

ABSTRACT
The normal function of PrPC, the cellular prion protein, has remained mysterious since its first description over 30 years ago. Amazingly, although complete deletion of the gene encoding PrPC has little phenotypic consequence, expression in transgenic mice of PrP molecules carrying certain internal deletions produces dramatic neurodegenerative phenotypes. In our recent paper,1 we have demonstrated that the flexible, N-terminal domain of PrPC possesses toxic effector functions, which are regulated by a docking interaction with the structured, C-terminal domain. Disruption of this inter-domain interaction, for example by deletions of the hinge region or by binding of antibodies to the C-terminal domain, results in abnormal ionic currents and degeneration of dendritic spines in cultured neuronal cells. This mechanism may contribute to the neurotoxicity of PrPSc and possibly other protein aggregates, and could play a role in the physiological activity of PrPC. These results also provide a warning about the potential toxic side effects of PrP-directed antibody therapies for prion and Alzheimer's diseases.
KEYWORDS: prion; neurotoxicity; neurodegeneration; ion; current; antibody, patch clamp; dendrite; nuclear magnetic resonance
INTRODUCTION: WHAT IS THE NORMAL FUNCTION OF PRPC?
This Extra View is related to our recent paper describing certain toxic activities of the prion protein, and how these are regulated by a novel intramolecular switch mechanism.1
It is now widely accepted that the infectious agent responsible for transmissible spongiform encephalopathies such as kuru and Creutzfeldt-Jakob disease is a protein molecule denoted PrPSc. PrPSc serves as a template for the misfolding of an endogenous version of the same protein, denoted PrPC, thereby generating additional molecules of PrPSc.2 An analogous mechanism involving propagation of protein conformation is thought to play a role in the intracerebral spread of misfolded proteins in other neurodegenerative diseases, including Alzheimer's and Parkinson's diseases,3 as well as in several physiological processes, including memory formation,4 signal transduction,5 and the formation of membrane-less organelles.6
Although a great deal is now understood about the molecular details of the PrPC-PrPSc conversion process, the normal, physiological function of PrPC has remained enigmatic. PrPC is present in all vertebrates, and is widely expressed throughout the central nervous system in both neuronal and non-neuronal cells. PrPC is a cell-surface glycoprotein, and is localized to the outer leaflet of the plasma membrane, where it is attached via a glycosyl-phosphatidylinositol (GPI) anchor.
PrPC on the cell surface serves as a receptor to which exogenous PrPSc initially binds as the first step in the generation of PrPSc.7 There is compelling evidence that this interaction plays a critical role in the downstream neurotoxic effects of PrPSc. Expression of cell-surface PrPC is necessary for full manifestation of prion-induced pathology, as neurons lacking PrPC are relatively resistant to the toxic effects of externally supplied PrPSc.8–11 Recent data also suggest that PrPC may serve as a toxicity-transducing receptor for other protein aggregates such as the Alzheimer's Aβ peptide.12 Thus, understanding the physiological activity of PrPC and how this activity is modulated by binding to pathological protein ligands is of great importance for both basic biology and medicine.
A potential clue to the function of PrPC is the identity of other cellular proteins with which it interacts. A number of protein-protein association studies have been performed using several different techniques, and a plethora of potential PrPC interacting partners have been published.13–15 Some of these identified interactors are cell surface receptors or adhesion molecules that may bind directly to PrPC. One example of such an interactor is the neural cell adhesion molecule, NCAM, which may participate with PrPC in regulating cellular morphogenesis.16 In contrast, a number of previously identified PrPC interactors are cytoplasmic proteins, which are unlikely to associate directly with PrPC, whose polypeptide chain is entirely extracellular. It is possible that these PrPC-associated molecules are components of a multi-protein complex that includes a transmembrane linker protein. One example of this class is the src-family protein kinase, fyn, which is thought to be linked to PrPC via the transmembrane receptor, mGluR5.17 Alternatively, intracellular proteins could interact with rare cytoplasmic variants of PrPC.18 Taken together, the available evidence suggests that, if PrPC does interact with partner proteins as part of its physiological function, it does so only transiently in a sub-stoichiometric ratio. Of course, this poses significant challenges in experimentally identifying these interactors.
In principle, a straightforward approach to uncovering the function of a protein is to knock down its expression and then look for phenotypic changes. However, when Prn-p, the gene encoding PrPC, is knocked out in mice, no overt anatomical or developmental defects are observed,19 although the mice are resistant to PrPSc infection as predicted by the prion hypothesis.20 More detailed examination of PrP knockout mice has revealed several subtle abnormalities, including a demyelinating polyneuropathy that appears with age.21 Cells cultured from PrP knock-out mice have been reported to display several kinds of alterations, such as reduced resistance to oxidative stress (reviewed in22). In all of these cases, however, the mechanistic role of PrPC in the observed phenotypes is poorly understood. Knock-down of the two PrP homologues in zebrafish produces dramatic developmental abnormalities23; however, some of these may be artifacts of the morpholino-modified RNAs used to knock down expression, based on the milder phenotypes observed with gene-editing techniques.24
SPONTANEOUS NEURODEGENERATION AND ABNORMAL IONIC CURRENTS INDUCED BY DELETION OF A CRITICAL REGION OF PRPC
Amazingly, while completely abolishing expression of PrPC produces little observable effect in mice, expression of certain mutant forms of the protein harboring deletions within the flexible N-terminal domain produces dramatic neurodegenerative phenotypes. This phenomenon was originally described in 1998 in transgenic mice expressing PrP molecules harboring a deletion of residues 32–121 or 32–134.25 These animals displayed early death, associated with massive degeneration of cerebellar granule neurons and white matter vacuolation. Importantly, these pathologies were strongly suppressed by co-expression of endogenous, wild-type PrPC, implying that the toxic effects of the mutant protein are antagonized by the wild-type protein. At a molecular level, this might be because the mutant and wild-type proteins physically interact with each other in a way that titrates out the toxic activity of the latter, or because the two proteins compete for binding to a common site. Subsequent studies from our laboratory examining shorter deletions of the N-terminal domain have revealed that residues 105–125 represent a critical toxicity-related region. Mice expressing PrP missing residues 105–125 die within the neonatal period, and require five-fold over-expression of wild-type PrPC from a second transgene to substantially suppress the phenotype.26
The lethal phenotype of the N-terminal deletion mutants has the potential to unmask how the physiological function of PrPC can be subverted to produce toxic effects, and how binding of PrPSc and other protein aggregates to cell-surface PrPC might engage pathological signal transduction mechanisms. Therefore, our laboratory has devoted considerable effort to understanding the molecular mechanisms underlying the toxicity of the deletion mutants, focusing particularly on the Δ105–125 version (referred to as ΔCR, for deletion of the Central Region) since this produces the strongest phenotype in mice. One potentially important clue was our discovery that expression of ΔCR PrP in transfected cell lines as well as neurons in culture and in brain slices produces large, spontaneous ionic currents that can be recorded by patch-clamping techniques.27,28 These currents, which are only observed at hyperpolarized holding potentials (<−50 mV), are very different in character from the typical currents associated with traditional ligand- or voltage-gated ion channels, in terms of their magnitude and sporadic nature. Ion substitution experiments have revealed that these currents are caused by relatively non-selective cation channels or pores. Importantly, the spontaneous currents produced by ΔCR PrP are suppressed by co-expression of wild-type PrP, paralleling observations on the toxicity of ΔCR PrP in transgenic mice. These observations strongly suggest that the spontaneous ionic currents observed in vitro cause the neurodegenerative phenotype seen in vivo.
THE EFFECTOR FUNCTION OF THE N-TERMINAL DOMAIN
Interestingly, deletion of the entire N-terminal domain of the mature prion protein (residues 23–134), is benign to mice,29 in contrast to the original deletion of residues 32–134, which caused a neurodegenerative phenotype.25 Since these two constructs differ only by the presence of nine amino acids (residues 23–31), these results directly implicate residues 23–31 as being essential for the toxic effects of central region deletion mutants. Consistent with this conclusion, deletion of residues 23–31,28 or substitution of the basic residues within this region with acidic residues,1 also abolishes the spontaneous ionic current activity associated with ΔCR PrP. One kind of molecular model to explain the role of residues 23–31 in ΔCR PrP-induced currents and toxicity is that this positively-charged region acts as a protein transduction domain. Such domains, exemplified by a polybasic region of the HIV Tat protein,30 are thought to interact with negatively charged phospholipids in the plasma membrane to create transient pores. Alternatively, this region may bind to other membrane proteins to induce currents indirectly.
Several additional observations in our recent paper1 support the notion that the flexible N-terminal domain of PrPC can act as a toxic effector. Ligands that bind to this region strongly inhibit the current activity associated with ΔCR PrP. Such ligands include the negatively charged glycosaminoglycan, pentosan polysulfate; Cu2+ ions, which bind to histidine residues within the octapeptide repeats; and antibodies recognizing the octapeptide repeats. Presumably, these ligands interfere with the ability of the N-terminus to interact with the lipid bilayer or protein partners to induce currents.
In order to test directly the role of the N-terminal domain as a toxic effector, we expressed this domain in the absence of the normal C-terminal domain.1 In order to ensure correct delivery of the protein to the cell surface, it was necessary to fuse the PrPC N-terminal domain to an unrelated protein, EGFP, which carried a GPI addition signal.31 Consistent with our predictions, the resulting fusion protein, PrP(N)-EGFP-GPI, induced spontaneous currents similar to ΔCR PrP when expressed in transfected cells (Fig. 1A, B). Furthermore, these currents could be silenced by addition of pentosan polysulfate, Cu2+ ions, and anti-octapeptide antibodies. Deletion of the polybasic region (residues 23–31) also suppressed the currents.
FIGURE 1.
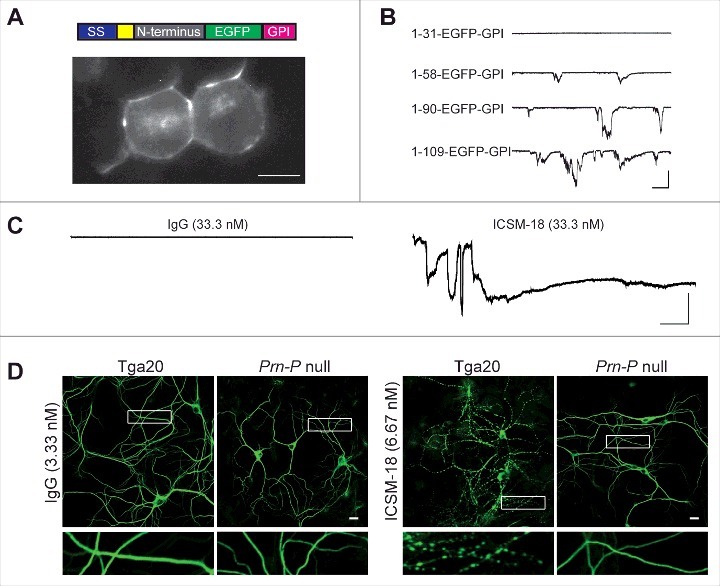
The isolated N-terminal domain of PrPC induces ionic currents, and anti-PrP antibodies induce currents and dendritic degeneration. (A) Top, schematic of PrP(N)-EGFP-GPI constructs containing the N-terminus of PrP fused to EGFP and the PrP GPI attachment sequence. The colored blocks represent the signal sequence (purple), polybasic residues 23–31 (yellow), different portions of the N-terminus (grey), EGFP (green), and the GPI attachment sequence (magenta). Bottom, fluorescence image of N2a cells expressing PrP(1-109)-EGFP-GPI, showing localization of the protein on the cell surface. Scale bar = 10 μm. (B) Representative traces of currents recorded from N2a cells expressing constructs with different lengths of the N-terminus (1–31, 1–58, 1–90 and 1–109). Scale bars: 500 pA, 30 s. (C) Representative traces of currents recorded from cells expressing wild-type PrP in the presence of non-specific IgG or ICSM18. Scale bars: 500 pA, 30 s. (D) Representative images showing dendrite morphology of cultured hippocampal neurons from Tga20 mice (which over-express wild-type PrPC) or Prn-p null mice after treatment for 48 hrs with anti-PrP antibody ICSM18 (6.67 nM) or non-specific IgG (33.3 nM). The cells were stained with an antibody to MAP2 to visualize dendrites. Boxed areas are enlarged below each image. Scale bar = 10 µm.
ANTIBODIES INDUCE CHANGES IN PRPC FUNCTION
The studies described thus far show that deletions encompassing a critical region in the central, hinge region of PrPC endow the protein with powerful toxic effects that are mediated by the flexible, N-terminal domain. Surprisingly, recent studies demonstrate that wild-type PrPC can also produce toxic effects when bound to antibodies targeting specific regions of the globular, C-terminal domain. Aguzzi and colleagues reported that treatment of cerebellar slices for days to weeks with antibodies targeting helix 1 in the C-terminal domain caused massive death of granule neurons.32 This effect could be prevented by simultaneous exposure to antibodies that recognized the octapeptide repeats in the N-terminal domain, consistent with a role of the latter as a toxic effector in this paradigm as well. In earlier studies, neurotoxic effects had also been observed when C-terminally directed antibodies were injected intracerebrally into mice.33,34
We wondered whether the neurotoxicity of anti-PrP antibodies observed in these experiments might be due to their ability to induce ionic currents, similar to the way that ΔCR PrP causes ionic currents in cultured cells27,28 and neuronal death in transgenic mice.26 When N2a cells expressing wild-type PrPC were treated with the antibody D18, which recognizes residues 132–156 in helix 1 of the C-terminal domain, robust inward currents were observed by patch clamping the cells at a holding potential of −70 mV.1 Antibodies POM1 and ICSM 18, which share overlapping epitopes with D18, produced similar currents (Fig. 1C). Paralleling the observations of Aguzzi and colleagues,32 antibodies recognizing the N-terminal octarepeats blocked the spontaneous currents generated by D18 and POM1.1 Other N-terminal ligands, including pentosan polysulfate and Cu2+ ions also silenced D18-induced currents.1
We found that, in addition to inducing spontaneous currents, C-terminally directed antibodies also caused severe degenerative changes in cultured neurons.1 Hippocampal neurons treated with D18, POM1, and ICSM 18 antibodies for 48 hours underwent significant dendritic degeneration marked by beading of the dendrites, a pathology often seen in glutamate-induced excitotoxic processes (Fig. 1D). Hippocampal neurons cultured from mice expressing Δ23–31 or Δ23–111 forms of PrP were morphologically unaffected by D18, POM1 or ICSM 18, indicating again a critical role for the N-terminal domain in PrPC-mediated toxicity.
It is possible that the dendritic degeneration produced by anti-PrP antibodies is a direct result of the abnormal ionic currents induced by these antibodies, although the two effects could represent parallel outputs of an upstream effector activity of the PrPC N-terminal domain. Aguzzi and colleagues have reported that apoptosis of granule neurons induced by chronic treatment of cerebellar slices with anti-PrP antibodies is accompanied by generation of reactive oxygen species, calpain activation, and stimulation of the PERK arm of the unfolded protein response.32,35 We suspect that the more subtle dendritic changes we observe in cultured neurons after shorter antibody treatments may involve different pathways.
A NOVEL STRUCTURAL MECHANISM REGULATING THE TOXIC ACTIVITY OF PRPC
Taken together, the findings described above suggest that the flexible, N-terminal domain of PrPC acts as a toxic effector, which, under normal physiological conditions, is regulated and inhibited by the presence of the structured, C-terminal domain (Fig. 2A). In this model, C-terminally directed antibodies like D18, POM1, and ICSM 18 would disrupt the regulatory activity of the C-terminal domain, thereby freeing the N-terminal domain to produce spontaneous ionic currents and neurodegenerative changes (Fig. 2B). Deletions of the hinge region connecting the N- and C-terminal domains, such as ΔCR, would also be expected to interfere with the regulatory interaction between these two domains (Fig. 2C), as would expression of the N-terminal domain in the absence of the C-terminal domain (Fig. 2D); in both cases, abnormal currents and toxicity would ensue. Ligands targeting the N-terminal domain, as well as deletion or mutation of the polybasic domain (residues 23–31), would abrogate these toxic activities.
FIGURE 2.
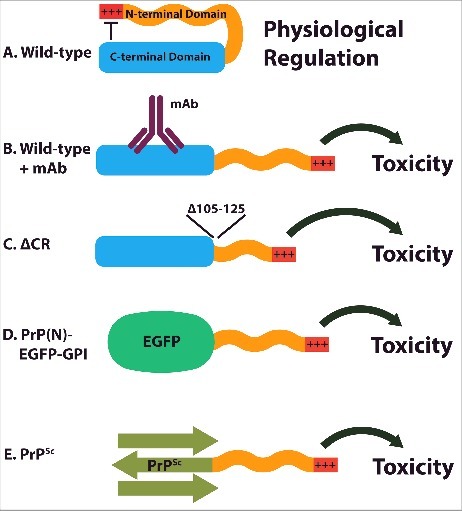
Model for regulation and dysregulation of PrPC. (A) Under normal physiological conditions, the C-terminal domain of PrPC inhibits the activity of the N-terminal domain. (B) Antibodies recognizing epitopes on the C-terminal domain interfere with its ability to regulate the N-terminal domain, resulting in toxicity. (C) Deletion of the hinge region connecting the N- and C-terminal domains also disrupts inter-domain regulation. (D) When the C-terminal domain is replaced with an unrelated protein (EGFP), the N-terminal domain is liberated to transduce toxic effects. (E) As a result of is altered conformation, the C-terminal domain of PrPSc may be incapable of interacting with the N-terminal domain to inhibit its toxic activity.
A plausible mechanism by which the C-terminal domain of PrPC could regulate the activity of the N-terminal domain is through a direct, intramolecular interaction between the two domains. Previous NMR studies had shown that Cu2+ ions, which are physiological ligands of the octapeptide repeats, as well as Zn2+ ions, drive the N-terminus of PrPC to associate with the C-terminal domain.36,37 Using these studies as a starting point, we performed NMR experiments in which we compared the 1H-15N HSQC spectra of wild-type and ΔCR PrP in the presence and absence of Cu2+ ions.1 As expected, addition of Cu+2 to wild-type PrPC caused paramagnetic-relaxation induced broadening of the HSQC cross-peaks of a number of C-terminal residues, which together comprise an electronegative patch on the surface of helices two and three. This result confirmed that Cu2+ enhances interaction between the N- and C-terminal domains. However when the same experiment was performed on ΔCR PrP, many fewer C-terminal residues were affected by the presence of Cu2+, suggesting diminished N-terminal/C-terminal interaction. Together, these data support the hypothesis that an inter-domain-interaction within PrPC is important for regulating the functional activity of the protein, and that deletions of the hinge region (such as ΔCR) as well as C-terminal ligands (such as helix 1-directed antibodies) that disrupt this interaction lead to abnormal ionic currents and neurodegeneration.
PATHOLOGICAL AND PHYSIOLOGICAL IMPLICATIONS OF THE MODEL
What is the relevance of this auto-regulatory interaction in terms of the pathological or physiological activities of PrPC? With regard to disease mechanisms, it is possible that conformational changes occurring in the C-terminal domain of PrPC upon its conversion to PrPSc could disrupt N-C regulation, leading to downstream neurotoxic effects mediated by the effector activity of the N-terminal domain (Fig. 2E). A similar effect might occur upon binding of protein aggregates such as Aβ oligomers to cell surface PrPC. These scenarios are consistent with the requirement for full-length, cell-surface PrPC for some of the synaptotoxic activities of PrPSc and Aβ. For example, the ability of PrPSc to induce retraction of dendritic spines on cultured hippocampal neurons requires expression of PrPC with an intact N-terminal domain.8 The fact that mice expressing N-terminally truncated PrPC ultimately remain susceptible to prion diseases after an extended incubation period38,39 argues against this model in its simplest form, although it does not rule out a contributory role for the N-terminal domain in the pathogenic process.
It is also possible that regulatory interactions of the N- and C-terminal domains play a role in the physiological activity of PrPC. For example, natural ligands, including proteins, small molecules or ions, may exist, whose binding to PrPC alters N-C interaction, thereby modulating an effector activity of the N-terminal domain. Such ligands may bind to the N-terminal domain, like copper ions, or to the C-terminal domain, analogous to helix 1-specific antibodies.
POTENTIAL SIDE-EFFECTS OF PRP ANTIBODY THERAPEUTICS
There are currently no effective treatments or cures for prion diseases. A primary therapeutic strategy has been to prevent accumulation of PrPSc in the brain, by either inhibiting its production or enhancing its clearance. One approach to achieving this has been to use anti-PrPC antibodies, which presumably act by stabilizing the conformation of PrPC or by preventing its interaction with PrPSc. A number of antibodies targeting different regions of PrPC have been shown to reduce PrPSc levels in prion-infected cell cultures, and some of these have a modest therapeutic effect in animal models when administered early in the disease course.40,41 One of these antibodies, ICSM18, directed against residues 143–156 on the outer surface of helix 1, has been converted into a humanized version, PRN100, with the goal of using it in human prion disease trials. Anti-PrP antibodies have also been investigated as treatments for Alzheimer's disease, since they can prevent binding of Aβ oligomers to PrPC.42
Our recent study highlighted in this review,1 in conjunction with other published studies,32,33,35,43 suggest that certain anti-PrP antibodies, particularly those targeting epitopes in the C-terminal domain (D18, POM1, ICSM18) or hinge region (D13), can produce dramatic neurotoxic effects when administered to cultured neurons and brain slices, or when injected intracerebrally into brain tissue. Although one published study argues that D13 and ICSM18 are non-toxic,44 the accumulated results, taken together, should serve as a warning about potentially serious side effects of adapting anti-PrP antibodies to clinical use. As PRN100 is being moved into human trials, it is important that the full nature of the antibody's potential toxicity be understood in order to minimize risk to patients.
One factor that should be considered in evaluating studies of the toxicity of anti-PrP antibodies is the effective dose of antibody being used. We found that ICSM18 produced spontaneous ionic currents in N2a cells at a concentration of 33.3 nM, and dendritic degeneration in cultured hippocampal neurons at a concentration of 6.67 nM.1 For comparison, a recent in vivo study in rats evaluated the ability of PRN100, the humanized version of ICSM18, to cross the blood-brain barrier after peripheral administration as a potential therapy for Alzheimer's disease.42 One hour after intravascular injection of a single 6 mg dose of PRN100, the concentration of the antibody in CSF was found to be 1.7 nM, only slightly below the concentration of ICSM18 that produced severe dendritic degeneration in cultured neurons. In this context, it is important to recognize that even sub-nanomolar concentrations of antibody could produce significant functional changes in synapses in the absence of observable morphological abnormalities.
Taken together, these considerations underscore the potential risks of using anti-PrP antibodies in a clinical setting. One possible solution to this problem would be to employ antibodies directed against the N-terminal domain of PrP, which do not appear to have toxic effects on their own and protect against the toxic effects of C-terminal antibodies.
CONCLUSIONS
Our recent publication1 demonstrates that the flexible, N-terminal domain of PrPC possesses toxic effector activities, including induction of abnormal ionic currents and degeneration of dendritic spines, which are normally held in check by a regulatory interaction with the structured C-terminal domain. Disruption of this inter-domain regulation, which can be caused by deletions within the hinge region or by binding to antibodies to the C-terminal domain, unleashes the effector activities of the N-terminal domain. This mechanism may contribute to the pathogenicity of PrPSc and Aβ oligomers, and may also play a role in the normal, physiological function of PrPC. It will be essential to consider these results when assessing the possible neurotoxic side effects of anti-PrP antibody therapies for prion and Alzheimer's diseases.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
FUNDING
Work in the Harris laboratory was supported by NIH grants NS065244 and NS101659.
REFERENCES
- 1.Wu B, McDonald AJ, Markham K, Rich CB, McHugh KP, Tatzelt J, Colby DW, Millhauser GL, Harris DA. The N-terminus of the prion protein is a toxic effector regulated by the C-terminus. Elife. 2017;6:e23473. doi: 10.7554/eLife.23473. PMID:28527237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. PMID:9811807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stopschinski BE, Diamond MI. The prion model for progression and diversity of neurodegenerative diseases. Lancet Neurol. 2017;16(4):323–32. doi: 10.1016/S1474-4422(17)30037-6. PMID:28238712 [DOI] [PubMed] [Google Scholar]
- 4.Rayman JB, Kandel ER. Functional Prions in the Brain. Cold Spring Harb Perspect Biol. 2017;9(1):a023671. doi: 10.1101/cshperspect.a023671. PMID:28049644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146(3):448–61. doi: 10.1016/j.cell.2011.06.041. PMID:21782231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163(1):123–33. doi: 10.1016/j.cell.2015.09.015. PMID:26406374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, Clarke AR, Schiavo G, Jat P, Collinge J, et al.. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun. 2011;2:281. doi: 10.1038/ncomms1282. PMID:21505437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang C, Imberdis T, Garza MC, Wille H, Harris DA. A neuronal culture system to detect prion synaptotoxicity. PLoS Pathog. 2016;12(5):e1005623. doi: 10.1371/journal.ppat.1005623. PMID:27227882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–43. doi: 10.1038/379339a0. PMID:8552188 [DOI] [PubMed] [Google Scholar]
- 10.Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302(5646):871–4. doi: 10.1126/science.1090187. PMID:14593181 [DOI] [PubMed] [Google Scholar]
- 11.Biasini E, Turnbaugh JA, Unterberger U, Harris DA. Prion protein at the crossroads of physiology and disease. Trends Neurosci. 2012;35(2):92–103. doi: 10.1016/j.tins.2011.10.002. PMID:22137337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salazar SV, Strittmatter SM. Cellular prion protein as a receptor for amyloid-beta oligomers in Alzheimer's disease. Biochem Biophys Res Commun. 2017;483(4):1143–47. doi: 10.1016/j.bbrc.2016.09.062. PMID:27639648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watts JC, Huo H, Bai Y, Ehsani S, Jeon AH, Shi T, Daude N, Lau A, Young R, Xu L, et al.. Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog. 2009;5(10):e1000608. doi: 10.1371/annotation/9eb11869-6acb-49b0-978e-abedc3cc545a . PMID:19798432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rutishauser D, Mertz KD, Moos R, Brunner E, Rülicke T, Calella AM, Aguzzi A. The comprehensive native interactome of a fully functional tagged prion protein. PLoS One. 2009;4(2):e4446. doi: 10.1371/journal.pone.0004446. PMID:19209230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, et al.. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol. 2001;314(5):1209–25. doi: 10.1006/jmbi.2000.5183. PMID:11743735 [DOI] [PubMed] [Google Scholar]
- 16.Mehrabian M, Brethour D, Wang H, Xi Z, Rogaeva E, Schmitt-Ulms G. The prion protein controls polysialylation of neural cell adhesion molecule 1 during cellular morphogenesis. PLoS One. 2015;10(8):e0133741. doi: 10.1371/journal.pone.0133741. PMID:26288071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, et al.. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer Aβ oligomer bound to cellular prion protein. Neuron. 2013;79(5):887–902. doi: 10.1016/j.neuron.2013.06.036. PMID:24012003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roucou X. Prion protein and RNA: A view from the cytoplasm. Front Biosci (Landmark Ed). 2009;14:5157–64. doi: 10.2741/3592. PMID:19482610 [DOI] [PubMed] [Google Scholar]
- 19.Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–82. doi: 10.1038/356577a0. PMID:1373228 [DOI] [PubMed] [Google Scholar]
- 20.Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73(7):1339–47. doi: 10.1016/0092-8674(93)90360-3. PMID:8100741 [DOI] [PubMed] [Google Scholar]
- 21.Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, et al.. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13:310–8. doi: 10.1038/nn.2483. PMID:20098419 [DOI] [PubMed] [Google Scholar]
- 22.Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrPC): Its physiological function and role in disease. Biochim Biophys Acta. 2007;1772:629–44. doi: 10.1016/j.bbadis.2007.02.011. PMID:17451912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Málaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CA. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009;7(3):e55. doi: 10.1371/journal.pbio.1000055. PMID:19278297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huc-Brandt S, Hieu N, Imberdis T, Cubedo N, Silhol M, Leighton PL, Domaschke T, Allison WT, Perrier V, Rossel M. Zebrafish prion protein PrP2 controls collective migration process during lateral line sensory system development. PLoS One. 2014;9(12):e113331. doi: 10.1371/journal.pone.0113331. PMID:25436888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, et al.. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93:203–14. doi: 10.1016/S0092-8674(00)81572-X. PMID:9568713 [DOI] [PubMed] [Google Scholar]
- 26.Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007;26:548–58. doi: 10.1038/sj.emboj.7601507. PMID:17245437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Solomon IH, Huettner JE, Harris DA. Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. J Biol Chem. 2010;285:26719–26726. doi: 10.1074/jbc.M110.134619. PMID:20573963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solomon IH, Khatri N, Biasini E, Massignan T, Huettner JE, Harris DA. An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J Biol Chem. 2011;286(16):14724–36. doi: 10.1074/jbc.M110.214973. PMID:21385869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westergard L, Turnbaugh JA, Harris DA. A nine amino acid domain is essential for mutant prion protein toxicity. J Neurosci. 2011;31(39):14005–17. doi: 10.1523/JNEUROSCI.1243-11.2011. PMID:21957261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wadia JS, Schaller M, Williamson RA, Dowdy SF. Pathologic prion protein infects cells by lipid-raft dependent macropinocytosis. PLoS One. 2008;3:e3314. doi: 10.1371/journal.pone.0003314. PMID:19390657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heske J, Heller U, Winklhofer KF, Tatzelt J. The C-terminal globular domain of the prion protein is necessary and sufficient for import into the endoplasmic reticulum. J Biol Chem. 2004;279(7):5435–43. doi: 10.1074/jbc.M309570200. PMID:14645231 [DOI] [PubMed] [Google Scholar]
- 32.Sonati T, Reimann RR, Falsig J, Baral PK, O'Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, et al.. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature. 2013;501(7465):102–6. doi: 10.1038/nature12402. PMID:23903654 [DOI] [PubMed] [Google Scholar]
- 33.Solforosi L, Criado JR, McGavern DB, Wirz S, Sánchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, et al.. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303(5663):1514–6. doi: 10.1126/science.1094273. PMID:14752167 [DOI] [PubMed] [Google Scholar]
- 34.Lefebvre-Roque M, Kremmer E, Gilch S, Zou WQ, Féraudet C, Gilles CM, Salès N, Grassi J, Gambetti P, Baron T, et al.. Toxic effects of intracerebral PrP antibody administration during the course of BSE infection in mice. Prion. 2007;1(3):198–206. doi: 10.4161/pri.1.3.4870. PMID:19164902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herrmann US, Sonati T, Falsig J, Reimann RR, Dametto P, O'Connor T, Li B, Lau A, Hornemann S, Sorce S, et al.. Prion infections and anti-PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog. 2015;11(2):e1004662. doi: 10.1371/journal.ppat.1004662. PMID:25710374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spevacek AR, Evans EG, Miller JL, Meyer HC, Pelton JG, Millhauser GL. Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure. 2013;21(2):236–46. doi: 10.1016/j.str.2012.12.002. PMID:23290724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans EG, Pushie MJ, Markham KA, Lee HW, Millhauser GL. Interaction between prion protein's copper-bound octarepeat domain and a charged C-terminal pocket suggests a mechanism for N-terminal regulation. Structure. 2016;24(7):1057–67. doi: 10.1016/j.str.2016.04.017. PMID:27265848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Supattapone S, Muramoto T, Legname G, Mehlhorn I, Cohen FE, DeArmond SJ, Prusiner SB, Scott MR. Identification of two prion protein regions that modify scrapie incubation time. J Virol. 2001;75(3):1408–13. doi: 10.1128/JVI.75.3.1408-1413.2001. PMID:11152514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turnbaugh JA, Unterberger U, Saá P, Massignan T, Fluharty BR, Bowman FP, Miller MB, Supattapone S, Biasini E, Harris DA. The N-terminal, polybasic region of PrPC dictates the efficiency of prion propagation by binding to PrPSc. J Neurosci. 2012;32(26):8817–30. doi: 10.1523/JNEUROSCI.1103-12.2012. PMID:22745483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422(6927):80–3. doi: 10.1038/nature01457. PMID:12621436 [DOI] [PubMed] [Google Scholar]
- 41.Carswell C, Khalili-Shirazi A, Brandner S, Martins S, Drynda R, Collinge J, Mead S, Clarke A. PAW35 Anti-prion protein monoclonal antibodies at low doses effectively treat prion disease in mice without side-effects. J Neurol Neurosurg Psychiatry. 2010;81(11):e33. doi: 10.1136/jnnp.2010.226340.63 [DOI] [Google Scholar]
- 42.Klyubin I, Nicoll AJ, Khalili-Shirazi A, Farmer M, Canning S, Mably A, Linehan J, Brown A, Wakeling M, Brandner S, et al.. Peripheral administration of a humanized anti-PrP antibody blocks Alzheimer's disease Aβ synaptotoxicity. J Neurosci. 2014;34(18):61405. doi: 10.1523/JNEUROSCI.3526-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reimann RR, Sonati T, Hornemann S, Herrmann US, Arand M, Hawke S, Aguzzi A. Differential toxicity of antibodies to the prion protein. PLoS Pathog. 2016;12(1):e1005401. doi: 10.1371/journal.ppat.1005401. PMID:26821311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klöhn PC, Farmer M, Linehan JM, O'Malley C, Fernandez de Marco M, Taylor W, Farrow M, Khalili-Shirazi A, Brandner S, Collinge J. PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science. 2012;335(6064):52. doi: 10.1126/science.1215579. PMID:22223800 [DOI] [PubMed] [Google Scholar]