

Abstract
Glycogen Synthase Kinase-3β (GSK-3β), a serine/threonine protein kinase, is an emerging therapeutic target in the treatment of human breast cancer. In this study, we demonstrate that the pharmacological inhibition of GSK-3 by two novel small molecule GSK-3 inhibitors, 9-ING-41 and 9-ING-87, reduced the viability of breast cancer cells but had little effect on non-tumorigenic cell growth. Moreover, treatment with 9-ING-41 enhanced the antitumor effect of irinotecan (CPT-11) against breast cancer cells in vitro. We next established two patient-derived xenograft tumor models (BC-1 and BC-2) from metastatic pleural effusions obtained from patients with progressive, chemorefractory breast cancer and demonstrated that 9-ING-41 also potentiated the effect of the chemotherapeutic drug CPT-11 in vivo, leading to regression of established BC-1 and BC-2 tumors in mice. Our results suggest that the inhibition of GSK-3 is a promising therapeutic approach to overcome chemoresistance in human breast cancer, and identify the GSK-3 inhibitor 9-ING-41 as a candidate targeted agent for metastatic breast cancer therapy.
Keywords: Breast cancer, GSK-3, Chemoresistance, 9-ING-41, Drug development
Introduction
Breast cancer is the leading cause of cancer deaths in women worldwide [1]. Despite enormous advances in early diagnosis and surgical treatment of breast cancer, as well as improvements in neoadjuvant and adjuvant therapy, approximately 30% of patients with breast cancer will develop incurable metastatic disease [2]. Treatment with conventional chemotherapeutic drugs has had little impact on metastatic disease progression. Metastatic breast cancer remains an incurable disease with median survival times ranging from one to four years depending on the subtype [2,3], and thus represents a significant unmet medical need. The identification of new targeted therapeutic agents is urgently needed.
Glycogen Synthase Kinase-3 (GSK-3), a serine/threonine protein kinase, was initially described as a key enzyme involved in glycogen metabolism [4,5] but is now recognized as a regulator of diverse cellular functions [6]. GSK-3 phosphorylates and thereby regulates the activity of many metabolic, signaling, and structural proteins [6]. There are two highly homologous forms of GSK-3 in mammals, GSK-3α and GSK-3β [6]. Historically, GSK-3β has been thought of as a potential tumor suppressor due to its ability to phosphorylate and thereby target pro-oncogenic molecules including c-Jun [7], c-Myc [8], cyclin D1 [9] and β-catenin [10] for ubiquitin-dependent proteosomal degradation. However, recent reports have suggested that GSK-3β is a positive regulator of cancer cell proliferation and survival [11–22] providing further support for GSK-3β as a therapeutic target in cancer. Previously, we identified GSK-3β as a novel therapeutic target in human leukemia, pancreatic, colon, bladder and renal cancer [11,14–17]. In human breast carcinoma, it has been shown that overexpression of GSK-3β was associated with several indicators of poor prognosis and breast cancer patients with GSK-3β expression in the highest quartile (246 of 1686 cases) had a 2.7 and 1.7-fold increased risk of distant relapse 5 and 10 years after tumor resection, respectively [23]. A recent study demonstrated that GSK-3β knockdown significantly inhibited breast cancer cell proliferation whereas GSK-3α knockdown had only a minor effect in four breast cancer cell lines further credentialing GSK-3β as a viable therapeutic target for the treatment of breast cancer [24]. We recently identified two lead ATP-competitive GSK-3β inhibitors, 9-ING-41 and 9-ING-87, based on the potency of their antiproliferative activity against pancreatic and ovarian cancer cells in vitro as well as supportive ADMET and PK studies [13,25]. Here, we describe the anti-tumor activity of these novel inhibitors of GSK-3 in both breast cancer cell lines and in novel patient-derived xenograft (PDX) models of metastatic chemorefractory breast cancer. We conclude that the inhibition of GSK-3 is a promising therapeutic approach to overcome chemoresistance in metastatic breast cancer.
Materials and methods
Cell culture and reagents
Breast cancer cell lines MDA-MB-468, SKBR3 and MCF10A were obtained from American Type Culture Collection (ATCC, Manassas, VA). MDA-MB-231/LM2-4 breast cancer cell line was a gift from Drs. Giulio Francia (University of Texas–El Paso) and Robert Kerbel (Sunnybrook Research Institute). GSK-3 inhibitors 9-ING-41 and 9-ING-87 were synthesized in the laboratory of Dr. Kozikowski as previously described [13]. All other chemicals were obtained from Sigma (St. Louis, MO).
Measurement of cell viability
The relative number of viable cancer cells was determined by measuring the optical density using CellTiter 96 Aqueous One Solution Cell Proliferation Assay kit [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS] according to the manufacturer’s instructions (Promega, Madison, WI). GI50 value for each compound was calculated with a non-linear regression model of standard slope using GraphPad Prism 6.0 software (GraphPad, San Diego, CA).
Immunoblot analysis and antibodies
For immunoblots, cells were lysed and whole protein extract from cells was prepared as described previously [11]. Protein sample concentrations were determined by Bradford protein assay and equal amounts (50 μg) of protein were loaded in each well of SDS-polyacrylamide gel. Cell extracts were separated by 10% SDS-PAGE, transferred to PVDF membrane and probed as indicated. The following antibodies were used for immunoblot analysis: GSK-3β, phospho-Glycogen Synthase (Ser641), Bcl-2, PARP, X-linked inhibitor of apoptosis protein (XIAP), GAPDH (Cell Signaling). Bound antibodies were detected as described previously [11].
Patient-derived xenograft (PDX) tumor models
The following research protocol was approved by Northwestern University institutional review board, and all patients provided appropriate informed consent. Metastatic pleural effusion samples were obtained from breast cancer patients and de-identified. Metastatic pleural effusion cells were engrafted into the mammary fat pad of non-obese diabetic/severe combined immunodeficient (NOD/SCID) gamma (NSG) mice (Jackson Laboratory). When a PDX tumor reached 1 cm in its largest dimension, the mouse was euthanized and freshly resected 2 mm × 2 mm tumor pieces were re-transplanted to NSG mice for in vivo studies. A piece of PDX tumor was fixed in 10% formalin and processed to paraffin-embedding. Paraffin sections (5 μm) were stained with hematoxylin and eosin (H&E) for histopathological evaluation. Expression of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) in human primary and PDX breast tumors was detected by immunohistochemical staining in the Pathology Core Facility (PCF), Robert H. Lurie Comprehensive Cancer Center using a DAKO automated immunostainer and standard En-Vision-HRP kit (DAKO, Carpinteria, CA).
Immunohistochemical staining
Immunohistochemical (IHC) staining was performed on paraffin sections of xenograft tumors. Paraffin sections were deparaffinized, and antigen retrieval was carried out in citric buffer in microwave for 10 min. The sections were incubated in 1% hydrogen peroxidase for 10 minutes to quench endogenous tissue peroxidase. Tissue sections were then incubated with the anti-β-catenin (BD Biosciences, San Jose, CA), anti-GSK-3β or anti-phospho-Glycogen Synthase (Ser641) antibody (Cell Signaling, Danvers, MA) overnight at +4 °C. The slides were stained using a standard EnVision+ System-HRP kit (DAKO, Carpinteria, CA) according to the manufacturer’s protocol. Immunohistochemical reactions were developed with diaminobenzidine as the chromogenic peroxidase substrate, and slides were counterstained with hematoxylin. Negative control samples included replacement of the primary antibody with nonimmune IgG1 (Dako).
Statistical analysis
All values are presented as mean ± SE. Cell viability assay and PDX tumor data were analyzed with one-way ANOVA. P values less than 0.05 are considered significant. Statistical analysis was performed using GraphPad Prism 6.0 software.
Results
Pharmacological inhibition of GSK-3 decreases viability of breast cancer cells
The two lead ATP-competitive GSK-3β inhibitors, 9-ING-41 and 9-ING-87, exhibit potent antiproliferative activity [13,25]. It should be noted that the two isoforms of GSK-3, α and β, are 98% homologous in the kinase domain and thus all known competitive inhibitors of GSK-3 inhibit both isoforms, and are thus referred to as GSK-3 inhibitors rather than GSK-3β inhibitors [13,26,27]. Compounds 9-ING-41 and 9-ING-87 are relatively selective for GSK-3 over ~320 other related kinases by at least one order of magnitude, including closely related serine/threonine kinases such as CDKs, PDKs, PKA, Akt, and PKCs [13]. Using immunoblotting, we found GSK-3β expression in three breast cancer cell lines and non-tumorigenic MCF10A cells (Fig. 1A). Next, we found that the inhibition of GSK-3 by 9-ING-41 and 9-ING-87 decreased MDA-MB-468 (ER−/PR−/HER2−) and SKBR3 (ER−/PR−/HER2+) breast cancer cell viability (Fig. 1B). Using immunoblotting, we found that the treatment of MDA-MB-468 and SKBR3 breast cancer cell lines with 9-ING-41 or 9-ING-87 resulted in inhibition of GSK-3 activity, as measured by the decreased expression of phospho-Glycogen Synthase, a downstream target of GSK-3 (Fig. 1C). We also found that the inhibition of GSK-3 resulted in a significant decrease in the expression of anti-apoptotic proteins Bcl-2 and XIAP and led to an induction of apoptosis in breast cancer cells as shown by PARP cleavage and Hoechst staining (Fig. 1C and D).
Fig. 1.
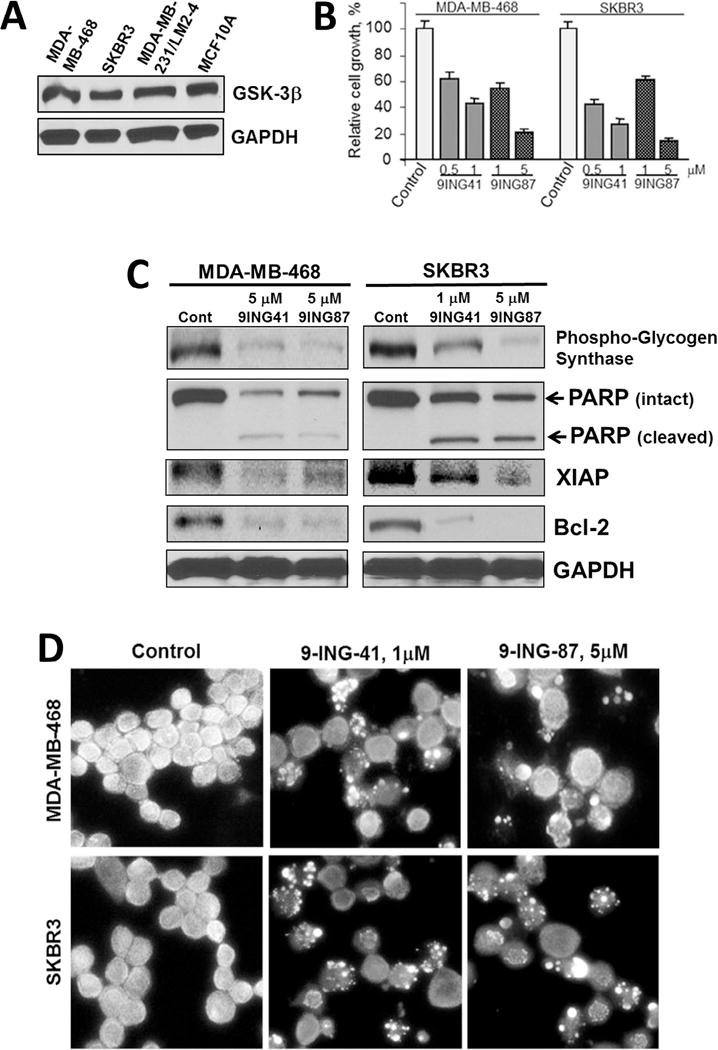
Pharmacological inhibition of GSK-3 suppresses viability of breast cancer cells. (A) Whole cell lysate was prepared from breast cancer cells, separated by SDS-PAGE (50 μg/well), transferred to PVDF membrane, and immunoblotted as indicated. (B) Relative cell growth was measured by MTS assay in MDA-MB-468 and SKBR3 breast cancer cells treated with 9-ING-41 (9ING41) and 9-ING-87 (9ING87) for 72 hours as indicated. (C) Breast cancer cells were treated with GSK-3 inhibitors 9-ING-41 (9ING41) and 9-ING-87 (9ING87) for 36 hours and Western immunoblotting was performed as indicated. (D) Representative pictures of Hoechst staining of breast cancer cells treated with 9-ING-41 and 9-ING-87 as indicated for 48 hours. Fragmented apoptotic nuclei show intense Hoechst staining.
We then benchmarked 9-ING-41 and 9-ING-87 against the toolkit GSK-3 inhibitors AR-A014418 [26] and SB-216763 [27], as well as LY2090314 (developed by Eli Lilly) using a cell viability assay and found that the GI50 of 9-ING-41 and 9-ING-87 for inhibition of breast cancer cell growth is significantly lower than the GI50 of other GSK-3 inhibitors, including LY2090314 (Fig. 2A). To determine whether GSK-3 inhibitors affect non-transformed mammary epithelial cells, we treated MCF10A cells with several of the GSK-3 inhibitors (Fig. 2B). MCF10A are non-tumorigenic cells derived from benign proliferative breast tissue that were spontaneously immortalized and are suggested as a benign breast epithelial cell model [28]. We found that pharmacological inhibition of GSK-3 did not significantly affect the growth of MCF10A cells, whereas growth of SKBR3 and various breast cancer cell lines at the same concentrations was suppressed by the GSK-3 inhibitors AR-A014418, 9-ING-41 and 9-ING-87 (Figs. 1B and 2B). Taken together, our in vitro results support GSK-3 as a mediator of breast cancer cell survival, and identify 9-ING-41 as a candidiate for the targeted treatment of human breast cancer.
Fig. 2.
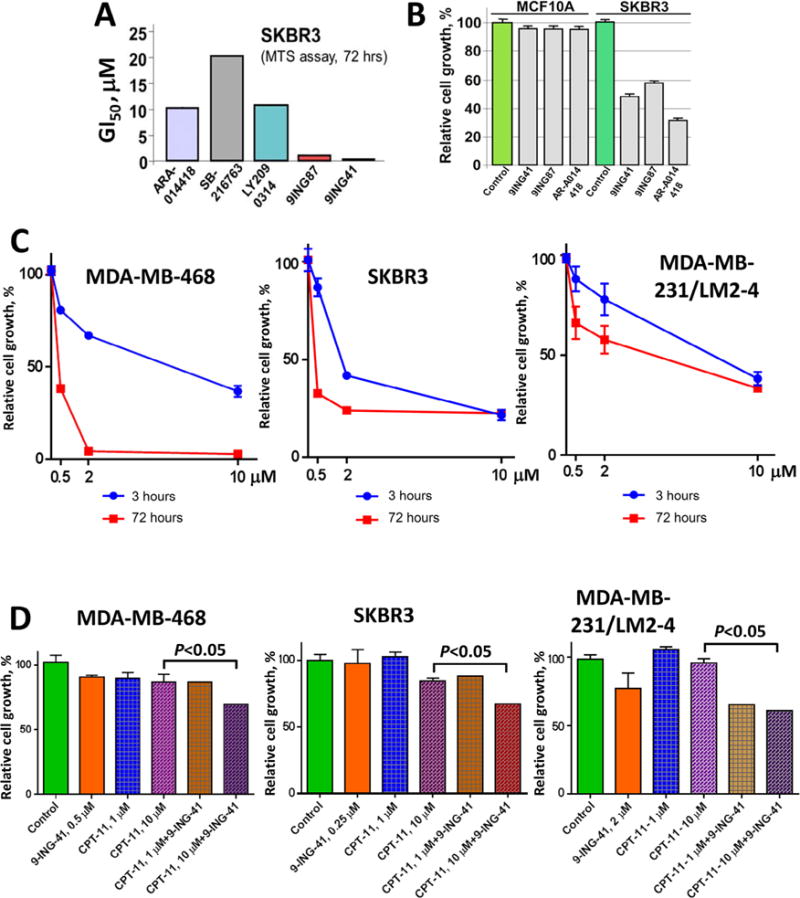
Pharmacological inhibition of GSK-3 potentiates the effect of conventional chemotherapeutic drugs in breast cancer cells. (A) GSK-3 inhibitors 9-ING-87 and 9-ING-41 show lowest GI50 in growth inhibition of SKBR3 breast cancer cells as compared to other GSK-3 inhibitors AR-A014418, SB-216763 and LY2090314. (B) Relative cell growth was measured by MTS assay in breast malignant (SKBR3) and benign (MCF10A) cells treated with 9-ING-41 (0.5 μM), 9-ING-87 (1 μM) and AR-A014418 (20 μM) for 72 hours as indicated. (C) Relative cell growth was measured by MTS assay in breast cancer cell lines treated with 0.5 μM, 2 μM and 10 μM of 9-ING-41 for 3 hours and 72 hours as indicated. (D) Breast cancer cell lines were treated with 9-ING-41, CPT-11 or combination of 9-ING-41 with CPT-11 for 3 hours at indicated. After the treatment, drugs were replaced with fresh media and relative cell growth was measured by MTS assay after 72 hours. Columns, mean; bars, SE.
GSK-3 inhibitor 9-ING-41 potentiates antitumor effect of conventional chemodrugs against breast cancer cell lines in vitro
We next tested our hypothesis that inhibition of GSK-3 may overcome resistance to the chemotherapeutic drug CPT-11 (irinotecan) using three distinct breast cancer cell lines. Traditional approaches for synergy analysis typically used with cytotoxic drugs are difficult to apply to small molecule enzyme inhibitors such as 9-ING-41. Synergy analysis requires the evaluation of inhibition across a range of low to high concentrations of drugs. However, there is a minimal threshold (minimal effective concentration) of GSK-3 inhibition that must be achieved in order to see effects on breast cancer cell proliferation that prevents the observation of inhibition at concentrations of 9-ING-41 below the minimal effective concentration thereby impeding synergy analysis. Further, the prolonged continuous treatment (48–72 hours) of breast cancer cell lines typically used in synergy analysis leads to significant apoptosis using 9-ING-41 or 9-ING-87 as a single agent (Fig. 1C and D) or to a significant decrease of cell viability using cytotoxic agents (data not shown). Continuous exposure of cancer cell lines to the same concentration of drug for 72 hours is also not clinically translatable to a patient scenario, where tumor exposure is variable over time after drug administration as the drug is cleared. Therefore, we modified our combination studies using pulses of drug exposure to breast cancer cell lines. Breast cancer cell lines were treated with 9-ING-41, CPT-11 or CPT-11 + 9-ING-41 for 3 hours. Post-treatment, cells were washed and all test compounds were replaced with fresh compound-free cell culture media. Breast cancer cells were then allowed to continue growing for 72 hours and relative cell growth was measured by a cell viability assay after 72 hours of growth. We compared the effect of 9-ING-41 on cell growth after 3 hour exposure to a 72 hour continuous exposure to compound (Fig. 2C). Varying responses were observed when breast cancer lines were exposed continuously for 72 hours to 9-ING-41 ranging from complete (e.g. MDA-MB-468) to partial (e.g. MDA-MB-231) inhibition of growth whereas only partial inhibition was observed across all three breast cancer cell lines tested after 3 hour exposure followed by washout (Fig. 2C). Based on the response of individual breast cancer cell line to 9-ING-41 treatment, we selected minimally effective concentration for 9-ING-41 of 0.25 μM, 0.5 μM and 2 μM for SKBR3, MDA-MB-468 and MDA-MB-231/LM2-4 breast cancer cell lines, respectively, for evaluation in combination experiments. Using the approach of short-term treatment (3 hours) to mimic transient drug exposure in vivo, we tested 9-ING-41 in combination with 1 μM and 10 μM of CPT-11 (Fig. 2D). CPT-11 at 10 μM exceeds the Cmax observed in patients when administered intravenously at a dose of 100 mg/m2 [29], which approximates a clinically active dose. Using short-term treatment (3 hours), we demonstrated that 9-ING-41 potentiates the antitumor effects of 10 μM CPT-11 against MDA-MB-468 (P < 0.05), SKBR3 (P < 0.05) and MDA-MB-231/LM2-4 (P < 0.05) breast cancer cell lines (Fig. 2D).
Development and characterization of breast cancer patient-derived xenograft (PDX) models BC-1 and BC-2
We established two metastatic breast cancer PDX models, BC-1 and BC-2, by inoculating cells obtained from metastatic pleural effusions from breast cancer patients into NSG mice. BC-1 and BC-2 were derived from patients whose primary tumors were ER+/PR+/HER2− and maintained this molecular profile in PDX tumors as confirmed by immunohistochemical staining in serial passages in vivo. Usually, 17β-estradiol pellet supplementation is required to grow ER-positive breast xenograft tumors in mice. However, we found that ER-positive BC-1 and BC-2 PDX tumors grew without 17β-estradiol pellet supplementation in NSG mice. Although ESR1 (the gene encoding the ER) mutations have not been found in treatment-naive human breast carcinomas, recently published studies identified ESR1 mutations affecting the ligand-binding domain (LBD) of ER in advanced ER-positive hormone-resistant breast cancer, particularly in metastatic lesions from women who took estrogen-lowering drugs such as aromatase inhibitors [30,31]. The patients from whom BC-1 and BC-2 were derived were heavily pretreated and progressed through multiple cycles of treatment including aromatase inhibitors and DNA damaging drugs prior to developing metastatic pleural effusions (Figs. 3 and 4). DNA-sequencing results for the ESR1 gene from the BC-1 PDX tumor reveal a Leu536Arg mutation in the ligand-binding domain of the ER whereas the BC-2 PDX tumor has a Tyr537Ser mutation. Although the Leu536Arg mutation has not been previously reported, a Leu536Gln mutation in ER-positive tumors has been shown to lead to ligand-independent ER signaling [30]. Moreover, the Tyr537Ser mutation has been reported in metastatic breast carcinomas to cause a conformational change in the ER, also leading to ligand-independent ER signaling and resistance to hormone therapy [30,31]. Our in vivo results and mutational analysis of ESR1 suggest that BC-1 and BC-2 are ER-positive hormone-resistant breast PDX tumor models.
Fig. 3.
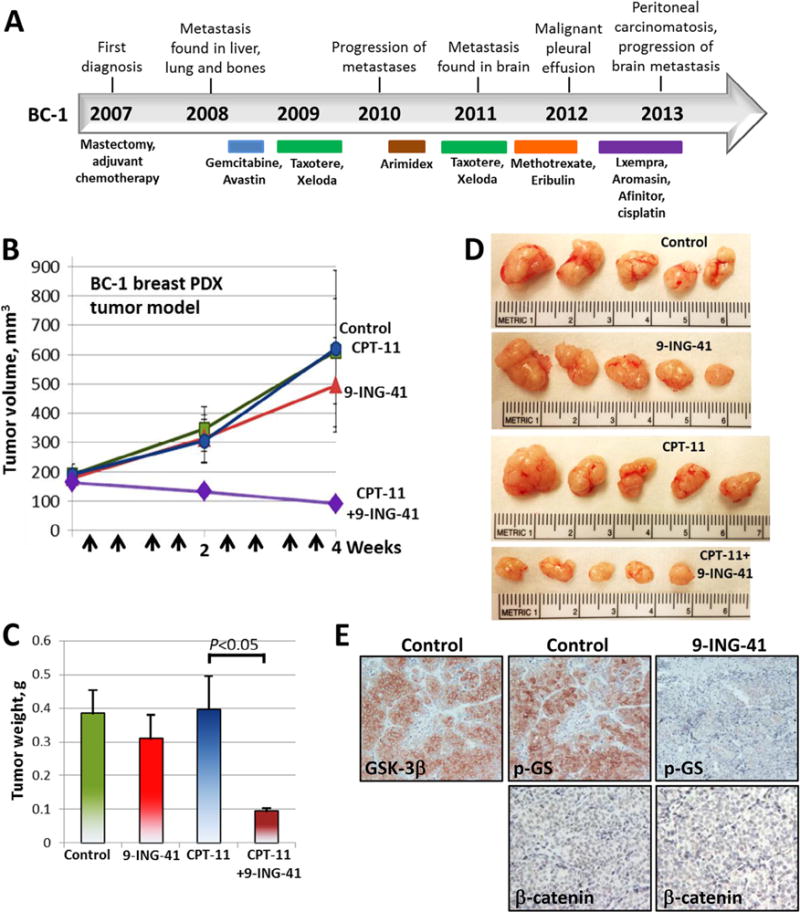
Treatment with 9-ING-41 enhances the antitumor effect of CPT-11 in breast BC-1 PDX (ER+/PR+/HER2−) tumors. (A) Treatment history of breast cancer patient (case BC-1). Breast PDX tumor model was established after orthotopic injection of metastatic pleural effusion cells (obtained from breast cancer patient) in NSG mouse. Breast PDX tumor pieces were re-transplanted orthotopically to 20 mice (1 tumor per mouse). Tumors were size matched and mice were randomized into 4 treatment groups: control (DMSO; n = 4 mice), CPT-11 (5 mg/kg, n = 5 mice), 9-ING-41 (70 mg/kg, n = 5 mice) and CPT-11 + 9-ING-41 (n = 5 mice). (B) Vehicle or drugs were injected as indicated by arrows. Points, mean tumor volume; bars, SE. (C) Weight of resected tumors was measured. Columns, mean tumor weight; bars, SE. (D) Representative pictures of PDX subQ tumors from each group of animals. (E) Representative pictures of GSK-3β, phospho-Glycogen Synthase and β-catenin expression in control and 9-ING-41-treated BC-1 PDX tumors.
Fig. 4.
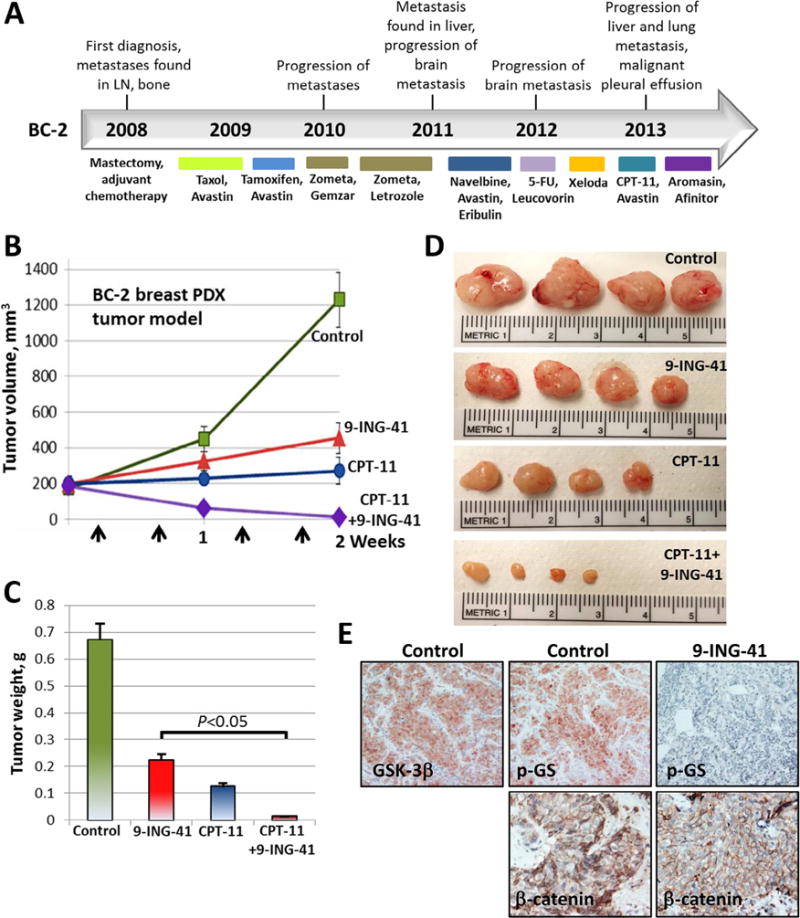
Treatment with CPT-11 + 9-ING-41 leads to a regression of breast BC-2 PDX (ER+/PR+/HER2−) tumors. (A) Treatment history of breast cancer patient (case BC-2). Breast PDX tumor model was established after orthotopic injection of metastatic pleural effusion cells (obtained from breast cancer patient) in NSG mouse. Breast PDX tumor pieces were re-transplanted orthotopically to 16 mice (1 tumor per mouse). Tumors were size matched and mice were randomized into 4 treatment groups: control (DMSO; n = 4 mice), CPT-11 (20 mg/kg, n = 4 mice), 9-ING-41 (70 mg/kg, n = 4 mice) and CPT-11 + 9-ING-41 (n = 4 mice). (B) Vehicle or drugs were injected as indicated by arrows. Points, mean tumor volume; bars, SE. (C) Weight of resected tumors was measured. Columns, mean tumor weight; bars, SE. (D) Representative pictures of PDX subQ tumors from each group of animals. (E) Representative pictures of GSK-3β, phospho-Glycogen Synthase and β-catenin expression in control and 9-ING-41-treated BC-2 PDX tumors.
The GSK-3 inhibitor 9-ING-41 potentiates the effects of CPT-11 on breast cancer PDX growth in vivo
We used the BC-1 and BC-2 breast PDX tumor models to evaluate the antitumor effect of 9-ING-41 alone and in combination with CPT-11. 9-ING-41 has completed extensive pre-clinical ADMET evaluation [25] and is currently being advanced through IND enabling development as a clinical candidate. Although we have established and grown BC-1 and BC-2 PDX tumors in mouse mammary fat pad (orthotopic site) for this study, we consider these to be models of metastatic, chemoresistant breast cancer based on the number of treatments that these tumors were exposed to in patients as well as the site of origin (metastatic pleural effusion) (Figs. 3 and 4). By immunohistochemical staining, we found that GSK-3β was ex-pressed in BC-1 and BC-2 PDX tumors (Figs. 3E and 4E). We also found that expression of phosphorylated glycogen synthase (p-GS), a downstream target of GSK-3, was downregulated in 9-ING-41-treated BC-1 and BC-2 PDX tumors indicating target engagement in the PDX models (Figs. 3E and 4E). We found that β-catenin expression was not affected by the treatment with 9-ING-41 in BC-1 (loss of β-catenin expression) and BC-2 (β-catenin membranous staining) PDX tumors (Figs. 3E and 4E). Based on the broad enhancement of antitumor effects when 9-ING-41 is added to CPT-11 in breast cancer cell lines (Fig. 2D) and our observation that the patient tumors from which BC-1 and BC-2 were derived were highly chemorefractory in the clinic, we evaluated the ability of 9-ING-41 treatment to enhance tumor response in the breast cancer PDX models when combined with chemotherapy (CPT-11). Tumors in mice bearing orthotopic BC-1 or BC-2 were staged to approximately 200 mm3 prior to initiation of treatments and randomized to 4 treatment groups: control, 9-ING-41, CPT-11 and CPT-11 + 9-ING-41. Vehicle (DMSO) or drugs were injected intraperitoneally using the schedules indicated (Figs. 3 and 4). We found that CPT-11 or 9-ING-41 monotherapy did not significantly inhibit BC-1 PDX tumor growth, whereas combined CPT-11 and 9-ING-41 therapy led to regression of BC-1 tumors (Fig. 3). Statistical analysis of PDX tumor weight suggests that 9-ING-41 potentiates the effect of CPT-11 on the inhibition of BC-1 tumor growth, leading to tumor regression in the CPT-11 + 9-ING-41 group (P < 0.05) (Fig. 3C). In contrast to the completely chemoresistant BC-1 PDX tumors, which did not respond to either 9-ING-41 or CPT-11 monotherapy, we found that the growth of BC-2 PDX tumors was significantly inhibited by 9-ING-41 or CPT-11 monotherapy (Fig. 4B–D). However, similar to the BC-1 model, only the combination of 9-ING-41 and CPT-11 treatments led to tumor regression (P < 0.05) (Fig. 4B and C).
Discussion
Recently, GSK-3β has been credentialed as a potential therapeutic target in human breast cancer [23,24]. GSK-3β knockdown significantly inhibited breast cancer cell proliferation whereas GSK-3α knockdown had only a minor effect in four breast cancer cell lines, providing further support for GSK-3βas a breast cancer therapeutic target [24]. In a recently published study of 1686 cases of breast cancer, overexpression of GSK-3β was associated with several indicators of poor prognosis including lymph node metastasis, increased tumor size, high pathological grade, ER-negative disease, PR-negative disease, increased proliferation (measured by Ki-67) and HER2 overexpression [23]. Moreover, breast cancer patients with GSK-3β expression in the highest quartile (246 of 1686 cases) had a 2.7 and 1.7-fold increased risk of distant relapse 5 and 10 years after tumor resection, respectively [23]. In the present study, we describe a novel GSK-3 inhibitor 9-ING-41 which shows robust antitumor activity in vitro and in vivo and possesses drug-like properties [13,25,32]. Our in vitro results demonstrate that 9-ING-41 is a more potent inhibitor of breast cancer cell growth than other available GSK-3 inhibitors including the clinical stage compound LY2090314. We demonstrate that inhibition of GSK-3 by 9-ING-41 decreases the survival of breast cancer cells in vitro, consistent with previously published studies in other tumor cell types [11–22,25,32–36]. Our previous studies in leukemic cells showed that the inhibition of GSK-3 using a toolkit inhibitor suppressed NF-κB transcriptional activity and decreased the expression of the antiapoptotic proteins (XIAP, Bcl-2), leading to enhaced cancer cell apoptosis [15]. Given that NF-κB-mediated chemoresistance has been shown to be a major driver of breast cancer progression [37,38], we examined inhibition of GSK-3 by 9-ING-41 in breast cancer cells in vitro and found it decreased the expression of anti-apoptotic molecules NF-κB target genes Bcl-2 and XIAP inducing apoptosis. We then tested whether 9-ING-41 could enhance the response of the chemoresistant metastatic breast cancer cell lines to the chemotherapeutic drug CPT-11 in vitro. Chemoresistance of cancer cells in vitro is defined by many factors including drug concentration and duration of exposure in cell culture. Because continuous exposure (72–96 hours) of breast cancer cells to either 9-ING-41 or a chemotherapeutic drug significantly suppresses cell viability and does not mimic tumor exposure to a drug in vivo, we carried out short term (3 hours) exposures of breast cancer cell lines using a minimal effective inhibitory concentration of 9-ING-41 in combination with a chemotherapeutic drug followed by washout with fresh drug-free cell culture media and then allowed the cells to grow for 72 hours. Under these conditions, we found that 9-ING-41 potentiates the antitumor effects of CPT-11 on breast cancer cell growth. Our in vitro results suggested that inhibition of GSK-3 by 9-ING-41 sensitized metastatic breast cancer cells to CPT-11 chemotherapy, a potent drug that has otherwise limited clinical activity in this indication.
A number of in vivo studies have investigated the antitumor activity of distinct GSK-3 inhibitors in a variety of cancer cell line-derived tumor xenograft models [14,21,22,24,33,39,40]. These studies utilized toolkit GSK-3 inhibitors as monotherapies. Although inhibition of tumor growth was reported, the antitumor effects were modest and the compounds used in these studies were not amenable for clinical development. In order to evaluate 9-ING-41 in combination with chemotherapy in vivo, we established two PDX models of metastatic breast cancer, BC-1 and BC-2, using metastatic pleural effusion cells obtained from breast cancer patients who had failed multiple cycles of treatment in the clinic. PDX tumor models have emerged as a promising approach to evaluate the effects of cancer drugs on patient tumors and provide a new tool for evaluating new oncology drug candidates under development [41]. Comparison of tumors from patients and corresponding PDX tumors have demonstrated consistency in histological appearance, growth, mutations and gene expression patterns when PDX models are passaged in vivo [41–43]. This preservation of molecular and histological characteristics in PDX models suggests that PDX models may be superior to traditional cell line xenografts and better predictive of treatment response of novel anticancer drugs, a hypothesis that has gained strong support from a recently published high throughput drug evaluation study using PDX models [41]. Although we found that BC-1 and BC-2 PDX tumors express ER, surprisingly, these tumors grow without estradiol supplementation in immunodeficient mice. We found that both of these PDX tumors carry activating mutations in ESR1 which confer ligand-independent activity to ER and make them resistant to tamoxifen and fulvestrant treatment [30,31]. These mechanistic insights are consistent with the clinical course and responses of the BC-1 and BC-2 patient donors. We found that treatment with the combination of CPT-11 and 9-ING-41, in contrast to either agent used alone, led to regression of established BC-1 tumors. These data are consistent with results of the in vitro studies showing that 9-ING-41 potentiates the antitumor effect of CPT-11 in breast cancer cells after short-term exposure. In contrast to CPT-11-resistant BC-1 PDX tumors, which were also resistant to 9-ING-41 monotherapy at the dose and schedule evaluated in vivo, we found that the growth of BC-2 PDX tumors was partially inhibited by either 9-ING-41 or CPT-11 monotherapy. Tumor regressions were only observed in the BC-1 and BC-2 PDX models when 9-ING-41 and CPT-11 were used in combination. Thus, our in vivo results provide a rationale for combining 9-ING-41 and CPT-11 as a novel therapeutic approach for the treatment of metastatic breast cancer as well as exploring additional combinations of 9-ING-41 with chemotherapy more extensively in metastatic breast cancer PDX tumor models as part of future pre-clinical development.
The GSK-3 inhibitor 9-ING-41 is now being advanced as a clinical candidate and a recently completed pre-IND meeting has helped to elucidate an IND enabling development plan for 9-ING-41, with an IND submission anticipated in early 2017. Taken together, our findings provide critical evidence that inhibition of GSK-3 may be a promising therapeutic approach to overcome chemoresistance in metastatic breast cancer and provide the first rationale for the clinical development of 9-ING-41 in this indication.
Acknowledgments
This study was supported by a generous donation from the Baskes Family to the Robert H. Lurie Comprehensive Cancer Center of Northwestern University and by Cancer Center Support Grant 2 P30 CA060553-19 (APM, AU) to the Robert H. Lurie Comprehensive Cancer Center of Northwestern University. We thank Jian-Jun Wei, Jeremy Mathews, Demirkan Gursel, Stephen Rohan, Bernice Frederick and Bella Shmaltsuyev (Pathology Core Facility, Robert H. Lurie Comprehensive Cancer Center, Northwestern University) and Gennadiy Bondarenko (Center for Developmental Therapeutics, Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Evanston, IL) for providing us clinical samples of breast cancer and technical assistance. We express our gratitude to Jody Hirsh for editorial assistance.
Abbreviations
- ADMET
absorption, distribution, metabolism, excretion, toxicity
- PK
pharmacokinetic
- CDK
cyclin-dependent kinase
- PKA
protein kinase A
- Akt
protein kinase B
- PKC
protein kinase C
- GLP
good laboratory practice
- IND
investigational new drug
Footnotes
Conflict of interest
9-ING-41 has been licensed to Actuate Therapeutics, Inc. Alan Kozikowski, Andrey Ugolkov, Thomas O’Halloran and Andrew Mazar hold an equity interest in Actuate Therapeutics, Inc. Alan Kozikowski and Irina Gaisina are inventors on the 9-ING-41 patent.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Santa-Maria CA, Gradishar WJ. Changing treatment paradigms in metastatic breast cancer: lessons learned. JAMA Oncol. 2015;1:528–534. doi: 10.1001/jamaoncol.2015.1198. [DOI] [PubMed] [Google Scholar]
- 3.O’Shaughnessy J. Extending survival with chemotherapy in metastatic breast cancer. Oncologist. 2005;10:20–29. doi: 10.1634/theoncologist.10-90003-20. [DOI] [PubMed] [Google Scholar]
- 4.Woodgett J. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welsh G, Proud C. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J. 1993;294:625–629. doi: 10.1042/bj2940625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 7.de Groot R, Auwerx J, Bourouis M, Sassone-Corsi P. Negative regulation of Jun/AP-1: conserved function of glycogen synthase kinase 3 and the Drosophila kinase shaggy. Oncogene. 1993;8:841–847. [PubMed] [Google Scholar]
- 8.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diehl J, Cheng M, Roussel M, Sherr C. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubinfeld B, Albert I, Porfir E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 11.Ougolkov A, Fernandez-Zapico M, Savoy D, Urrutia R, Billadeau D. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005;65:2076–2081. doi: 10.1158/0008-5472.CAN-04-3642. [DOI] [PubMed] [Google Scholar]
- 12.Shakoori A, Ougolkov A, Zhang B, Modarressi M, Billadeau D, Mai M, et al. Deregulated GSK3beta activity in colorectal cancer: its association with tumor cell survival and proliferation. Biochem Biophys Res Commun. 2005;334:1365–1373. doi: 10.1016/j.bbrc.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 13.Gaisina I, Gallier F, Ougolkov A, Kim K, Kurome T, Guo S, et al. From a natural product lead to the identification of potent and selective benzofuran-3-yl-(indol-3-yl)maleimides as glycogen synthase kinase 3beta inhibitors that suppress proliferation and survival of pancreatic cancer cells. J Med Chem. 2009;52:1853–1863. doi: 10.1021/jm801317h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ougolkov A, Fernandez-Zapico M, Bilim V, Smyrk T, Chari S, Billadeau D. Aberrant nuclear accumulation of glycogen synthase kinase-3beta in human pancreatic cancer: association with kinase activity and tumor dedifferentiation, Clin. Cancer Res. 2006;12:5074–5081. doi: 10.1158/1078-0432.CCR-06-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ougolkov A, Bone N, Fernandez-Zapico M, Kay N, Billadeau D. Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor kappaB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood. 2007;110:735–742. doi: 10.1182/blood-2006-12-060947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bilim V, Ougolkov A, Yuuki K, Naito S, Kawazoe H, Muto A, et al. Glycogen synthase kinase-3: a new therapeutic target in renal cell carcinoma. Br J Cancer. 2009;101:2005–2014. doi: 10.1038/sj.bjc.6605437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naito S, Bilim V, Yuuki K, Ugolkov A, Motoyama T, Nagaoka A, et al. Glycogen synthase kinase-3beta: a prognostic marker and a potential therapeutic target in human bladder cancer. Clin Cancer Res. 2010;16:5124–5132. doi: 10.1158/1078-0432.CCR-10-0275. [DOI] [PubMed] [Google Scholar]
- 18.Cao Q, Lu X, Feng Y. Glycogen synthase kinase-3beta positively regulates the proliferation of human ovarian cancer cells. Cell Res. 2006;16:671–677. doi: 10.1038/sj.cr.7310078. [DOI] [PubMed] [Google Scholar]
- 19.Kunnimalaiyaan M, Vaccaro A, Ndiaye M, Chen H. Inactivation of glycogen synthase kinase-3beta, a downstream target of the raf-1 pathway, is associated with growth suppression in medullary thyroid cancer cells. Mol Cancer Ther. 2007;6:1151–1158. doi: 10.1158/1535-7163.MCT-06-0665. [DOI] [PubMed] [Google Scholar]
- 20.Miyashita K, Kawakami K, Nakada M, Mai W, Shakoori A, Fujisawa H, et al. Potential therapeutic effect of glycogen synthase kinase 3beta inhibition against human glioblastoma. Clin Cancer Res. 2009;15:887–897. doi: 10.1158/1078-0432.CCR-08-0760. [DOI] [PubMed] [Google Scholar]
- 21.Zhu Q, Yang J, Han S, Liu J, Holzbeierlein J, Thrashe J, et al. Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo. Prostate. 2011;71:835–845. doi: 10.1002/pros.21300. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Smith KS, Murphy M, Piloto O, Somervaille T, Cleary M. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature. 2008;455:1205–1209. doi: 10.1038/nature07284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quintayo MA, Munro AF, Thomas J, Kunkler IH, Jack W, Kerr GR, et al. GSK3β and cyclin D1 expression predicts outcome in early breast cancer patients. Breast Cancer Res Treat. 2012;136:161–168. doi: 10.1007/s10549-012-2229-8. [DOI] [PubMed] [Google Scholar]
- 24.Shin S, Wolgamott L, Tcherkezian J, Vallabhapurapu S, Yu Y, Roux PP, et al. Glycogen synthase kinase-3β positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene. 2014;33:1690–1699. doi: 10.1038/onc.2013.113. [DOI] [PubMed] [Google Scholar]
- 25.Hilliard T, Gaisina I, Muehlbauer A, Gaisin A, Gallier F, Burdette J. Glycogen synthase kinase 3beta inhibitors induce apoptosis in ovarian cancer cells and inhibit in-vivo tumor growth. Anticancer Drugs. 2011;22:978–985. doi: 10.1097/CAD.0b013e32834ac8fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhat R, Xue Y, Berg S, Hellberg S, Ormö M, Nilsson Y, et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278:45937–45945. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- 27.Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000;7:793–803. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- 28.Qu Y, Han B, Yu Y, Yao W, Bose S, Karlan BY, et al. Evaluation of MCF10A as a reliable model for normal human mammary epithelial cells. PLoS ONE. 2015;10:e0131285. doi: 10.1371/journal.pone.0131285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miya T, Goya T, Fujii H, Ohtsu T, Itoh K, Igarashi T, et al. Factors affecting the pharmacokinetics of CPT-11: the body mass index, age and sex are independent predictors of pharmacokinetic parameters of CPT-11, Invest. New Drugs. 2001;19:61–67. doi: 10.1023/a:1006456717846. [DOI] [PubMed] [Google Scholar]
- 30.Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pal K, Cao Y, Gaisina IN, Bhattacharya S, Dutta SK, Wang E, et al. Inhibition of GSK-3 induces differentiation and impaired glucose metabolism in renal cancer. Mol Cancer Ther. 2014;13:285–296. doi: 10.1158/1535-7163.MCT-13-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazor M, Kawano Y, Zhu H, Waxman J, Kypta RM. Inhibition of glycogen synthase kinase-3 represses androgen receptor activity and prostate cancer cell growth. Oncogene. 2004;23:7882–7892. doi: 10.1038/sj.onc.1208068. [DOI] [PubMed] [Google Scholar]
- 34.Zeng J, Liu D, Qiu Z, Huang Y, Chen B, Wang L, et al. GSK3β overexpression indicates poor prognosis and its inhibition reduces cell proliferation and survival of non-small cell lung cancer cells. PLoS ONE. 2014;9:e91231. doi: 10.1371/journal.pone.0091231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carter YM, Kunnimalaiyaan S, Chen H, Gamblin TC, Kunnimalaiyaan M. Specific glycogen synthase kinase-3 inhibition reduces neuroendocrine markers and suppresses neuroblastoma cell growth. Cancer Biol Ther. 2014;15:510–515. doi: 10.4161/cbt.28015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kotliarova S, Pastorino S, Kovell LC, Kotliarov Y, Song H, Zhang W, et al. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-kappaB, and glucose regulation. Cancer Res. 2008;68:6643–6651. doi: 10.1158/0008-5472.CAN-08-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buchholz T, Garg A, Chakravarti N, Aggarwal B, Esteva F, Kuerer H, et al. The nuclear transcription factor kappaB/bcl-2 pathway correlates with pathologic complete response to doxorubicin-based neoadjuvant chemotherapy in human breast cancer. Clin Cancer Res. 2005;11:8398–8402. doi: 10.1158/1078-0432.CCR-05-0885. [DOI] [PubMed] [Google Scholar]
- 38.Nakshatri H, Bhat-Nakshatri P, Martin D, Goulet RJ, Jr, Sledge GW., Jr Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol. 1997;17:3629–3639. doi: 10.1128/mcb.17.7.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimasaki T, Ishigaki Y, Nakamura Y, Takata T, Nakaya N, Nakajima H, et al. Glycogen synthase kinase 3β inhibition sensitizes pancreatic cancer cells to gemcitabine. J Gastroenterol. 2012;47:321–333. doi: 10.1007/s00535-011-0484-9. [DOI] [PubMed] [Google Scholar]
- 40.Shakoori A, Mai W, Miyashita K, Yasumoto K, Takahashi Y, Ooi A, et al. Inhibition of GSK-3 beta activity attenuates proliferation of human colon cancer cells in rodents. Cancer Sci. 2007;98:1388–1393. doi: 10.1111/j.1349-7006.2007.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318–1325. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- 42.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MY, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17:1514–1520. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73:4885–4897. doi: 10.1158/0008-5472.CAN-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]