

Abstract
Background and Purpose
Amphetamine is a releaser of dopamine stored in synaptic terminals, which can suppress appetite by changing the expression levels of neuropeptide Y (NPY) and proopiomelanocortin (POMC) in the hypothalamus. This study explored whether ERKs are involved in appetite control mediated by cAMP response element binding protein (CREB), NPY and POMC in amphetamine‐treated rats.
Experimental Approach
Rats were given amphetamine for 4 days, and changes in feeding behaviour and expression levels of phosphorylated‐ERK (pERK), pCREB, NPY and melanocortin MC3 receptors were examined and compared.
Key Results
Following amphetamine treatment, food intake, body weight and NPY expression decreased, whereas the expression of pERK, pCREB, MC3 receptors and pCREB/DNA binding activity increased. In amphetamine‐treated rats, both cerebral ERK knockdown and pretreatment with a peripheral dopamine receptor antagonist decreased NPY but increased pERK, pCREB and MC3 receptor expression. Moreover, the immunofluorescence of hypothalamic pERK increased following amphetamine treatment.
Conclusions and Implications
These results suggest that ERK/CREB signalling participates in the effects mediated by dopamine receptor/NPY/POMC on appetite control in rats treated with amphetamine. These findings advance the knowledge on the involvement of ERK/CREB signalling in the reciprocal regulation by NPY and POMC of appetite after amphetamine treatment.
Abbreviations
- ARC
arcuate nucleus
- ChIP
chromatin immunoprecipitation
- IHC
immunohistochemistry
- NPY
neuropeptide Y
- ODN
oligodeoxynucleotides
- pCREB
phospho‐cAMP response element binding protein
- pERK
phospho‐ERK
- POMC
proopiomelanocortin
- PVN
paraventricular nucleus
- S‐ODN
phosphorothioate oligodeoxynucleotides
Introduction
Many neuronal populations are known to affect feeding behaviour in the hypothalamus. These include the orexigenic signals neuropeptide Y (NPY) and agouti‐related peptide, and the anorexigenic signals proopiomelanocortin (POMC), and cocaine‐ and amphetamine‐related transcription. Several compounds and drugs that modulate satiety exert their effects by inhibiting NPY or activating POMC neurons (Gilbert et al., 2014; Rocha et al., 2014). Amphetamine is a well‐known appetite suppressant. Its anorectic effect is associated with the central release of dopamine, which in turn can decrease NPY but increase POMC expression in the hypothalamus (Gillard et al., 1993; Chen et al., 2001; Hsieh et al., 2014). In addition, NPY and POMC may function reciprocally in the control of amphetamine‐mediated appetite suppression (Fioramonti et al., 2007; Hsieh et al., 2011, 2013a; Chu et al., 2014). The melanocortin receptor (MC receptor) belongs to the family of POMC system, which can regulate feeding behaviour and energy expenditure (Butler, 2006). The MC receptor has five subtypes, that is, MC1 through to MC5 receptors. In the CNS, MC3 and MC4 receptors are involved in the regulation of appetite (Cone, 2005) and participate in the control of amphetamine‐mediated anorexia (Hsieh et al., 2011; Chu et al., 2014). In rodent animals, obesity can result from deletion of either MC4 (Huszar et al., 1997) or MC3 receptors (Butler et al., 2000).
MAPKs, which include ERK, JNK and p38 MAPK, are important mediators responding to extracellullar stimuli and cellular stress (Ichijo, 1999; Mielke and Herdegen, 2000; Chang and Karin, 2001). In the brain, ERK1/2 is associated with the drug‐mediated psychomotor activity, reward behaviour and drug‐seeking relapse behaviours (Valjent et al., 2000; Sun et al., 2016). Previous studies revealed that hypothalamic phosphorylated‐ERK (pERK) signalling is required for the induction of cAMP response element binding protein (CREB) and NPY genes (Olsson and Nanberg, 2001; Barnea et al., 2004). CREB is downstream of ERK and is required for dopamine‐dependent gene expression (Andersson et al., 2001), alcohol consumption‐associated NPY gene expression (Pandey, 2003; Moonat and Pandey, 2012) and feeding behaviours (Hsieh et al., 2008). In the brain, previous studies examined the involvement of ERK/CREB signalling in different brain regions for measuring different behavioural responses. For example, (i) the pERK/ pCREB pathway in the hippocampus was shown to be involved in improved cognitive function (Lim et al., 2014) and memory (Xiang et al., 2016), and (ii) alterations in the ERK/CREB signalling in the mesocorticolimbic system attenuated nicotine‐mediated locomotor sensitization (Zhu et al., 2015). It is unclear whether pERK‐pCREB signalling is involved in the anorectic effects of amphetamine. Previous studies suggested that activation of pERK/pCREB signalling in the hypothalamus contributed to a fasting response (Morikawa et al., 2004) and that inhibition of ERK/CREB signalling in the striatum decreases amphetamine‐induced hyperlocomotion and the expression of neuropeptide genes (Shi and McGinty, 2007; McGinty and Mendelson, 2011). Thus, it is possible that hypothalamic pERK/pCREB signalling is involved in the reciprocal regulation by NPY/POMC of amphetamine‐mediated appetite control.
Methods
Animals
All of the procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health (8th edition, 2011). All animal surgeries were performed under sodium pentobarbital (35–40 mg·kg−1; i.p.) anaesthesia. The used dose would be added half the volume if rats felt pain or wake up. Every effort was made to minimize suffering in these animals. A total of 255 animals were used in the experiments described here. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Male Wistar rats weighing 200–300 g were obtained from the National Laboratory Animal Centre (Taipei, Taiwan). The animals were housed individually in cages and maintained at a temperature of 22 ± 2°C in a room with a 12 h light–dark cycle (lights on at 06:00 h). The rats were also habituated to frequent handling. Drugs were administered, and food intake was determined every day at the beginning of the dark phase (18:00 h). Water and chow (LabDiet, PMI Nutrition International, Brentwood, MO, USA) were freely available throughout the experiment. Food intake data points above 40 g·day−1 were discarded because they indicated food spillage.
Animal treatment
To examine the effect of amphetamine on feeding behaviour and body weight, rats (n = 8 in each drug‐treated group; total of four groups) were injected i.p. with amphetamine (1, 2 or 4 mg·kg−1) once a day for 4 days. Amphetamine was first injected at the end of Day 0 (i.e. at 18:00 h), which is also the beginning of Day 1. Food intake and body weight data were calculated with respect to that on the previous day. After the administration of amphetamine for 40 min, rats were anaesthetized with pentobarbital (35–40 mg·kg−1; i.p.) and decapitated. As described in our previous report, amphetamine can decrease hypothalamic NPY expression at about 40–50 min following drug treatment (Hsieh et al., 2005).
To determine the effect of daily amphetamine (2 mg·kg−1; i.p.) on changes in hypothalamic total ERK1/2 (tERK1/2), total CREB (tCREB), pERK1/2, pCREB, NPY, POMC and MC3 receptor expression, rats were injected with the drug once a day for 1, 2, 3 or 4 days, depending on the group (n = 8 for each daily treatment groups; total of five groups). Rats were treated with amphetamine (2 mg·kg−1; i.p.) 40–50 min before being killed to enhance the effects of the drug on hypothalamic protein expression. The rats were anaesthetized and decapitated. Following decapitation, the hypothalamic portions were immediately removed from the brain and subjected to analysis to detect protein (described above) expression or stored at −80°C until future use.
To determine the effect of amphetamine on pCREB/3DNA binding activity, rats were given amphetamine (4 mg·kg−1; i.p.; n = 6 for each daily treatment group; total of five groups) daily for 4 days at the beginning of dark phase (at 18:00 h). The hypothalamus was removed to determine pCREB/DNA binding activity by a technique of chromatin immunoprecipitation (ChIP) assay 40–50 min after amphetamine treatment.
To assess the effect of pretreatment with an ERK antisense oligodeoxynucleotide (ODN) on the response to amphetamine (4 mg·kg−1; i.p.), rats (n = 6 per group; total of seven groups) were given ERK antisense (20 μg in a 10 μL vehicle) i.c.v. 1 h before amphetamine treatment daily for 4 days. Before amphetamine treatment, rats were administered a similar dose of ERK antisense (i.c.v.) daily for 2–3 days until the feeding behavioural response slightly increased. As reported previously, either continuous or repeated i.c.v. injections of antisense may be necessary to maximize behavioural effects and importantly to block the synthesis of a constitutively active gene product (Zhang and Creese, 1993; Ogawa and Pfaff, 1998).
To determine the effect of haloperidol pretreatment on amphetamine‐induced feeding behaviour (n = 8 each group; total of four groups) and hypothalamic protein expression on Day 1, rats (n = 8 each group; total of four groups) were injected with haloperidol (1 mg·kg−1; i.p.) 40 min before amphetamine (4 mg·kg−1) treatment. Haloperidol is a non‐selective dopamine D1/D2 receptor antagonist, which can block approximately 90% of the response of amphetamine‐induced anorexia (Chen et al., 2001). Rats received haloperidol and/or amphetamine at 40 min prior to being anaesthetized and then decapitated immediately to remove the hypothalamus. Haloperidol was first dissolved in DMSO and then diluted with 0.9% saline. This mixed solution was used as a vehicle. The hypothalamic protein detected included pERK1/2, pCREB, NPY, POMC and MC3 receptors.
To examine the effect of amphetamine on the expression of pERK1/2 immunofluorescence in the hypothalamic arcuate nucleus (ARC) and paraventricular nucleus (PVN), rats were given amphetamine (4 mg·kg−1; n = 5 each group; total of three groups) once a day for 2 days. Forty minutes after the second day of amphetamine treatment, rats were killed, under ether anaesthesia, by transaortic perfusion with a fixative solution, as described below.
Chromatin immunoprecipitation (ChIP) assay
ChIP analysis was performed as described previously (Chien et al., 2012). Chromatin isolation and the ChIP assay were performed by using the EZ‐ChIP kit (Millipore, Bedford, MA, USA) according to the manufacturer's instructions. For a detailed description of the procedure, please see our previous publication (Hsieh et al., 2014). The sequences of the PCR primers used for CREB are as follows: 5′‐GAAAGCAGTGACTGAGGAGCTTGTA‐3′ (forward) and 5′‐ GGGCTAAGCAGTTGGTGGTGCAGGATGCA‐3′ (backward). Extracted DNA (2 μL) was used for 45 cycles of amplification in 50 μL of reaction mixture under the following conditions: 95°C for 30 s, 58°C for 60 s and 72°C for 30 s. The PCR products were analysed by 2% agarose gel electrophoresis. Equal amounts of the soluble cross‐linked chromatins present in each PCR were confirmed by the product for input. Rabbit polyclonal IgG was used as a negative control. The concentration of CREB/DNA binding activity is presented as a percentage of the control group.
Lateral ventricular cannulation
Stereotaxic surgery (Kopf Model 900, Tujunga, CA, USA) was performed on each rat under pentobarbital anaesthesia (30 mg·kg−1, i.p.). A cannula was inserted near the junction of the right lateral ventricle and the third ventricle (coordinates: 0.8 mm posterior to the Bregma, 1.5 mm from the midline and 3.5–4.0 mm below the dura) (Paxinos and Watson, 1986). For a detailed description of the procedure, please see our previous report (Kuo et al., 2011). For all experiments, the cannula placement was verified by histochemistry of the brain section and by the administration of angiotensin II (100 ng per rat). Angiotensin II reliably induces water drinking in non‐deprived rats when administered into the cerebroventricles (Ritter et al., 1981). Only data from rats that drank more than 10 mL of water in 30 min were included in this study. Behavioural testing of drinking began about 1 week after the cannulation surgery and the restoration of feeding behaviour, and then angiotensin II was administered to confirm the cannula placement. About 2 days after the treatment with angiotensin II, to confirm the restoration of normal drinking behaviour, the amphetamine treatment experiments were started.
Cerebral infusion of ERK antisense
The sequence of ERK antisense ODN was designed as reported previously (Sale et al., 1995): antisense: 5′‐GCCGCCGCCGCCGCCAT‐3′, directed against the initiation of the translation start site of rat ERK1 and ERK2 mRNAs, missense: 5′‐CGCGCGCTCGCGCACCC‐3′. The efficacy of the antisense ODN in inhibiting ERK expression was confirmed at the protein level by Western blotting. Twelve hours after the last administration of the antisense ODN, the expression of the total ERK protein was reduced by approximately 60% in the rat spinal cord (Sale et al., 1995). Rats were handled and injected i.c.v. with vehicle 4 days prior to the experimental injections to accustom them to the procedure. One hour before amphetamine (4 mg·kg−1·day−1, i.p.) treatment, ERK antisense ODN, 20 μg (in 10 μL)·day−1, was administered to eight rats daily for 4 days. An equivalent dose of missense ODN was administered to each of the eight rats that served as the control. Food intake and body weight changes were recorded daily. We used ERK antisense that was phosphorothioate‐ODNs (S‐ODNs) modified only on the three terminal bases of both the 5′ and 3′ ends, because these S‐ODNs can improve hybridization affinity and nuclease resistance and were regarded as a well‐established agent in several vertebrate systems (Ogawa and Pfaff, 1998). Both antisense and missense S‐ODNs were dissolved in artificial CSF containing 140 mM NaCl, 3.35 mM KCl, 1.15 mM MgCl2, 1.26 mM CaCl2, 1.2 mM Na2HPO4 and 0.3 mM NaH2PO4; pH 7.4.
Western blotting
Hypothalamic protein levels were measured in a block of mediobasal hypothalamic tissue as described previously (Morris, 1989). The block of mediobasal hypothalamic tissue was dissected rostral‐caudally from the optic chiasma to the mammillary body and extended laterally from the midline of hypothalamus to the perihypothalamic nucleus and was superior to the anterior commissure. Proteins were separated on a 12.5% SDS polyacrylamide gel (for NPY, MC3 receptors, pERK1/2, pCREB and β‐actin), which were transferred onto a nitrocellulose membrane, and incubated with specific antibodies against NPY (1:1000 dilution), pERK1/2 (1:1000), pCREB (1:500 dilution), MC3 receptor (1:1000 dilution) and β‐actin (1:1000 dilution). After incubation with horseradish peroxidase goat anti‐rabbit IgG, the colour signal was developed using 4‐chloro‐1‐naphthol/3,3′‐diaminobenzidine and 0.9% (w.v‐1) NaCl in Tris–HCl. The relative photographic density was quantified by scanning the photographic negative film on a Gel Documentation and Analysis System (AlphaImager 2000, Alpha Innotech Corporation, San Leandro, CA, USA).
Immunohistochemistry (IHC)
The rats were killed under ether anaesthesia by transaortic perfusion with 50 mL heparin‐treated saline and then treated with 250 mL of fixative (4% paraformaldehyde in 0.1 M phosphate buffer; pH 7.4). For details, please see our previous report (Kuo et al., 2011). Sections were incubated at 4°C for 24 h with primary antibody anti‐pERK1/2 at a 1:50 dilution. After being washed with Tris‐buffered saline with Tween 20 (TBST), all sections were incubated at 37°C for 1 h using a rhodamine red‐X‐conjugated goat anti‐mouse IgG (1:50 dilution) to stain pERK and using DAPI (0.5 μg·mL−1) to stain the nucleus. Slides were rinsed with TBST for a total of three washes. Tissues were then covered with mounting media (Vector Laboratories, Burlingame, CA, USA) and sealed with cover slips. Images were visualized by an upright fluorescent microscope (ZEISS AXioskop2, Carl Zeiss Microscopy, Thornwood, NY, USA) and captured digitally. Fluorescence intensities were quantified by ImageJ software (National Institute of Health, Bethesda, MD, USA). Global calibration, Global scale and Threshold were set to remove the background and to select the area for quantification during the procedure.
In the procedure of double immunofluorecent staining, primary antibodies of anti‐pERK1/2 and anti‐POMC (1:100, Cell Signalling Technology, Danvers, MA, USA) were applied on the sections following 24 h incubation at 4°C. The next day, all sections were further incubated with DAPI and corresponding secondary antibodies: fluorescein isothiocyanate‐conjugated goat anti‐mouse IgG (1:50, Jackson ImmunoResearch) for anti‐pERK1/2 and rhodamine‐conjugated goat anti‐rabbit IgG (1:50, Jackson ImmunoResearch) for anti‐POMC, at 37°C for 1 h in the dark. Results were then observed under a confocal microscope (ZEISS LSM 510 META).
Statistical analysis
A one‐way or two‐way ANOVA followed by Dunnett's test (P < 0.05) was used to detect significant differences in or between the groups. The statistical analyses with one‐way ANOVA was performed to assess the results shown in Figures 2, 3, 4, 5. The two‐way ANOVA was performed to analyse the results in Figure 1. Student's t‐test was used to detect the significant differences in the IHC experiment. Data are presented as the means ± SEM. Statistical significance was set at P < 0.05. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Figure 2.
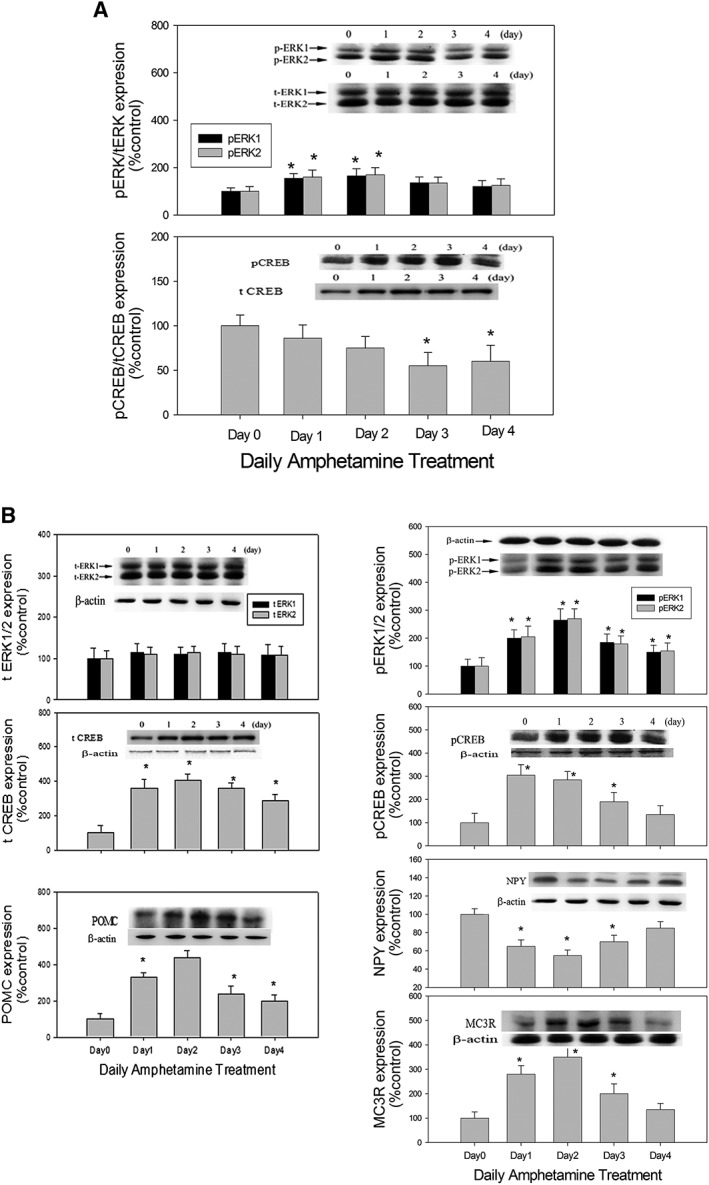
The effect of daily amphetamine (2 mg·kg−1; i.p.) on the ratios of (A) hypothalamic pERK/tERK and pCREB/tCREB expression (upper panel), and on (B) NPY, POMC, pERK1/2, pCREB and MC3 receptor (MC3R) (lower panel) expression over a 4 day period. Inserted images showed the results of Western blots, and the columns represent their relative densitometric values. Contents of protein in AMPH‐treated groups were indicated as the percentage of the control group. Bars are the means ± SEM; n = 8 for each daily treatment group (from Day 0 to Day 4; total of five day‐dependent groups). *P < 0.05 versus control.
Figure 3.
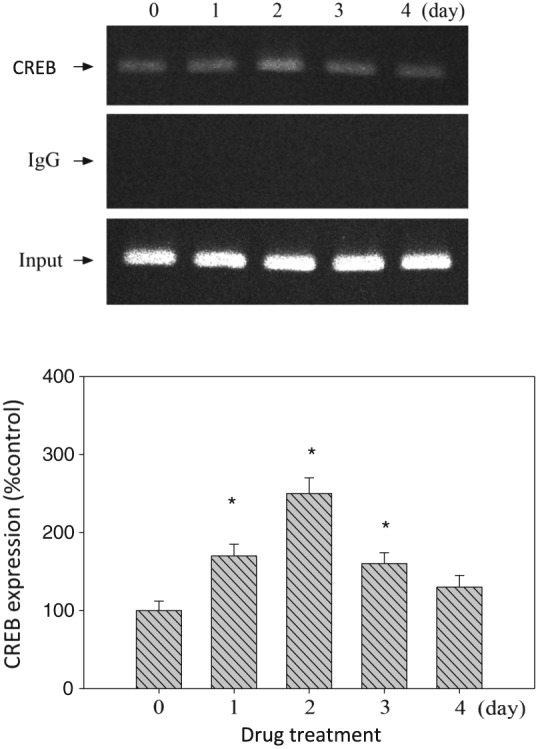
The hypothalamus tissue was prepared for the ChIP assay using antibodies specific for CREB. PCR amplification using primers designed against CREB‐binding sites was performed. Equal amounts of the soluble cross‐linked chromatins present in each PCR were confirmed by the product for input. Rabbit polyclonal IgG was used as a negative control. Input, 1% of sonicated cross‐linked chromatins. Upper panel: the result of ChIP analysis of CREB/DNA binding activity. Lower panel: relative densitometric values for ChIP assay. Concentrations of CREB/DNA binding activity were indicated as the percentage of the control group. Bars are the means ± SEM; n = 6 for each group (from Day 0 to Day 4; total of five day‐dependent groups). *P < 0.05 versus control.
Figure 4.
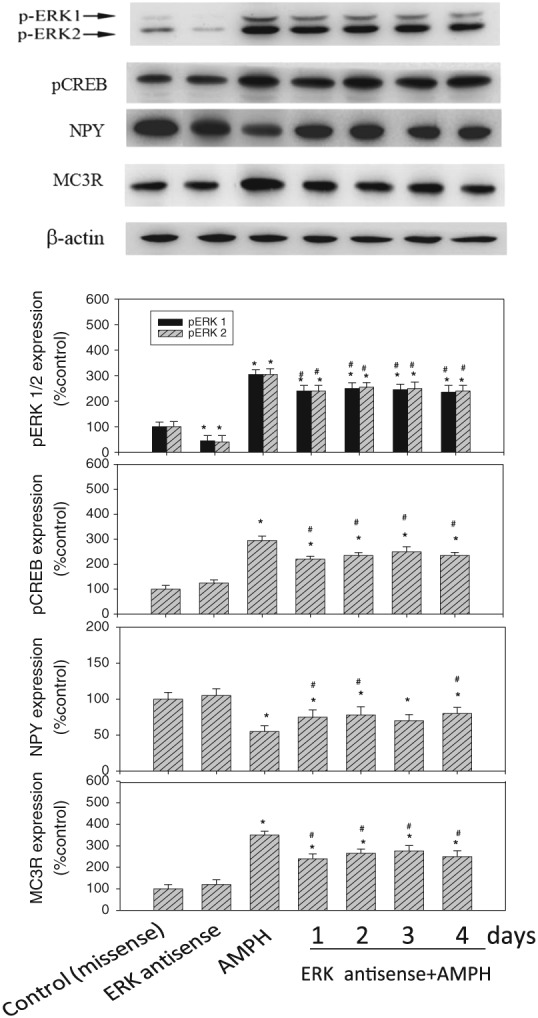
The effect of ERK antisense/amphetamine (AMPH) co‐administration on the changes in hypothalamic pERK1/2, pCREB, NPY, POMC and MC3 receptor (MC3R) expression during a 4 day period of drug treatment. Upper panel: each of the protein levels was measured by Western blot. Lower panel: the protein levels in drug‐treated groups were indicated as the percentage of the control group (Day 0). Each densitometric value represents the mean ± SEM; n = 6 for each group (total of seven drug‐treated groups). *P < 0.05 versus the control (missense‐treated) group. # P < 0.05 versus the AMPH‐treated group.
Figure 5.
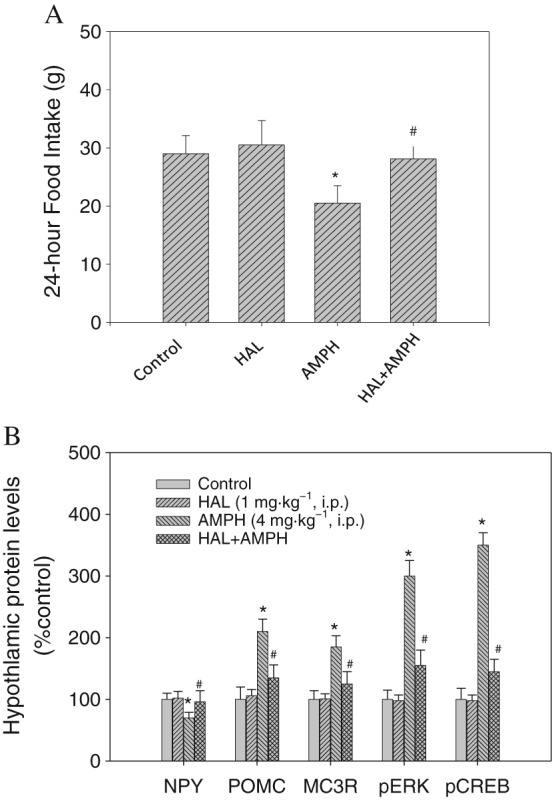
(A) The effect of haloperidol/amphetamine (AMPH) co‐administration on food intake on Day 1 (n = 8 for each group; total of four groups). (B) The effect of haloperidol/AMPH co‐administration on the changes in hypothalamic pERK1/2, pCREB, NPY, POMC and MC3 receptor (MC3R) expression on Day 1. The protein levels in drug‐treated groups (n = 8 for each group; total of four groups) were indicated as the percentage of the control group (Day 0). Each densitometric value represents the mean ± SEM. *P < 0.05 versus the control group. # P < 0.05 versus the AMPH‐treated group.
Figure 1.
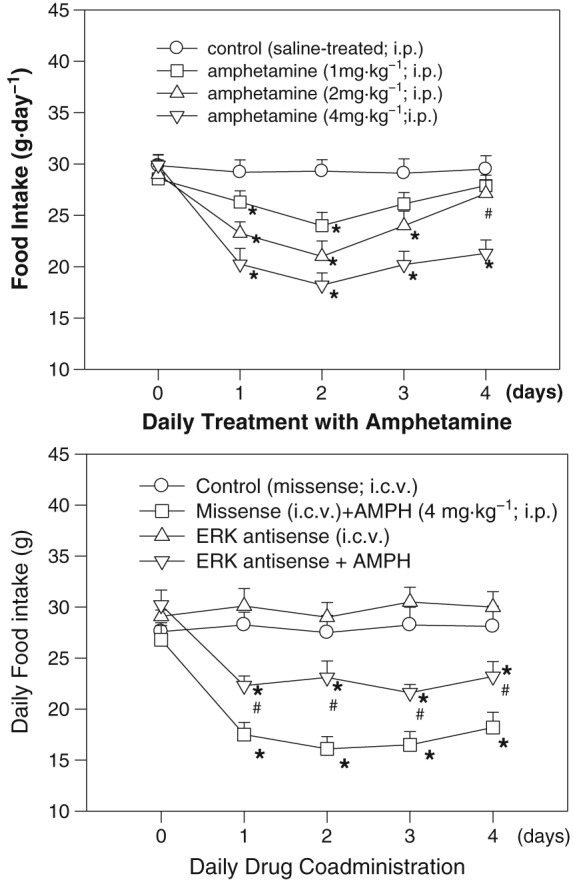
Upper panel: The effect of daily amphetamine (AMPH) treatment on food intake over a 4 day period. Amphetamine (1, 2 or 4 mg·kg−1; i.p.) was administered to rats once a day (at 18:00 h of each day) for 4 days. The first injection of amphetamine was conducted at the end of Day 0. Each point represents the mean ± SEM; n = 8 for each group (total of four groups). *P < 0.05 versus the control group. # P < 0.05 versus the amphetamine‐treated (2 mg·kg−1) group on Day 2. Lower panel: Effects of daily ERK antisense/amphetamine co‐administration on daily food intake over a 4 day period. Daily ERK antisense (or missense) treatment [20 μg·(10 μL·day−1)−1, i.c.v.] was administered 1 h before daily amphetamine (4 mg·kg−1; i.p.) treatment. *P < 0.05 versus the missense groups. # P < 0.05 versus the amphetamine‐treated groups of each treatment day. Bars are the means ± SEM; n = 8 for each group (total of four drug‐treated groups).
Drugs, chemicals and reagents
D‐amphetamine (sulphate salt), angiotensin II (powder), haloperidol (powder), MC3 receptors, pentobarbital (sodium salt), DAPI, and Tris–HCl were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Antibodies against NPY, tERK1/2, pERK1/2 and β‐actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The MC3 receptor antibody was from Gibco BRL, Life Technologies, Inc. (Rockville, MD, USA). POMC, tCREB, pCREB antibody were purchased from Cell Signaling Technology (Beverly, MA, USA). Rhodamine red‐X‐conjugated goat anti‐mouse IgG was from Jackson ImmunoResearch, (West Grove, PA, USA). TRIZOL reagent (Life Technologies, Inc., Grand Island, USA) was used in tissue homogenization. ERK antisense and missense were synthesized by Proligo Pty Ltd (Singapore).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
The effect of amphetamine administration or pERK antisense/amphetamine co‐administration on feeding behaviour and body weight
The change of food intake following amphetamine treatment is shown in the upper panel of Figure 1. Statistical analysis by two‐way ANOVA followed by Dunnett's test revealed significant treatment‐dependent and time‐dependent effects, but there was no interaction effect. Results showed that (i) amphetamine‐induced anorectic responses were dose‐dependent; (ii) amphetamine (2 mg·kg−1) decreased food intake during drug treatment with the maximum decrease on Day 2 (i.e. anorectic effect) and this reduction was reversed to normal level on Day 4 (i.e. tolerant effect) compared to that on Day 2; and (iii) amphetamine (4 mg·kg−1) induced a continuous anorectic response from Day 1 to Day 4 with no tolerant effect. Based on these findings, amphetamine (2 mg·kg−1) was employed for the study of hypothalamic protein analysis since there was a drug tolerance, while amphetamine (4 mg·kg−1) was used for the studies of ERK antisense/amphetamine co‐administration, effect of pretreatment with haloperidol and IHC experiment to detect pERK immunofluorescence since it could exert a larger anorectic response and thus was more clear to detect behavioural responses and pERK particles expression.
As shown in the lower panel of Figure 1, ERK antisense can partially block amphetamine‐induced anorexia, indicating the involvement of ERK signalling during amphetamine treatment. Using two‐way ANOVA to measure the effect of ERK antisense, significant drug‐dependent and time‐dependent effects were revealed, but there was no interaction effect. Comparing the food intake between antisense/amphetamine‐treated and amphetamine‐treated rats, it revealed significant effects from Day 1 to Day 4. The feeding response in missense‐treated rats was similar to that in saline‐treated rats (showed in the upper panel of Figure 1). Moreover, the anorectic response in missense/amphetamine‐treated rats was not changed when compared to that in amphetamine‐treated rats. These results revealed the noninterference of missense treatment in this study and also revealed that ERK knockdown could modify the feeding responses in amphetamine‐treated rats.
Changes in body weight in rats receiving amphetamine and/or ERK antisense treatment are shown in Table 1. The results revealed that decreases in daily body weight in amphetamine‐ with or without antisense‐treated rats followed a pattern similar to the decreases in daily food intake. ERK knockdown could result in a restoration of food intake and body weight change, revealing the involvement of ERK signalling in the regulation of amphetamine‐induced reduction of body weight.
Table 1.
Changes of daily body weight in rats treated with amphetamine (AMPH), ERK antisense/amphetamine and haloperidol (HAL)/amphetamine over a 4 day drug treatment period
| Groups | Daily body weight change | ||||
|---|---|---|---|---|---|
| Day 0 | Day 1 | Day 2 | Day 3 | Day 4 | |
| Exp A: The effect of AMPH administration | |||||
| Control (saline; i.p.; n = 8) | 6.4 ± 0.5 | 6.1 ± 0.4 | 6.3 ± 0.5 | 6.0 ± 0.4 | 6.2 ± 0.5 |
| AMPH (1 mg·kg−1; i.p.; n = 8) | 6.5 ± 0.4 | 5.1 ± 0.4 | 4.5 ± 0.6* | 4.9 ± 0.5 | 5.8 ± 0.5 |
| AMPH (2 mg·kg−1; i.p.; n = 8) | 6.4 ± 0.6 | 4.8 ± 0.4* | 4.1 ± 0.6* | 4.7 ± 0.5* | 5.6 ± 0.6 |
| AMPH (4 mg·kg−1; i.p.; n = 8) | 6.2 ± 0.4 | 3.4 ± 0.6* | 3.0 ± 0.7* | 3.5 ± 0.5* | 2.9 ± 0.6* |
| Exp B: The effect of ERK antisense/AMPH co‐administration | |||||
| Control (missense; i.c.v.; n = 6) | 6.5 ± 0.5 | 6.2 ± 0.3 | 6.1 ± 0.4 | 6.2 ± 0.5 | 6.5 ± 0.4 |
| AMPH (4 mg·kg−1; i.p.; n = 6) | 6.6 ± 0.3 | 4.5 ± 0.4* | 3.7 ± 0.2* | 4.4 ± 0.5* | 5.1 ± 0.5* |
| ERK antisense (i.c.v.; n = 6) | 5.5 ± 0.5 | 5.3 ± 0.3 | 5.2 ± 0.5 | 4.9 ± 0.4 | 5.4 ± 0.4 |
| ERK antisense/AMPH (n = 6) | 6.2 ± 0.3 | 4.9 ± 0.5*, # | 4.7 ± 0.3*, # | 5.0 ± 0.5*, # | 4.8 ± 0.4*, # |
| Exp C: The effect of HAL/AMPH co‐administration | |||||
| Control (vehicle; i.p.; n = 8) | 6.2 ± 0.5 | 5.8 ± 0.3 | 6.0 ± 0.4 | 6.2 ± 0.5 | 6.3 ± 0.4 |
| AMPH (4 mg·kg−1; i.p.; n = 8) | 6.6 ± 0.5 | 4.7 ± 0.3* | 4.8 ± 0.1* | 5.0 ± 0.4* | 5.1 ± 0.6* |
| HAL(1 mg·kg−1; i.p.; n = 8) | 6.1 ± 0.5 | 6.2 ± 0.4 | 5.8 ± 0.5 | 6.0 ± 0.5 | 5.8 ± 0.6 |
| HAL/AMPH (n = 8) | 6.3 ± 0.3 | 5.9 ± 0.5 | 5.6 ± 0.5 | 5.4 ± 0.6 | 6.0 ± 0.5 |
The change in body weight was calculated with respect to the body weight of the previous day.
Each result is expressed as mean ± SEM (n = 8) for each group in Experiments A and C, while n = 6 for each group in Experiment B.
indicates P < 0.05 compared to the control group.
indicates P < 0.05 versus AMPH‐treated group.
The effect of amphetamine on tERK, pERK, tCREB, pCREB, NPY, POMC and MC3 receptor expression
Results shown in the upper panel of Figure 2 revealed that daily amphetamine increased tCREB, pCREB and pERK expression with the maximum response on Day 2. Moreover, total ERK1/2 (tERK1/2) expression was not changed, and pERK/tERK ratio increased on Day 1 and Day 2 following amphetamine treatment. However, both total CREB (tCREB) and pCREB expression were elevated in a similar pattern from Day 1 to Day 4. Therefore, pCREB/tCREB ratio was almost equal to one from Day 1 to Day 4 following amphetamine treatment (upper panel of Figure 2). Analysis with one‐way ANOVA revealed a significant difference in expression of pERK/tERK on Day 1 and Day 2 and tCREB/pCREB on Day 3 and Day 4.
Results shown in the lower panel of Figure 2 revealed that daily amphetamine increased POMC and MC3 receptor expression with the maximum response on Day 2, which was opposite to the decrease in NPY expression with the biggest reduction on Day 2. Using β‐actin as the internal standard, the ratio of NPY, POMC,pERK, tERK, pCREB, tCREB and MC3 receptor over β‐actin in each group was calculated and compared. Analysis with one‐way ANOVA revealed a significant difference in the expression of NPY, POMC, pERK1/2, pCREB and MC3 receptors compared to each control group.
The effect of amphetamine on CREB/DNA binding activity
Results shown in Figure 3 reveal that amphetamine can increase pCREB and DNA binding activity in the hypothalamus. Analysis with one‐way ANOVA from Day 0 to Day 4 revealed the increases in pCREB from Day 1 to Day 3 compared to the control. This result indicates that pCREB/DNA binding activity increased with the maximum response on Day 2 during amphetamine treatment.
The effect of ERK antisense/amphetamine co‐administration on pERK1/2, pCREB, NPY, POMC and MC3 receptor expression
As shown in Figure 4, ERK antisense pretreatment could modulate pCREB, NPY, POMC and MC3 receptor expression in amphetamine‐treated rats. Moreover, ERK antisense by itself could significantly reduce pERK1/2 expression (about 60%) but showed no significant effect on pCREB, NPY, POMC and MC3 receptor expression compared to the control (missense‐treated) group, revealing a specific effect of ERK antisense on the decrease of pERK1/2 expression. Using β‐actin as the internal standard, the ratio of pERK1/2, pCREB, NPY, POMC and MC3 receptors to β‐actin in each group was calculated and compared. Analysis by one‐way ANOVA followed by Dunnett's test revealed that ERK antisense/amphetamine co‐administration could decrease pERK1/2 expression by approximately 28%, pCREB expression by approximately 40%, POMC expression by approximately 30% and MC3 receptor expression by approximately 38% from Day 1 to Day 4, compared to the amphetamine only‐treated group. However, ERK antisense/amphetamine co‐administration increased NPY expression by approximately 30% compared to the amphetamine‐treated groups. Taken together, these results revealed that a pretreatment with ERK antisense of the amphetamine‐treated group resulted in partial reversal of the increased pERK1/2, pCREB, NPY, POMC and MC3 receptor expression when compared to the amphetamine‐treated group.
Effects of haloperidol pretreatment on food intake and expression levels of pERK, pCREB, NPY, POMC and MC3 receptors
Results shown in the upper panel of Figure 5 revealed that pretreatment with haloperidol before amphetamine could restore the amphetamine‐induced anorexia to a normal level. Pretreatment with haloperidol reverse food intake by 90% compared to that in the amphetamine‐treated group. The food intake in vehicle‐treated rats was similar to that in saline‐treated rats, revealing the noninterference of vehicle in this study. Moreover, the expression of feeding in haloperidol‐treated rats was not significantly changed if compared to that in vehicle‐treated rats, revealing that haloperidol had no significant effect on basal food intake in a 24 h testing period.
Results shown in the lower panel of Figure 5 revealed that haloperidol pretreatment had significant effects on pERK/pCREB/NPY/POMC/MC3 receptor expression compared to those in amphetamine‐treated groups. Using β‐actin as the internal standard, the protein ratio of pERK, pCREB, NPY, POMC and MC3 receptors to β‐actin in each group was calculated and compared. One‐way ANOVA followed by Dunnett's test, significant effects on NPY, POMC, MC3 receptor, pERK and pCREB levels were revealed in the amphetamine‐treated group compared to the control group. Moreover, pretreatment with haloperidol of amphetamine‐treated rats significantly inhibited the effects of amphetamine on pERK (about 80%), pCREB (about 85%), NPY (about 90%) and MC3 receptor (about 88%) expression. Haloperidol treatment alone did not alter pERK/pCREB/NPY/MC3 receptor expression levels compared to that in control group.
The effect of amphetamine on pERK1/2 and POMC immunofluorescent expression
Results of IHC staining shown in Figure 6 indicate significant influences of amphetamine on the fluorescent intensity of pERK1/2 in hypothalamic ARC and PVN (n = 5 for each group; total of three groups). The expression of pERK1/2‐positive cells appeared to be increased in the region of PVN (about 290%) and ARC (about 150%) in rats receiving 4 mg·kg−1 amphetamine compared to the control groups (t‐test, P < 0.05). Double staining of the pERK and nucleus revealed that pERK1/2 immunofluorescent intensity was located mainly at the perinuclear regions, which might include cytoplasm or nerve terminals, in the PVN and ARC in the amphetamine‐treated group (Figure 6G–I). Results of double immunofluorecent staining in ARC area, which are shown in Figure 7, indicated the co‐localization of pERK/POMC image by merging pERK1/2 (green) and POMC (red) images, which resulted in a yellow colour shown in the merged image panel. However, similar double staining procedure in PVN area showed no yellow colour (data not shown).
Figure 6.
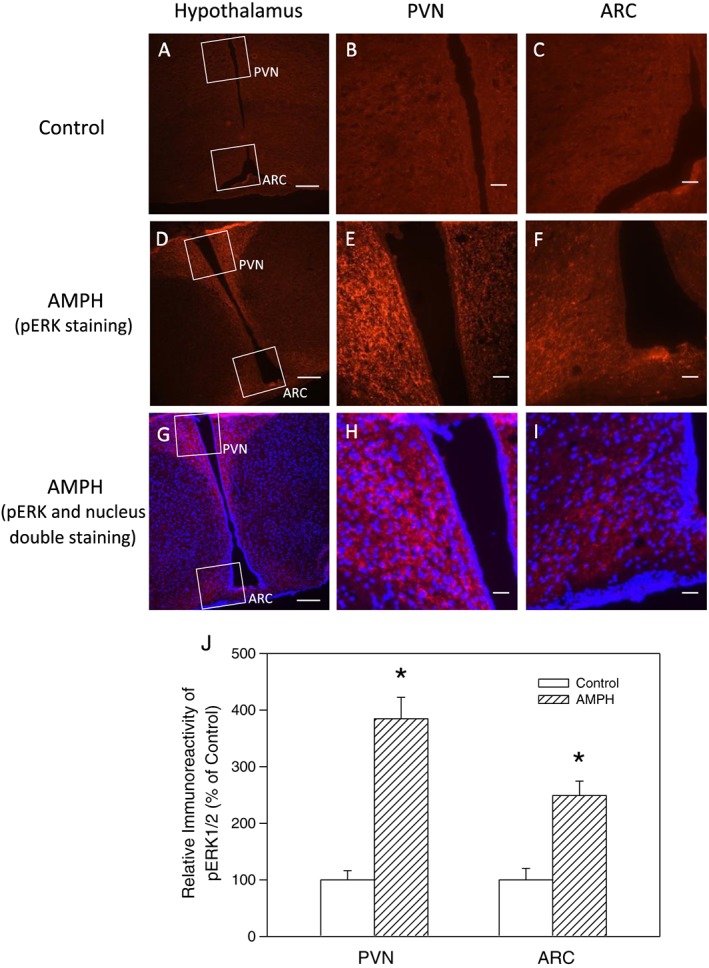
Immunofluorescent staining of pERK1/2 in hypothalamic ARC and PVN regions in rats receiving saline or amphetamine (AMPH; 4 mg·kg−1; i.p.) treatment. Frontal sections through the median eminence level were obtained. Upper panel: photo images of (A–C) show sections in control group, photo images of (D–F) show pERK fluorescence in AMPH‐treated group and photo images of (G–I) show pERK (red) and nucleus (blue) fluorescence of double staining in AMPH‐treated group. Scale bars in images of (A, D and G) are 100 μm (100×), whereas those in (B, C, E, F, H and I) are 20 μm (400×). Lower panel: results of relative densitometric values (n = 5 for each group; total of three groups) for pERK fluorescence intensity in PVN and ARC regions compared to the control group in AMPH‐treated rat. Each value represents mean ± SEM. *P < 0.05 versus control (t‐test).
Figure 7.
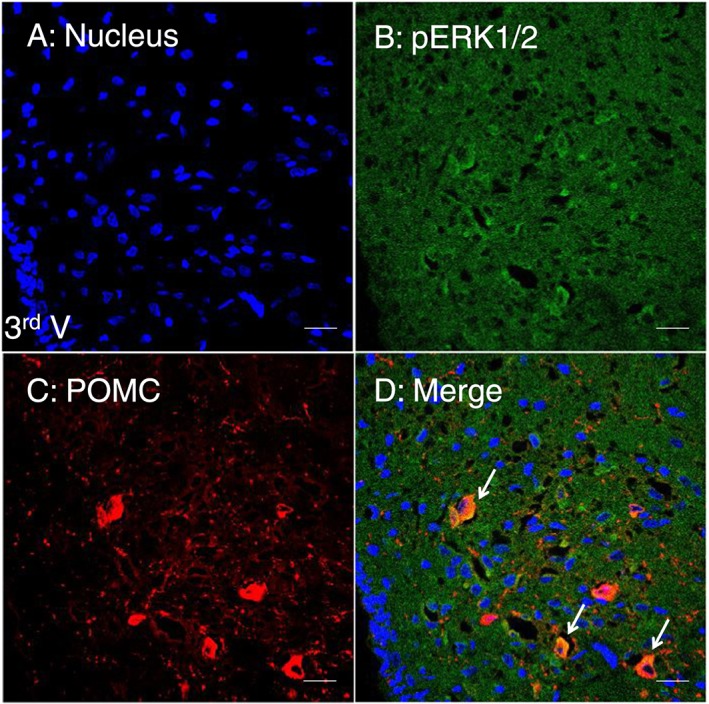
Double immunofluorescent staining showing the co‐localization of pERK1/2 and POMC proteins in the ARC of amphetamine‐treated rats. Frozen sections of the brain were plated on slides and stained with anti‐pERK1/2 and anti‐POCM antibodies in the hypothalamus. Under the analysis of confocal microscopy, the co‐localization image was seen by the merge of pERK1/2 (green) and POMC (red) immunofluorescent images, giving a yellow colour in the merged image panel. Blue: nucleus. Scale bar: 20 μm. 3rd V: the third ventricle.
Discussions and conclusions
POMC and MC3 receptor expression was found to be increased as compared to that of NPY in amphetamine‐treated rats, revealing reciprocal regulation by NPY‐ and POMC‐containing neurons. In addition, the changes in pERK and pCREB expression and pCREB/DNA binding activity were similar to that of POMC and MC3 receptors, indicating that the activation of pERK/pCREB signalling might occur in POMC‐containing neurons. Furthermore, pERK1/2 immunoreactivity increased in the ARC and PVN, demonstrating the involvement of hypothalamic ERK signalling in the anorectic effects of amphetamine in rats. Finally, the dopamine receptor was shown to be involved in the expression of pERK/pCREB/NPY/POMC signalling in the amphetamine‐treated rats. The present findings suggest that hypothalamic dopamine receptor/pERK/pCREB signalling participates in the reciprocal regulation by POMC and NPY of appetite control in amphetamine‐treated rats.
The results of the present study showed that, in amphetamine‐treated rats, feeding behaviour and NPY expression markedly decreased on Day 1 and Day 2 (i.e. an anorectic effect), with the maximum decrease on Day 2. This anorectic effect was associated with satiety or a positive energy balance. By contrast, MC3 receptor expression increased, with the maximum increase on Day 2. The expression levels had gradually returned to control by Day 3 and Day 4 (i.e. a tolerant effect) due to hunger or a negative energy balance (Kuo et al., 2011). The present results confirm that POMC (or MC3 receptors) might function reciprocally with that of NPY during amphetamine treatment (Hsieh et al., 2007).
We ruled out the possibility that the changes in NPY level were simply secondary to reduced feeding, rather than the specific action of amphetamine (a dopamine agonist) on hypothalamic NPY. Our previous report revealed that (i) pair‐fed (non‐drug‐treated) animals showed no change in NPY level compared to those in normal‐fed animals, and (ii) from the time course of a 24 h (which was separated into four intervals, i.e. 0–6 h, 6–12 h, 12–18 h and 18–24 h) feeding behaviour, we found that an amphetamine‐induced anorectic effect was short‐lived, that is, only in the interval of 0–6 h (Kuo, 2002; 2006). Our experiment was carried out on a normal 12–12 h light–dark cycle with amphetamine administered at 18:00 h, which is the beginning of the dark period.
The ability of NPY to inhibit POMC neurons via the release of GABA (Chee et al., 2010) or unidirectional inhibitory inputs from NPY to POMC neurons (Hsieh et al., 2007; Gong et al., 2008; Hsieh et al., 2013a) underlies the existence of reciprocal regulation by NPY and POMC neurons as described previously. In addition to these reports, we recently found that the increased oxidative stress on Day 1 and Day 2, but not on Day 3 and Day 4, may play a functional role in the reciprocal regulation of NPY and POMC during amphetamine treatment (Chu et al., 2016). It is because the increased oxidative stress may interrupt the increasing effect of corticosterone on NPY expression during amphetamine treatment. Corticosterone may increase NPY expression in fasting mice (a condition of negative energy balance), which is independent of MAPK, c‐FOS and CREB expression (Morikawa et al., 2004). However, we found that this increasing effect of corticosterone on NPY expression on Day 1 and Day 2 may be interrupted because of the increased oxidative stress in amphetamine‐treated animals (Chu et al., 2016).
Focusing on the viewpoint of energy balance, the present study revealed that pERK/pCREB/MC3 receptor expression increased and NPY expression decreased on Day 1 and Day 2 (i.e. during positive energy balance), whereas both pERK/pCREB/MC3 receptor and NPY expression returned to control levels on Day 3 and Day 4 (i.e. during negative energy balance) in amphetamine‐treated rats. However, these results were not consistent with those from a previous report indicating that both pERK/pCREB and NPY expression increased in fasting mice (negative energy balance), while pERK/pCREB expression decreased but NPY expression increased in fasting ob/ob mice (a model of negative energy balance) (Morikawa et al., 2004). These results are complex, however, we concluded that hypothalamic NPY‐containing neurons are always activated in response to negative energy balance. It is suggested that during the regulation of hypothalamic NPY/POMC‐mediated appetite control, pERK/pCREB signalling can be inhibited or activated, depending on the conditions of energy balance.
To investigate further the possible involvement of ERK in NPY/POMC‐mediated appetite controls, knockdown of the expression of ERK by cerebral infusion with ERK antisense was used. In the present study, ERK knockdown attenuated the reduction in the expression of NPY but increased the expression of MC3 receptors and CREB in the antisense/amphetamine‐treated rats compared to the amphetamine‐treated rats. These results demonstrated that ERK knockdown in the brain might modulate hypothalamic CREB/NPY/POMC‐mediated appetite control.
Amphetamine can change pERK1/2 expression, while tERK1/2 expression keeps constant during daily treatment. Activated pERK1/2 appeared to evoke the downstream signal since pERK1/2 was expressed in a way similar to that of pCREB, a transcription factor. Amphetamine treatment can change both pCREB and tCREB expression, which changed in a similar manner to that of POMC and MC3 receptors during drug treatment. Although the pCREB/tCREB ration remained constant on Day 1 and Day 2, both tCREB and pCREB expression increased and thus increased POMC and MC3 receptor gene expression. Therefore, pERK1/2 and pCREB is involved in the amphetamine‐mediated appetite control.
The inhibitory effect of haloperidol on amphetamine‐induced suppression of feeding might involve both central and peripheral antagonism of dopamine receptors. The present results revealed that, following haloperidol/amphetamine co‐administration, the return of food intake and pERK/pCERB/NPY/POMC/MC3 receptor expression were about 80–90% compared to each control level, revealing the major role of dopamine receptors in the `regulation of amphetamine‐mediated anorexia (Kuo, 2006). However, following the co‐administration of ERK antisense/amphetamine, the partial reversal of pERK/pCREB/NPY/POMC/MC3 receptor expression was about 30–50% compared to normal levels, implying the possibilities that (i) antisense ERK did not delete ERK completely (only 60% reduction), and (ii) ERK‐CREB signalling was not the only pathway involved in the induction of NPY/POMC gene expression in the amphetamine‐treated rats. Previous reports revealed that some transcription factors, such as AP‐1 (Hsieh et al., 2006), NF‐κB (Kuo et al., 2012) and STAT3 (Chu et al., 2014), and some cytoplasmic factors, such as PI3K (Chu et al., 2014), anti‐oxidative enzymes (Hsieh et al., 2013b), PKC (Kuo et al., 2011) and PKA (Hsieh et al., 2007), were also activated and involved in regulating NPY/POMC gene expression during amphetamine treatment. Some previous reports revealed that ERK co‐operates with other transcription factors in the brain in the control of behaviour‐associated diseases. For example, ERK/NF‐κB down‐regulation participated in improvements in Parkinson's disease (Lee et al., 2014; Lv et al., 2015), and ERK/PI3K activation participated in the neuro‐protective effects in Alzheimer's disease (Li et al., 2015). Thus, it is clear that ERK might co‐operate with other transcription factors in the regulation of amphetamine‐mediated appetite suppression.
In the present study, the results of immunofluorescent staining provided evidence that pERK1/2 immunoreactivity increased in the hypothalamic ARC and PVN regions in amphetamine‐treated rats, revealing that ERK signalling was activated in the regions of ARC and PVN (shown in Figure 6). However, using double immunofluorescent staining (pERK/POMC) (shown in Figure 7), it was revealed that only POMC neurons in the area of ARC, but not PVN (data not shown), showed pERK1/2 immunofluorescence. These results suggest that the pERK/pCREB/POMC signal pathway was activated in the hypothalamic ARC region in amphetamine‐treated rats.
Drugs targeting ERK, CREB, NPY and POMC expression have been suggested as clinical pharmacotherapy for the improvement of some brain diseases and obesity. Previous findings indicated that ERK/CREB signalling participated in regulating anxiety‐like behaviour induced by the anorectic neuropeptide nesfatin‐1 (Ge et al., 2015) and that the activation of the dopamine D1 receptor/ERK/CREB signal pathway in the nucleus accumbens played a critical role in the control of reward‐seeking behaviour (Kirschmann et al., 2014). Thus, ERK/CREB signalling may be a target for the improvement of some brain‐associated behavioural disorders. Moreover, NPY and POMC have been added to the list of feeding‐behaviour‐related neuropeptides and are regarded as endogenous satiety factors in the treatment of obesity (Aronne and Thornton‐Jones, 2007; Parker and Bloom, 2012).
In conclusion, the present results suggest that dopamine receptors and hypothalamic ERK/CREB signalling participate in the reciprocal regulation by NPY/POMC neurons of amphetamine‐induced appetite suppression (Figure 8).
Figure 8.
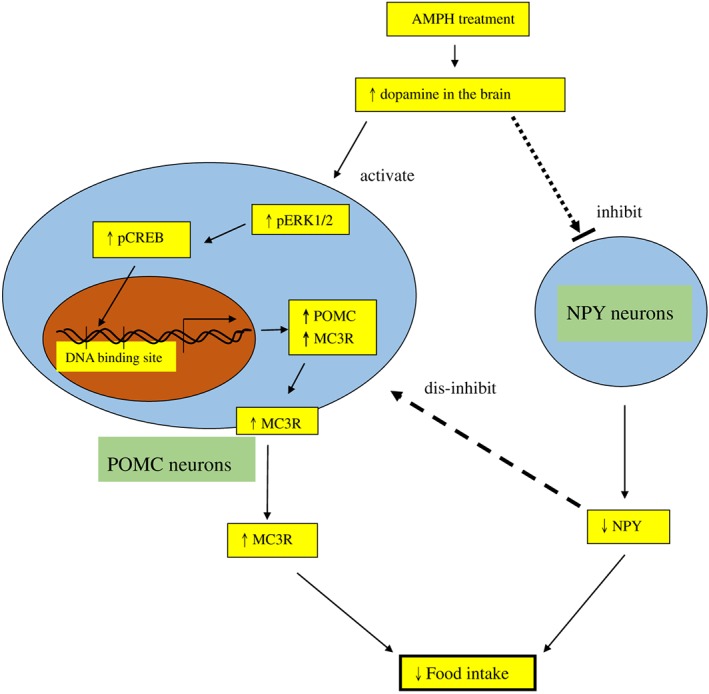
Schematic illustration showing the flow in the suppressive action of amphetamine (AMPH). Amphetamine treatment activates dopamine release in the brain, which may in turn activate hypothalamic POMC expression, inhibit hypothalamic NPY expression and finally decrease food intake. The activation of POMC neurons, which is partly due to the disinhibition of NPY neurons, may result in the increased expression of pERK1/2, pCREB, POMC and MC3 receptor (MC3R). Finally, the decrease in NPY or the increase in MC3 receptors may contribute to the reduction of food intake. The thinner arrows indicate the flow of amphetamine treatment in the induction of anorexia. The solid black arrow indicates the activation, the dotted arrows indicate the inhibition and the dashed arrow represents the disinhibition (or activation). The upward and downward arrows in the squares indicate the responses of increase and decrease.
Author contributions
D.Y.K. conceived and designed the study, performed experiments, analysed data and wrote the paper. C.H.Y. performed some of the experimental work and data analysis. Y.S.H. and P.N.C. performed some of the experimental work and discussed some of the results. J.R.C. helped with the double immunofluprecent staining.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
Confocal microscopy was performed in the Instrument Centre of Chung Shan Medical University, which is supported by National Science Council, Ministry of Education and Chung Shan Medical University. This study was supported by a grant from the cooperation of National Chung Hsing University and Chung Shan Medical University (NCHU‐CSMU‐10607) in Taiwan, ROC.
Yu, C.‐H. , Hsieh, Y.‐S. , Chen, P.‐N. , Chen, J.‐R. , and Kuo, D.‐Y. (2018) Knockdown of the transcript of ERK in the brain modulates hypothalamic neuropeptide‐mediated appetite control in amphetamine‐treated rats. British Journal of Pharmacology, 175: 726–739. doi: 10.1111/bph.14120.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M, Konradi C, Cenci MA (2001). cAMP response element‐binding protein is required for dopamine‐dependent gene expression in the intact but not the dopamine‐denervated striatum. J Neurosci 21: 9930–9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronne LJ, Thornton‐Jones ZD (2007). New targets for obesity pharmacotherapy. Clin Pharmacol Ther 81: 748–752. [DOI] [PubMed] [Google Scholar]
- Barnea A, Roberts J, Croll SD (2004). Continuous exposure to brain‐derived neurotrophic factor is required for persistent activation of TrkB receptor, the ERK signaling pathway, and the induction of neuropeptide Y production in cortical cultures. Brain Res 1020: 106–117. [DOI] [PubMed] [Google Scholar]
- Butler AA (2006). The melanocortin system and energy balance. Peptides 27: 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J et al (2000). A unique metabolic syndrome causes obesity in the melanocortin‐3 receptor‐deficient mouse. Endocrinology 141: 3518–3521. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M (2001). Mammalian MAP kinase signalling cascades. Nature 410: 37–40. [DOI] [PubMed] [Google Scholar]
- Chee MJ, Myers MG Jr, Price CJ, Colmers WF (2010). Neuropeptide Y suppresses anorexigenic output from the ventromedial nucleus of the hypothalamus. J Neurosci 30: 3380–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TY, Duh SL, Huang CC, Lin TB, Kuo DY (2001). Evidence for the involvement of dopamine D1 and D2 receptors in mediating the decrease of food intake during repeated treatment with amphetamine. J Biomed Sci 8: 462–466. [DOI] [PubMed] [Google Scholar]
- Chien MH, Ying TH, Hsieh YS, Chang YC, Yeh CM, Ko JL et al (2012). Dioscorea nipponica Makino inhibits migration and invasion of human oral cancer HSC‐3 cells by transcriptional inhibition of matrix metalloproteinase‐2 through modulation of CREB and AP‐1 activity. Food Chem Toxicol 50: 558–566. [DOI] [PubMed] [Google Scholar]
- Chu SC, Chen PN, Hsieh YS, Yu CH, Lin MH, Lin YH et al (2014). Involvement of hypothalamic PI3K‐STAT3 signalling in regulating amphetamine‐mediated appetite suppression. Br J Pharmacol 171: 3223–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu SC, Yu CH, Chen PN, Ho YJ, Hsieh YS, Kuo DY (2016). Role of oxidative stress in disrupting the function of negative glucocorticoid response element in daily amphetamine‐treated rats. Psychoneuroendocrinology 71: 1–11. [DOI] [PubMed] [Google Scholar]
- Cone RD (2005). Anatomy and regulation of the central melanocortin system. Nat Neurosci 8: 571–578. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioramonti X, Contié S, Song Z, Routh VH, Lorsignol A, Pénicaud L (2007). Characterization of glucosensing neuron subpopulations in the arcuate nucleus: integration in neuropeptide Y and pro‐opio‐melanocortin networks? Diabetes 56: 1219–1227. [DOI] [PubMed] [Google Scholar]
- Ge JF, Xu YY, Qin G, Pan XY, Cheng JQ, Chen FH (2015). Nesfatin‐1, a potent anorexic agent, decreases exploration and induces anxiety‐like behavior in rats without altering learning or memory. Brain Res 1629: 171–181. [DOI] [PubMed] [Google Scholar]
- Gilbert WK, Jieru EL, Erik SB, Scott AW (2014). Anti‐obesity pharmacotherapy: new drugs and emerging targets. Clin Pharmacol Ther 95: 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillard ER, Dang DQ, Stanley BG (1993). Evidence that neuropeptide Y and dopamine in the perifornical hypothalamus interact antagonistically in the control of food intake. Brain Res 628: 128–136. [DOI] [PubMed] [Google Scholar]
- Hsieh YH, Chen PN, Yu CH, Liao JM, Kuo DY (2013a). Inhibiting neuropeptide Y Y1 receptor modulates melanocortin receptor‐ and NFkB‐mediated feeding behavior in phenylpropanolamine‐treated rats. Horm Behav 64: 95–102. [DOI] [PubMed] [Google Scholar]
- Hsieh YS, Yang SF, Kuo DY (2005). Amphetamine, an appetite suppressant, decreases neuropeptide Y immunoreactivity in rat hypothalamic paraventriculum. Regul Pept 127: 169–176. [DOI] [PubMed] [Google Scholar]
- Hsieh YS, Yang SF, Chiou HL, Kuo DY (2006). Activations of c‐fos/c‐jun signaling are involved in the modulation of hypothalamic superoxide dismutase (SOD) and neuropeptide Y (NPY) gene expression in amphetamine‐mediated appetite suppression. Toxicol Appl Pharmacol 212: 99–109. [DOI] [PubMed] [Google Scholar]
- Hsieh YS, Yang SF, Kuo DY (2007). Intracerebral administration of protein kinase A (PKA) or c‐AMP response element binding protein (CREB) antisense oligonucleiotide can modulate amphetamine‐mediated appetite suppression in free‐moving rats. Am J Physiol Endocrinol Metab 292: 123–131. [DOI] [PubMed] [Google Scholar]
- Hsieh YS, Yang SF, Chu SC, Ho YJ, Kuo CS, Kuo DY (2008). Transcriptional interruption of cAMP response element binding protein modulates superoxide dismutase and neuropeptide Y‐mediated feeding behavior in freely moving rats. J Neurochem 105: 1438–1449. [DOI] [PubMed] [Google Scholar]
- Hsieh YS, Yang SF, Chen PN, Chu SC, Chen CH, Kuo DY (2011). Knocking down the transcript of protein kinase C‐lambda modulates hypothalamic glutathione peroxidase, melanocortin receptor and neuropeptide Y gene expression in amphetamine‐treated rats. J Psychopharmacol 25: 982–994. [DOI] [PubMed] [Google Scholar]
- Hsieh YS, Chen PN, Kuo MS, Kuo DY (2013b). Neuropeptide Y Y1 receptor knockdown can modify glutathione peroxidase and c‐AMP response element binding protein in phenylpropanolamine‐treated rats. Arch Toxicol 87: 469–479. [DOI] [PubMed] [Google Scholar]
- Hsieh YS, Chen PN, Yu CH, Kuo DY (2014). Central dopamine action modulates neuropeptide‐controlled appetite via the hypothalamic PI3K/NFκB‐dependent mechanism. Genes Brain Behav 13: 784–793. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild‐Huntress V, Dunmore JH, Fang Q, Berkemeier LR et al (1997). Targeted disruption of the melanocortin‐4 receptor results in obesity in mice. Cell 88: 131–141. [DOI] [PubMed] [Google Scholar]
- Ichijo H (1999). From receptors to stress‐activated MAP kinases. Oncogene 18: 6087–6093. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschmann EK, Mauna JC, Willis CM, Foster RL, Chipman AM, Thiels E (2014). Appetitive cue‐evoked ERK signaling in the nucleus accumbens requires NMDA and D1 dopamine receptor activation and regulates CREB phosphorylation. Learn Mem 21: 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo DY (2002). Co‐administration of dopamine D1 and D2 agonists additively decreases daily food intake, body weight and hypothalamic neuropeptide Y level. J Biomed Sci 9: 126–132. [DOI] [PubMed] [Google Scholar]
- Kuo DY (2006). Hypothalamic neuropeptide Y (NPY) and the attenuation of hyperphagia in streptozotocin diabetic rats treated with dopamine D1/D2 agonists. Br J Pharmacol 148: 640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo DY, Chen PN, Yang SF, Chu SC, Chen CH, Kuo MS et al (2011). Role of reactive oxygen species‐related enzymes in neuropeptide Y and proopiomelanocortin‐mediated appetite control: A study using atypical protein kinase C knockdown. Antioxid Redox Signal 15: 2147–2159. [DOI] [PubMed] [Google Scholar]
- Kuo DY, Chen PN, Kuo MH, Chen CH, Hsieh YS, Chu SC (2012). NF‐kappaB knockdown can modulate amphetamine‐mediated feeding response. Neuropharmacology 62: 1684–1694. [DOI] [PubMed] [Google Scholar]
- Lee E, Park HR, Ji ST, Lee Y, Lee J (2014). Baicalein attenuates astroglial activation in the 1‐methyl‐4‐phenyl‐1,2,3,4‐tetrahydropyridine‐induced Parkinson's disease model by downregulating the activations of nuclear factor‐κB, ERK, and JNK. J Neurosci Res 92: 130–139. [DOI] [PubMed] [Google Scholar]
- Li J, Ding X, Zhang R, Jiang W, Sun X, Xia Z et al (2015). Harpagoside ameliorates the amyloid‐β‐induced cognitive impairment in rats via up‐regulating BDNF expression and MAPK/PI3K pathways. Neuroscience 303: 103–114. [DOI] [PubMed] [Google Scholar]
- Lim S, Moon M, Oh H, Kim HG, Kim SY, Oh MS (2014). Ginger improves cognitive function via NGF‐induced ERK/CREB activation in the hippocampus of the mouse. J Nutr Biochem 25: 1058–1065. [DOI] [PubMed] [Google Scholar]
- Lv Y, Zhang Z, Hou L, Zhang L, Zhang J, Wang Y et al (2015). Phytic acid attenuates inflammatory responses and the levels of NF‐κB and p‐ERK in MPTP‐induced Parkinson's disease model of mice. Neurosci Lett 597: 132–136. [DOI] [PubMed] [Google Scholar]
- McGinty JF, Mendelson JE (2011). Is brain‐derived neurotrophic factor a selective biomarker that predicts cocaine relapse outcomes? Biol Psychiatry 70: 700–701. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielke K, Herdegen T (2000). JNK and p38 stresskinases ‐degenerative effectors of signal‐transduction‐cascades in the nervous system. Prog Neurobiol 61: 45–60. [DOI] [PubMed] [Google Scholar]
- Moonat S, Pandey SC (2012). Stress, epigenetics, and alcoholism. Alcohol Res 34: 495–505. [PMC free article] [PubMed] [Google Scholar]
- Morikawa Y, Ueyama E, Senba E (2004). Fasting‐induced activation of mitogen‐activated protein kinases (ERK/p38) in the mouse hypothalamus. J Neuroendocrinol 16: 105–112. [DOI] [PubMed] [Google Scholar]
- Morris BJ (1989). Neuronal localization of neuropeptide Y gene expression in rat brain. J Comp Neurol 290: 358–368. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Pfaff DW (1998). Current status of antisense DNA methods in behavioral studies. Chem Senses 23: 249–255. [DOI] [PubMed] [Google Scholar]
- Olsson AK, Nanberg E (2001). A functional role for ERK in gene induction, but not in neurite outgrowth in differentiating neuroblastoma cells. Exp Cell Res 265: 21–30. [DOI] [PubMed] [Google Scholar]
- Pandey SC (2003). Anxiety and alcohol abuse disorders: a common role for CREB and its target, the neuropeptide Y gene. Trends Pharmacol Sci 24: 456–460. [DOI] [PubMed] [Google Scholar]
- Parker JA, Bloom SR (2012). Hypothalamic neuropeptides and the regulation of appetite. Neuropharmacology 63: 18–30. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986). The rat brain in stereotaxic coordinates, 2nd edn. Academic Press: Sydney, Australia. [Google Scholar]
- Ritter RC, Slusser PG, Stone S (1981). Glucoreceptors controlling feeding and blood glucose: location in the hindbrain. Science 213: 451–452. [DOI] [PubMed] [Google Scholar]
- Rocha ML, Fernandes PP, Lotufo BM, Manhães AC, Barradas PC, Tenorio F (2014). Undernutrition during early life alters neuropeptide Y distribution along the arcuate/paraventricular pathway. Neuroscience 256: 379–391. [DOI] [PubMed] [Google Scholar]
- Sale EM, Atkinson PG, Sale GJ (1995). The requirement of MAP kinase for the differentiation of fibroblasts to adipocytes, for insulin activation of p90 S6 kinase and for insulin or serum stimulation of DNA synthesis. J Eur Mol Biol Org 14: 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, McGinty JF (2007). Repeated amphetamine treatment increases phosphorylation of extracellular signal‐regulated kinase, protein kinase B, and cyclase response element‐binding protein in the rat striatum. J Neurochem 103: 706–713. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WL, Quizon PM, Zhu J (2016). Molecular mechanism: ERK signaling, drug addiction, and behavioral effects. Prog Mol Biol Transl Sci 137: 1–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J (2000). Involvement of the extracellular signal‐regulated kinase cascade for cocaine‐rewarding properties. J Neurosci 20: 8701–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang L, Cao XL, Xing TY, Mori D, Tang RQ, Li J et al (2016). Mixture of peanut skin extract and fish oil improves memory in mice via modulation of anti‐oxidative stress and regulation of BDNF/ERK/CREB signaling pathways. Forum Nutr 8: E256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Creese I (1993). Antisense oligodeoxynucleotide reduces brain dopamine D2 receptors: behavioral correlates. Neurosci Lett 161: 223–226. [DOI] [PubMed] [Google Scholar]
- Zhu J, Midde NM, Gomez AM, Sun WL, Harrod SB (2015). Intra‐ventral tegmental area HIV‐1 Tat1‐86 attenuates nicotine‐mediated locomotor sensitization and alters mesocorticolimbic ERK and CREB signaling in rats. Front Microbiol 6: 540. [DOI] [PMC free article] [PubMed] [Google Scholar]