatment led to attenuation of sarpogrelate-mediated neuroprotection. LE caused significant changes in the expression of genes involved in iron metabolism, oxidative stress, and apoptosis. These changes were prevented by sarpogrelate treatment.
Conclusions
Sarpogrelate-mediated retinal protection involves a transient activation of the MAPK/ERK pathway, although this pathway alone does not account for the full effect of neuroprotection.
Keywords: sarpogrelate, MAPK/ERK, retinal degeneration, light damage, 5-HT2A
Retinitis pigmentosa (RP), an inherited retinal dystrophy that results in debilitating vision loss, is genetically heterogeneous with mutations in over 60 genes that lead to progressive photoreceptor death.1 Despite the diverse function of various genes that cause RP, common pathways mediate photoreceptor cell death.2,3 Researchers have long sought gene-independent treatments for RP that can cease or slow photoreceptor cell death.4 One such approach, systems pharmacology, encompasses the use of a combination of two or more drugs to achieve a positive therapeutic effect by affecting downstream common effectors or signaling pathways.5–7 This strategy allows for a rational therapeutic design based off basic knowledge of signaling pathways.5 Since the pathways that cause photoreceptor cell death are complex and multifactorial, targeting multiple signaling pathways is a more effective approach for treating such a complex disease.3,5
Increasing evidence suggests that multiple G protein (guanine nucleotide–binding protein)–coupled receptors (GPCRs) are expressed in the retina and modulation of these receptors can protect the retina from outer retinal degeneration.6–12 A retinal transcriptome analysis has demonstrated expression of serotonergic (5-HT), adrenergic, and dopaminergic GPCRs in the mammalian retina.6 Activation and inhibition of these common GPCRs are known to modulate diverse secondary pathways including adenylyl cyclase, small GTPase Ras homologs, phospholipase C (PLC), and regulation of ion channels, which are important for regulating cellular functions including metabolism, growth, proliferation, survival, transcription, and protein synthesis.13–15 However, it has yet to be determined if these pathways are activated/inhibited in the retina and account for the neuroprotection achieved with GPCR agonists/antagonists.
To further our ability to rationally develop therapies for photoreceptor cell death, we sought to investigate the cell signaling associated with sarpogrelate-mediated neuroprotection. Sarpogrelate, marketed as ANPLAG, is a 5-HT2A antagonist that has been used to treat patients with peripheral arterial disease in Japan, China, and South Korea.16,17 We have recently characterized sarpogrelate's neuroprotective effects in the retina and observed a dose-dependent protection against light-induced retinopathy.10 Many light-induced retinopathy methods have been established.4 Some include the use of fluorescent white light, broadband green light, or narrowband blue or green light.4 Depending on the intensity and duration of the varying light sources, degeneration rates and induction of cell death pathways can differ.4,18,19 For our studies we have consistently used an acute bright white light exposure, which has been well characterized as a model for studying apoptosis associated with retinal degeneration.4 In the current study, phosphorylation changes of proteins in two main GPCR pathways, mitogen-activated protein kinase/extracellular signal–related kinase (MAPK/ERK) and phosphoinositide 3-kinase/phosphoinositide-dependent kinase 1/protein kinase B (PI3K/PDK/AKT), were investigated to determine the signaling pathways playing a role in sarpogrelate-mediated neuroprotection.
Methods
Animals
All experiments and animal handling procedures were approved by and performed in compliance with the policies of the Institutional Animal Care and Use Committee at Oregon Health & Science University and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Male albino BALBc/J mice (The Jackson Laboratory, Bar Harbor, ME, USA) between 2 to 3 months of age were used. Mice were housed in a 12-hour alternating light/dark cycle room. The light cycle (∼15 lux) occurred from 9:00 PM to 9:00 AM, and dark cycle from 9:00 AM to 9:00 PM.
Injections
At 11:00 AM, 2 hours after the start of the dark cycle, mice were administered an intraperitoneal injection of 25 mg/kg sarpogrelate (dissolved in saline) (Tocris Bioscience, Bristol, UK) or equivalent volume of saline immediately before 1 hour of bright light exposure (LE).10,11 For MEK inhibitor experiments, mice were administered an intraperitoneal injection of 5 mg/kg PD0325901 (dissolved in distilled water with 1% dimethyl sulfoxide) (SelleckChem, Houston, TX, USA) or vehicle 15 minutes before sarpogrelate or saline injection. After LE, mice were returned to the dark until the appropriate time of retinal harvest, or returned back to the 12-hour alternating light/dark cycle room for future in vivo imaging and electrophysiology studies.
Light-Induced Retinopathy
A custom light box was built for the induction of retinopathy.10,11 It was able to hold up to 16 mice and produced approximately 10,000 lux of uniform light. After 2 hours of dark adaptation, albino BALB/c mice were exposed to 1 hour of light emitted by four compact fluorescent lamps (42 watts, 6500 K color temperature), which give off fluorescent white light having an emission spectrum similar to daylight.4,10,11 Room temperature was maintained within the box by using an air conditioning unit. For each experiment, mice injected with 0.9% sodium chloride were placed in the light box to serve as controls and to ensure retinopathy was induced.
Phosphorylation Antibody Array
At 3 hours post LE, phosphorylation of signaling proteins was analyzed with a high-throughput cAMP responsive element–binding protein (CREB) phosphorylation antibody array (PCR174; Full Moon Biosystems, Sunnyvale, CA, USA). Protease and phosphatase inhibitors (Thermo Scientific, Rockford, IL, USA) were added to the provided protein extraction buffer. Protein lysate was extracted, purified, and quantified by using UV absorbance spectroscopy (NanoDrop 2000 Spectrophotometer; Thermo Scientific). Protein samples underwent biotinylation while the array was immersed in blocking solution. A protein coupling mix was used to bind proteins to array. Finally, the array was fluorescently labeled with Cy3-streptavidin and scanned by the manufacturer (532 nm) (Full Moon Biosystems). Images were quantified with ImageJ MicroArray Plugin (OptiNav, Inc.; www.optinav.info/MicroArray_Profile.htm, in the public domain). For data analysis, background signals were removed from all measurements. A ratio, comparing phosphorylated protein to total protein, was calculated. Then experimental groups were compared to control groups to determine ratio changes in phosphorylation due to LE or sarpogrelate treatment. The experiments were performed independently two times, three animals per group.
Western Blot
Temporal phosphorylation changes were assessed by Western blot. For our experiments evaluating sarpogrelate without LE, retinas were collected 5 minutes, 20 minutes, 30 minutes, 1 hour 20 minutes, 6 hours, 12 hours, 24 hours, and 48 hours post sarpogrelate or saline injection. For our experiments evaluating sarpogrelate-mediated neuroprotection with LE, retinas were collected immediately after 20 minutes of LE, 1 hour of LE; or at 20 minutes, 1 hour, 3 hours, 7 hours, 24 hours, 48 hours, or 96 hours post LE. Retinas were harvested and sonicated in phosphate-buffered saline (1X PBS) containing protease and phosphatase inhibitors (Thermo Scientific). Loaded samples (200 μg) were separated on a 12% acrylamide gel and transferred onto a membrane overnight at 4°. The next day, membranes were incubated in Odyssey infrared imaging system blocking buffer (Li-Cor, Lincoln, NE, USA) for 1 hour. Blots were then labeled with the following rabbit polyclonal antibodies diluted at 1:1000: phospho-PKCβ Ser661 (A8169; Assay Biotechnology [AB], Sunnyvale, CA, USA), phospho-PKCα/βII Thr638/641 (9375; Cell Signaling Technology [CST], Danvers, MA, USA), phospho-MEK1/2 Ser217/221 (9121; CST), MEK1/2 (9122; CST), phospho-p44/42 MAPK (pERK1/2) Thr202/Tyr204 (9101; CST), p44/42 MAPK (ERK1/2) (9102; CST), phospho-PDK1 Ser241 (3438; CST), phospho-Akt Ser473 (9271; CST), Akt (9272; CST), phospho-CREB Ser133 (A7053; AB), B-cell lymphoma 2 (Bcl2, 2870; CST), and phospho-Fos Thr232 (A8226; AB). After overnight incubation in primary antibodies, blots were incubated in a goat anti-rabbit IRDye 680RD secondary antibody (Li-Cor) diluted at 1:15000 for 1 hour at room temperature. Finally, blots were imaged with an infrared imaging system (Odyssey, Odyssey 2.1 software; Li-Cor). As an internal control for protein loading, membranes were also probed with GAPDH (2118; CST) or eIF4E (9742; CST). Using optical density measurements obtained with Odyssey software, each sample was normalized to the loading control. Then experimental groups were compared to control groups to determine fold changes in phosphorylation due to LE or sarpogrelate treatment. The experiments were performed independently two times, two to three animals per group.
qPCR Array
Changes in gene expression were assessed with a customized RT2 Profiler PCR Array (SABiosciences, Germantown, MD, USA) that consisted of 84 genes involved in oxidative stress and cell death (see Supplementary Table S2). Briefly, retinas were harvested at 48 hours post LE and RNA was extracted from retina by using an RNeasy Mini Kit (QIAGEN, Germantown, MD, USA). Total RNA (0.5 μg) was converted to cDNA by using the RT2 first strand kit (SABiosciences). All samples were checked for DNA contamination and impurities that would affect PCR amplification by using the RT2 RNA QC PCR array (SABiosciences). Resulting cDNA was mixed with RT2 SYBR Green master mix (SABiosciences) and dispersed into the custom 96-well PCR array. Using a QuantStudio 3 Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) samples were initially denatured at 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. Gene expression presented was calculated in the QIAGEN GeneGlobe data analysis center (www.SAbiosciences.com/pcrarraydataanalysis.php, in the public domain). The data were also verified through manual ΔΔCT calculations.20 β-Actin was used as the internal control. The experiments were performed independently two times, three animals per group.
In Vivo Spectral-Domain Optical Coherence Tomography (SD-OCT)
Seven days following drug treatment and LE, in vivo SD-OCT imaging was performed as previously reported.10,11,21 Retinal layers were segmented for quantification of outer retinal thickness by using a custom-designed SD-OCT segmentation program built in IGOR Pro (IGOR Pro 6.37; WaveMetrics, Inc., Lake Oswego, OR, USA). Outer retinal thickness is defined as the thickness from Bruch's membrane to the inner nuclear layer/outer plexiform layer interface.
In Vivo Electrophysiology
Two to 7 days after SD-OCT acquisition, ERGs were performed as previously reported.10,11 Light intensities ranged from −4.34 to 3.55 log cd•s/m2 and ERG traces were averaged and analyzed by using custom software.
Statistical Analysis
All temporal Western blot data were plotted in Prism (Prism 6.0; GraphPad Software, Inc., LaJolla, CA, USA). To determine significant differences between groups at different time points, a 2-way ANOVA multiple comparisons test was performed. Average outer retinal thickness (REC+) was calculated for each animal and plotted in Prism as well. A 1-way ANOVA was used to compare REC+ in different treatment groups. For all analysis, a P value less than 0.05 was considered significant.
Results
Phosphorylation Changes Associated With Sarpogrelate Treatment
We have previously demonstrated the neuroprotective effects of the 5-HT2A antagonist, sarpogrelate, against bright light exposure.10 With this study, we sought to investigate the cell signaling associated with sarpogrelate-mediated neuroprotection (see Fig. 1 for experimental schematic). The 5-HT2A receptor is known to modulate both the MAPK/ERK and the PI3K/PDK/AKT pathway, which play prominent roles in cell survival.22–27 To investigate if these complex signaling changes also occur in the retina in association to neuroprotection, a high-throughput phosphorylation antibody array was used to quantify 63 specific phosphorylation sites on proteins important to the activation and deactivation of common signaling pathways (Table; Supplementary Table S1). Retinas from three groups of mice, namely, (1) saline injected, unexposed to light (Saline+No LE), (2) saline injected, exposed to light (Saline+LE), and (3) sarpogrelate injected, exposed to light (Sarp+LE), were harvested 3 hours after light exposure (LE, ∼10,000 lux, 1 hour) and used to calculate protein phosphorylation changes specifically due to LE and sarpogrelate treatment. At 3 hours post LE, sarpogrelate treatment led to an increase in phosphorylation of proteins critical for activation of the MAPK/ERK pathway, including PKCα/βII, PKCβ, c-Raf, Raf1, MEK1, ERK1/2, and SHC-transforming protein 1 (Shc) (Table). Downstream transcription factors including CREB and serum response factor (SRF) also showed increased phosphorylation of their main activation sites at 3 hours post LE (Table), indicating activation of these transcription factors. In most cases, sarpogrelate treatment reversed phosphorylation events driven by LE (Supplementary Table S1).
Figure 1.

Experimental methods schematic. A schematic of the experimental methods, illustrating the timing of the animals' light cycles, bright light exposure, and experimental interventions including sarpogrelate administration, pretreatment with MEKi, retina collection, and in vivo testing. Animals are on a 12-hour light-dark cycle. At 2 hours after the start of their dark cycle, sarpogrelate was administered while maintaining dark-adapted conditions (green arrow) immediately before LE. In the appropriate experiments, pretreatment with a MEKi (red arrow) was administered 30 minutes before sarpogrelate treatment also under maintained dark conditions. Bright LE (yellow arrow and bar) lasted 1 hour with 10,000 lux of light, and occurred 2 hours after the start of their dark cycle. Retinas were collected at the following time points for Western blot analyses (light blue bar): immediately after 20 minutes of LE, after the 1 hour of LE, and at +20 minutes, +1 hour, +3 hours, +7 hours, +24 hours, +48 hours, and +96 hours post LE. Retinas were collected at +3 hours post LE for phosphorylation array (dark blue bar, PhosphoArray) and +48 hours post LE for qPCR array (maroon bar). In other studies, animals were maintained for in vivo OCT and ERG testing, which occurred 7 to 10 days after LE (purple bar). // indicates time lapse; white and gray arrow bar indicates 12-hour light/dark cycle. Admin, administration; Sarp, sarpogrelate; OCT, optical coherence tomography.
Table.
Phosphorylation Changes Mediated by Sarpogrelate at 3 Hours Post Light Exposure
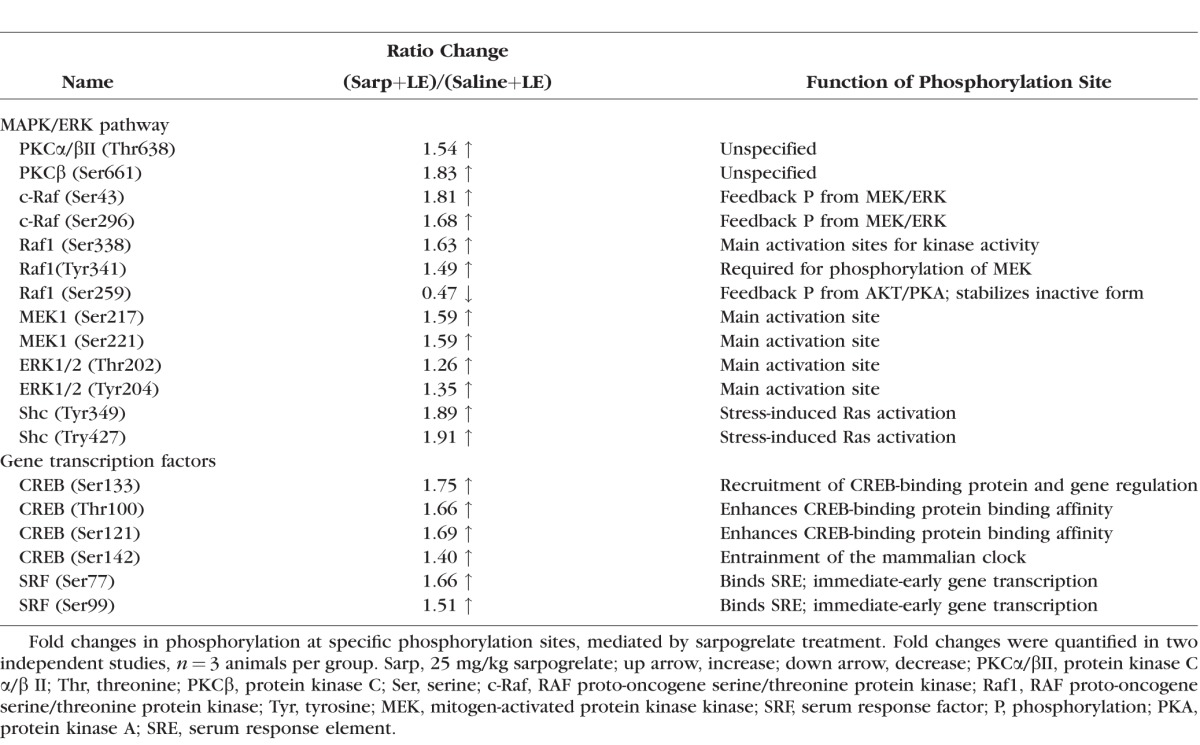
To confirm findings from the phosphorylation array and to assess temporal changes in cell signaling, Western blots for selected phosphorylated proteins were performed (Fig. 2, Supplementary Fig. S1). In addition, effects of sarpogrelate were assessed in mice not exposed to light (Fig. 2A, Supplementary Fig. S1A) and in mice that were exposed to light (Fig. 2B, Supplementary Fig. S1B) to determine if sarpogrelate-mediated cell signaling changes were affected by stress. In the absence of LE, sarpogrelate treatment caused an increase in phosphorylation of PKCβ Ser661 at 5 minutes post injection, an increase in phosphorylation of MEK Ser217/222 at 5 minutes, 20 minutes, and 1 hour 20 minutes post injection, and an increase in phosphorylation of ERK1/2 Thr202/Tyr204 at 1 hour 20 minutes post injection (P < 0.05; Fig. 2A). Further downstream, phosphorylation of CREB Ser133 was significantly increased at 24 hours post injection and total Bcl2 protein was increased at 48 hours post injection (P < 0.05; Fig. 2A).
Figure 2.

Sarpogrelate treatment results in transient MAPK/ERK activation. Line graphs showing average fold change in phosphorylation of key proteins in the MAPK/ERK pathway at multiple times post injection (A) and post light exposure (B). Data points are presented as mean ± standard error. A 2-way ANOVA multiple comparisons test was used to compare fold changes between Sarp+No LE and Saline+No LE groups in (A) and also used to compare fold change difference between Sarp+LE and Saline+LE in (B). *P ≤ 0.05. n = 3 to 5 animals per group. Sarp, 25 mg/kg sarpogrelate; p, phosphorylated; PKC, protein kinase C; MEK, mitogen-activated protein kinase kinase; ERK1/2, extracellular signal–related kinase 1/2.
Figure 2.

Continued.
Temporal effects of sarpogrelate differed with LE (Fig. 2B). Sarpogrelate treatment with LE did not show the immediate increase in phosphorylation of PKCβ Ser661, but rather a late increase at 7 hours post LE (P < 0.05; Fig. 2B). In addition, sarpogrelate treatment led to significantly decreased phosphorylation of MEK Ser217/222 at 20 minutes and 1 hour into LE, followed by a peak increase at 3 hours post LE (P < 0.05; Fig. 2B). Peak phosphorylated MEK levels returned to Saline+No LE baseline control levels by 7 hours through 96 hours post LE. Phosphorylation of ERK1/2 Thr202/Tyr204 followed the same temporal pattern as phosphorylation of MEK Ser217/222 with early inhibition, peak activation at 3 hours post LE, followed by return to baseline after 7 hours post LE (Fig. 2B). LE alone increased phosphorylation of ERK1/2 Thr202/Tyr204 at 3, 24, and 48 hours post LE. At 24 hours post LE, sarpogrelate treatment significantly reduced this increased phosphorylation of ERK1/2, restoring pERK1/2 back to control levels (P < 0.05; Fig. 2B). Further downstream, sarpogrelate treatment led to increased phosphorylated CREB Ser133 as compared to both Saline+LE mice and Saline+No LE mice at 3 hours and 96 hours post LE (P < 0.05; Fig. 2B). At the 48-hour time point, LE alone caused a significant decrease in phosphorylation of CREB Ser133, but sarpogrelate treatment increased phosphorylation, restoring pCREB levels to those observed in normal, nondamaged mice (P < 0.05; Fig. 2B).
Data from the phosphorylation antibody array suggested that PI3K and Akt were activated at 3 hours post LE (Supplementary Table S1). However, temporal Western blot analysis did not confirm these findings (Supplementary Fig. S2). For the most part, phosphorylated PDK1 Ser241 and AKT Ser473 levels stayed consistent with controls over time (Supplementary Fig. S2). Owing to this discrepancy, we cannot confirm activation of the PI3K/PDK/AKT pathway.
Functional Significance of the MAPK/ERK Pathway
To determine the neuroprotective role of the observed transient MAPK/ERK activation, ERK1/2 phosphorylation was blocked with a MEK inhibitor (MEKi), PD0325901 (Fig. 3). Pretreatment with MEKi effectively reduced levels of phosphorylated ERK1/2 by approximately 80% at 3 hours post LE, the time at which peak levels of phosphorylated ERK1/2 were observed (Fig. 3A). MEKi treatment did not affect phosphorylation of other signaling proteins such as protein kinase A regulatory subunit R2B (PKA-R2B) and Akt (Fig. 3A). Pretreatment with the MEKi (MEKi+Sarp+LE) led to a decrease in the neuroprotection achieved with sarpogrelate treatment (Vehicle+Sarp+LE) as assessed by measurements of outer retinal thickness, particularly evident in the superior and temporal quadrants of the retina (P < 0.05; Figs. 3B, 3C). The statistically significant reduction in retinal layer preservation, however, did not lead to a significant decrease in retinal function as measured by ERGs in MEKi-pretreated mice (Fig. 3D).
Figure 3.

MEKi treatment reduces sarpogrelate's neuroprotective effects. (A) Western blot analysis showing that MEKi treatment reduces phosphorylation of ERK1/2 by 80% at 3 hours post light exposure. (B) Representative SD-OCT images of superior retina from mice in each group. (C) Spider graphs of average outer retinal thickness demonstrate that MEKi treatment reduces preservation in the temporal and superior retinal quadrants. The gray area indicates ± 2 standard deviations of the Vehicle+Saline+No LE averaged data. Averaged quadrant data demonstrated that MEKi treatment resulted in a significant reduction in outer retinal thickness when compared to mice injected with sarpogrelate alone. Group averages are represented as mean ± standard error. An ordinary 1-way ANOVA was used to compare groups. *P ≤ 0.05. n = 4 to 9 animals per group. (D) ERG traces, average a-wave amplitudes, and average b-wave amplitudes show that sarpogrelate's effects on retinal function are not altered by MEKi treatment. Data points are represented as mean ± standard error. n = 4 to 9 animals per group. Sarp, 25 mg/kg sarpogrelate; PKA-R2B, protein kinase A regulatory subunit R2B.
Gene Expression Changes Associated With Sarpogrelate Treatment
Since pretreatment with the MEKi decreased sarpogrelate's neuroprotective effects, we hypothesized that MEKi treatment may also alter gene expression associated with cell death, linking upstream phosphorylation changes with downstream gene expression. Thus, a qPCR array was used to capture the expression of genes involved in oxidative stress, iron metabolism, apoptosis, autophagy, and necrosis at 48 hours post LE (Fig. 4; Supplementary Table S2). Pretreatment with MEKi alone did not alter gene expression (Supplementary Table S2). However, LE significantly altered the regulation of several genes associated with oxidative stress (Aox1, Cybb, Mb, Tpo, Ucp3), apoptosis (Bcl2, Bcl2a1a, Casp1, Casp7, Tnfrs1a, Xiap, Parp2), and iron metabolism (CP, Lcn2, Ppef2, Steap4, Trf) (P < 0.05, fold change > 2; Fig. 4). Treatment with sarpogrelate before LE reversed gene expression changes caused by LE (P < 0.05, fold change > 2; Fig. 4).
Figure 4.

Sarpogrelate treatment reverses gene expression changes associated with light exposure. Bar graphs reporting retinal oxidative stress (A), apoptosis (B), and iron metabolism (C) gene expression changes caused by light exposure and sarpogrelate treatment. The comparative CT method was used to determine gene expression changes associated with light exposure by comparing data from Vehicle+Saline+No LE and Vehicle+Saline+LE groups (black bar). To determine gene expression changes associated with sarpogrelate treatment, data from the Vehicle+Saline+LE and Vehicle+Sarp+LE groups were compared (striped bar). QIAGEN GeneGlobe data analysis center–calculated fold-regulation changes. Fold regulation greater than 2 represents a biologically significant change in gene expression. n = 6 animals per group. Sarp, 25 mg/kg sarpogrelate; Aox1, aldehyde oxidase 1; Cybb, cytochrome b-245 heavy chain; Mb, myoglobin; Tpo, thyroid peroxidase; Ucp3, mitochondrial uncoupling protein 3; Bcl2a1a, B-cell lymphoma 2–related protein a1; Casp1, caspase 1; Casp7, caspase 7; Tnfrsf1a, tumor necrosis factor receptor superfamily member 1a; Xiap, X-linked inhibitor of apoptosis protein; Parp2, poly [ADP-ribose] polymerase 2; CP, ceruloplasmin; Lcn2, lipocalin-2; Ppef2, protein phosphatase with EF-hand domain 2; Steap4, six-transmembrane epithelial antigen of prostate 4; Trf, transferrin.
Discussion
We have previously demonstrated the neuroprotective benefits of sarpogrelate in protecting against light-induced retinopathy.10 To further the development of such therapies for retinal degeneration, it is important to understand the neuroprotective mechanisms mediating cell survival. In this study, we examined the GPCR signaling pathways activated by sarpogrelate in a light-induced retinopathy model. Results from both phosphorylation antibody arrays and temporal Western blots demonstrated that sarpogrelate treatment results in a transient activation of multiple proteins in the MAPK/ERK pathway and consistent activation of CREB, a converging effector transcription factor highly relevant in neuronal survival and plasticity. Inhibition of ERK1/2 activation reduced but did not eliminate sarpogrelate's retinal neuroprotective effects. This suggests that the MAPK/ERK pathway plays an important role in neuroprotection, but that this pathway alone does not account for the full protective effect.
Previous studies have demonstrated that activation of 5-HT2A receptors with serotonin or a 5-HT2A agonist leads to activation of the MAPK/ERK pathway (Fig. 5).22,23 One study has evaluated signaling pathways in human embryonic kidney cells stably expressing 5-HT2A receptors. Upon activation with 5-HT2A receptor agonists, a sustained phosphorylation of ERK1/2 is observed, but when cells are treated with a 5-HT2A antagonist, phosphorylation of ERK1/2 becomes transient.23 In our in vivo studies, treatment with sarpogrelate, a 5-HT2A antagonist, also resulted in transient MAPK/ERK activation. Temporal dynamics following delivery of 25 mg/kg sarpogrelate before LE demonstrated a biphasic pattern of MEK/ERK1/2 phosphorylation (pMEK/pERK1/2). There was an initial reduction in pMEK/pERK1/2 at 20 minutes into LE, followed by a reversal with maximum phosphorylation at 3 hours post LE. By 7 hours post LE, pMEK/pERK1/2 returned to control levels (Fig. 2B).
Figure 5.

5-HT2A signaling pathways. Activation of the 5-HT2A receptor induces several pathways via the Gαq and Gβγ subunits. Green arrow indicates activation; small dashed green arrow indicates multiple signaling pathways not shown; large dashed green arrow indicates activation of transcription; red bar indicates inhibition. Light blue ovals signify proteins in the Akt pathway; orange ovals signify proteins in the MAPK/ERK pathway; yellow ovals signify proteins in the ROS pathway; purple ovals signify proteins in the DAG/IP3/Ca+2/CamK pathway. Purple squares in the nucleus signify transcription factors, apoptotic and antiapoptotic proteins. 5-HT, serotonin; 5-HT2A, serotonin 2A receptor; PIP2, phosphatidylinositol 4,5-biphosphate; IP3, inositol 1,4,5-trisphosphate; Ca+2, calcium; CamK, calmodulin-dependent protein kinase; Raf, mitogen-activated protein kinase kinase kinase; p90RSK, 90-kDa ribosomal S6 kinase; NADPH, nicotinamide adenine dinucleotide phosphate; ROS, reactive oxygen species; PI3K, phsophotidylinositol-3 kinase; PIP3, phosphatidylinositol 3,4,5-triphosphate; PDK1, phosphoinositide-dependent kinase 1; NFκB, nuclear factor κ-light-chain-enhancer of activated B cells; CRE, cAMP response element; SRF, serum response factor; SRE, serum response element; Bcl-XL, B-cell lymphoma–extra large; Bax, bcl-2–like protein 4; Bak, bcl-2 homologous antagonist/killer.
Activation of ERK1/2 has been extensively studied for its role in neuronal cell death and survival.24–27 Ultimately, studies suggest that the magnitude and the duration of ERK1/2 activation determines cellular outcomes.25 In our study, sarpogrelate-treated mice showed a transient ERK1/2 activation with onset at approximately 3 hours post LE and return to basal levels at 7 hours post LE, whereas LE alone showed a low magnitude, long duration of ERK1/2 activation with onset approximately 3 hours post LE and return to baseline at 96 hours post LE. We hypothesize that this sarpogrelate-mediated transient ERK1/2 activation plays a role in retinal neuroprotection, altering the ERK1/2 signaling that occurs in response to stress alone. In addition to our data, studies have shown that transient ERK1/2 activation reflects a prosurvival response.24–27 For example, in cultured hippocampal neurons, brain-derived neurotrophic factor–mediated neuroprotection involves increased phosphorylation of ERK1/2 from 2.5 minutes to 1 hour post administration and a decrease in ERK1/2 phosphorylation to basal levels 6 to 24 hours post administration.27
5-HT2A receptors are expressed in a variety of cell types. Thus, multiple studies have attempted to map 5-HT2A signaling in these various cell types including the muscular/vascular system, neuronal tissue, and kidney tissue with differences observed in varying cells.22 These studies22,23 have found at least five different 5-HT2A modalities to activate MAPK/ERK, which are demonstrated in Figure 5: (1) PLC/PKC-dependent raf activation (Fig. 5, orange pathway), (2) PLC/Ca+2CamK-dependent ERK activation (Fig. 5, purple pathway), (3) ROS-dependent MEK activation (Fig. 5, yellow pathway), (4) direct G protein activation of ras (Fig. 5, orange pathway from Gβγ), and (5) tyrosine kinase (src/shc)–dependent activation of ras (Fig. 5, blue pathway).22,23
The classical 5-HT2A activation of MAPK/ERK initiates through Gαq activation of PLC, which then hydrolyzes phosphatidylinositol 4,5-biphosphate (PIP2) to inositol triphosphate (IP3) and diacylglycerol (DAG), ultimately resulting in a release of intracellular calcium stores. The released calcium binds PKC, allowing for PKC docking at DAG and activation of raf. Activation of raf subsequently activates MEK1/2 and ERK1/2 (Fig. 5). Our phosphorylation array and Western blot data demonstrated activation of PKCβ Ser661 at time points consistent with MEK/ERK1/2 activation. This supports our hypothesis that transient MAPK/ERK activation occurs in a PLC/PKC-dependent manner (Fig. 2; Table). Our phosphorylation array data also showed activation of CamK2, CamK4, and shc, which support calmodulin- or tyrosine kinase–dependent activation of MAPK/ERK (Supplementary Table S1). These data are consistent with a study that has demonstrated PKC-independent and calmodulin- and tyrosine kinase–dependent MAPK/ERK activation in a well-characterized neuronal cell line.28
Once activated, potentially through multiple pathways, ERK1/2 may control cell survival or apoptosis by regulating the activity of anti- and pro-apoptotic transcription factors.26 ERK1/2-activated p90 ribosomal S6 kinase (p90RSK) phosphorylates CREB Ser133, which strongly enhances CREB-dependent transcription. Activated CREB can promote cell survival, whereas inhibition of CREB phosphorylation at Ser133 triggers neuronal apoptosis.26 We demonstrated that sarpogrelate significantly increases phosphorylation of CREB Ser133 at 3 hours and 96 hours post LE, indicating sustained CREB activation (Fig. 2B). Additionally, at 48 hours post LE, Western blot analysis indicated that light exposure inhibits phosphorylation of CREB Ser133, which could trigger apoptosis. Sarpogrelate treatment reverted phosphorylation of CREB levels back to control levels, which could prevent apoptotic signaling (Fig. 2B). Our data suggest that activation of CREB plays an important role in sarpogrelate-mediated neuroprotection.
CREB is a converging effector transcription factor that can be activated from multiple GPCR signaling pathways. In our study, when ERK1/2 activation was inhibited with MEK inhibitor PD0325901, sarpogrelate's neuroprotective effects were reduced, but not completely suppressed, suggesting that either (1) phosphorylation of CREB may occur through both ERK1/2-dependent and ERK1/2-independent pathways, or (2) additional important neuroprotective pathways may occur independently from ERK1/2 and CREB activation. We chose a second-generation PD0325901 inhibitor, which reduced ERK1/2 activation by 80%, significantly more potent than first-generation inhibitors.29 Although ERK1/2 activation was significantly reduced, outer retinal degeneration decreased by only 8%. We hypothesize that additional pathways are playing a role in neuroprotection. However, it is also possible that the remaining 20% of ERK1/2 activation was sufficient to elicit neuroprotective effects. Further inhibition of ERK1/2 was not possible without causing retinal toxicity (data not shown).
CREB Ser133 activation is further supported by downstream gene expression changes. At 24 hours post treatment and LE, genes regulated by CREB or that contain multiple CREB binding sites, including Bcl2, X-linked inhibitor of apoptosis protein (XIAP), and mitochondrial uncoupling protein 3 (UCP3), were significantly increased with sarpogrelate treatment (Fig. 4).30,31
While there are differences in cell death signaling and apoptotic pathways between light-induced retinopathy models depending on the quality, duration, and intensity of light and the animal model,4,18 our findings resulting from acute bright light exposure have been implicated in other light-induced retinopathy and inherited retinal degeneration studies.4 Bcl2 and XIAP both play important roles in inhibiting apoptosis, whereas UCP3 protects the mitochondria from induced oxidative stress. Bcl2, which localizes to the outer membrane of mitochondria, binds and inhibits proapoptotic proteins Bax and Bak, which usually promote apoptosis by altering mitochondrial functions and activating the release of downstream apoptogenic factors.32 Treatment with Bcl2 has been shown to protect mice from light-induced retinopathy and slows retinal degeneration in rd1, rd2, and S334ter RP mouse models.4 XIAP binds and inhibits caspases 3, 7, and 9 and further possesses E3 ubiquitin ligase activity that can promote the ubiquitination and degradation of caspases.33 Treatment with XIAP has been able to protect photoreceptors in both P23H and S334ter rhodopsin transgenic rat models of retinitis pigmentosa.4,33
In summary, we showed that saprogrelate-mediated neuroprotection involves a transient activation of the MAPK/ERK pathway and sustained activation of CREB, leading to transcription of antiapoptotic genes Bcl2, Xiap, and Ucp3. Future studies will continue to map changes in signaling proteins with GPCR activation/inhibition and downstream gene expression changes. This method will not only provide specific and novel therapeutic targets for retinal cell death, but also aid in developing systems pharmacology therapeutics for retinal degeneration.
Supplementary Material
Acknowledgments
Supported by an unrestricted departmental funding from Research to Prevent Blindness (New York, NY, USA), Grant P30 EY010572 from the National Institutes of Health (Bethesda, MD, USA), K08 Career Development Award (K08 EY021186, MEP), Alcon Young Investigator Award (MEP), Foundation Fighting Blindness Enhanced Research and Clinical Training Award (CD-NMT-0914-0659-OHSU, MEP), Career Development Award from Research to Prevent Blindness (MEP), and Knights Templar Eye Foundation Career Starter Grant (RCR).
Disclosure: C.A. Ku, None; R.C. Ryals, None; D. Jiang, None; A.S. Coyner, None; K.K. Weller, None; W. Sinha, None; B.M. Robb, None; P. Yang, None; M.E. Pennesi, None
References
- 1. Daiger SP,, Bowne SJ,, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007; 125: 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pierce EA. Pathways to photoreceptor cell death in inherited retinal degenerations. Bioessays. 2001; 23: 605–618. [DOI] [PubMed] [Google Scholar]
- 3. Sancho-Pelluz J, Arango-Gonzalez B, Kustermann S, et al. et al . Photoreceptor cell death mechanisms in inherited retinal degeneration. Mol Neurobiol. 2008; 38: 253–269. [DOI] [PubMed] [Google Scholar]
- 4. Wenzel A,, Grimm C,, Samardzija M,, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005; 24: 275–306. [DOI] [PubMed] [Google Scholar]
- 5. Chen Y,, Palczewski K. Systems pharmacology links GPCRs with retinal degenerative disorders. Annu Rev Pharmacol Toxicol. 2016; 56: 273–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Y, Palczewska G, Mustafi D, et al. et al . Systems pharmacology identifies drug targets for Stargardt disease-associated retinal degeneration. J Clin Invest. 2013; 123: 5119–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Y, Palczewska G, Masuho I, et al. et al . Synergistically acting agonists and antagonists of G protein-coupled receptors prevent photoreceptor cell degeneration. Sci Signal. 2016; 9: ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Collier RJ, Patel Y, Martin EA, et al. et al . Agonists at the serotonin receptor (5-HT(1A)) protect the retina from severe photo-oxidative stress. Invest Ophthalmol Vis Sci. 2011; 52: 2118–2126. [DOI] [PubMed] [Google Scholar]
- 9. Thampi P, Rao HV, Mitter SK, et al. et al . The 5HT1a receptor agonist 8-Oh DPAT induces protection from lipofuscin accumulation and oxidative stress in the retinal pigment epithelium. PLoS One. 2012; 7: e34468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tullis BE, Ryals RC, Coyner AS, et al. et al . Sarpogrelate, a 5-HT2A receptor antagonist, protects the retina from light-induced retinopathy. Invest Ophthalmol Vis Sci. 2015; 56: 4560–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coyner AS, Ryals RC, Ku CA, et al. et al . Retinal neuroprotective effects of flibanserin, an FDA-approved dual serotonin receptor agonist-antagonist. PLoS One. 2016; 11: e0159776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biswal MR, Ahmed CM, Ildefonso CJ, et al. et al . Systemic treatment with a 5HT1a agonist induces anti-oxidant protection and preserves the retina from mitochondrial oxidative stress. Exp Eye Res. 2015; 140: 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oldham WM,, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008; 9: 60–71. [DOI] [PubMed] [Google Scholar]
- 14. Gilman AG. G proteins and dual control of adenylate cyclase. Cell. 1984; 36: 577–579. [DOI] [PubMed] [Google Scholar]
- 15. Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987; 56: 615–649. [DOI] [PubMed] [Google Scholar]
- 16. Shinohara Y, Nishimaru K, Sawada T, et al. et al . Sarpogrelate-aspirin comparative clinical study for efficacy and safety in secondary prevention of cerebral infarction (S-ACCESS): a randomized, double-blind, aspirin-controlled trial. Stroke. 2008; 39: 1827–1833. [DOI] [PubMed] [Google Scholar]
- 17. Uchiyama S,, Ozaki Y,, Satoh K,, Kondo K,, Nishimaru K. Effect of sarpogrelate, a 5-HT(2A) antagonist, on platelet aggregation in patients with ischemic stroke: clinical-pharmacological dose-response study. Cerebrovasc Dis. 2007; 24: 264–270. [DOI] [PubMed] [Google Scholar]
- 18. Hao W, Wenzel A, Obin MS, et al. et al . Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat Genet. 2002; 32: 254–260. [DOI] [PubMed] [Google Scholar]
- 19. Wenzel A,, Reme CE,, Williams TP,, Hafezi F,, Grimm C. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci. 2001; 21: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmittgen TD,, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008; 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- 21. Pennesi ME, Michaels KV, Magee SS, et al. et al . Long-term characterization of retinal degeneration in rd1 and rd10 mice using spectral domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2012; 53: 4644–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Masson J,, Emerit MB,, Hamon M,, Darmon M. Serotonergic signaling: multiple effectors and pleiotropic effects. WIREs Membr Transp Signal. 2012; 1: 685–713. [Google Scholar]
- 23. Chang CW,, Poteet E,, Schetz JA,, Gumus ZH,, Weinstein H. Toward a quantitative representation of the cell signaling mechanisms of hallucinogens: measurement and mathematical modeling of 5-HT1A and 5-HT2A receptor-mediated ERK1/2 activation. Neuropharmacology. 2009; 56 (suppl 1): 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roskoski R., Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012; 66: 105–143. [DOI] [PubMed] [Google Scholar]
- 25. Subramaniam S,, Unsicker K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010; 277: 22–29. [DOI] [PubMed] [Google Scholar]
- 26. Lu Z,, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006; 58: 621–631. [DOI] [PubMed] [Google Scholar]
- 27. Almeida RD, Manadas BJ, Melo CV, et al. et al . Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005; 12: 1329–1343. [DOI] [PubMed] [Google Scholar]
- 28. Quinn JC,, Johnson-Farley NN,, Yoon J,, Cowen DS. Activation of extracellular-regulated kinase by 5-hydroxytryptamine(2A) receptors in PC12 cells is protein kinase C-independent and requires calmodulin and tyrosine kinases. J Pharmacol Exp Ther. 2002; 303: 746–752. [DOI] [PubMed] [Google Scholar]
- 29. Iverson C, Larson G, Lai C, et al. et al . RDEA119/BAY 869766: a potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res. 2009; 69: 6839–6847. [DOI] [PubMed] [Google Scholar]
- 30. Mayr B,, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001; 2: 599–609. [DOI] [PubMed] [Google Scholar]
- 31. Zhang X, Odom DT, Koo SH, et al. et al . Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005; 102: 4459–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cottet S,, Schorderet DF. Triggering of Bcl-2-related pathway is associated with apoptosis of photoreceptors in Rpe65-/- mouse model of Leber's congenital amaurosis. Apoptosis. 2008; 13: 329–342. [DOI] [PubMed] [Google Scholar]
- 33. Leonard KC, Petrin D, Coupland SG, et al. et al . XIAP protection of photoreceptors in animal models of retinitis pigmentosa. PLoS One. 2007; 2: e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.