

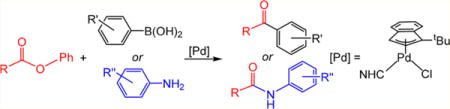
Abstract
Esters are valuable electrophiles for cross-coupling due to their ubiquity and ease of synthesis. However, harsh conditions are traditionally required for the effective cross-coupling of ester substrates. Utilizing a recently discovered precatalyst, Pd-catalyzed Suzuki–Miyaura and Buchwald–Hartwig reactions involving cleavage of the C(acyl)–O bond of aryl esters that proceed under mild conditions are reported. The Pd(II) precatalyst is highly active because it is reduced to the Pd(0) active species more rapidly than previous precatalysts.
Graphical abstract
Pd-catalyzed cross-coupling is one of the most powerful synthetic methods and is widely utilized in the synthesis of both pharmaceuticals and agrochemicals.1 Aryl halides or pseudo halides are typically utilized as the electrophile in cross-coupling reactions, and many catalysts that operate under mild conditions have been developed for these substrates.1 Nevertheless, to increase the applicability of the method there is interest in extending cross-coupling reactions to a broader range of electrophiles. For example Suzuki–Miyaura reactions involving triflates,2 sulfonates,3 thioesters,4 sulfonyl chlorides,5 perfluorinated arenes,6 diazonium and trimethylammonium salts,7 aryl methyl ethers,8 amides,9 and nitroarenes10 have all been reported. In particular, in the past decade it has been demonstrated that aryl esters can be used as the electrophile in cross-coupling reactions.11 Unactivated aryl esters are valuable substrates because they can readily be synthesized from phenols12 or carboxylic acids,13 are bench stable, and are common intermediates in organic synthesis. However, the use of aryl esters as electrophiles can lead to selectivity problems as either the C(aryl)–O bond14 or C(acyl)–O bond15–17 can potentially be cleaved (Figure 1).18 Additionally, cleavage of the C(acyl)–O bond followed by decarbonylation provides another alternative pathway.19 Catalysts that are selective for all three possible reactions have now been developed, but in general the conditions required for aryl ester cross-coupling are harsh.20 For example, elevated temperatures (80–130 °C), high catalyst loadings (3–15 mol %), and a significant excess of nucleophile and base are typically required. This limits the practicality of these reactions.
Figure 1.
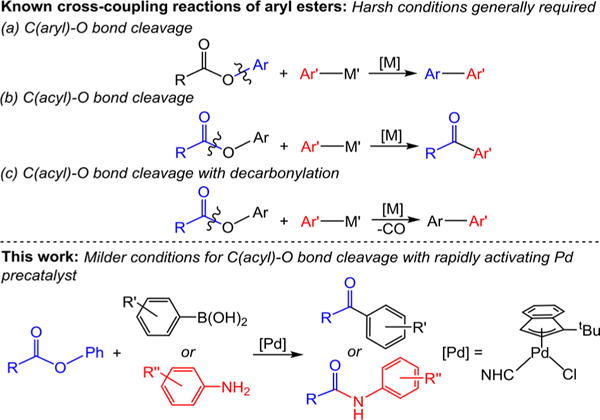
Summary of previous work on cross-coupling reactions of aryl esters and comparison to this work.
Suzuki–Miyaura reactions involving cleavage of the C-(acyl)–O bond in aryl esters represent a straightforward method for the synthesis of ketones from stable carboxylic acid derivatives. They offer significant advantages in terms of chemoselectivity, functional group tolerance, and atom economy compared to standard stoichiometric routes for the synthesis of aryl ketones, such as the addition of organometallic nucleophiles to Weinreb amides.21 Similarly, Buchwald– Hartwig reactions between aryl esters and amines to generate amides, especially those that involve non-nucleophilic amines, provide advantages over conventional routes to these biologically relevant molecules.22 In seminal work in 2017, Newman and co-workers reported the first Suzuki–Miyaura reactions of aryl esters.16a Using 3 mol % of (η3-cinnamyl)Pd-(IPr)(Cl) (IPr = 1,3-bis(2,6-diisopropyl-phenyl)-1,3-dihydro-2H-imidazol-2-ylidene) as the precatalyst, they were able to couple a range of aryl esters at 90 °C. Subsequently, the same group extended their work to Buchwald–Hartwig reactions between aryl esters and aniline-based substrates.16b In this case, 3 mol % of (η3-allyl)Pd(IPr)(Cl) was used as the precatalyst, along with elevated temperatures (110 °C), and extended times (16 h). In related work, Szostak and co-workers have demonstrated both Suzuki–Miyaura and Buchwald–Hartwig reactions of aryl esters using Pd-PEPPSI-type precatalysts.16e,f These initial examples demonstrate a new type of selectivity in cross-coupling reactions involving aryl esters, but milder conditions and lower catalyst loadings would make the reactions more synthetically accessible.
Recently, we described new bench-stable and commercially available precatalysts for cross-coupling based on the (η3-1-tBu-indenyl)Pd(L)(Cl) scaffold, which is compatible with both state-of-the-art N-heterocyclic carbene (NHC) and phosphine ligands.23 These precatalysts are typically more active for cross-coupling reactions than related precatalysts of the form (η3-allyl)Pd(L)(Cl) and (η3-cinnamyl)Pd(L)(Cl) because they activate to monoligated L-Pd(0) more rapidly and do not form inactive Pd(I) dimers.23,24 Here, we demonstrate that due to their rapid activation, precatalysts of the form (η3-1-tBu-indenyl)Pd(NHC)(Cl) catalyze Suzuki–Miyaura and Buchwald–Hartwig reactions involving cleavage of the C(acyl)–O bond of aryl esters under mild conditions. In fact, Suzuki– Miyaura reactions can be performed at room temperature using just 1 mol % catalyst loading, which are conditions comparable to reactions involving aryl halides.1a,b,d,1,1g,25 We note that while our work was in progress Szostak and co-workers reported the use of our precatalyst to perform room temperature Suzuki–Miyaura reactions of aryl esters, but the conditions described in this work are milder in regard to catalyst loading, time, and equivalents of both nucleophile and base.16d
Initially, we tested (η3-1-tBu-indenyl)Pd(IPr)(Cl) as a precatalyst for the coupling of phenyl benzoate with phenyl-boronic acid under the same conditions used by Newman et al. (3 mol % [Pd], 90 °C, THF) for (η3-cinnamyl)Pd(IPr)(Cl)16a and observed excellent activity (see Supporting Information, SI). Subsequently, we optimized the conditions for ester coupling using (η3-1-tBu-indenyl)Pd(IPr)(Cl) as the precatalyst by varying the catalyst loading, solvent, base, and equivalents of boronic acid (see SI). Using a 1 mol % loading of (η3-1-tBu-indenyl)Pd(IPr)(Cl), a 4:1 mixture of THF/H2O as the solvent, and 2 equiv of KOH as the base, we were able to quantitatively couple phenyl benzoate and phenylboronic acid to benzophenone at room temperature in 6 h. Other bases, such as K2CO3 or K3PO4, were also compatible with the reaction, but required longer reaction times (see SI). We also evaluated other ancillary ligands on precatalysts of the form (η3-1-tBu-indenyl)Pd(L)(Cl). Although the best results were obtained with IPr, some other NHC ligands, such as SIPr, also resulted in conversion to product (see SI). In contrast, no product was observed with phosphine ligands (see SI).
Using our optimized conditions, we expanded the substrate scope to other boronic acids (Scheme 1). We were able to couple a variety of arylboronic acids with phenyl benzoate in good to excellent yield at room temperature (1a–1g). The isolated yield of benzophenone (1a) was lower than expected based on the quantitative GC yield (see SI), presumably due to loss of product during workup.26 The coupling reaction involving the formation of 1d was performed on a 1 mmol scale, indicating that the reaction can be executed on a synthetically useful scale. The reaction was tolerant of sterically more demanding arylboronic acids with substituents in the 2-position of the aryl ring (1e, 1f). Although a substrate with an electron-donating substituent on the arylboronic acid was coupled in high yield (1b), a lower yield was observed when an electron-withdrawing group was present on the arylboronic acid (1g). Additionally, a very low yield of product was observed by GC when 4-nitrophenylboronic acid was used as a substrate with phenyl benzoate, and no attempt was made to isolate the product from this reaction. Heteroarylboronic acids, including 2-heteroaryl compounds, which readily undergo protodeborylation,27 also afforded product in good yield (1h–1j).
Scheme 1. Isolated Yields of Products in Suzuki–Miyaura Reactions Using Aryl Estersb.

aPerformed on 1 mmol scale in relation to benzoate. Yields are the average of two runs. bConditions: aryl ester (0.50 mmol), boronic acid (0.75 mmol), KOH (1.0 mmol), (η3-1-tBu-indenyl)Pd(IPr)(Cl) precatalyst (0.005 mmol), THF (2 mL), water (0.5 mL).
We also varied the aryl ester substrate in coupling reactions with 4-methoxyphenylboronic acid (Scheme 1, 1k–1n). When the aryl ring of the acyl group contained moderately electron-deficient or electron-neutral substituents good yields were obtained. However, GC analysis indicated that a lower yield was obtained when an electron-rich aryl ester substrate, such as phenyl 4-methoxybenzoate, was utilized and no attempts were made to isolate the product from this reaction. Similarly, no product was observed when the highly electron deficient substrate phenyl 4-nitrobenzoate was utilized and a low yield when methyl phenyl terephthalate was used (1m). A reduced yield was also observed with phenyl 2-naphthoate (1n) but we were able to isolate the product from this reaction in 50% yield. By GC the mixed alkyl aryl ketone 4′-methoxyacetophenone was formed in high yield using phenyl acetate as the starting ester, but attempts to isolate this product were unsuccessful.
Our results demonstrate that (η3-1-tBu-indenyl)Pd(IPr)(Cl) is a much more active precatalyst for Suzuki–Miyaura reactions of aryl esters compared to (η3-cinnamyl)Pd(IPr)(Cl). Furthermore, our conditions require a significantly lower number of equivalents of boronic acid (1.5 equiv vs 4.5 equiv) and base (2.0 equiv vs 7.2 equiv), lower catalyst loading (1 mol % vs 3 mol %), and shorter reaction time (6 h versus 15 h) compared to those very recently reported by Szostak and co-workers using our precatalyst.16d We propose that our use of water as a cosolvent greatly assists the reaction by solubilizing the base and boronic acid and increasing the rate of precatalyst activation (vide infra). Our results show that if the conditions and precatalyst are optimized, aryl esters can be coupled under conditions comparable to those normally used for aryl halides. It has been proposed that elevated temperatures are required for cross-coupling reactions involving aryl esters because the oxidative addition of the C(acyl)–O bond to the metal is challenging.20 Given that the active species in our system, monoligated IPr-Pd(0), is likely the same as in the Newman system,16a our results provide strong evidence that the elementary steps in the catalytic cycle are unlikely to have been the reason for the harsh conditions. Instead, precatalyst activation to form the active Pd(0) species may have been the problem. To probe the rate of precatalyst activation, we compared the rate of reduction of (η3-1-tBu-indenyl)Pd(IPr)-(Cl) and (η3-cinnamyl)Pd(IPr)(Cl) to Pd(0) with KOH (the base in catalysis) in a 4:1 THF/H2O mixture (our optimized solvent mixture). Using the chelating olefin 1,3-divinyl-1,1,3,3-tetramethyldisiloxane (dvds) as a trap for Pd(0), all of the (η3-1-tBu-indenyl)Pd(IPr)(Cl) was reduced to Pd(0) in just 2 h (eq 1). In contrast, for (η3-cinnamyl)Pd(IPr)(Cl) only 35%
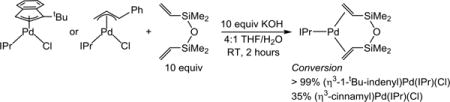 |
(eq 1) |
conversion to Pd(0) was achieved after 2 h, with the rest of the Pd still in the form of the starting precatalyst. This is consistent with our hypothesis that (η3-1-tBu-indenyl)Pd(IPr)(Cl) is a more efficient precatalyst because it activates rapidly. Addition-ally, slower activation of (η3-1-tBu-indenyl)Pd(IPr)(Cl) was observed when THF was used as the sole solvent, supporting our hypothesis that the use of water as a cosolvent increases the rate of activation (see SI). Previously, we have described how protic solvents can play a large role in the activation of precatalysts of the type (η3-1-tBu-indenyl)Pd(L)(Cl), and our observations here are in agreement with those results.24
Given the exceptional activity of (η3-1-tBu-indenyl)Pd(IPr)-(Cl) for Suzuki–Miyaura reactions involving aryl esters, we explored systems based on the (η3-1-tBu-indenyl)Pd(L)(Cl) framework for Buchwald–Hartwig couplings. Using 1 mol % (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (SIPr = 1,3-bis(2,6-diisoprop-yl-phenyl)imidazolidin-2-ylidene) as the precatalyst we were able to couple phenyl benzoate and aniline in essentially quantitative yield at 40 °C, in 4 h, using a 4:1 H2O/THF solvent mixture (Table 1). A number of different bases, including Cs2CO3, K3PO4, K2CO3, and Na2CO3, could be used with no significant change in activity. The reaction was extremely sensitive to the ancillary ligand, and under our optimized conditions no activity was observed when SIPr was replaced with other common NHC or phosphine ligands. Additionally, under our standard conditions (1 mol % precatalyst, 40 °C, 4:1 H2O/THF, 2 equiv Cs2CO3), no activity was seen using precatalysts of the type (η3-allyl)Pd-(L)(Cl) (L = IPr or SIPr), which have previously been used for this reaction with more forcing conditions.16b Control experiments indicated that, in an analogous fashion to the Suzuki– Miyaura reaction, this was due to slow activation under our optimized conditions, whereas (η3-1-tBu-indenyl)Pd(SIPr)(Cl) activated rapidly (see SI).
Table 1.
Yields for Screening of Pd-Catalyzed Buchwald–Hartwig Reactions of Phenyl Benzoatea
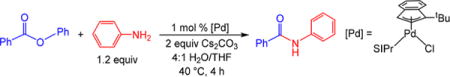
| ||
|---|---|---|
| entry | deviation from optimized conditions | yield (%)b |
| 1 | none | >99 |
| 2 | (η3-allyl)Pd(SIPr)(Cl) instead of (η3-1-tBu-indenyl) Pd(SIPr)(Cl) | 0 |
| 3 | (η3-allyl)Pd(IPr)(Cl) instead of (η3-1-tBu-indenyl) Pd(SIPr)(Cl) | 0 |
| 4 | K3PO4 instead of Cs2CO3 | >99 |
| 5 | K2CO3 instead of Cs2CO3 | >99 |
| 6 | Na2CO3 instead of Cs2CO3 | >99 |
| 7 | IPr instead of SIPr | 0 |
| 8 | SIMes instead of SIPr | 0 |
| 9 | IPr*OMe instead of SIPr | 0 |
| 10 | XPhos instead of SIPr | 0 |
| 11 | P(o-tol)3 instead of SIPr | 0 |
| 12 | PtBu3 instead of SIPr | 0 |
Conditions: phenyl benzoate (0.20 mmol), aniline (0.24 mmol), Cs2CO3 (0.3 mmol), (η3-1-tBu-indenyl)Pd(L)(Cl) (0.002 mmol), solvent (1 mL).
Yields are the average of two runs and were determined by GC conversion. SIMes = 1,3-bis(2,4,6-trimethylphen-yl)-4,5-dihydroimidazol-2-ylidene. IPr*OMe = 1,3-bis(2,6-bis-(diphenylmethyl)-4-methoxyphenyl)imidazol-2-ylidene. XPhos = 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl.
The substrate scope for the coupling of 4-methoxyaniline with a variety of phenyl esters was examined (Scheme 2). Phenyl benzoates containing both electron rich and electron poor substituents on the aryl ring of the acyl group were successfully coupled (2a–2e). In some cases slight modifications to our optimized conditions were required to achieve isolated yields of approximately 90%, but there were no clear trends based on the electronic properties of the acyl group. Nevertheless, we did not observe any product when 4-nitrobenzophenone or methyl phenyl terephthalate, both of which contain electron withdrawing groups, were used as substrates. Phenyl 2-methylbenzoate (2f), which contains an ortho-substituent on the aryl ring of the acyl group, and phenyl 2-naphthoate (2g) were also coupled in high yield. However, presumably due to the increased steric congestion, no product was observed when phenyl 2,6-dimethylbenzoate was used as a substrate. The reaction was tolerant to oxygen containing heterocycles on the ester as phenyl furan-2-carboxylate (2h) was successfully coupled. In contrast, no reaction was observed using phenyl picolinate, likely due to coordination of the nitrogen atom of the pyridine ring to the catalyst. The reaction is also compatible with phenyl esters containing alkyl acyl groups. Phenyl isobutyrate (2i) and phenyl pivalate (2j) were coupled with yields of 67 and 80%, respectively. It is noteworthy that all of the esters used in in this work are relatively unactivated, and as shown by Newman, will not form amides under basic conditions without a catalyst.16b
Scheme 2. Isolated Yields of Products in Buchwald–Hartwig Reactions Using Aryl Esters and 4-Methoxyanilineb.
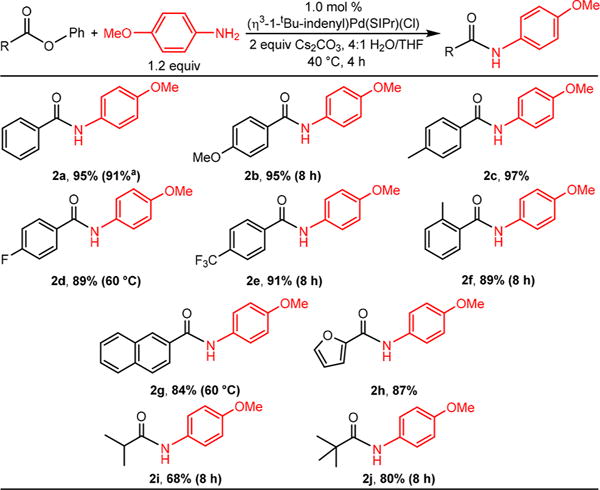
aPerformed on 1 mmol scale in relation to aryl ester. Yields are the average of two runs. bConditions: aryl ester (0.50 mmol), 4-methoxyaniline (0.60 mmol), Cs2CO3 (0.75 mmol), (η3-1-tBu-indenyl)Pd(SIPr)(Cl) precatalyst (0.005 mmol), water (2 mL), THF (0.5 mL).
We also explored the substrate scope for the coupling of phenyl benzoate with different substituted aniline nucleophiles (Scheme 3). The reaction was tolerant to anilines with electron donating and electron withdrawing substituents (3a–3h). However, lower yields or more forcing conditions were required with substrates with more electron withdrawing substituents, such as 4-trifluoromethylaniline (3f) and methyl 4-aminobenzoate (3g). The steric properties of the aniline affected the reaction, and significantly reduced yields were observed with 2,6-dimethylaniline (3d). Both 1- and 2-naphthylamine (3j, 3j) were coupled in high yield, but the reaction was not very tolerant to anilines with heteroatoms. For example, 2-aminopyridine (3k) was coupled, but more forcing conditions were required, and the yield was lowered significantly. Additionally, no product was observed when 4-aminopyridine was used as the nucleophile. Overall, our conditions represent a substantial improvement over those previously described in the literature in terms of catalyst loading (1 mol % vs 3 mol %), temperature (40 °C vs 110 °C), and time (4 h vs 16 h). These results indicate that Buchwald-Hartwig reactions of aryl esters can be performed at temperatures only slightly above room temperature, which presents the opportunity to increase the synthetic applications of this reaction.
Scheme 3. Isolated Yields of Products in Buchwald–Hartwig Reactions Using Phenyl Benzoate and Different Substituted Anilinesb.
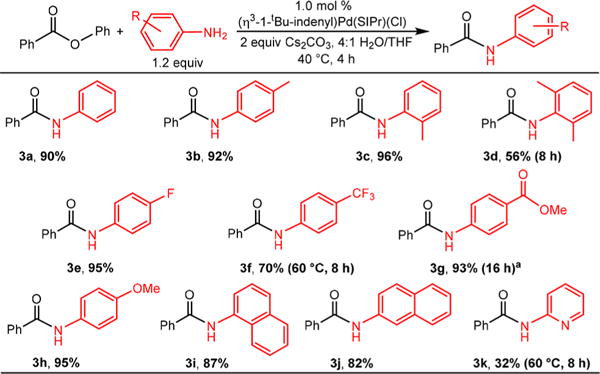
a2.5 equiv of amine and 3 equiv of Cs2CO3 were utilized. bConditions: aryl ester (0.50 mmol), substituted aniline (0.60 mmol), Cs2CO3 (0.75 mmol), (η3-1-tBu-indenyl)Pd(SIPr)(Cl) precatalyst (0.005 mmol), water (2 mL), THF (0.5 mL). Yields are the average of two runs.
In conclusion, we have demonstrated that, because our (1-tBu-indenyl)Pd(L)(Cl) precatalysts activate rapidly, they can be used for Suzuki–Miyaura and Buchwald–Hartwig couplings involving aryl ester substrates without the need for forcing conditions. Our conditions are the mildest reported to date for these synthetically relevant reactions and indicate that the barriers for the elementary steps in catalysis do not require elevated temperatures.20 In fact, cross-coupling reactions involving aryl ester electrophiles may actually be as facile as those involving aryl halides. In future work we will attempt to use our fast activating systems to improve other reactions where precatalyst activation is the limiting factor in catalysis, which may be a more common problem than is currently realized.
EXPERIMENTAL SECTION
General
Experiments were performed under a dinitrogen atmosphere in an M-Braun drybox or using standard Schlenk techniques unless otherwise stated. Under standard glovebox conditions purging was not performed between uses of diethyl ether, pentane, benzene, toluene, and THF; thus when any of these solvents were used, traces of all these solvents were in the atmosphere and could be found intermixed in the solvent bottles. Moisture- and air-sensitive liquids were transferred by stainless steel cannula on a Schlenk line or in a drybox. NMR spectra were recorded on Agilent-400, -500, and -600 spectrometers at ambient probe temperatures unless noted. Chemical shifts are reported with respect to residual internal protio solvent for 1H and 13C NMR spectra. Gas chromatography (GC) analyses were performed on a Shimadzu GC-2010 Plus apparatus equipped with a flame ionization detector and a Shimadzu SHRXI-5MS column (30 m, 250 μm inner diameter, film: 0.25 μm). The following conditions were utilized for GC analyses: flow rate 1.23 mL/min constant flow, column temperature 50 °C (held for 5 min), 20 °C/min increase to 300 °C (held for 5 min), total time 22.5 min. THF was dried by passage through a column of activated alumina followed by storage under dinitrogen. H2O was degassed by sparging with dinitrogen for 1 h and stored under dinitrogen. All commercial chemicals were used as received except where noted. Ethyl acetate and hexanes (Fisher Scientific) were used as received. SIPr, IPr*OMe, IMes, and SIMes were purchased from Strem Chemicals. SIPr was stored in a −35 °C fridge in dinitrogen-filled glovebox. IPr was either synthesized according to a literature procedure,28 or purchased from Strem Chemicals. XPhos, RuPhos, and PtBu3 were purchased from Sigma-Aldrich and stored in a dinitrogen-filled glovebox; P(o-tolyl)3 was purchased from Strem Chemicals and stored in a dinitrogen-filled glovebox. Esters were all prepared from acyl chlorides or carboxylic acids using literature procedures.16a Palladium precatalysts were synthesized by methods previously reported.23,29
Substrate Scope for Pd-Catalyzed Suzuki–Miyaura Reactions (Scheme 1)
The following general procedure was used to determine the substrate scope unless otherwise stated:
Electrophile (0.5 mmol), boronic acid (0.75 mmol), KOH (1.0 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) were added to a 4 dram vial equipped with a magnetic stir bar. THF (2 mL) and H2O (0.5 mL) were added in a glovebox. The vial was stirred at rt for 6 h. At this time, the vial was opened to air and the crude mixture was concentrated under vacuum, dissolved in toluene (1 mL), and 3 M KOHaq (3 mL) was added and stirred for 1 h at rt. The organic layer was extracted with ethyl acetate (3 × 5 mL), washed with brine, dried (MgSO4), and concentrated. Purification was performed using silica column chromatography.
Benzophenone (1a)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), phenylboronic acid (91.4 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 1% EtOAc/99% hexanes. The average of two runs provided a yield of 51% (46.5 mg). 1H and 13C NMR data were consistent with that published in the literature.16a
4-Methoxyphenyl(phenyl)methanone (1b)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), 4-methoxyphenylboronic acid (114.0 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. No column purification was needed. The average of two runs provided a yield of 86% (91.2 mg). 1H and 13C NMR data were consistent with that published in the literature.30
Naphthalen-1-yl(phenyl)methanone (1c)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), naphthalen-1-ylboronic acid (129.0 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. At this time, 0.1 M KOHaq (2 mL) was added and the mixture was stirred at 60 °C for 2 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 1% EtOAc/99% hexanes. The average of two runs provided a yield of 91% (105.7 mg). 1H and 13C NMR data were consistent with that published in the literature.31
Naphthalen-2-yl(phenyl)methanone (1d)
Following the general procedure above, a mixture of phenyl benzoate (198.2 mg, 1.00 mmol), naphthalen-2-ylboronic acid (258.0 mg, 1.50 mmol), KOH (112.2 mg, 2.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (7.0 mg, 0.01 mmol) in THF (4.0 mL) and H2O (1.0 mL) were stirred at rt for 6 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 1% EtOAc/99% hexanes. The average of two runs provided a yield of 65% (152.0 mg). 1H and 13C NMR data were consistent with that published in the literature.32
(2-Methoxyphenyl)(phenyl)methanone (1e)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), 2-methoxyphenylboronic acid (114.0 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. At this time, 0.1 M KOHaq (2 mL) was added and the mixture was stirred at 60 °C for 2 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 1% EtOAc/99% hexanes. The average of two runs provided a yield of 88% (93.4 mg). 1H and 13C NMR data were consistent with that published in the literature.16a
Phenyl(o-tolyl)methanone (1f)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), o-tolylboronic acid (102.0 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. No column purification was needed. The average of two runs provided a yield of 86% (84.4 mg). 1H and 13C NMR data were consistent with that published in the literature.16a
Phenyl(4-(trifluoromethyl)phenyl)methanone (1g)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), (4-(trifluoromethyl)phenyl)boronic acid (142.4 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)-(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. The crude mixture was concentrated under vacuum, dissolved in toluene (1 mL) and 3 mL of 3 M KOHaq was added to the reaction mixture and stirred for 1 h at rt. The residue was concentrated under reduced pressure, dissolved in 1:1 THF/KOHaq (0.1M) and stirred at 60 °C for 1 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 5% EtOAc/95% hexanes. The average of two runs provided a yield of 67% (83.8 mg). 1H and 13C NMR data were consistent with that published in the literature.16a
Furan-2-yl(phenyl)methanone (1h)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), furan-2-ylboronic acid (83.9 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. At this time, the vial was opened to air and the organic layer was extracted with ethyl acetate (3 × 5 mL), washed with brine, dried (MgSO4), and concentrated. The crude mixture was dissolved in 1 mL of CH2Cl2, and 2 mL of KOHaq (3M) was added and stirred for 1 h at rt. The organic layer was extracted with CH2Cl2 (3 × 5 mL), washed with brine, dried (MgSO4), and concentrated. No further purification was needed. The average of two runs provided a yield of 83% (71 mg). 1H and 13C NMR data were consistent with that published in the literature.33
Furan-3-yl(phenyl)methanone (1j)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), furan-3-ylboronic acid (83.9 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 5% EtOAc/95% hexanes. The average of two runs provided a yield of 74% (63.7 mg). 1H and 13C NMR data were consistent with that published in the literature.16a
Phenyl(thiophen-2-yl)methanone (1j)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), thiophen-2-ylboronic acid (96.0 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at 50 °C for 6 h. At this time, the vial was opened to air and the organic layer was extracted with ethyl acetate (3 × 5 mL), washed with brine, dried (MgSO4), and concentrated. The crude mixture was dissolved in 1 mL of CH2Cl2, and 2 mL of KOHaq (3M) was added and stirred for 1 h at rt. The organic layer was extracted with CH2Cl2 (3 × 5 mL), washed with brine, dried (MgSO4), and concentrated. No further purification was needed. The average of two runs provided a yield of 81% (76.0 mg). 1H and 13C NMR data were consistent with that published in the literature.34
(4-Methoxyphenyl)(p-tolyl)methanone (1k)
Following the general procedure above, a mixture of phenyl(4-methyl)benzoate (106.1 mg, 0.5 mmol), (4-methoxyphenyl)boronic acid (114.0 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 10% EtOAc/99% hexanes. The average of two runs provided a yield of 64% (72.1 mg). 1H and 13C NMR data were consistent with that published in the literature.35
(4-Methoxyphenyl)(4-trifluoromethylphenyl)methanone (1l)
Following the general procedure above, a mixture of phenyl(4-trifluoromethyl)benzoate (133.1 mg, 0.5 mmol), (4-methoxyphenyl)-boronic acid (114.0 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 10% EtOAc/90% hexanes. The average of two runs provided a yield of 64% (89.3 mg). 1H and 13C NMR data were consistent with that published in the literature.36
Methyl 4-(4-Methoxybenzoyl)benzoate (1m)
Following the general procedure above, a mixture of methyl phenyl terephthalate (128.1 mg, 0.5 mmol), (4-methoxyphenyl)boronic acid (114.0 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol, and (η3-1-tBu-indenyl)Pd-(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 16 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 5% EtOAc/95% hexanes. The average of two runs provided a yield of 32% (43.9 mg). 1H and 13C NMR data were consistent with that published in the literature.37
(4-Methoxyphenyl)(naphthalene-2-yl)methanone (1n)
Following the general procedure above, a mixture of phenyl(2-naphthoate) (124.1 mg, 0.5 mmol), (4-methoxyphenyl)boronic acid (114.0 mg, 0.75 mmol), KOH (56.1 mg, 1.00 mmol), and (η3-1-tBu-indenyl)Pd-(IPr)(Cl) (3.5 mg, 0.005 mmol) in THF (2.0 mL) and H2O (0.5 mL) were stirred at rt for 6 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 1% EtOAc/99% hexanes. Organic solvents were evaporated under reduced pressure and the residue was dissolved in 1 mL of CH2Cl2, and 2 mL of KOHaq (3M) was added and stirred for 1 h at rt. The organic layer was extracted with CH2Cl2 (3 × 5 mL), washed with brine, dried (MgSO4), and concentrated. The average of two runs provided a yield of 50% (65 mg). 1H and 13C NMR data were consistent with that published in the literature.38
Bis(4-methoxyphenyl) Methanone
Following the general proce-dure above, a mixture of phenyl(4-methoxy)benzoate (45.6 mg, 0.2 mmol), (4-methoxyphenyl)boronic acid (36.5 mg, 0.3 mmol), KOH (22.4 mg, 0.4 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (1.4 mg, 0.002 mmol) in THF (0.8 mL) and H2O (0.2 mL) were stirred at rt for 6 h. Comparison of the GC responses of phenyl(4-methoxy)-benzoate starting material and bis(4-methoxyphenyl) methanone product indicated 60% conversion. Isolation of this product was not attempted.
1-(4-Methoxyphenyl)ethan-1-one
Following the general procedure above, a mixture of phenyl acetate (27.2 mg, 0.2 mmol), (4-methoxyphenyl)boronic acid (45.6 mg, 0.3 mmol), K2CO3 (55.3 mg, 0.4 mmol), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) (1.4 mg, 0.001 mmol) in THF (0.8 mL) and H2O (0.2 mL) were stirred at rt for 6 h. Comparison of the GC responses of phenyl(4-methoxy)benzoate starting material and bis(4-methoxyphenyl) methanone product indicated >99% conversion. Successful isolation of this product was not achieved.
Screening Conditions for Pd-Catalyzed Buchwald–Hartwig Reactions (Table 1)
Phenyl benzoate (39.6 mg, 0.2 mmol), aniline (22 μL, 0.24 mmol), base (0.3 mmol), and (η3-1-tBu-indenyl)Pd(L)-(Cl) (0.002 mmol) were added to a 1 dram vial equipped with a stir bar. THF (0.2 mL) and H2O (0.8 mL) were added in a glovebox. The vial was stirred at 40 °C for 4 h. At this time, the vial was opened to air, extracted with ethyl acetate, and filtered through a silica plug. Conversion was determined by comparison of the GC responses of phenyl benzoate starting material and benzanilide product.
Substrate Scope for Pd-Catalyzed Buchwald–Hartwig Reactions (Schemes 2 and 3)
The following general procedure was used to determine the substrate scope unless otherwise stated:
Electrophile (0.5 mmol), amine (0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) were added to a 2 dram vial equipped with a magnetic stir bar. THF (0.5 mL) and H2O (2 mL) were added in a glovebox. The vial was stirred at 40 °C for 4 h. After this time, the vial was opened to air and the aqueous layer was extracted with EtOAc (5 × 4 mL) and concentrated under vacuum. The resulting solid was dissolved in toluene (2–4 mL) and stirred at rt with 1 M HClaq (4 mL) for 30 min. The aqueous layer was extracted with EtOAc (5 × 4 mL). The combined organic layer was washed with NaHCO3(aq) and concentrated. Purification was performed using silica column chromatography.
N-(4-Methoxyphenyl)benzamide (2a, 3h)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), 4-methoxyaniline (73.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 20% EtOAc/80% hexanes. The average of two runs provided a yield of 95% (107.6 mg). 1H and 13C NMR data were consistent with that published in the literature.39
N-(4-Methoxyphenyl)benzamide (2a, 3h)
N-(4-Methoxyphenyl)-benzamide was also isolated on a 1 mmol scale of phenyl benzoate. Following the general procedure above, a mixture of phenyl benzoate (198.2 mg, 1.0 mmol), 4-methoxyaniline (147.8 mg, 1.2 mmol), Cs2CO3 (488.8 mg, 1.5 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (7.0 mg, 0.01 mmol) in THF (1 mL) and H2O (4 mL) were stirred at 40 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 20% EtOAc/80% hexanes. The average of two runs provided a yield of 91% (206.0 mg). 1H and 13C NMR data were consistent with that published in the literature.39
N-(4-Methoxyphenyl)-4-methoxybenzamide (2b)
Following the general procedure above, a mixture of phenyl 4-methoxy benzoate (114.1 mg, 0.5 mmol), 4-methoxyaniline (73.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 8 h. Purification was performed by filtering with minimal EtOAc and collecting the solid. The average of two runs provided a yield of 95% (122.4 mg). 1H and 13C NMR data were consistent with that published in the literature.40
N-(4-Methoxyphenyl)-4-methylbenzamide (2c)
Following the general procedure above, a mixture of phenyl 4-methyl benzoate (106.1 mg, 0.5 mmol), 4-methoxyaniline (73.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 20% EtOAc/80% hexanes. The average of two runs provided a yield of 97% (117.3 mg). 1H and 13C NMR data were consistent with that published in the literature.41
N-(4-Methoxyphenyl)-4-fluorobenzamide (2d)
Following the general procedure above, a mixture of phenyl 4-fluoro benzoate (108.1 mg, 0.5 mmol), 4-methoxyaniline (73.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 60 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 20% EtOAc/80% hexanes. The average of two runs provided a yield of 89% (109.0 mg). 1H and 13C NMR data were consistent with that published in the literature.42
N-(4-Methoxyphenyl)-4-trifluoromethylbenzamide (2e)
Following the general procedure above, a mixture of phenyl 4-trifluoromethyl benzoate (114.1 mg, 0.5 mmol), 4-methoxyaniline (73.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd-(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 8 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 50% EtOAc/50% hexanes. The average of two runs provided a yield of 91% (133.9 mg). 1H and 13C NMR data were consistent with that published in the literature.43
N-(4-Methoxyphenyl)-2-methylbenzamide (2f)
Following the general procedure above, a mixture of phenyl 2-methylbenzoate (106.1 mg, 0.5 mmol), 2-methylaniline (73.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 8 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 20% EtOAc/80% hexanes. The average of two runs provided a yield of 89% (107.8 mg). 1H and 13C NMR data were consistent with that published in the literature.44
N-(4-Methoxyphenyl)-2-naphthylamide (2g)
Following the gen-eral procedure above, a mixture of phenyl 2-naphthoate (124.1 mg, 0.5 mmol), 4-methoxyaniline (73.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 60 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 30% EtOAc/70% hexanes. The average of two runs provided a yield of 84% (116.2 mg). 1H and 13C NMR data were consistent with that published in the literature.45
N-(4-Methoxyphenyl)-2-furylamide (2h)
Following the general procedure above, a mixture of phenyl furan 2-carboxylate (94.1 mg, 0.5 mmol), 4-methoxyaniline (73.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 30% EtOAc/70% hexanes. The average of two runs provided a yield of 87% (94.6 mg). 1H and 13C NMR data were consistent with that published in the literature.46
N-(4-Methoxyphenyl)-isobutyramide (2i)
Following the general procedure above, a mixture of phenyl isobutyrate (82.0 mg, 0.5 mmol), 4-methoxyaniline (73.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 8 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 20% EtOAc/80% hexanes. The average of two runs provided a yield of 68% (65.6 mg). 1H and 13C NMR data were consistent with that published in the literature.47
N-(4-Methoxyphenyl)-tertbutyramide (2j)
Following the general procedure above, a mixture of phenyl tertbutyrate (89.1 mg, 0.5 mmol), 4-methoxyaniline (73.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 8 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 20% EtOAc/80% hexanes. The average of two runs provided a yield of 80% (82.5 mg). 1H and 13C NMR data were consistent with that published in the literature.48
N-Phenylbenzamide (3a)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), aniline (55.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 20% EtOAc/80% hexanes. The average of two runs provided a yield of 90% (88.7 mg). 1H and 13C NMR data were consistent with that published in the literature.39
N-(4-Methylphenyl)benzamide (3b)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), p-toluidine (64.3 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 50% EtOAc/50% hexanes. The average of two runs provided a yield of 92% (96.8 mg). 1H and 13C NMR data were consistent with that published in the literature.39
N-(2-Methylphenyl)benzamide (3c)
Following the general proce-dure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), o-toluidine (64.3 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 50% EtOAc/50% hexanes. The average of two runs provided a yield of 96% (101.2 mg). 1H and 13C NMR data were consistent with that published in the literature.39
N-(2,6-Dimethylphenyl)benzamide (3d)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), 2,6-dimethylaniline (72.7 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 8 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 30% EtOAc/70% hexanes. The average of two runs provided a yield of 56% (63.5 mg). 1H and 13C NMR data were consistent with that published in the literature.49
N-(p-Fluorophenyl)benzamide (3e)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), 4-fluoroaniline (66.7 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 50% EtOAc/50% hexanes. The average of two runs provided a yield of 95% (102.1 mg). 1H and 13C NMR data were consistent with that published in the literature.39
N-(p-Trifluoromethylphenyl)benzamide (3f)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), 4-trifluoromethylaniline (96.7 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 60 °C for 8 h. After this time, the vial was opened to air and the aqueous layer was extracted with EtOAc (5 × 4 mL) and concentrated under vacuum. The resulting solid was dissolved in toluene (2 mL) and stirred at rt with 1 N HClaq (4 mL) for 2 h. The aqueous layer was extracted with EtOAc (5 × 4 mL). The combined organic layer was washed with NaHCO3(aq) and concentrated. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 50% EtOAc/50% hexanes. The average of two runs provided a yield of 70% (92.2 mg). 1H and 13C NMR data were consistent with that published in the literature.50
Methyl 4-Benzamidobenzoate (3g)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), methyl 4-aminobenzoate (189.5 mg, 1.25 mmol), Cs2CO3 (488.7 mg, 1.5 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 16 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 30% EtOAc/70% hexanes. The average of two runs provided a yield of 93% (119.0 mg). 1H and 13C NMR data were consistent with that published in the literature.51
N-(1-Naphthyl)benzamide (3i)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), 1-naphthylamine (85.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 50% EtOAc/50% hexanes. The average of two runs provided a yield of 87% (107.6 mg). 1H and 13C NMR data were consistent with that published in the literature.49
N-(2-Naphthyl)benzamide (3j)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), 2-naphthylamine (85.9 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 40 °C for 4 h. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 50% EtOAc/50% hexanes. The average of two runs provided a yield of 82% (101.4 mg). 1H and 13C NMR data were consistent with that published in the literature.52
N-(2-Pyridyl)benzamide (3k)
Following the general procedure above, a mixture of phenyl benzoate (99.1 mg, 0.5 mmol), 2-pyridylamine (56.5 mg, 0.6 mmol), Cs2CO3 (244.4 mg, 0.75 mmol), and (η3-1-tBu-indenyl)Pd(SIPr)(Cl) (3.5 mg, 0.005 mmol) in THF (0.5 mL) and H2O (2 mL) were stirred at 60 °C for 8 h. The resulting solid was not stirred with HClaq. Purification was performed using silica column chromatography by using a gradient of 100% hexanes to 30% EtOAc/70% hexanes. The average of two runs provided a yield of 32% (31.6 mg). 1H and 13C NMR data were consistent with that published in the literature.53
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the NIHGMS under Award Number R01GM120162. R.M.D., P.R.M., and M.M. thank the NSF for support as Graduate Research Fellows.
Footnotes
ORCID
Nilay Hazari: 0000-0001-8337-198X
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b02588.
Additional details of screening and activation experiments and NMR spectra (PDF)
References
- 1.For recent reviews about cross-coupling see:; (a) Magano J, Dunetz JR. Chem Rev. 2011;111:2177–2250. doi: 10.1021/cr100346g. [DOI] [PubMed] [Google Scholar]; (b) Valente C, Calimsiz S, Hoi KH, Mallik D, Sayah M, Organ MG. Angew Chem, Int Ed. 2012;51:3314–3332. doi: 10.1002/anie.201106131. [DOI] [PubMed] [Google Scholar]; (c) Johansson Seechurn CC, Kitching MO, Colacot TJ, Snieckus V. Angew Chem, Int Ed. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]; (d) Colacot TJ, editor. New Trends in Cross-Coupling: Theory and Applications. The Royal Society of Chemistry; Cambridge UK: 2015. pp. 1–864. (RSC Catalysis Series No. 21). [Google Scholar]; (e) Gildner PG, Colacot TJ. Organometallics. 2015;34:5497. [Google Scholar]; (f) Chem Rev. 2016;116:12564–12649. doi: 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Hazari N, Melvin PR, Mohadjer Beromi M. Nat Rev Chem. 2017;1:0025. doi: 10.1038/s41570-017-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huth A, Beetz I, Schumann I. Tetrahedron. 1989;45:6679–6682. [Google Scholar]
- 3.Nguyen HN, Huang X, Buchwald SL. J Am Chem Soc. 2003;125:11818–11819. doi: 10.1021/ja036947t. [DOI] [PubMed] [Google Scholar]
- 4.Liebeskind LS, Srogl J. Org Lett. 2002;4:979–981. doi: 10.1021/ol0200091. [DOI] [PubMed] [Google Scholar]
- 5.Dubbaka SR, Vogel P. Org Lett. 2004;6:95–98. doi: 10.1021/ol036131x. [DOI] [PubMed] [Google Scholar]
- 6.Schaub T, Backes M, Radius U. J Am Chem Soc. 2006;128:15964–15965. doi: 10.1021/ja064068b. [DOI] [PubMed] [Google Scholar]
- 7.(a) Darses S, Jeffery T, Genet JP, Brayer JL, Demoute JP. Tetrahedron Lett. 1996;37:3857–3860. [Google Scholar]; (b) Blakey SB, MacMillan DWC. J Am Chem Soc. 2003;125:6046–6047. doi: 10.1021/ja034908b. [DOI] [PubMed] [Google Scholar]
- 8.Tobisu M, Shimasaki T, Chatani N. Angew Chem, Int Ed. 2008;47:4866–4869. doi: 10.1002/anie.200801447. [DOI] [PubMed] [Google Scholar]
- 9.Weires NA, Baker EL, Garg NK. Nat Chem. 2015;8:75–79. doi: 10.1038/nchem.2388. [DOI] [PubMed] [Google Scholar]
- 10.Yadav MR, Nagaoka M, Kashihara M, Zhong RL, Miyazaki T, Sakaki S, Nakao Y. J Am Chem Soc. 2017;139:9423–9426. doi: 10.1021/jacs.7b03159. [DOI] [PubMed] [Google Scholar]
- 11.(a) Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V. Chem Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mesganaw T, Garg NK. Org Process Res Dev. 2013;17:29–39. [Google Scholar]; (c) Tobisu M, Chatani N. Top Curr Chem. 2016;374:1–28. doi: 10.1007/s41061-016-0043-1. [DOI] [PubMed] [Google Scholar]; (d) Mondal M, Begum T, Bora U. Org Chem Front. 2017;4:1430–1434. [Google Scholar]
- 12.Rappoport Z. The Chemistry of Phenols. John Wiley & Sons; Chichester: 2003. [Google Scholar]
- 13.Li J. Fischer-Speier Esterification. In: Li JJ, Corey EJ, editors. Name Reactions for Functional Group Transformations. John Wiley & Sons; NJ: 2007. pp. 458–461. [Google Scholar]
- 14.(a) Quasdorf KW, Tian X, Garg NK. J Am Chem Soc. 2008;130:14422–14423. doi: 10.1021/ja806244b. [DOI] [PubMed] [Google Scholar]; (b) Guan BT, Wang Y, Li BJ, Yu DG, Shi ZJ. J Am Chem Soc. 2008;130:14468–14470. doi: 10.1021/ja8056503. [DOI] [PubMed] [Google Scholar]; (c) Li BJ, Li YZ, Lu XY, Liu J, Guan BT, Shi ZJ. Angew Chem, Int Ed. 2008;47:10124–10127. doi: 10.1002/anie.200803814. [DOI] [PubMed] [Google Scholar]; (d) Ehle AR, Zhou Q, Watson MP. Org Lett. 2012;14:1202–1205. doi: 10.1021/ol203322v. [DOI] [PubMed] [Google Scholar]; (e) Shimasaki T, Tobisu M, Chatani N. Angew Chem, Int Ed. 2010;49:2929–2932. doi: 10.1002/anie.200907287. [DOI] [PubMed] [Google Scholar]; (f) Cornella J, Jackson EP, Martin R. Angew Chem, Int Ed. 2015;54:4075–4078. doi: 10.1002/anie.201412051. [DOI] [PubMed] [Google Scholar]
- 15.For examples containing only 2-pyridyl esters see:; Tatamidani H, Kakiuchi F, Chatani N. Org Lett. 2004;6:3597–3599. doi: 10.1021/ol048513o. [DOI] [PubMed] [Google Scholar]
- 16.For examples containing unactivated aryl esters see:; (a) Ben Halima T, Zhang W, Yalaoui I, Hong X, Yang YF, Houk KN, Newman SG. J Am Chem Soc. 2017;139:1311–1318. doi: 10.1021/jacs.6b12329. [DOI] [PubMed] [Google Scholar]; (b) Ben Halima T, Vandavasi JK, Shkoor M, Newman SG. ACS Catal. 2017;7:2176–2180. [Google Scholar]; (c) Malapit CA, Caldwell DR, Sassu N, Milbin S, Howell AR. Org Lett. 2017;19:1966–1969. doi: 10.1021/acs.orglett.7b00494. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lei P, Meng G, Shi S, Ling Y, An J, Szostak R, Szostak M. Chem Sci. 2017;8:6525–6530. doi: 10.1039/c7sc02692g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shi S, Lei P, Szostak M. Organometallics. 2017;36:3784–3789. [Google Scholar]; (f) Shi S, Szostak M. Chem Commun. 2017;53:10584–10587. doi: 10.1039/c7cc06186b. [DOI] [PubMed] [Google Scholar]
- 17.For examples containing methyl esters see:; Hie L, Fine Nathel NF, Hong X, Yang Y-F, Houk KN, Garg NK. Angew Chem, Int Ed. 2016;55:2810–2814. doi: 10.1002/anie.201511486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong X, Liang Y, Houk KN. J Am Chem Soc. 2014;136:2017–2025. doi: 10.1021/ja4118413. [DOI] [PubMed] [Google Scholar]
- 19.(a) Amaike K, Muto K, Yamaguchi J, Itami K. J Am Chem Soc. 2012;134:13573–13576. doi: 10.1021/ja306062c. [DOI] [PubMed] [Google Scholar]; (b) Muto K, Yamaguchi J, Musaev DG, Itami K. Nat Commun. 2015;6:7508. doi: 10.1038/ncomms8508. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) LaBerge NA, Love JA. Eur J Org Chem. 2015;2015:5546–5553. [Google Scholar]; (d) Guo L, Chatupheeraphat A, Rueping M. Angew Chem, Int Ed. 2016;55:11810–11813. doi: 10.1002/anie.201604696. [DOI] [PubMed] [Google Scholar]; (e) Pu X, Hu J, Zhao Y, Shi Z. ACS Catal. 2016;6:6692–6698. [Google Scholar]; (f) Muto K, Hatakeyama T, Itami K, Yamaguchi J. Org Lett. 2016;18:5106–5109. doi: 10.1021/acs.orglett.6b02556. [DOI] [PubMed] [Google Scholar]; (g) Liu X, Jia J, Rueping M. ACS Catal. 2017;7:4491–4496. [Google Scholar]
- 20.Takise R, Muto K, Yamaguchi J. Chem Soc Rev. 2017;46:5864–5888. doi: 10.1039/c7cs00182g. [DOI] [PubMed] [Google Scholar]
- 21.Balasubramaniam S, Aidhen IS. Synthesis. 2008;2008:3707–3738. [Google Scholar]
- 22.Constable DJC, Dunn PJ, Hayler JD, Humphrey GR, Leazer JL, Linderman RJ, Lorenz K, Manley J, Pearlman BA, Wells A, Zaks A, Zhang TY. Green Chem. 2007;9:411–420. [Google Scholar]
- 23.Melvin PR, Nova A, Balcells D, Dai W, Hazari N, Hruszkewycz DP, Shah HP, Tudge MT. ACS Catal. 2015;5:3680–3688. [Google Scholar]
- 24.Melvin PR, Balcells D, Hazari N, Nova A. ACS Catal. 2015;5:5596–5606. [Google Scholar]
- 25.(a) Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; (b) Corbet JP, Mignani G. Chem Rev. 2006;106:2651–2710. doi: 10.1021/cr0505268. [DOI] [PubMed] [Google Scholar]
- 26.In the SI we have provided a comparison of GC yields before isolation and isolated yields for all reactions that were performed in this manuscript.
- 27.Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073–14075. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang P, Wang W, Ritter T. J Am Chem Soc. 2011;133:11482–11484. doi: 10.1021/ja2048072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Viciu MS, Germaneau RF, Navarro-Fernandez O, Stevens ED, Nolan SP. Organometallics. 2002;21:5470–5472. [Google Scholar]; (b) Marion N, Navarro O, Mei J, Stevens ED, Scott NM, Nolan SP. J Am Chem Soc. 2006;128:4101–4111. doi: 10.1021/ja057704z. [DOI] [PubMed] [Google Scholar]
- 30.Das T, Chakraborty A, Sarkar A. Tetrahedron Lett. 2014;55:7198–7202. [Google Scholar]
- 31.Yu A, Shen L, Cui X, Peng D, Wu Y. Tetrahedron. 2012;68:2283–2288. [Google Scholar]
- 32.Biju AT, Glorius F. Angew Chem, Int Ed. 2010;49:9761–9764. doi: 10.1002/anie.201005490. [DOI] [PubMed] [Google Scholar]
- 33.Ortiz P, del Hoyo AM, Harutyunyan SR. Eur J Org Chem. 2015;2015:72–76. [Google Scholar]
- 34.Gautam P, Dhiman M, Polshettiwar V, Bhanage BM. Green Chem. 2016;18:5890–5899. [Google Scholar]
- 35.Cheng WM, Shang R, Yu HZ, Fu Y. Chem – Eur J. 2015;21:13191–13195. doi: 10.1002/chem.201502286. [DOI] [PubMed] [Google Scholar]
- 36.Benischke AD, Leroux M, Knoll I, Knochel P. Org Lett. 2016;18:3626–3629. doi: 10.1021/acs.orglett.6b01677. [DOI] [PubMed] [Google Scholar]
- 37.Qi X, Jiang LB, Li HP, Wu XF. Chem – Eur J. 2015;21:17650–17656. doi: 10.1002/chem.201502943. [DOI] [PubMed] [Google Scholar]
- 38.Li H, Xu Y, Shi E, Wei W, Suo X, Wan X. Chem Commun. 2011;47:7880–7882. doi: 10.1039/c1cc12843d. [DOI] [PubMed] [Google Scholar]
- 39.Deshidi R, Rizvi MA, Shah BA. RSC Adv. 2015;5:90521–90524. [Google Scholar]
- 40.Xie F, Du C, Pang Y, Lian X, Xue C, Chen Y, Wang X, Cheng M, Guo C, Lin B, Liu Y. Tetrahedron Lett. 2016;57:5820–5824. [Google Scholar]
- 41.Sharma N, Sekar G. Adv Synth Catal. 2016;358:314–320. [Google Scholar]
- 42.Waisser K, Kuneš J, Kubicová L, Buděšínský M, Exner O. Magn Reson Chem. 1997;35:543–548. [Google Scholar]
- 43.Obata A, Ano Y, Chatani N. Chem Sci. 2017;8:6650–6655. doi: 10.1039/c7sc01750b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nozawa-Kumada K, Kadokawa J, Kameyama T, Kondo Y. Org Lett. 2015;17:4479–4481. doi: 10.1021/acs.orglett.5b02235. [DOI] [PubMed] [Google Scholar]
- 45.Gonec T, Bobal P, Sujan J, Pesko M, Guo J, Kralova K, Pavlacka L, Vesely L, Kreckova E, Kos J, Coffey A, Kollar P, Imramovsky A, Placek L, Jampilek J. Molecules. 2012;17:613–644. doi: 10.3390/molecules17010613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee CK, Yu JS, Ji YR. J Heterocycl Chem. 2002;39:1219–1227. [Google Scholar]
- 47.McPherson CG, Livingstone K, Jamieson C, Simpson I. Synlett. 2015;27:88–92. [Google Scholar]
- 48.Kathiravan S, Nicholls IA. Tetrahedron Lett. 2017;58:1–4. [Google Scholar]
- 49.Zhang L, Su S, Wu H, Wang S. Tetrahedron. 2009;65:10022–10024. [Google Scholar]
- 50.Whittaker AM, Dong VM. Angew Chem, Int Ed. 2015;54:1312–1315. doi: 10.1002/anie.201410322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaplaneris N, Koutoulogenis G, Raftopoulou M, Kokotos CG. J Org Chem. 2015;80:5464–5473. doi: 10.1021/acs.joc.5b00283. [DOI] [PubMed] [Google Scholar]
- 52.Chen CT, Kuo JH, Pawar VD, Munot YS, Weng SS, Ku CH, Liu CY. J Org Chem. 2005;70:1188–1197. doi: 10.1021/jo048363v. [DOI] [PubMed] [Google Scholar]
- 53.Rostamnia S, Doustkhah E, Golchin-Hosseini H, Zeynizadeh B, Xin H, Luque R. Catal Sci Technol. 2016;6:4124–4133. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.