

ABSTRACT
We report the case of a 76-year-old woman presenting with 47-month history of progressive dementia and cortical blindness with no family history. Antibodies against thyroid glands and the N-terminus of α-enolase (NAE) were detected in her serum. Neurological examination revealed progressive dementia, frontal signs, visual disturbance, and exaggerated bilateral tendon reflexes in both legs. Diffusion MRI showed cortical hyper-intensities in the bilateral occipital and parietal, and the left frontal and temporal cortices. 99mTc-ethyl cysteinate dimer-single photon emission computed tomography indicated decreased regional cerebral blood flow throughout the bilateral parietal lobes and partially in the left frontal and temporal lobes. PRNP gene analysis showed no mutations with methionine homozygosity at codon 129 in peripheral blood. Cerebrospinal fluid examination, including 14-3-3 and total tau protein detection, revealed normal levels; however, prion proteins were amplified by the real-time quaking-induced conversion method. Hashimoto's encephalopathy was excluded on the basis of unresponsiveness to corticosteroids. The symptoms progressed slowly. Periodic sharp-wave complexes were observed on electroencephalogram 36 months after the onset of symptoms; the patient reached a state of akinetic mutism at 47 months. This was a probable case of MM2-cortical-type sCJD with anti-NAE antibodies based on the World Health Organization (WHO) diagnostic criteria for sCJD, genetic information, and the slowly progressive course. However, this case did not meet with the probable WHO diagnostic criteria until 3 years after symptom onset, highlighting the difficulty of diagnosing a living case of the MM2-type of sCJD. Therefore, establishment of clinical diagnostic criteria for MM2-type of sCJD is required.
KEYWORDS: anti-N-terminus of α-enolase antibody, corticosteroid, Hashimoto encephalopathy, MM2-cortical-type, MRI, real-time quaking-induced conversion assay, sporadic Creutzfeldt-Jakob disease
INTRODUCTION
MM2-cortical-type sporadic Creutzfeldt-Jakob disease (sCJD) is a fatal dementia that presents with relatively slow progression. The frequency of MM2-cortical-type sCJD is reported to be 2% of sCJD cases in the Caucasian population and 6.7% of sCJD in the Japanese population.1 Early clinical symptoms of CJD may overlap with those of Hashimoto's encephalopathy, an autoimmune-mediated encephalopathy. Therefore, Hashimoto's encephalopathy is an important differential diagnosis in treating CJD. Presence of anti-N-terminus of α-enolase antibodies (anti-NAE Abs) has been reported to be a diagnostic marker of Hashimoto's encephalopathy.2,3 In addition, 3% of patients with Hashimoto's encephalopathy with anti-NAE Abs present with progressive dementia mimicking CJD, the so called CJD-type Hashimoto's encephalopathy.4 In contrast, low titer of neuronal antibodies associated with immune-mediated encephalopathy (anti-voltage gated potassium channel-complex [VGKC complex], anti-N-Methyl-D-Aspartate Receptor, or anti-glycine receptor [GlyR] antibodies) were detected in several cases of sCJD.5 Neuronal antibodies occur rarely in patients with suspected sCJD, and when present, this diagnosis should be interpreted with caution. Therefore, clinical follow-up and responsiveness to immunotherapy are crucial. Herein, we report the clinical findings of a probable case of MM2-cortical-type sCJD with anti-NAE Abs.
METHODS AND RESULTS
Patient Characteristics and Clinical Course
A 76-year-old Japanese woman was admitted to our hospital with a 5-month history of dementia. She had no familial history of central nervous system disease including prion disease. Neurological examination revealed a progressive dementia with a revised Hasegawa dementia scale (HDS-R) score of 6/30. The results of routine laboratory tests were normal, and endocrine tests showed a free T3 level of 2.17 pg/ml, free T4 level of 1.03 ng/dl, and TSH level of 1.06 μIU/ml. Two months later, she reported blurred vision on admission. Brain diffusion-weighted MRI showed hyper-intense areas in the bilateral occipital and parietal cortices, and left temporal and frontal cortices (Fig. 1A). An easy Z-score (eZIS) analysis of 99mTc-ethyl cysteinate dimer-single photon emission computed tomography (99mTc-ECD-SPECT) revealed (Fujifilm RI Pharma, Tokyo, Japan) decreased regional cerebral blood flow (rCBF) in the bilateral parietal lobes with left-sided predominance; decreases were also observed in a portion of the left temporal and frontal lobes (Fig. 1B and C).
FIGURE 1.
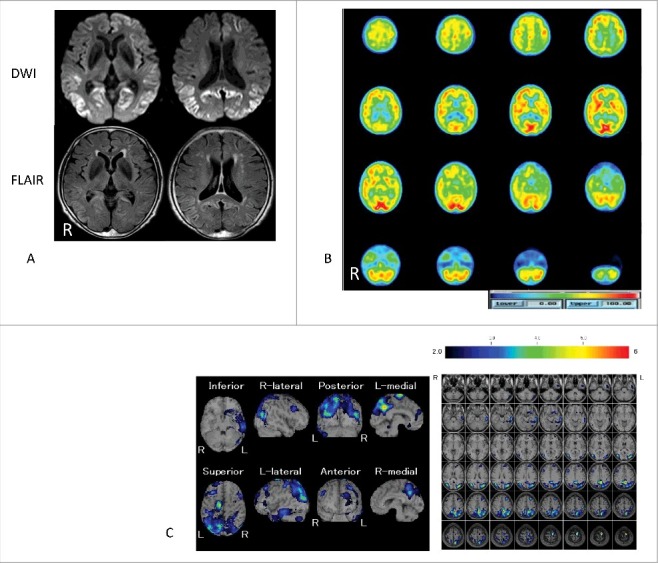
Images of MR and 99mTc-ethyl cysteinate dimer-single photon emission computed tomography (99mTc-ECD-SPECT) 7 months after symptom onset. Panel A: Diffusion-weighted images (DWI) and fluid-attenuated inversion recovery (FLAIR) images of MR; Panels B and C: Plain images and easy Z-score analysis images of 99mTc-ECD-SPECT. In panel A, DWI shows apparent hyper-intensity areas in the bilateral occipital, parietal, and partial left frontal and temporal cortices. Additionally, FLAIR images showed slightly hyper-intense area in these cortices. In panel B, the scale bar from 0 to 100 is indicated by the blue to red (higher regional cerebral blood flow [rCBF]) color gradient. In panel C, easy Z-score analysis images of 99mTc-ECD-SPECT reveal decreased rCBF bilaterally in the parietal lobes with left-sided predominance and partial decreases in the left temporal and frontal lobes. A higher Z-score scale indicates a lower rCBF. The Z-score scale of 2 to 6 is indicated by the black to red (lower rCBF) color gradient.
The patient was readmitted to our hospital 8 months after the onset of initial symptoms. Neurological examination revealed progressive dementia (HDS-R of 4/30), bilateral forced grasping, cortical visual disturbance, and exaggerated bilateral tendon reflexes in the lower extremities without Babinski's signs. Convulsions were not observed. Endocrine tests revealed a free T3 level of 1.78 pg/ml, free T4 level of 0.89 ng/dl, and TSH level of 2.35 μIU/ml. Anti-thyroid peroxidase (17.50 U/ml, NR < 3.2 U/ml) and anti-thyroglobulin (2.8 U/ml, NR < 0.3 U/ml) antibodies were positive. These anti-thyroid glands antibodies were analyzed by electro chemiluminescence immunoassay (SRL Inc., Tokyo, Japan). Thyroid aspiration biopsy confirmed subclinical Hashimoto thyroiditis with mild lymphocyte infiltration. Brain diffusion-weighted images (DWI) of MR showed cortical hyper-intense areas bilaterally in the occipital and parietal cortices, and also in the left frontal and temporal cortices at 8 months after symptom onset. An eZIS analysis of 99mTc-ECD-SPECT revealed an additional decrease in rCBF in the parietal lobes from those in the initial examination. Periodic sharp-wave complexes (PSWCs) were not observed on the electroencephalogram (EEG). Routine cerebrospinal fluid (CSF) analysis revealed normal cell (1 /μl), protein (40 mg/dl), and glucose levels (72 mg/dl). CSF work-up for prion disease was negative for both 14-3-3 and total tau proteins. PRNP gene analysis of peripheral blood DNA revealed methionine homozygosity at codon 129, with no mutations.
Detection of Anti-NAE Abs and Immunotherapy
Immunoblotting analysis of serum anti-NAE Abs was performed using NAE recombinant protein expressed in human cultured cells, as described previously.3 Briefly, recombinant NAE was expressed in cultured HEK 293 cells and purified through a His column.3 This analysis was evaluated using 1/320 diluted serum from the patient, because anti-NAE Abs titer in normal control was below 1/80. Immunoblot analysis with anti-NAE Abs obtained from the patient's serum at the first admission revealed a weak positive band at 29 kDa (Fig. 2). On the basis of detecting anti-NAE Abs in her serum, we suspected that the patient had Hashimoto encephalitis. Despite the patient receiving two courses of steroid pulse therapy (methylprednisolone 1,000 mg/d, 3d per course) and tapering from 40 mg/d to off within 4 weeks, her symptoms gradually deteriorated. Given this unresponsiveness to corticosteroids, the diagnosis of Hashimoto's encephalopathy was excluded.
FIGURE 2.
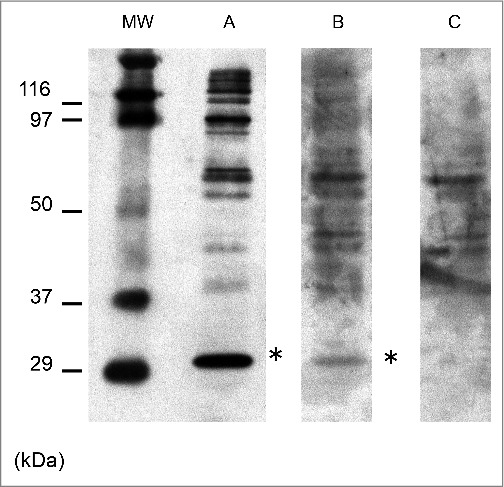
Immunoblot analysis of the anti-N-terminus of α-enolase (NAE) antibodies. Anti- NAE antibodies (*) were detected in serum obtained from the patient 8 months after symptom onset. MW: the molecular weight marker; A: the serum obtained from Hashimoto encephalitis with anti- NAE antibodies as a positive control; B: the serum obtained from the present case 8 months after the onset; C: the serum obtained from the anti-NAE antibodies negative patient as a negative control. The bands at 97 kDa or above indicate immunological reactivity with derivatives from HEK293 cells, which were used to generate recombinant NAE protein. Bands greater than 97 kDa correspond to non-specific reactions with sera and appeared variously across samples. These bands were confirmed to be non-immunologically reactive with anti-His antibody, which could be used to verify expression of the recombinant NAE protein.2,3
Result of Real-time Quaking-Induced Conversion and Long-Term Follow-Up
Later, we performed a real-time quaking-induced conversion (RT-QUIC) assay using a CSF sample obtained 7 months after the onset of the initial symptom. The RT-QUIC assay involved the use of soluble recombinant PrP as a substrate, which was seeded with PrPsc and then subjected to intermittent automated shaking as described previously.12 We obtained a positive result (Fig. 3).
FIGURE 3.
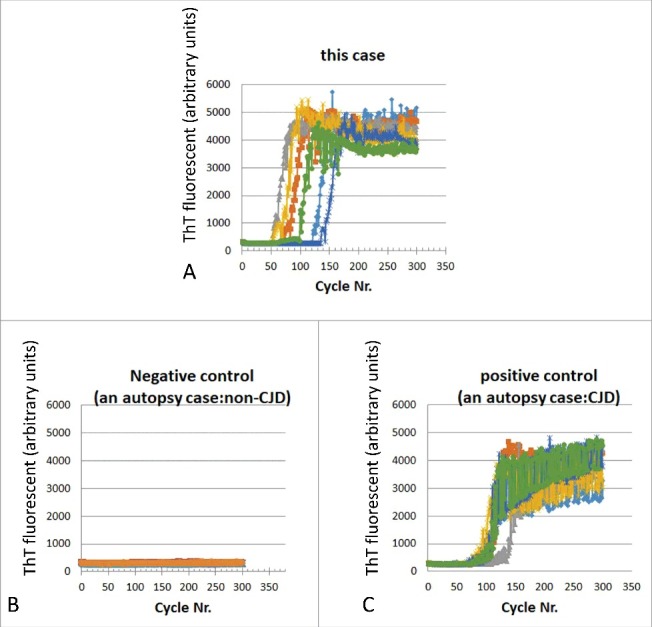
Real-time quaking-induced conversion (RT-QUIC) assay. (A) RT-QUIC cerebrospinal fluid sample obtained 7 months after the onset of initial symptoms from the present case, (B) Negative control (autopsy-verified case of non-CJD), and (C) Positive control (autopsy-verified case of CJD).
The patient was discharged from our hospital. Thereafter, we carried out a long-term follow-up the patient using a multidisciplinary approach described previously.6 The patient received chronic care services in a nursing home, and later entered into a chronic care hospital. Serial MRI revealed progressive brain atrophy. Serial diffusion-weighted MR images showed that cortical hyper-intense areas extended to frontal and temporal cortices and bilateral basal ganglia from 7 months to 37 months post-symptom onset. DWI signals were diminished in the occipital cortices at 37 months compared with those at 7 and 18 months. In the frontal and temporal cortices, signals were diminished at 39 months compared with those at 37 months (Fig. 4). Additionally, serial EEG revealed PSWCs from 36–46 months after the onset of symptoms (the terminal-phase of her disease course) (Fig. 5), but none were found in the early-phase. Convulsions or non-convulsion epileptic seizures were not observed in her clinical course. She could take fully-supported diet orally until 46 months after the onset of symptoms. Finally, she reached a state of akinetic mutism 47 months after the onset of symptoms, and she died of aspiration pneumonia. Unfortunately, consent to postmortem analysis could not be obtained.
FIGURE 4.
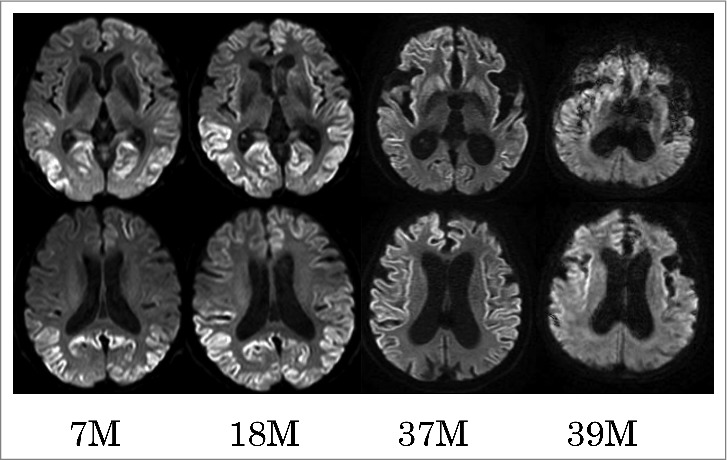
Serial diffusion-weighted images of MR. Serial diffusion-weighted images (DWI) obtained at 7, 18, 37, and 39 months (M) after the onset of initial symptoms. The upper panel shows serial MR images at the level of the basal ganglia, and the lower panel shows those at the slice of corona radiata. Serial DWI showed that cortical hyper-intense area extended across frontal and temporal cortices and bilateral basal ganglia from 7 months to 37 months. DWI signals were diminished in the occipital cortices at 37 months, compared with those at 7 or 18 months, and in the frontal and temporal cortices at 39 months versus 37 months.
FIGURE 5.
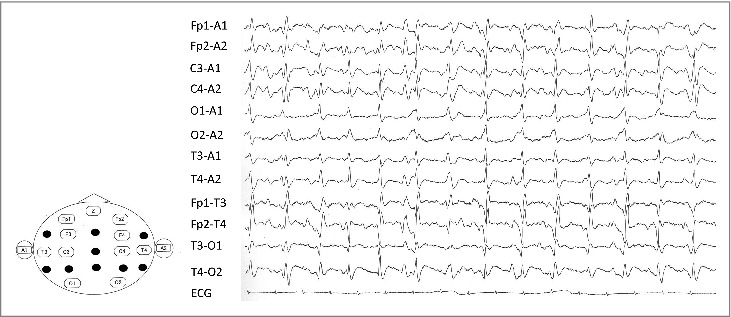
Electroencephalogram (EEG) 36 months after the onset. Periodic sharp-wave complexes, which observed on the EEG 36 months after the onset of initial symptoms.
DISCUSSION
We reviewed 4 reported cases of CJD-type Hashimoto's encephalopathy and our case (Table 1).7-10 Responsiveness to corticosteroids is a pivotal differentiator between CJD-type Hashimoto's encephalopathy and CJD with anti-NAE Abs. A work-up that includes thyroid hormone tests and anti-thyroid glands antibodies is a crucial first step diagnosing progressive dementia including Hashimoto's encephalopathy. If clinicians encounter a patient with progressive dementia having positive anti-thyroid glands antibodies, confirmation of steroid-responsiveness and long-term follow-up are required. Although detection of anti-NAE Abs has high specificity as a diagnostic marker for Hashimoto's encephalitis, this diagnosis was excluded in the current case because of unresponsiveness to corticosteroids. Several similar cases of CJD with other autoantibodies associated with immune-mediated encephalopathy have been reported.5 The frequency of such cases was estimated to be < 5% of sCJD, and the titer of autoantibodies in prion disease were lower than in autoimmune-mediated encephalopathy.5 The mechanism of appearance of autoantibodies remains unknown, but it postulated to result in extensive and rapid neuronal destruction, as indicated by a sCJD case in which both anti-VGKC-complex and anti-GlyR antibodies were identified.5 This study also highlighted that neuronal antibodies occur only rarely in patients with suspected sCJD, and when present, the diagnosis should be interpreted with caution.5
TABLE 1.
Clinical findings of CJD-type of Hashimoto's encephalopathy and CJD with anti-NAE antibodies.
| Author (year) | Age of onset (y) / sex | Consciouness disturbance/ progressive dementia | Myoclonus | Convulsion/ seizure | Serum anti-thyroid Abs | Serum anti-NAE Abs | EEG | Hyper-intense area on DWI | 14-3-3 protein/ total tau protein in the CSF | Result of RT-QUIC assay of the CSF | PRNP gene information | Reponsivenss to corticosteroids | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HE 1 | Wilhelm-Gössling (1998) | 58/F | +/N.A. | + | +/+ | TPO+, Tg+ | N.A. | Triphagic wave | N.A. | N.A. | N.A. | N.A. | + | |
| HE 2 | Sakurai (2007) | 76/F | −/+ → +/+ (early-phase) | + | +/+ | TPO+, Tg+ | + | PSWCs | − | −/− | N.A. | No muration, codon 129 M/M, 219 E/E | + | |
| HE 3 | de Cerqueria (2008) | 68/F | +/+ | + | +/+ | TPO+ | N.A. | Triphagic wave | − | N.A. | N.A. | N.A. | + | |
| HE 4 | Muramatsu (2013) | 57/M | +/+ | + | +/+ | TPO+, Tg+ | + | PSWCs | Bilateral frontal cortices and thalamus | +/N.A. | N.A. | No mutation | + | |
| CJD with anti-NAE Abs |
Present case |
76/F |
−/+ |
− (early-phase) → + (terminal-phase) |
−/− |
TPO+, Tg+ |
+ |
slow wave (early-phase) → PSWCs (terminal-phase) |
Bilateral occipital, parietal, left frontal and temporal cortices |
−/− |
+ |
No mutation, codon 129 M/M, 219 E/E |
− |
|
| MM2C sCJD | 64.3 (49-77)1) | −/+(relatively slow progressive course) → +/+ | + (67%)1) | N.A. | N.A. | N.A. | +or − (0-42%)1,20,23) | + (~100%)11,20) | + (30-91%)11,20, 21, 23, 24)/ + (40%)21) | + (44.4%)21) | no mutation, codon 129 M/M | − |
Abs: antibodies; anti-NAE Abs: anti-N-terminal alpha enolase antibodies; CJD: Creutzfeldt-Jakob disease; CSF: cerebrospinal fluid; DWI: diffusion-weighted image; EEG: electroencephalogram; F: female; HE: Hashimoto's encephalopathy; M: male; N.A.; not available or no discription; PSWCs: periodic sharp-wave complexes; RT-QUIC: real-time quaking-induced conversion; y: year-old; +: positive; -: negative.
Even today, prion disease is untreatable and fatal. If an autoimmune-mediated encephalopathy case is misdiagnosed as a prion disease, the patient's symptoms would not be improved due to under-treatment. Therefore, when clinicians encounter a patient presenting progressive neuropsychiatric symptoms with a low-titer of neuronal antibodies, at least first-line immunotherapy should be administered, and patient responsiveness assessed before conclusively making a diagnosis of prion disease. In our case, long-term follow-up demonstrated that the patient's clinical course was similar to MM2-cortical-type sCJD.1,11 However, information from pathological analysis and/or western blot analysis of prion protein was not obtained Ultimately, we clinically diagnosed the patient as a probable case of MM2-cortical-type sCJD based on the slowly progressive course, the results of gene analysis, and the WHO diagnostic criteria for sCJD.
Retrospectively, we found that the RT-QUIC results specifically supported the diagnosis of sCJD in the early-phase of the disease. The RT-QUIC assay is relatively new,12 and reportedly provides a sensitivity of 85% and specificity of 99% for diagnosing CJD.13 False-positives resulting from this assay are rarely reported,13-16 and are often unaccompanied by cortical hyper-intensities on DWI except in convulsion cases.13,16 Cortical hyper-intensities in DWI caused by convulsions or seizures often present as edematous and are usually diminished within one month.17 In contrast, our case did not show such a reversible radiological course (Fig. 3). Long-term follow-up studies including serial MRI never supported a clinical diagnosis of frontotemporal dementia, Alzheimer's disease, dementia with Lewy bodies, or encephalopathy with convulsion. Additionally, findings from our patient's serial MRI and SPECT studies indicating initial and widespread occipital involvement were different from those observed in MM2-thalamic-type sCJD18 or V180I genetic CJD.19
Finally, we summarize the case from initial symptoms to the major clinical symptoms manifested over the course of the disease. The initial symptoms indicative of dementia were the only neuropsychiatric symptom in the initial 6 months after onset, and later was accompanied by blurred vision at 7 months after onset of symptoms. Pyramidal signs were detected at 8 months after onset, extrapyramidal signs were detected by 24 months after onset, and myoclonus was finally observed in 36 months after onset. PSWCs were not observed until 36 months after onset. This case did not satisfy the WHO diagnostic criteria for sCJD in the initial 6-month of the disease course because of isolated progressive dementia. In addition, the case did not comply as a probable item on the WHO diagnostic criteria for sCJD until 3 years after the onset of the initial symptoms. Clinical characteristics of MM2-cortical type sCJD for this and previous cases are summarized in Table 1.1,11,20-24 As a rule, clinical progression of MM2-type sCJD is relatively slower than that of MM1-type sCJD.1,11,20 In the early-phase of MM2-cortical type sCJD, we have often encountered such an isolated progressive dementia with supportive MRI findings; however, a results of CSF work-ups were not supportive of a diagnosis of prion disease. Clinicians often have difficulty in making an early diagnosis of MM2-type sCJD. Therefore, establishment of specific clinical diagnostic criteria for MM2-type sCJD is required.
ABBREVIATIONS
- anti-NAE Abs
anti-N-terminus of α-enolase antibodies
- CJD
Creutzfeldt-Jakob disease
- CSF
cerebrospinal fluid
- DW
diffusion-weighted
- EEG
electronic encephalogram
- FLAIR
fluid-attenuated inversion recovery
- NSE
neuron-specific enolase
- PSWCs
periodic sharp-wave complexes
- RT-QUIC
real-time quaking-induced conversion
- sCJD
sporadic Creutzfeldt-Jakob disease.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
ACKNOWLEDGMENTS
We thank Dr. Masumi Nawata, Gihoku Kousei Hospital for providing chronic care to the patient, and Dr. Nobuaki Yoshikura, Department of Neurology and Geriatrics, Gifu University Graduate School of Medicine for technical advice.
FUNDING
This work was supported by grants-in-aid from the Research Committee of Prion Disease and Slow Virus infection (for TK, and KS), and from the Research Committee of Prion Disease Surveillance (for TK, KS, and TI), the Ministry of Health, Labour and Welfare of Japan.
REFERENCES
- 1.Iwasaki Y. Creutzfeldt-Jakob disease. Neuropathology. 2017;37:174-88. doi: 10.1111/neup.12355. PMID:28028861 [DOI] [PubMed] [Google Scholar]
- 2.Yoneda M, Fujii A, Ito A, Yokoyama H, Nakagawa H, Kuriyama M. High prevalence of serum autoantibodies against the amino terminal of alpha-enolase in Hashimoto's encephalitis. J Neuroimmunol. 2007;185:195-200. doi: 10.1016/j.jneuroim.2007.01.018. PMID:17335908 [DOI] [PubMed] [Google Scholar]
- 3.Fujii A, Yoneda M, Ito T, Yamamura O, Satomi S, Higa H, Kimura M, Suzuki M, Yamashita M, Yuasa T, et al.. Autoantibodies against the amino terminal of alpha-enolase are a useful diagnostic marker of Hashimoto's encephalitis. J Neuroimmunol. 2005;162:130-6. doi: 10.1016/j.jneuroim.2005.02.004. PMID:15833368 [DOI] [PubMed] [Google Scholar]
- 4.Yoneda M. Hashimoto's encephalopathy and autoantibodies. Brain Nerve. 2013;65:365-76. PMID:23568984 [PubMed] [Google Scholar]
- 5.Rossi M, Mead S, Collinge J, Rudge P, Vincent A. Neuronal antibodies in patients with suspected or confirmed sporadic Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiartry. 2015;86:692-4. doi: 10.1136/jnnp-2014-308695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayashi Y, Inuzuka T. A multidisciplinary medical network approach is crucial for increasing the number of autopsies for prion disease [Reply to: How can we increase the numbers of autopsies for prion disease? A model system in Japan]. J Neurol Sci. 2017;377:95-6. doi: 10.1016/j.jns.2017.04.004. PMID:28477717 [DOI] [PubMed] [Google Scholar]
- 7.Sakurai T, Tanaka Y, Koumura A, Hayashi Y, Kimura A, Hozumi I, Yoneda M, Inuzuka T. Case report of a patient with Hashimoto's encephalopathy associated with Basedow's disease mimicking Creutzfeldt-Jakob disease. Brain Nerve. 2008;60:559-65. PMID:18516979 [PubMed] [Google Scholar]
- 8.Wilhelm-Gössling C, Weckbecker K, Brabant EG, Dengler R. Autoimmune encephalopathy in Hashimoto's thyroiditis. A differential diagnosis in progressive dementia syndrome. Dtsch Med Wochenschr. 1998;123:279-84. doi: 10.1055/s-2007-1023949. PMID:9528645 [DOI] [PubMed] [Google Scholar]
- 9.de Cerqueria AC, Bezerra JM, de Magalhaes GC, Rozenthal M, Nardi AE. Hashimoto's encephalopathy with clinical features similar to those of Creutzfeldt-Jakob disease. Arq Neuropsiquiatr. 2008;66:903-5. doi: 10.1590/S0004-282X2008000600029. PMID:19099139 [DOI] [PubMed] [Google Scholar]
- 10.Muramatsu T, Hamano T, Shirafuji N, Matsunaga A, Ikawa M, Yoneda M. Hashimoto's encephalopathy presenting periodic synchronous discharge, as a differential diagnosis for Creutzfeldt-Jakob disease. Rinsho Shinkeigaku. 2013;53:716-20. doi: 10.5692/clinicalneurol.53.716. PMID:24097320 [DOI] [PubMed] [Google Scholar]
- 11.Hamaguchi T, Kitamoto T, Sato T, Mizusawa H, Nakamura Y, Noguchi M, Furukawa Y, Ishida C, Kuji I, Mitani K, et al.. Clinical diagnosis of MM2-type sporadic Creutzfeldt-Jakob disease. Neurology. 2005;64:643-8. doi: 10.1212/01.WNL.0000151847.57956.FA. PMID:15728285 [DOI] [PubMed] [Google Scholar]
- 12.Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D, Matsubara T, Nakagaki T, Yamanaka H, Shirabe S, et al.. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med. 2011;17:175-8. doi: 10.1038/nm.2294. PMID:21278748 [DOI] [PubMed] [Google Scholar]
- 13.Cramm M, Schmitz M, Karch A, Mitrova E, Kuhn F, Schroeder B, Raeber A, Varges D, Kim YS, Satoh K, et al.. Stability and reproducibility underscore utility of RT-QuIC for diagnosis of Creutzfeldt-Jakob disease. Mol Neurobiol. 2016;53:1896-904. doi: 10.1007/s12035-015-9133-2. PMID:25823511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayashi Y, Iwasaki Y, Takekoshi A, Yoshikura N, Asano T, Mimuro M, Kimura A, Satoh K, Kitamoto T, Yoshida M, et al.. An autopsy-verified case of FTLD-TDP type A with upper motor neuron disease mimicking MM2-thalamic-type sporadic Creutzfeldt-Jakob disease. Prion. 2016;10:492-501. doi: 10.1080/19336896.2016.1243192. PMID:27929803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foutz A, Appleby BS, Hamlin C, Liu X, Yang S, Cohen Y, Chen W, Blevins J, Wang H, Gambetti P, et al.. Diagnostic and prognostic value of human prion detection in cerebrospinal fluid. Ann Neurol. 2017;81:79-92. doi: 10.1002/ana.24833. PMID:27893164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi Y, Iwasaki Y, Yoshikura N, Asano T, Mimuro M, Kimura A, Satoh K, Kitamoto T, Yoshida M, Inuzuka T. An autopsy-verified case of steroid-responsive encephalopathy with convulsion and a false-positive result from the real-time quaking-induced conversion assay. Prion. 2017;4:284-92. doi: 10.1080/19336896.2017.1345416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jabeen SA, Cherukuri P, Mridula R, Harshavardhana KR, Gaddamanugu P, Sarva S, Meena AK, Borgohain R, Rani YJ. A prospective study of diffusion weighted magnetic resonance imaging abnormalities in patients with cluster of seizures and status epilepticus. Clin Neurol Neruosurg. 2017;155:70-4. doi: 10.1016/j.clineuro.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi Y, Iwasaki Y, Yoshikura N, Asano T, Hatano T, Tatsumi S, Satoh K, Kimura A, Kitamoto T, Yoshida M, et al.. Decreased regional cerebral blood flow in the bilateral thalami and medulla oblongata determined by an easy Z-score (eZIS) analysis of99mTc-ECD-SPECT images in a case of MM2-thalamic-type sporadic Creutzfeldt-Jakob disease. J Neurol Sci. 2015;358:447-52. doi: 10.1016/j.jns.2015.09.356. PMID:26421831 [DOI] [PubMed] [Google Scholar]
- 19.Hayashi Y, Yoshikura N, Takekoshi A, Yamada M, Asano T, Kimura A, Satoh K, Kitamoto T, Inuzuka T. Preserved regional cerebral blood flow in the occipital cortices, brainstem, and cerebellum of patients with V180I-129M genetic Creutzfeldt-Jakob disease in serial SPECT studies. J Neurol Sci. 2016;370:145-51. doi: 10.1016/j.jns.2016.09.043. PMID:27772745 [DOI] [PubMed] [Google Scholar]
- 20.Nozaki I, Hamaguchi T, Sanjo N, Noguchi-Shinohara M, Sakai K, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Moriwaka F, et al.. Prospective 10-year surveillance of human prion disease in Japan. Brain. 2010;133:3043-57. doi: 10.1093/brain/awq216. PMID:20855418 [DOI] [PubMed] [Google Scholar]
- 21.Lattanzio F, Abu-Rumeileh S, Franceschini A, Kai H, Amore G, Poggiolini I, Rossi M, Baiardi S, McGuire L, Ladogana A, et al.. Prion-specific and surrogate CSF biomarkers in Creutzfeldt-Jakob disease: diagnostic accuracy in relation to molecular subtypes and analysis of neuropathological correlates of p-tau and Aβ42 levels. Acta Neuropathol. 2017;133:559-78. doi: 10.1007/s00401-017-1683-0. PMID:28205010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirose K, Iwasaki Y, Izumi M, Yoshida M, Hashizume Y, Kitamoto T, Sahashi K. MM2-thalamic-type sporadic Creutzfeldt-Jakob disease with widespread neocortical pathology. Acta Neuropathol. 2006;112:503-11. doi: 10.1007/s00401-006-0131-3. PMID:16957926 [DOI] [PubMed] [Google Scholar]
- 23.Krasnianski A, Meissner B, Schultz-Schaeffer W, Kallenberg K, Barti M, Heinemann U, Varges D, Kretzschmar HA, Zerr I. Clinical features and diagnosis of the MM2 cortical subtype of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2006;63:876-80. doi: 10.1001/archneur.63.6.876. PMID:16769870 [DOI] [PubMed] [Google Scholar]
- 24.Castellani RJ, Colucci M, Xie Z, Zou W, Li C, Parchi P, Capellari S, Pastore M, Rahbar MH, Chen SG, et al.. Sensitivity of 14-3-3 protein test varies in subtypes of sporadic Creutzfeldt-Jakob disease. Neurology. 2004;96:436-42. doi: 10.1212/01.WNL.0000135153.96325.3B. [DOI] [PubMed] [Google Scholar]