

Abstract
Background and Purpose
Benzodiazepines, non‐selective positive allosteric modulators (PAMs) of GABAA receptors, have significant side effects that limit their clinical utility. As many of these side effects are mediated by the α1 subunit, there has been a concerted effort to develop α2/3 subtype‐selective PAMs.
Experimental Approach
In vitro screening assays were used to identify molecules with functional selectivity for receptors containing α2/3 subunits over those containing α1 subunits. In vivo receptor occupancy (RO) was conducted, prior to confirmation of in vivo α2/3 and α1 pharmacology through quantitative EEG (qEEG) beta frequency and zolpidem drug discrimination in rats respectively. PF‐06372865 was then progressed to Phase 1 clinical trials.
Key Results
PF‐06372865 exhibited functional selectivity for those receptors containing α2/3/5 subunits, with significant positive allosteric modulation (90–140%) but negligible activity (≤20%) at GABAA receptors containing α1 subunits. PF‐06372865 exhibited concentration‐dependent occupancy of GABAA receptors in preclinical species. There was an occupancy‐dependent increase in qEEG beta frequency and no generalization to a GABAA α1 cue in the drug‐discrimination assay, clearly demonstrating the lack of modulation at the GABAA receptors containing an α1 subtype. In a Phase 1 single ascending dose study in healthy volunteers, evaluation of the pharmacodynamics of PF‐06372865 demonstrated a robust increase in saccadic peak velocity (a marker of α2/3 pharmacology), increases in beta frequency qEEG and a slight saturating increase in body sway.
Conclusions and Implications
PF‐06372865 has a unique clinical pharmacology profile and a highly predictive translational data package from preclinical species to the clinical setting.
Abbreviations
- BZD
benzodiazepine
- CI
confidence interval
- dpm
disintegrations per minute
- LSmean
least squares mean
- MTD
maximum tolerated dose
- NEAA
non‐essential amino acid
- PAM
positive allosteric modulator
- PDs
pharmacodynamics
- PK
pharmacokinetic
- qEEG
quantitative EEG
- RO
receptor occupancy
- SPV
saccadic peak velocity
- TAC
time activity curve
Introduction
GABAA receptors are heteropentameric inhibitory ligand‐gated ion channels, which are important drug targets (Rudolph and Knoflach, 2011; Engin et al., 2012). The majority of GABAA receptors present in the CNS contain two α, two β and a single γ subunit and are sensitive to benzodiazepines (BZDs), which are allosteric modulators. That is, they exhibit no intrinsic activity of their own but potentiate or inhibit the effects of GABA at receptors that contain either an α1, α2, α3 or α5 subunit in conjunction with a γ2 subunit (McKernan and Whiting, 1996). GABA activation of GABAA receptors leads to the opening of their integrated chloride channels, leading to chloride influx, hyperpolarizing neurones and, therefore, decreasing the probability of firing.
BZDs have been used clinically for over 50 years but, although they are effective anxiolytics and anticonvulsants, their use is severely limited by their side effect profile. Elegant molecular studies, in which GABAA receptors containing specific α subunits have been rendered unresponsive to diazepam, have been able to define the contribution of those subunits to different aspects of the in vivo pharmacology. These studies, together with subtype‐selective pharmacological tool compounds, have assigned the sedative effects of BZDs to α1 activity (McKernan et al., 2015), anxiolytic and analgesic activity to α2/3 subunits (Dias et al., 2005; Atack et al., 2006; Knabl et al., 2008) and some of the effects on memory function to α5 receptors (Dawson et al., 2006).
Some compounds, which exhibit varying degrees of subtype selectivity, have previously been evaluated clinically. The first of these compounds was MK‐0343, which is a partial positive allosteric modulator (PAM), with some subtype selectivity α3 > α2 = α5 > α1. Despite the lower α1 activity of this compound (~20% of diazepam), it still caused appreciable sedation in clinical studies where the levels of receptor occupancy (RO) were below the limits of detection (de Haas et al., 2008). This compound was followed by two others from Merck, TPA023 and TPA023B, both of which exhibited lower α1 activity than MK‐0343 and had reduced sedative liability in clinical trials (de Haas et al., 2007; Atack, 2009; Atack et al., 2011a). However, dose‐limiting adverse effects including drowsiness meant that neither of these compounds could be dosed higher than approximately 60% RO. More recently, AZD7325 has been evaluated in clinical trials, high levels of RO (>80% at 10 mg) were obtained, but only very small pharmacodynamic (PD) effects were observed (Chen et al., 2014). The NeuroSearch compound NS11821 has also been evaluated recently (Zuiker et al., 2016), but the RO levels achieved with this compound are not reported.
Therefore, although many improvements have been made in understanding the preclinical to clinical translation of subtype‐selective GABAA receptor PAMs, there are still some key gaps in understanding the relationship between in vitro α1 activity and clinical adverse events (AEs) and the preclinical to clinical translation of α2/3 activity. Herein, we have identified a novel GABAA subtype‐selective modulator PF‐06372865 (7‐ethyl‐4‐(4′‐(ethylsulfonyl)‐6‐fluoro‐2′‐methoxybiphenyl‐3‐yl)‐7H‐imidazo[4,5‐c]pyridazine) using a high‐throughput electrophysiological assay. We have built a biomarker‐based approach, following the latter two of the three pillars of survival (Morgan et al., 2012), to enable us to understand target binding and expression of pharmacology both preclinically and clinically. Using this approach, we show that the lack of preclinical in vivo α1 activity determined using a rodent zolpidem drug‐discrimination model and the α2/3 activity measured using quantitative EEG (qEEG) translate through to the clinical setting.
Methods
Cell culture
HEK293 cell lines expressing either human GABAA α1β3γ2, α3β3γ2, α4β3γ2 or α6β3γ2 were cultured in DMEM/F12 containing 10% FBS, 2 mM l‐glutamine, 1% non‐essential amino acids (NEAAs), 800 μg·mL−1 G418, 200 μg·mL−1 hygromycin B and 0.8 μM puromycin. HEK293 cells expressing α2β2γ2 and α5β2γ2 were cultured in Eagle's minimal essential medium containing 2 mM glutamax, 1% sodium pyruvate, 1% NEAAs, 800 μg·mL−1 G418, 200 μg·mL−1 hygromycin and 10 μg·mL−1 zeocin. CHO cells stably expressing rat GABAA α1β3γ2 or expressing rat GABAA α2β3γ2 under a tetracycline‐inducible promoter were cultured in DMEM containing 10% FBS, 2 mM glutamax, 1% sodium pyruvate, 1% NEAAs, 800 μg·mL−1 G418, 200 μg·mL−1 hygromycin and 10 μg·mL−1 zeocin. All cells were kept under 80% confluency during routine cell culture to maintain expression of the GABAA receptor at sufficient levels for electrophysiological recordings. For the rat GABAA α2β3γ2 cells expression was induced 24 h prior to experimentation or membrane preparation by the addition of 1 μg·mL−1 doxycycline to the culture media.
Membrane preparation
Cells were grown in T225 flasks up to 80% confluency in full growth medium. The cell layer was washed three times with PBS, and cells were detached from the flasks using enzyme‐free cell dissociation buffer, resuspended in full growth medium and centrifuged for 5 min at 1000× g before being washed once with PBS. Cells were resuspended in ice‐cold buffer (50 mM Tris‐HCl) and homogenized at 4°C using an Ultra‐Turrax® (6 × 5 s blasts on the maximal setting). The homogenate was centrifuged for 20 min at 1000× g, and the supernatant was collected and then centrifuged at 55 000× g (4°C) for 45 min. The resulting pellet was resuspended in buffer, and aliquots were stored at −80°C. Protein concentration was determined using the Bradford assay (Sigma‐Aldrich, Gillingham, UK), using BSA as a standard.
Radioligand binding assay for GABAA receptors containing either α1, α2, α3 or α5 subunits
Saturation binding was performed by incubating 5 μg membrane in assay buffer (50 mM Tris‐Cl and 0.5% F127 pluronic acid) containing between 0.5 and 50 nM [3H]‐flumazenil, in a final assay volume of 200 μL. Non‐specific binding (NSB) was determined using 0.9 μM bretazenil. The reaction was initiated by addition of membrane and after 2 h of incubation at room temperature, was terminated by rapid filtration using a vacuum harvester with four 0.8 mL washes of ice‐cold wash buffer (50 mM Tris‐Cl, pH 7.4), followed by liquid scintillation counting. Data were fitted to a one‐site hyperbolic equation in GraphPad Prism, and K d values were determined as follows: human α1β3γ2 (4.51 nM), α2β2γ2 (7.60 nM), α3β3γ2 (4.75 nM), α5β2γ2 (2.52 nM), rat α1β3γ2 (5.60 nM) and α2β3γ2 (16.07 nM).
For competition‐binding experiments, compounds were incubated with the following amounts of membrane preparations human α1β3γ2, α5β2γ2 and rat α1β3γ2 (4 μg), human α2β2γ2 (10 μg) and, human α3β3γ2 and rat α2β3γ2 (8 μg) and the following concentrations of [3H]‐flumazenil human α1β3γ2, α3β3γ2 and rat α1β3γ2 (5 nM), human α2β2γ2 (10 nM), α5β2γ2 (3 nM) and rat α2β3γ2 (15 nM) in a final assay volume of 201 μL. Reactions were initiated, incubated and terminated as above. Raw data were analysed using SiGHTS proprietary software, using a four‐parameter logistic equation to determine IC50, and K i values were determined using the Cheng–Prusoff equation (Cheng and Prusoff, 1973).
Radioligand binding assay for GABAA receptors containing either α4 or α6 subunits
Saturation binding was performed by incubating 5 μg membrane in assay buffer (50 mM Tris‐HCl and 250 mM KCl) containing 0.8 to 80 nM [3H]‐Ro15‐4513 in a final assay volume of 200 μL. DMCM (methyl‐6,7‐dimethoxy‐4‐ethyl‐β‐carboline‐3‐carboxylate; 12.4 μM) was used to determine NSB. The assay was initiated, incubated, terminated and counted as above. Data were fitted to a one‐site hyperbolic equation in GraphPad Prism, and K d values were determined as follows: GABAA human α4β3γ2 (8.74 nM) and α6β2γ2 (7.60 nM).
For competition‐binding experiments, compounds were incubated with the following amounts of membrane preparations and [3H]‐Ro15–4513, respectively, and α4β3γ2 (8 μg, 6 nM) and α6β2γ2 (10 μg, 10 nM) in a final assay volume of 201 μL. Reactions were initiated, incubated and terminated, and K i values were determined as above.
QPatch determination of functional activity
The QPatch automated electrophysiology assay was run on QPatch HT instruments (Barcelona, Spain). The recording solutions used in these experiments were as follows: extracellular solution (in mM) 137 NaCl, 4 KCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES, 10 glucose, pH 7.4 with NaOH and osmolarity 303–308 mOsm; intracellular solution (in mM) 90 KCl, 50 KF, 1 MgCl2, 10 HEPES, 11 EGTA, 2 Mg‐ATP, pH 7.35 with KOH, and osmolarity 295–300 mOsm.
An open‐channel assay format was used for generating these data, in which GABA was first applied in the presence of 0.1% DMSO for 9 s to allow the GABA current to stabilize. This was followed by the addition of a PAM in the presence of the same GABA concentration for 15 s. This application was washed off using the extracellular solution containing 0.1% DMSO. The following GABA concentrations were used: α2‐, α3‐ and α5‐containing receptors 0.8–1 μM and α1‐containing receptors 0.4–0.5 μM. The following quality control criteria were applied to all raw data on the QPatch software: minimum current amplitude 80 pA (with leak current subtracted), maximum series resistance 16 MΩ, rundown 20% and maximum leak current 150 pA. The peak current amplitude for the PAM application was obtained using a third‐degree polynomial fit, or if this failed, the average of the current during the PAM application period.
Manual patch determination of functional activity
Currents were recorded using the whole‐cell configuration of the patch‐clamp technique (Hamill et al., 1981). Patch electrodes were pulled from 1.2 mm borosilicate glass on a Narishigi puller with patch pipette resistance between 3 and 5 MΩ when filled with the intracellular solution. Currents were recorded using a Multiclamp 700B amplifier (Axon Instruments) and pClamp10 software (Axon Instruments, Wokingham, UK). Currents were held at −60 mV, filtered at 2 kHz and digitized at 5 kHz using Digidata 1440 interface (Molecular Devices, Wokingham, UK). Extracellular solutions contained (mM) 140 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 10 HEPES and 10 glucose (pH 7.4; 320–330 mOsm). The pipette solution contained (mM): 140 KCl, 10 HEPES, 10 EGTA, 1 CaCl2, 2 MgCl2 and 2 MgATP (pH 7.3; 300 mOsm).
Cells were held at −60 mV using a gap‐free protocol at room temperature. The effect of compounds was tested using an open‐state protocol. Whole‐cell current was activated by a low concentration of GABA for 3 to 5 s and followed with the same concentration of GABA plus testing compounds for 3 to 5 s or until the current reached the peak. The GABA concentration used in this assay was the approximate EC5 to EC10 for each receptor subtype, to enable activation of the GABAA receptor at a low enough level to prevent rundown of the current.
Animals
All experiments performed in the UK were conducted in accordance with the Home Office Animals (Scientific Procedures) Act (1986) and were subject to local ethical review. All procedures involving animals in the USA were conducted with the approval of Pfizer and were compliant with US National Research Council's Guide for the Care and Use of Laboratory Animals, the US Public Health Service's Policy on Humane Care and Use of Laboratory Animals, and Guide for the Care and Use of Laboratory Animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Native tissue preparation and binding assays
Non‐human primate tissue samples were obtained from male animals being culled for other reasons and subject to local ethical review. Male caesarean‐derived rats (200–250 g) were killed using schedule 1 stun and decapitation. Rhesus monkey whole‐brain coronal slices and cerebellum and rat forebrain spinal cord and cerebellum were dissected and snap frozen in isopentane and then stored at −80°C until use. On the day of the assay, tissues were thawed and homogenized in 0.32 M sucrose (including complete protease inhibitor), and the homogenate was centrifuged at 3000× g for 10 min at 4°C. The resultant supernatant was centrifuged at 48 000× g for 30 min 4°C, and the resulting pellet was resuspended in assay buffer (50 mM Tris, pH 7.4, 4°C). For binding experiments, rhesus monkey forebrain or cerebella membranes (20 μg) or rat forebrain, spinal cord or cerebella membranes (25 μg) were incubated with between 0.5 and 50 nM [3H]‐flumazenil (saturation binding) or varying concentrations of PF‐06372865 and 1.8 nM [3H]‐flumazenil (competition binding) in a total volume of 250 μL assay buffer, for 30 min at room temperature. NSB was determined using 100 μM bretazenil. The reaction was terminated and counted, and K i values were determined as described above, using the following K d values: rhesus monkey forebrain, 2.75 nM; cerebellum, 3.90 nM; rat forebrain, 2.13 nM (Sullivan et al., 2004); cerebellum, 1.65 nM (Sullivan et al., 2004); and spinal cord, 3 nM (Atack et al., 2006).
Rat receptor occupancy studies
Sprague–Dawley rats have been previously used to assess RO of GABAA PAMs (Atack et al., 2009; Nickolls et al., 2011), and as this strain was also suitable for subsequent in vivo studies, it was also used to assess in vivo RO of PF‐06372865. In these studies, male Sprague–Dawley rats (250–300 g) received either vehicle control (17% solutol/18% glycerol formal/65% water) or PF‐06372865 (0.3, 1 or 10 mg·kg−1) p.o., with a 1 h pretreatment time. NSB was determined in a separate group of animals by administering 5 mg·kg−1 bretazenil i.p., with a 30 min pretreatment time. At 3 minbefore beling killed, animals were dosed i.v. with 10 μCi·kg−1 [3H]‐flumazenil. Following their death, trunk blood was collected in EDTA tubes and kept on ice until centrifugation at 1000× g for 10 min at 4°C. The resulting plasma was collected. In addition, the whole brain and whole spinal cord were removed, dissected and homogenized in 10 volumes of ice‐cold buffer (50 mM Tris‐HCl, pH 7.4, 4°C) using a polytron homogenizer. Three 300 μL aliquots of homogenate were filtered over 0.5% v.v‐1 polyethyleneimine‐soaked GF/B filters to separate the bound radioactivity from the free radioactivity and washed twice in 5 mL ice‐cold buffer. Filters were then placed in vials, scintillation fluid added and radioactivity counted. Plasma samples were also collected for pharmacokinetic (PK) analysis. The RO values of PF‐06372865 were determined by calculating the reduction in specific binding in drug‐treated rats relative to vehicle controls. Typically, vehicle levels of radioactivity were around 2000 dpm and non‐specific (bretazenil treated) levels were around 50 dpm. Occ50 values were generated by fitting data to a four‐parameter logistic equation in GraphPad Prism.
Bioanalysis
Plasma and tissue homogenate samples were analysed by protein precipitation with volumes of internal standard containing acetonitrile (5:1 ratio with sample), followed by mixing and centrifugation to pellet protein. Supernatant was then mixed (1:1) with water prior to analysis by LC‐MS/MS monitoring a multiple reaction monitoring transition for PF‐06372865: 441.4 > 348.2. Limits of quantification of 0.5 ng·mL−1 were achieved.
Zolpidem drug‐discrimination assay
Drug discrimination has previously been used by Pfizer to determine the pharmacology of cannabinoid ligands (Young et al., 2009). In this study, 12 male Sprague–Dawley rats (490–600 g, Charles River, Boston, MA, USA) were trained using an operant food‐maintained task, to discriminate between the presence and absence of 0.87 mg·kg−1 i.p. zolpidem (a GABAA α1‐selective PAM). The sample size was based on prior experience with the drug‐discrimination model in rats and confirmed as appropriate through the demonstration of lever discrimination following training with zolpidem. Operant responding was maintained by delivery of food pellets, with the correct response (left or right lever) being dependent on the presence or absence of the drug cue. Once rats were reliably discriminating the training drug cue from vehicle, as assessed by examining lever choice, the similarity of the cue induced by the p.o. administration of PF‐06372865 (0.3, 1, 3 and 10 mg·kg−1, vehicle 17% solutol/18% glycerol formal/65% water) was examined during generalization tests, in a randomized crossover design, with all rats receiving all doses of compound. Concurrently, locomotor activity was recorded. Immediately following generalization tests, rectal temperature was measured, and lateral tail vein blood samples were taken for PK analysis. Generalization session data were analysed using ANOVA with PF‐06372865 dose as the factor. Post hoc comparisons were made with vehicle using Dunnett's test. For confirmation of positive (training drug) and negative (training drug vehicle) controls, paired t‐tests were used. Due to poor performance in the training sessions, one rat did not complete the 3 and 10 mg·kg−1 doses and one rat did not complete the vehicle dose. Rectal temperature was not assessed in one of the rats when receiving the 3 mg·kg−1 dose and one of the rats when receiving the 0.3 mg·kg−1 dose.
Rat qEEG
Sprague–Dawley rats have previously been used to assess the qEEG profile of subtype‐selective GABAA PAMs (Nickolls et al., 2011) and, therefore, were considered a suitable species for these studies. Sample sizes were based on previous data and the assay capability tool (Miranda et al., 2014). Eight male Sprague–Dawley rats (~300 g, Charles River, Margate, Kent, UK) were anaesthetized using isoflurane (using 3% isoflurane anaesthesia in 100% O2 and were maintained using 1.5–2.0% isoflurane in 100% O2 as assessed by interdigital reflex), and impanted i.p. with radiotelemetric transmitters (TL11M2 F40‐EET, Data Sciences International, St. Paul, MN, USA) and with cortical EEG electrodes (stainless steel screw electrodes). These were implanted epidurally over the left parietal cortex (2.0 mm anterior and 2.0 mm lateral to lambda) and over the left frontal cortex (2.0 mm anterior and 2.0 mm lateral to bregma) for a frontal–parietal EEG recording (Kantor et al., 2002; Ivarsson et al., 2005). The cortical electrodes and accompanying leads were secured to the skull by covering with dental acrylic. Animals recovered in heated boxes before being returned to their home cages (from this point, animals were single housed). EEG studies were conducted a minimum of 2 weeks after surgery. At the beginning of the light phase, animals received either 1, 3 or 10 mg·kg−1 PF‐06372865 or vehicle control (17% solutol/18% glycerol formal/65% water) p.o. in a randomized, four‐way crossover design, so that all animals received all of the treatments, thus enabling within animal comparisons (n = 8). EEG data were then immediately recorded, sampling continuously at 500 Hz (bandpass filtered with cut‐off frequencies of 0.1 and 100 Hz) for 6 h with Data Sciences International hardware and Data Acquisition Gold version 3.01 software (Data Sciences International). Data were analysed using Spike 6 (CED, Cambridge, UK). For the EEG analysis, consecutive 12 s epochs were subjected to a fast Fourier transform and the EEG power density within five frequency bands (delta 0.75–4 Hz, theta 6–9 Hz, alpha 8–13 Hz, beta 13–40 Hz and gamma 40–80 Hz) were calculated. Epochs containing artefacts were excluded from analysis, but otherwise, data were integrated for each frequency band, as defined above, and mean values were computed for each. The data 0–6 h post‐administration were selected based on the PK properties of PF‐06372865. Data were analysed comparing PF‐06372865 treatment with the vehicle treatment using a one‐way ANOVA with the significance level set at P < 0.05.
Single ascending dose clinical study
This was a double‐blind, third‐party open (i.e. subject blind, investigator blind and sponsor open), randomized, placebo‐controlled, ascending single p.o. dose, three cohort design, with placebo substitution, crossover study of PF‐06372865 (Supporting Information Figure S1). In addition, a fourth cohort was run to further explore the PD of PF‐06372865 dosed alone, lorazepam alone and PF‐06372865 in combination with lorazepam (additional PD endpoints and combination data are not reported in this manuscript). All cohorts were conducted over a maximum of five treatment periods. Eligible subjects were healthy male subjects aged between 18 and 55 years, inclusive. Subjects were required to have a body mass index between 17.5 and 30.5 kg m−2 and a total body weight >50 kg. For all four cohorts, the randomization schedule was provided to the site by the sponsor. Cohorts 1 to 3 included 10 subjects, and in each period, eight subjects from the respective cohort received PF‐06372865, and the remaining two subjects received placebo. Cohorts 1 and 2 were run as an interleaving design. Cohort 3 continued after the end of cohort 2 and completed before the start of additional cohort 4. The fourth cohort explored 15 and 65 mg dose (p.o. suspension) of PF‐06372865 and 2 mg dose (p.o. tablet) of lorazepam. The doses explored in the first three cohorts ranged from 0.04 to 100 mg. All doses were administered as p.o. suspension except in one period in cohort 3, in which a 25 mg dose was also administered as a tablet formulation to investigate relative bioavailability between the two formulations (suspension and tablet).
NeuroCart
Measurements of saccadic eye movements were recorded as previously described (de Haas et al., 2008, 2009). Saccadic peak velocity (SPV) is closely related to the anxiolytic properties of BZDs (de Visser et al., 2003), and its measurement has been validated as the most sensitive biomarker for their effects (van Steveninck et al., 1991; 1992; 1999; de Haas et al., 2007). Two minute body sway measurements were performed as previously described (de Haas et al., 2009). Body sway is a measure of postural stability that has previously been shown to be sensitive to BZDs (van Steveninck et al., 1996). Pharmaco‐EEG recordings were performed as previously described (de Haas et al., 2010). EEG recordings were made at Fz, Cz, Pz and Oz. For each lead, fast Fourier transform analysis was performed to obtain the sum of amplitudes in the delta (0.5–4 Hz), theta (4–7.5 Hz), alpha (7.5–13.5 Hz) and beta (13.5–35 Hz) frequency ranges. The duration of EEG measurements was 64 s per session, and channels were bandpass filtered with cut‐off frequencies of 0.1 and 100 Hz (−6 dB). Change in amplitudes in the beta frequency band of the EEG was found to be a relevant measure of the pharmacological effect intensity of BZDs (Mandema et al., 1992).
Saccadic eye movements, body sway and EEG assessments were performed at pre‐dose/baseline (twice), 0.5, 1, 1.5, 2, 4, 6 and 12 h post‐dose.
Sample size
A sample size of 10 subjects in each of cohorts 1, 2 and 3 (with eight active and two placebo in each period), was chosen based on the need to minimize first exposure to humans of a new chemical entity and the requirement to provide adequate safety and toleration information at each dose level. Ten subjects were also considered sufficient to provide PD evaluation. A sample size of 15 subjects in cohort 4 was selected to ensure balance in the design and to provide sufficient precision and acceptable operating characteristics for internal decision‐making based on assumed within subject SDs of 37°·s−1 for SPV and 0.39 loge (mm) for body sway. These estimates were based on data from cohorts 1, 2 and 3 of the study.
Statistical analyses
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The PD analysis set was defined as all subjects randomized and treated who had at least one of the PD parameters of primary interest in at least one treatment period. Body sway was loge transformed prior to analysis. A mixed effects model was fitted to the SPV, body sway and EEG endpoints using data collected during the first 6 h of post‐dose, with data pooled for cohorts 1–3 but analysed separately for cohort 4. For the combined dataset on cohorts 1, 2 and 3, the fixed effects included in the model were baseline, time, treatment and treatment by time interaction. Time was included as a repeated effect within each subject * period. Baseline was included as two separate variables, the average baseline for the subject and the deviation of each treatment baseline from the average baseline for each subject (Kenward and Roger, 2010). For cohort 4, the fixed effects included in the model were baseline, period, time, treatment and treatment by time interaction. Time was fitted as a repeated effect within each subject * period. Baseline was included as two separate variables, the average baseline for the subject and the deviation of each period baseline from the average baseline for each subject. For all cohorts, the least squares means (LSmeans) together with 95% confidence intervals (CIs) were obtained for each treatment averaged across the first 6 h post‐dose and similar averaged effects for treatments compared with placebo. Statistical significance was based on a two‐sided P‐value <0.05. No adjustment for multiple testing was applied.
Human PET RO study
The PET study was a phase 1, open‐label, single‐centre and single p.o. dose study, to characterize the relationship between the GABAA RO in the brain as a function of the plasma concentration of PF‐06372865 in healthy adult subjects by PET imaging using the radioligand [11C]‐flumazenil. Two doses of PF‐06372865 were tested in this study, 10 mg (n = 3) and 65 mg (n = 2). Each subject was scanned three times with a baseline scan and two post‐dose scans. The baseline and the first post‐dose scans occurred on day 1 prior to and approximately 1.5 h after PF‐06372865 dosing respectively. The second post‐dose scan occurred on day 2 at approximately 24 h after PF‐06372865 dosing. The injected dose of [11C]‐flumazenil was 570 ± 141 MBq. PET images were acquired using an ECAT EXACT HR+ scanner (Siemens Medical Systems, Knoxville, TN, USA) in three‐dimensional mode. Each PET session lasted approximately 120 min. Venous blood samples were collected to assess the plasma concentration of PF‐06372865 during the PET scan period. Arterial blood samples were collected to measure the arterial input function and the unmetabolized fraction of the radiotracer. In addition, each subject had a baseline MRI scan of the head for use in image co‐registration and to screen subjects for possible anatomical abnormalities.
Dynamic PET scan data were reconstructed with corrections for attenuation, normalization, scatter, randoms and dead time using the ordered subset‐expectation maximization algorithm (4 iterations and 16 subsets). PET images were corrected for motion using a mutual information algorithm (FSL‐FLIRT). The automated anatomical labelling template (Tzourio‐Mazoyer et al., 2002) was applied to generate regional time activity curves (TACs) (amygdala, caudate, cerebellum, centrum semiovale, cingulum, hippocampus, frontal, insula, occipital, pallidum, putamen, temporal and thalamus) after co‐registration between the template and each subject's MRI and PET images. The TACs were fitted with the 1T compartment model using the arterial input function and metabolite correction. V T was estimated for each region. RO for the whole brain was computed with Lassen plots using baseline and blocking V T for each region (Lassen et al., 1995). The relationship between observed average plasma PF‐06372865 concentration during the PET scanning period and measured brain GABAA RO was fitted with an E max model using the following equation, where RO max was the maximum achievable % RO, C avg represented the average plasma concentration of PF‐06372865 during the PET scanning period and Occ50 was the plasma concentration corresponding to 50% RO max. Occ50,α1, Occ50,α2 and Occ50,α3 are the Occ50 for α1, α2 and α3 subunits respectively:
PF‐06372865 binding for GABAA subtypes α1, α2 and α3 in whole brain was also estimated using modelling as below.
In this model, the differences between the binding affinity for α1 (Occ50,α1), α2 (Occ50,α2) and α3 (Occ50,α3) in human brain (Occ50 ratios) were assumed to be same as the ratio of K i, which was obtained from in vitro binding assays. The abundance of α1, α2 and α3 receptors was approximated to 50, 25 and 25% based on total GABAA receptors in the whole brain (McKernan and Whiting, 1996). Other subtypes such as α4, α5 and α6 receptors have very low levels compared with α1, α2 and α3 in the whole brain; therefore, their contributions to the overall RO in whole brain were disregarded.
Materials
PF‐06372865 was synthesized at Pfizer Laboratories (Sandwich, UK) as described in WO2014091368 (Omoto et al., 2014). 3H‐ligands were obtained from PerkinElmer (Cambridge, UK). All other chemicals were supplied by Sigma‐Aldrich (Gillingham, UK). Human GABAA α1β3γ2, α3β3γ2, α4β3γ2 and α6β3γ2 cell lines were purchased from Millipore (Watford, UK) (now Eurofins), and all other cell lines were made in‐house. Cell culture media were purchased through Fisher Scientific (Loughborough, UK).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
We have conducted a high‐throughput screening campaign for α2/3 functionally selective GABAA receptor PAMs using radioligand binding and electrophysiological assays. To our knowledge, this is the first example of a high‐throughput QPatch electrophysiological screening assay being used to identify GABAA receptor PAMs. Previously, campaigns have been limited by the use of manual patch techniques to follow up hits from binding assays; however, the advance in high‐throughput electrophysiological screening allowed us to determine the functional activity of all the compounds tested in our initial binding assay. We chose to look for functionally selective rather than binding‐selective PAMs due to historical data suggesting that it is very difficult to obtain binding‐selective compounds (Atack et al., 2009). We identified a lead molecule, PF‐06372865 (Figure 1), which exhibited the desired in vitro pharmacological properties and was subsequently progressed through in vitro and in vivo preclinical screening, safety testing and into clinical studies.
Figure 1.
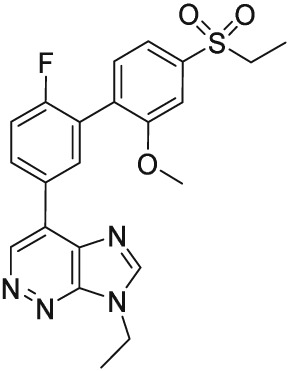
Chemical structure of PF‐06372865.
The affinity of PF‐06372865 for the BZD site of GABAA receptors was determined in competition‐binding experiments, versus [3H]‐flumazenil (receptors containing α1/2/3/5 subunits) or [3H]Ro15‐4513 (receptors containing α4/6 subunits), in membranes from recombinant cell lines expressing GABAA receptors containing specific α subunits (Table 1). PF‐06372865 was determined to be a high‐affinity ligand at GABAA receptors containing an α1, α2, α3 or α5 subunit (Figure 2A) but had no affinity for GABAA receptors containing an α4 or 6 subunit, which is typical of BZD site ligands. Interestingly, PF‐06372865 did not display identical affinity at α1/2/3/5 containing GABAA receptors and had a rank order of affinity α1 ˃ α3 ≈ α2 ˃ α5.
Table 1.
In vitro properties of PF‐06372865
| Affinity (nM) | EC50 QPatch (nM) | E max QPatch (% potentiation of GABA EC10) | E max manual patch (% potentiation of GABA EC10) | E max manual patch (normalized to diazepam) | |
|---|---|---|---|---|---|
| Human α1β3γ2 | 0.18 (0.09–0.35, n = 5) | 40.5 (11.0–149, n = 6) | 21.3 ± 2.0 (n = 21) | 20.2 ± 3.4 (n = 8) | 10.9 ± 2.5 (n = 8) |
| Human α2β2γ2 | 2.92 (1.03–8.24, n = 8) | 20.8 (15.7–27.6, n = 7) | 134.1 ± 2.9 (n = 34) | 123.7 ± 12.3 (n = 7) | 35.3 ± 1.5 (n = 7) |
| Human α3β3γ2 | 1.06 (0.56–2.00, n = 6) | 20.7 (7.8–55.1, n = 3) | 91.6 ± 5.4 (n = 22) | 144.6 ± 20.4 (n = 6) | 48.9 ± 8.4 (n = 6) |
| Human α4β3γ2 | >19 000 (n = 6) | nd | nd | nd | nd |
| Human α5β2γ2 | 18.04 (7.28–44.71, n = 6) | 30.9 (12.2–78.6, n = 4) | 90.9 ± 3.6 (n = 31) | 95.0 ± 11.5 (n = 7) | 69.8 ± 8.9 (n = 7) |
| Human α6β2γ2 | >19 000 (n = 6) | nd | nd | nd | nd |
| Rat α1β3γ2 | 0.34 (0.13–0.93, n = 5) | nd | 0.6 ± 3.4 (n = 11) | 6.0 ± 3.2 (n = 8) | 8.9 ± 7.2 (n = 8) |
| Rat α2β3γ2 | 4.58 (2.07–10.14, n = 6) | nd | 54.3 ± 6.0 (n = 12) | 49.6 ± 8.5 (n = 7) | 39.3 ± 2.4 (n = 7) |
The in vitro binding and functional profile of PF‐06372865 were measured as described in Methods. Binding affinity (K i) is shown in nM with geometric mean and 95% CIs. Functional activity is presented as mean ± SEM potentiation of a GABA EC5–20 concentration. The n numbers are given in parentheses. nd, not determined.
Figure 2.

(A) Concentration–response curves for PF‐06372865 in [3H]‐flumazenil competition‐binding assays to membranes containing GABAA receptors expressing different α subunits. Data are representative, duplicate, determinations from between 5 and 8 separate experiments. (B) Example of GABAA current response to the co‐application of GABA and 0.1% DMSO (first pipette mark), the co‐application of GABA and PF‐06372865 (second pipette mark), followed by a wash with extracellular solution (third pipette mark); peak recording is indicated by the line. (C) Example of GABAA current response to the co‐application of GABA and 0.1% DMSO (first pipette mark), followed by a second co‐application of GABA and 0.1% DMSO (second pipette mark), followed by a wash with extracellular solution (third pipette mark); peak recording is indicated by the line. (D) Concentration–response curves of QPatch functional response for human GABAA receptors containing different α subunits. Graphs are the mean ± SEM data from all experiments (n = 3–7).
The functional activity of PF‐06372865 was determined in electrophysiological experiments in the same recombinant cell lines. In screening mode QPatch assays and follow‐up concentration–response curves and manual patch assays, an open‐channel assay format was used. In this protocol, GABAA receptors were opened using an EC10 concentration of GABA, and then compound was applied and potentiation measured (Figure 2B, C). This protocol resulted in less rundown in the system and less data variability than traditional GABAA potentiation experiments, in which a GABA plus potentiator response is compared with a GABA alone response. The data generated determined that PF‐06372865 exhibited subtype selectivity for GABAA receptors containing α2, α3 and α5 subunits, with significant positive allosteric modulation (90–140%) but negligible activity (neutral modulation ≤20%) at GABAA receptors containing α1 subunits (Figure 2D). Therefore, although the higher affinity for PF‐06372865 at α1‐containing receptors will result in greater RO at these receptors than receptors containing α2/α3 or α5 subunits (for the given dose), this should result in little functional effect because of the very low level of allosteric modulation.
In order to determine whether PF‐06372865 exhibited any species selectivity, the affinity and functional activity of PF‐06372865 were also determined in recombinant cell lines expressing rat GABAA receptors containing either rat α1 or α2 subunits, using the same methodology as for the cell lines expressing human subunits. This demonstrated that there was no evidence that this compound displayed any species selectivity. Furthermore, the affinity of PF‐06372865 was determined in rat and rhesus monkey native membrane preparations in competition‐binding experiments versus [3H]‐flumazenil (Table 2). PF‐06372865 was determined to be a high‐affinity BZD site ligand in these preparations. The differences in affinity between cerebellum (α1 rich) and spinal cord (α2 rich) binding in both rats and monkeys are likely reflective of the higher affinity for α1‐containing versus α2‐containing GABAA receptors, as observed in the recombinant membrane preparations.
Table 2.
Binding affinity of PF‐06372865 in native membrane preparations
| Rat forebrain | Rat cerebellum | Rat spinal cord | Rhesus macaque forebrain | Rhesus macaque cerebellum | |
|---|---|---|---|---|---|
| K i (nM) (95% CI) | 1.06 (0.63–1.78), n = 7 | 0.47 (0.35–0.62), n = 6 | 2.49 (1.49–4.15), n = 7 | 1.26 (0.46–3.49), n = 4 | 0.55 (0.30–1.01), n = 6 |
The in vitro binding properties of PF‐06372865 were measured as described in Methods. Binding affinity (K i) is shown as nM with geometric mean and 95% CIs.
Due to its favourable in vitro profile, PF‐06372865 was progressed to in vivo experiments, where firstly in vivo RO was measured in rats using [3H]‐flumazenil. In vivo, PF‐06372865 showed dose‐dependent GABAA RO in rat forebrain, spinal cord and cerebellum. The dose, which occupied 50% of the receptors (Occ50), was 0.54, 0.99 and 0.26 mg·kg−1 in rat brain, spinal cord and cerebellum respectively (Figure 3A). The total concentrations of PF‐06372865, which gave 50% RO, were 51.8 ng·mL−1 and 32.3, 32.3, 30.9 and 7.6 ng·g−1 in plasma, forebrain, spinal cord and cerebellum, respectively, corresponding to an Occ50 unbound plasma concentration of 8.5 nM (Figure 3B). These data mimic the in vitro binding data and are an important consideration in translating to clinical data as whole brain PET occupancy measures will overestimate RO at α2‐containing receptors, as the majority of receptors (~50%) present in the brain contain an α1 subunit (McKernan and Whiting, 1996). The PK profile in rats was also determined with a t 1/2 of 5.5 h after 1 mg·kg−1 dosed p.o.
Figure 3.
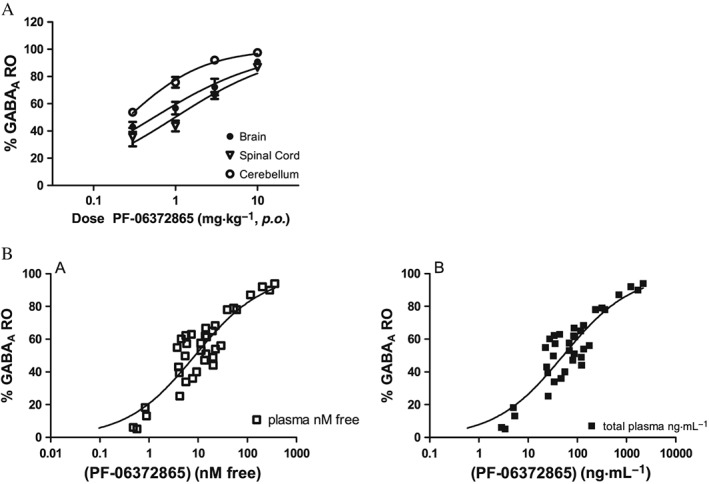
(A) In vivo GABAA receptor occupancy (RO) of PF‐06372865 in rats at 1 h post‐dose. Values are expressed as percentage inhibition of [3H]‐flumazenil binding relative to vehicle, shown as mean ± SEM (n = 4 per group); (B) relationship of total and free plasma to brain RO.
For subsequent in vivo experiments, doses were chosen based on RO. We have previously shown a dose‐dependent increase in qEEG beta frequency with the GABAA receptor PAMs L‐838,417 and TPA023 (Nickolls et al., 2011). Similarly, PF‐06372865 (1–10 mg·kg−1) also induced a dose‐dependent increase in qEEG beta frequency in rats implanted with a telemetry transmitter to record EEG (Figure 4), which was significant at 3 mg·kg−1 (~75% RO) and 10 mg·kg−1 (~100% RO). Significant changes in other qEEG parameters were also observed, including a significant decrease in theta, delta and alpha activity and an increase in gamma activity.
Figure 4.
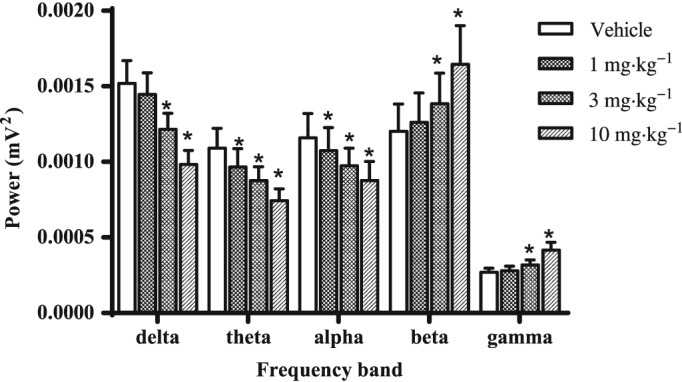
The effect of vehicle and 1, 3 and 10 mg·kg−1 PF‐06372865 0–6 h post‐treatment on qEEG recordings in the light phase. Data are expressed as mean ± SEM (n = 8) delta (0.75–4 Hz), theta (6–9 Hz), alpha (8–13 Hz), beta (14–40 Hz) and gamma (40–80 Hz) power (mV2) in the 6 h period post‐administration, and were analysed with a one‐way ANOVA with the significance level set at P < 0.05.
In zolpidem drug discrimination, PF‐06372865 (up to 10 mg·kg−1 (~100% RO) produced weak generalization (<20%) to zolpidem consistent with a lack of GABAA α1 activity in vivo (Figure 5A). Body temperature and locomotor activity were also measured in this assay, and neither was significantly affected by PF‐06372865 (Figure 5B, C).
Figure 5.
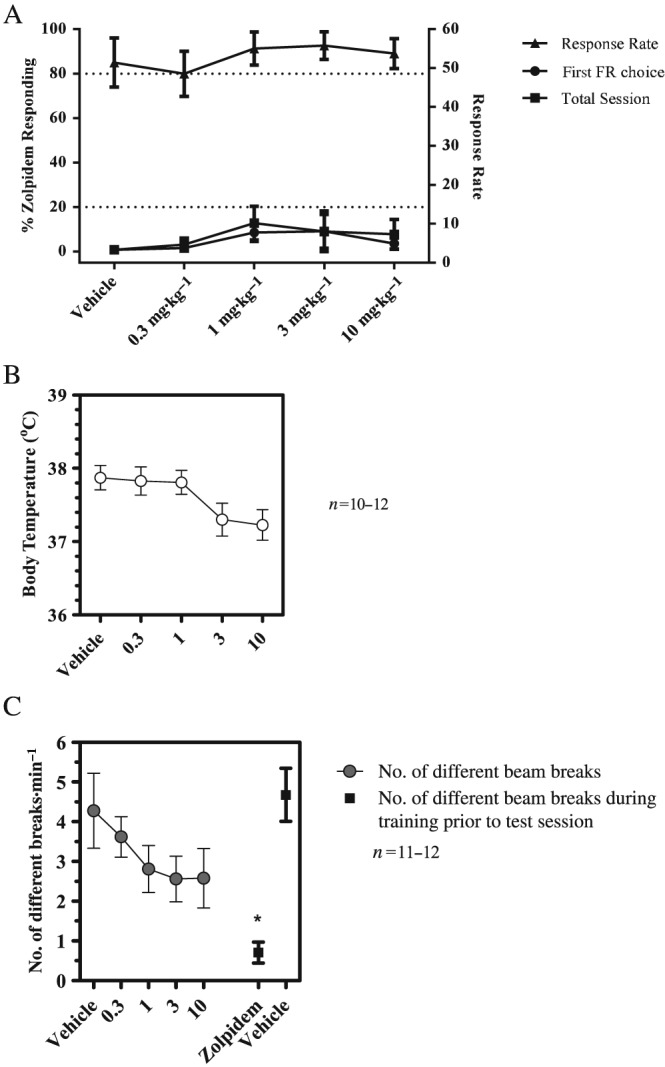
(A) Drug‐discrimination of PF‐06372865 in zolpidem‐trained rats. Data show mean percentage zolpidem‐appropriate responding (left axis) or responses.min‐1 (right axis) (±SEM) during training or generalization tests with PF‐06372865. (B) Effects of PF‐06372865 on core body temperature. Data show mean rectal temperature (±SEM) 65–75 min post‐dosing. (C) Effects of PF‐06372865 on locomotor activity during test session. Data show mean locomotor counts (±SEM) during training or generalization tests. FR, fixed ratio.
Clinical data
In this report, we disclose data from two Phase 1 studies: a single ascending dose (SAD) and a PET study. A total of 45 healthy subjects (44 male and 1 female) completed the SAD PK/PD study (Supporting Information Figure S2). There were 10 subjects in each of the first 3 cohorts and 15 subjects in cohort 4, with the age ranging from 27 to 33 years across the cohorts (Supporting Information Table S1). Their weight ranged from 73.1 to 77.6 kg, and the majority of subjects were white (37 out of 45). A total of five subjects (all males) completed the PET study, with the age ranging from 29 to 50 years. Weight ranged from 66.5 to 99.0 kg; two subjects were black and three were of other race.
Pharmacokinetics
A summary of plasma PK parameter values following single p.o. doses of PF‐06372865 is shown in Table 3. In summary, PF‐06372865 was absorbed rapidly following single doses of 0.04 to 100 mg as p.o. suspension with median T max of 1 to 4 h. Mean t 1/2 was similar across all doses, ranging from 6.0 to 8.9 h. In general, plasma PF‐06372865, AUCinf and C max appeared to increase proportionally across the entire dose range from 0.04 to 100 mg (Figure 6); however, this was not formally tested. The mean CL/F ranged from 17.35 to 26.86 L·h−1, and the mean Vz/F ranged from 194.7 to 260 L across all doses.
Table 3.
Summary of plasma PF‐06372865 PK parameter values following single p.o. doses
| Parameter (unit) | Parameter summary statisticsa for PF‐06372865 by treatment | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cohort 1 | Cohort 2 | Cohort 3 | |||||||||||||
| 0.04 mg | 0.2 mg | 0.8 mg | 4 mg | 10 mg | 0.1 mg | 0.4 mg | 1.6 mg | 6 mg | 15 mg | 25 mg | 40 mg | 65 mg | 100 mg | 25 mg tablet | |
| N (n) | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 | 8, 8 |
| C max (ng·mL−1) | 0.2287 (36) | 0.9714 (38) | 4.030 (31) | 18.88 (27) | 54.67 (36) | 0.4993 (23) | 1.692 (13) | 9.279 (25) | 30.46 (28) | 89.45 (33) | 128.5 (38) | 224.0 (38) | 285.1 (28) | 559.3 (39) | 118.7 (33) |
| T max (h) | 1.00 (0.500–1.52) | 1.50 (0.983–2.00) | 1.06 (0.500–2.00) | 1.00 (0.500–4.00) | 1.50 (0.500–2.00) | 2.00 (0.500–4.00) | 1.53 (1.03–2.02) | 1.50 (0.500–2.00) | 1.25 (0.500–2.00) | 1.00 (0.250–4.05) | 4.00 (1.50–4.02) | 1.50 (0.250–4.02) | 2.00 (0.250–4.00) | 4.00 (0.250–4.05) | 2.00 (0.983–4.08) |
| AUC24 (ng·h·mL−1) | 1.603 (44) | 6.946 (47) | 29.95 (39) | 140.9 (34) | 439.4 (49) | 4.386 (31) | 15.42 (20) | 78.03 (18) | 269.3 (32) | 747.2 (37) | 1150 (46) | 1632 (68) | 2439 (69) | 4975 (56) | 942.5 (68) |
| AUClast (ng·h·mL−1) | 1.599 (48) | 7.189 (53) | 32.24 (45) | 148.6 (38) | 487.4 (52) | 4.828 (40) | 17.18 (26) | 88.21 (25) | 297.6 (35) | 854.2 (48) | 1298 (54) | 1866 (83) | 2746 (80) | 5753 (70) | 1066 (83) |
| AUCinf (ng·h·mL−1) | 1.739 (46) | 7.439 (52) | 32.52 (45) | 148.9 (38) | 487.8 (52) | 5.024 (38) | 17.48 (27) | 88.64 (25) | 297.7 (35) | 857.5 (49) | 1300 (54) | 1868 (83) | 2748 (80) | 5763 (70) | 1066 (83) |
| t 1/2 (h) | 6.193 ± 2.163 | 6.006 ± 1.870 | 6.838 ± 2.113 | 6.195 ± 2.012 | 7.984 ± 1.895 | 8.466 ± 2.913 | 8.173 ± 2.578 | 8.401 ± 2.619 | 7.716 ± 1.215 | 8.880 ± 3.881 | 8.195 ± 2.619 | 7.728 ± 3.480 | 8.306 ± 2.987 | 8.675 ± 3.212 | 8.239 ± 3.628 |
| CL/F (L·h−1) | 22.98 (46) | 26.86 (52) | 24.61 (45) | 26.85 (38) | 20.51 (52) | 19.91 (38) | 22.89 (27) | 18.05 (25) | 20.13 (35) | 17.49 (49) | 19.26 (54) | 21.40 (83) | 23.64 (80) | 17.35 (70) | 23.45 (83) |
| Vz/F (L) | 194.7 (28) | 221.6 (27) | 232.4 (20) | 230.0 (26) | 229.9 (44) | 232.0 (21) | 260.0 (14) | 211.6 (15) | 221.4 (20) | 210.3 (27) | 215.1 (42) | 211.9 (27) | 258.1 (28) | 201.8 (33) | 251.4 (31) |
AUC24, area under the concentration–time profile from time zero to 24 h post‐dose; AUCinf, area under the plasma concentration–time profile from time zero extrapolated to infinite time; AUClast, area under the plasma concentration–time profile from time zero to the time of the last quantifiable concentration; CL/F, apparent oral clearance; C max, maximum observed plasma concentration; CV, coefficient of variation; N, number of subjects in the treatment group and contributing to the mean; n, number of subjects with reportable t 1/2, AUCinf, CL/F and Vz/F; T max, time for C max; Vz/F, apparent volume of distribution.
Geometric mean (% Geometric CV) for all except: median (range) for T max and arithmetic mean ± SD for t 1/2.
Figure 6.
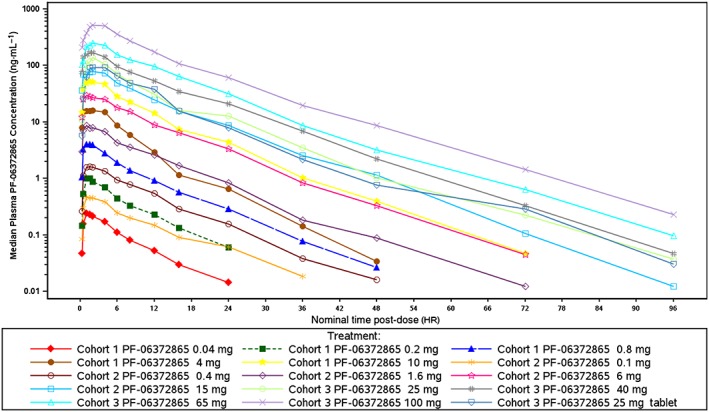
Median plasma PF‐06372865 concentration–time profiles following single p.o. doses (semi‐logarithmic plot).
Safety
There were no deaths, serious or severe AEs, discontinuations or temporary discontinuations due to AEs, in any cohort in the SAD study or PET study. Single p.o. doses of PF‐06372865 escalating from 0.04 to 100 mg were generally safe and well tolerated in the healthy subjects evaluated. Supporting Information Table S2 is a summary of treatment‐emergent AEs. The incidence and severity of AEs (all mild in severity) did not largely increase with increasing doses beyond 6 mg, as demonstrated in Figure 7 for dizziness and somnolence. A maximum tolerated dose (MTD) was not achieved with 100 mg.
Figure 7.

Incidence of dizziness and somnolence: treatment‐emergent adverse events. Plot of the percentage of subjects reporting a treatment‐emergent adverse event of dizziness or somnolence (all causality) by dose from both the SAD and PET studies. Shape represents the treatment, and colour represents the study/cohort. LRZ, lorazepam; (T), tablet.
Pharmacodynamics
Doses of 4 mg PF‐06372865 and higher were related to dose‐dependent, statistically significant reduction in SPV versus placebo. The effect of the 100 mg dose on SPV was similar to the 65 mg dose, with a reduction in the SPV averaged over the first 6 h post‐dose of approximately 130°·s−1 compared with placebo (Figure 8A). A single 2 mg dose of lorazepam decreased the average SPV by 38.6°·s−1 (95% CI: 11.0, 66.2) compared with placebo, similar to a 4 mg dose of PF‐06372865 (reduction of 37.7 [14.3, 61.0] versus placebo). The effects of dosing PF‐06372865 at 0.8 and 4 mg and the majority of higher doses on body sway were statistically significantly greater than placebo. There was an increase in the average body sway over the first 6 h post‐dose with increasing doses of PF‐06372865 up to 10 mg (Figure 8B). The effect of doses between 10 and 100 mg on body sway were similar (LSmean ratio versus placebo of 1.55 [95% CI: 1.25, 1.95] and 1.54 [95% CI: 1.22, 1.92] for the 10 and 100 mg doses, respectively). By comparison, a 2 mg dose of lorazepam demonstrated a greater increase in body sway versus placebo than PF‐06372865 (LSmean ratio versus placebo of 1.97 [95% CI: 1.60, 2.46]).
Figure 8.
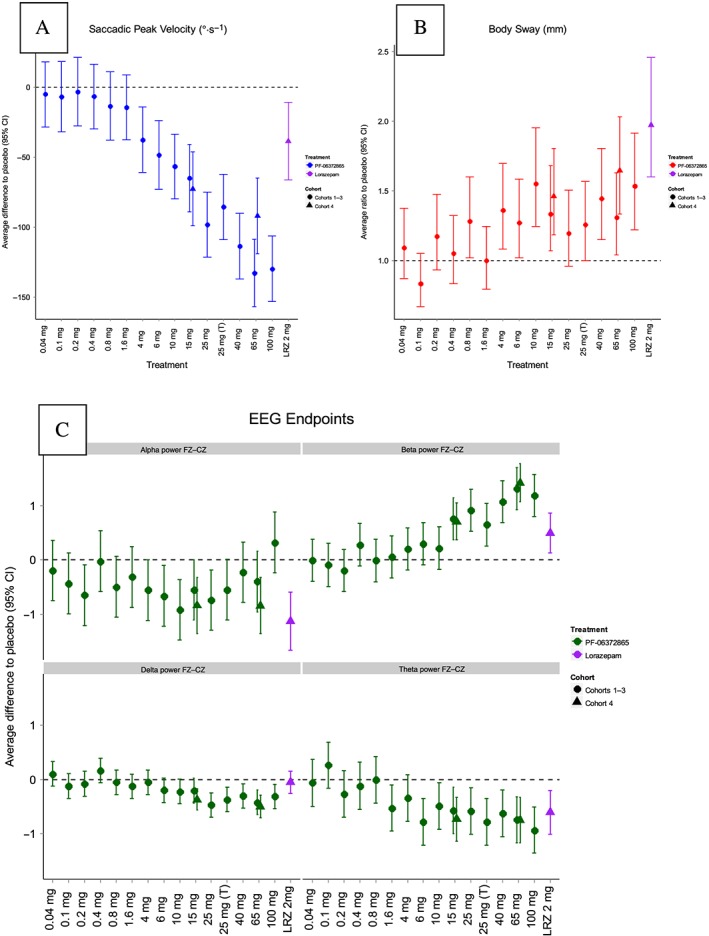
Plot of difference to placebo in overall LSmeans for (A) SPV, (B) body sway and (C) qEEG frequency band by dose (SAD study). LSmeans represent the average treatment effect versus placebo across the first 6 h post‐dose along with 95% CIs. Error bars that do not intersect the horizontal dashed line indicate a statistically significant effect versus placebo (P < 0.05). Shape represents the cohort, and colour represents the treatment. LRZ, lorazepam; (T), tablet.
In qEEG, there was a statistically significantly increase in beta power FZ–CZ means, compared with placebo, for 15 mg and all higher doses of PF‐06372865 (Figure 8C). There were also statistically significant decreases in delta power FZ–CZ and theta FZ–CZ. Administration of 2 mg lorazepam elicited statistically significant increases in beta and decreases in alpha and theta.
Further NeuroCart endpoints including saccadic eye movement, smooth pursuit eye movement, adaptive tracking, Visual Verbal Learning Test and subjective effects measured with Bond and Lader visual analogue scale will be reported in another manuscript.
PET
Following p.o. administration of single doses of PF‐06372865 10 and 65 mg, at the time of the first post‐dose PET scan (nominal time post‐dose 1.5 h), the median GABAA RO in the whole brain was 68.6 and 88.9% respectively. At the time of the second post‐dose PET scan (nominal time post‐dose: 24 h), the median GABAA RO in the whole brain was 29.3 and 75% at the PF‐06372865 10 and 65 mg dose levels respectively (Figure 9A). The relationship between GABAA RO in whole brain and plasma PF‐06372865 concentrations can be well described by a simple E max model (Figure 9B). Using this model, the RO max of the whole brain was estimated to be 88.4% (95% CI of 83.9–92.8%), and the corresponding Occ50 was estimated to be 2.4 nM unbound concentration, 95% CI of 1.9–3.0 (8.2 ng·mL−1 total concentration, 95% CI of 6.5–10.4).
Figure 9.
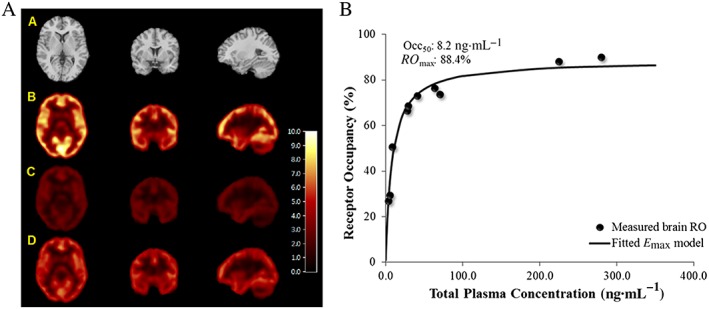
(A) Representative PET and MRI images in healthy human subjects. (A) MRI, (B) baseline PET, (C) first post‐drug PET at 1.5 h (~66% RO) and (D) second post‐drug PET at 24 h (~27% RO). All images were from the same subject who was dosed with 10 mg of PF‐06372865. Images are displayed along three orthogonal views (coronal, transverse and sagittal views from left to right columns). PET images are in V T units (mL blood.cm‐3 tissue). (B) PET RO versus plasma PF‐06372865 concentration relationship in human subjects. The individual dots are the brain GABAA RO measured, while the line is the fitted relationship with an E max model.
Further modelling was conducted to estimate the RO binding in whole brain at α1/α2/α3 subtype receptors. The data were described well by a simple E max model for the receptor binding at each subunit. The RO max was assumed the same for different α subunits and estimated to be 95.6% [relative standard error (RSE): 2.18%] by assuming the ratios of Occ50,α1/Occ50,α2/Occ50 and α3/Occ50,α2 were same as the K i ratios determined in vitro for α1/α2 (0.18/2.92) and α3/α2 (1.06/2.92) respectively. The Occ50,α2 was estimated to be 53.2 ng·mL−1 (RSE: 13.7%), and Occ50,α1 and Occ50,α3 were 3.28 and 19.3 ng·mL−1 respectively.
Discussion
Attrition in drug discovery is high, and increasing our ability to translate preclinical data into success in the clinic is paramount to reducing this. Therefore, in this programme, in which we identified a novel subtype‐selective GABAA PAM, we developed a biomarker‐based approach to enable us to translate our preclinical pharmacological profile into a clinical profile in a disease agnostic way. Our findings indicate that qEEG beta frequency increases are a translatable biomarker for α2/3 subtype‐selective PAMs and that zolpidem drug discrimination is a reliable determination of significant α1 activity in vivo. Clinically, PF‐06372865 was better tolerated than similar ligands tested previously, as an MTD was not identified and the effects on the SPV endpoint were much greater than a 2 mg dose of lorazepam.
Preclinically, PF‐06372865 was a high‐affinity ligand for the BZD site of the GABAA receptor, with functional selectivity, in vitro and in vivo, for receptors containing an α2/3 subunit compared with those containing an α1 subunit. Affinity values indicated binding selectivity for receptors containing an α1 subunit over those containing α2/3/5 subunits; this was not confirmed in functional assays. However, in accordance with receptor theory, we consider the binding K i values to be the translatable parameter. Additionally, unlike functional assays, the binding assays were performed at equilibrium, and also, due to the very low functional activity at receptors containing an α1 subunit, there was a large CI associated with the potency value.
Clinically, PF‐06372865 was well tolerated in the SAD studies (0.04 to 100 mg), and an MTD was not identified. The most frequently reported AEs were dizziness and somnolence, which were recorded as mild and occurred at frequencies greater than placebo at doses about 4 mg and higher. Escalation beyond 100 mg was not possible due to available safety margins based on regulatory toxicological studies (data not shown). Drug exposure was reasonably linear with dose (Table 3), and therefore, the wide therapeutic margin was not a product of poor drug exposure at higher doses. Indeed, the [11C]‐flumazenil PET study demonstrated that the majority of the occupancy curve had been examined with RO at 65 mg being ~75%, which allows us to compare PDs across the entire occupancy–response curve in both rats and humans.
Mechanistic studies using α1‐preferring ligands, such as zolpidem, in humans (de Haas et al., 2010), primates (Licata et al., 2009) and rodents (Bayley et al., 1996), and genetic studies in mice (Rudolph et al., 1999; McKernan et al., 2000) all indicate that α1‐containing receptors mediate motor coordination, balance and sedation. Previously, models such as beam walking or rotarod have been used to estimate these effects (Atack et al., 2006). Nevertheless, humans appear to be highly sensitive to α1‐mediated effects, which are apparent at low levels of RO (<10%), compared with preclinical species. Therefore, we choose not to look at motor deficits in rodents, but instead, we utilized a pharmacological approach and used drug discrimination to determine in vivo α1 receptor activity. The readout of drug discrimination relies on the interoceptive effect the drug produces in the trained animal and is therefore more sensitive to the PD effect on the brain compared with using crude deficits in motor performance. Herein, we have shown that even at full RO PF‐06372865 did not cause generalization to the sedative BZD zolpidem, confirming the minimal α1 activity observed in vitro. In comparison, in clinical studies, the body sway endpoint (assessing postural stability) was the most sensitive PD effect with small, but significant, increases occurring at submilligram doses. This was plateaued by 10 mg and did not significantly increase up to 100 mg despite marked increases in RO across this dose range. PF‐06372865 has a K i at α1‐containing receptors of 0.18 nM, whereas the K i at α2‐containing receptors is 16‐fold lower at 2.92 nM (Table 1). This means that α1‐containing receptors will be occupied preferentially at lower doses, with significant occupancy of α2‐containing receptors occurring at higher doses, whereas the PET study measures total RO (i.e. the sum of α1/2/3/5‐containing receptors). With this differential occupancy in mind, it is clear that the body sway effect is mediated by α1‐containing receptors, because this is the receptor subtype chiefly occupied at submilligram doses and approaches maximum occupancy at <10 mg. The overall effect size was less than that caused by a 2 mg dose of lorazepam (which is predicted to result in RO of approximately 11%; Lingford‐Hughes et al., 2005), indicating that PF‐06372865 has considerably less impairing effect on balance than a non‐selective BZD.
The NeuroCart battery also included tests that demonstrate desired pharmacology, foremost of which is reduction in SPV, which is thought to be closely related to the anxiolytic properties of BZDs (de Visser et al., 2003). The decrease in SPV observed with PF‐06372865 was approximately twice that of 2 mg lorazepam at the 65 or 100 mg doses (Figure 8A). This magnitude of SPV reduction has not been observed before with either non‐selective BZDs or any of the other subtype‐selective PAMs, which all exhibited a decrease in SPV less than or equivalent to that of 2 mg lorazepam at the maximum dose tested. The underlying reason behind the large effect on SPV caused by PF‐06372865 is likely related to both the high levels of α2 RO that can be achieved without dose‐limiting adverse events and the relatively high level of efficacy this compound possesses at α2‐containing receptors. Supporting Information Table S3 indicates that the peak potentiation in the QPatch assay for α2‐containing receptors is 134% for PF‐06372865 compared with 53% for TPA023 and 75% for TPA023B. Additionally, the data suggest that SPV effects are mediated across a wide range of α2 occupancies, as the SPV curve only flattens off around the 65 mg dose in line with the RO curve from the PET study. Thus, in the TPA023 and TPA023B studies, the dose‐limited adverse events probably capped the maximum possible RO well below that achieved in the current study. The RO for NS11821 has not been published, but the authors themselves comment that brain penetration appears to have been limited, which suggests the full dose–occupancy curve is yet to be explored (Zuiker et al., 2016). It is less clear why PF‐06372865 had a much larger effect size on SPV than AZD7325 because it achieved similarly high levels of RO compared with PF‐06372865 (Chen et al., 2014). It is possible that the greater differences in α1 versus α2 affinity (30‐fold vs. 16‐fold, Supporting Information Table S4) and the lower α2 efficacy (70 vs. 134%) explain the differences, but equally, other factors may play a role.
We also assessed the effect of PF‐06372865 on qEEG in both rats and humans. Recently, Christian and colleagues performed a comprehensive analysis of the effects of zolpidem, lorazepam, TPA023 (Atack et al., 2006) and TP003 (Dias et al., 2005) as well as AZD7325 and six other AstraZeneca compounds (Christian et al., 2015). They conclude that it is indeed the α2/3 activity that correlates with an increase in beta and gamma activity. This is in agreement with the significant increase in both beta and gamma power preclinically and significant increase in beta power clinically that we observed with PF‐06372865. The current pharmacological tools do not allow the relative contributions of α2 and α3 subunits to be dissociated; however, previous work with diazepam‐insensitive mutant mice indicates that the α2 subunit is primarily responsible for modulating these effects (Kopp et al., 2003; 2004). The clinical data obtained with PF‐06372865 support the use of qEEG as a translatable biomarker for GABAA receptors; the preclinical changes of alpha, beta, theta and delta power observed in rat qEEG were correlated with similar changes in the qEEG in the human subjects.
Previously, the Merck compounds, MK‐0343, TPA023 and TPA023B all exhibited dose‐limiting adverse events in clinical trials at receptor occupancies equivalent to approximately 10% (Atack et al., 2011b), 65% (Atack et al., 2010) and 65% (Van Laere et al., 2008) respectively. The α1 activity of MK‐0343 (57% in the QPatch assay; Supporting Information Table S3) was probably responsible for the dose‐limiting sedation observed in the clinic (Atack et al., 2011b). However, PF‐06372865, TPA023 and TPA023B possess similar α1 efficacy (21, 29 and 17%, respectively), and so, it is difficult to see how α1 efficacy could explain the difference in tolerability; likewise, the efficacy at α2‐, α3‐ or α5‐containing receptors does not suggest an obvious reason. It is possible that TPA023 and TPA023B share an unknown off‐target liability, but this seems highly unlikely as they are from different chemical series. Additionally, the observed effects are seen at low nM plasma concentrations and were all consistent with GABAA PAMs suggesting that GABA potentiation is the most likely cause. Currently, we have an incomplete understanding of the effects of PAMs on GABAA receptors at the biophysical level, as the in vitro assessment of activity was in recombinant cell lines and the level of allosteric modulation determined by the peak potentiation of a low concentration of GABA. In contrast, in vivo, GABA concentrations are very high in the synapse following action potential‐induced exocytosis of neurotransmitter. Hence, the chloride current potentiation is more related to changes in AUC of the IPSP than a change in the peak current. The rate at which the IPSP returns to baseline will depend on several factors, which GABAA PAMs could affect, including changes in the affinity of GABA for the receptor, the rate of desensitization or changes in channel gating kinetics such as opening probability or opening time. While studies of the effects of classical, non‐selective BZDs (e.g. lorazepam) have been conducted in detail (MacDonald and Twyman, 1992; McKernan and Whiting, 1996), such studies have not been published for these subtype‐selective compounds and could yield a more in‐depth understanding of why similar PAMs possess different clinical profiles.
In conclusion, we have demonstrated that PF‐06372865 has a pharmacological profile that translates well from preclinical assays to human studies. PF‐06372865 is well tolerated across the entire dose–occupancy range and possesses the largest functional effect on α2‐related PD endpoints for this class of compound, thus making it an ideal tool to further study the potential benefits of subtype‐selective GABAA PAMs.
Author contributions
R.G., G.V.A., J.K., L.C., A.R.B., C.S., C.W., C.H., R.M.O., A.P., R.L.F., E.D.M., G.F. and P.D. performed the research. S.A.N., R.G., A.M., C.W., R.M.O., L.C., E.D.M., M.W., D.G., J.V.G., D.S.R., P.D. and R.P.B. designed the research studies. R.G., J.K., L.C., A.R.B., A.M., C.H., S.A.N., C.W., R.M.O., D.S.R., R.L.F., A.P., R.Q., M.W., D.G. and P.D. analysed the data. S.A.N., R.G., D.S.R. and P.D. wrote the paper.
Conflict of interest
These studies were sponsored by Pfizer. S.A.N., R.G., J.H., L.C., A.R.B., C.S., A.M., C.W., C.H., R.M.O., A.P., R.L.F., L.C., R.Q., G.F., M.W., D.G., D.S.R., P.D. and R.P.B. are or were employees of Pfizer at the time of this research and may own stock in the company. G.V.A. and J.V.G are employees of the Centre of Human Drug Research. E.D.M. is an employee of Yale University. There are no other known conflicts of interest to declare.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Trial Design of SAD.
Figure S2 Disposition of subjects for the SAD and PET clinical studies.
Table S1 Demographic Characteristics of SAD Study (Cohorts 1–4).
Table S2 Incidence of Treatment‐Emergent Adverse Events, All Causality (Treatment‐Related) [Limited to AEs reported in 2 or more subjects].
Table S3 Functional activity of clinical comparators.
Table S4 In vitro binding affinity comparisons of clinical candidates.
Acknowledgements
The authors thank the healthy volunteers who participated in these trials. The authors acknowledge the Radiochemistry and the Imaging Team of the Yale PET Center for performing the imaging experiments. The authors also thank John Hutchison, Santiago Arroyo and Ruth McKernan for their support and critical review of the PF‐06372865 programme. Pfizer funded this body of work.
Nickolls, S. A. , Gurrell, R. , van Amerongen, G. , Kammonen, J. , Cao, L. , Brown, A. R. , Stead, C. , Mead, A. , Watson, C. , Hsu, C. , Owen, R. M. , Pike, A. , Fish, R. L. , Chen, L. , Qiu, R. , Morris, E. D. , Feng, G. , Whitlock, M. , Gorman, D. , van Gerven, J. , Reynolds, D. S. , Dua, P. , and Butt, R. P. (2018) Pharmacology in translation: the preclinical and early clinical profile of the novel α2/3 functionally selective GABAA receptor positive allosteric modulator PF‐06372865. British Journal of Pharmacology, 175: 708–725. doi: 10.1111/bph.14119.
Trial numbers: NCT01951144 and NCT02138500.
References
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atack JR (2009). Subtype‐selective GABAA receptor modulation yields a novel pharmacological profile: the design and development of TPA023. Adv Pharmacol (San Diego, Calif) 57: 137–185. [DOI] [PubMed] [Google Scholar]
- Atack JR, Eng WS, Gibson RE, Ryan C, Francis B, Sohal B et al (2009). The plasma-occupancy relationship of the novel GABAA receptor benzodiazepine site ligand, alpha5IA, is similar in rats and primates. Br J Pharmacol 157: 796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atack JR, Hallett DJ, Tye S, Wafford KA, Ryan C, Sanabria‐Bohorquez SM et al (2011a). Preclinical and clinical pharmacology of TPA023B, a GABAA receptor α2/α3 subtype‐selective partial agonist. J Psychopharmacol (Oxford, England) 25: 329–344. [DOI] [PubMed] [Google Scholar]
- Atack JR, Wafford KA, Street LJ, Dawson GR, Tye S, Van Laere K et al (2011b). MRK‐409 (MK‐0343), a GABAA receptor subtype‐selective partial agonist, is a non‐sedating anxiolytic in preclinical species but causes sedation in humans. J Psychopharmacol (Oxford, England) 25: 314–328. [DOI] [PubMed] [Google Scholar]
- Atack JR, Wafford KA, Tye SJ, Cook SM, Sohal B, Pike A et al (2006). TPA023 [7‐(1,1‐dimethylethyl)‐6‐(2‐ethyl‐2H‐1,2,4‐triazol‐3‐ylmethoxy)‐3‐(2‐fluorophenyl)‐1,2,4‐triazolo[4,3‐b]pyridazine], an agonist selective for α2‐ and α3‐containing GABAA receptors, is a nonsedating anxiolytic in rodents and primates. J Pharmacol Exp Ther 316: 410–422. [DOI] [PubMed] [Google Scholar]
- Atack JR, Wong DF, Fryer TD, Ryan C, Sanabria S, Zhou Y et al (2010). Benzodiazepine binding site occupancy by the novel GABAA receptor subtype‐selective drug 7‐(1,1‐dimethylethyl)‐6‐(2‐ethyl‐2H‐1,2,4‐triazol‐3‐ylmethoxy)‐3‐(2‐fluorophenyl)‐1,2,4‐triazolo[4,3‐b]pyridazine (TPA023) in rats, primates, and humans. J Pharmacol Exp Ther 332: 17–25. [DOI] [PubMed] [Google Scholar]
- Bayley PJ, Bentley GD, Jackson A, Williamson D, Dawson GR (1996). Comparison of benzodiazepine (BZ) receptor agonists in two rodent activity tests. J Psychopharmacol (Oxford, England) 10: 206–213. [DOI] [PubMed] [Google Scholar]
- Chen X, Jacobs G, de Kam M, Jaeger J, Lappalainen J, Maruff P et al (2014). The central nervous system effects of the partial GABA‐Aα2,3‐selective receptor modulator AZD7325 in comparison with lorazepam in healthy males. Br J Clin Pharmacol 78: 1298–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH (1973). Relationship between the inhibition constant (K I) and the concentration of inhibitor which causes 50 per cent inhibition (I 50) of an enzymatic reaction. Biochem Pharmacol 22: 3099–3108. [DOI] [PubMed] [Google Scholar]
- Christian EP, Snyder DH, Song W, Gurley DA, Smolka J, Maier DL et al (2015). EEG‐β/γ spectral power elevation in rat: a translatable biomarker elicited by GABAAα2/3‐positive allosteric modulators at nonsedating anxiolytic doses. J Neurophysiol 113: 116–131. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson GR, Maubach KA, Collinson N, Cobain M, Everitt BJ, MacLeod AM et al (2006). An inverse agonist selective for α5 subunit‐containing GABAA receptors enhances cognition. J Pharmacol Exp Ther 316: 1335–1345. [DOI] [PubMed] [Google Scholar]
- Dias R, Sheppard WF, Fradley RL, Garrett EM, Stanley JL, Tye SJ et al (2005). Evidence for a significant role of α3‐containing GABAA receptors in mediating the anxiolytic effects of benzodiazepines. J Neurosci 25: 10682–10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin E, Liu J, Rudolph U (2012). α2‐Containing GABAA receptors: a target for the development of novel treatment strategies for CNS disorders. Pharmacol Ther 136: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haas SL, de Visser SJ, van der Post JP, de Smet M, Schoemaker RC, Rijnbeek B et al (2007). Pharmacodynamic and pharmacokinetic effects of TPA023, a GABAA α2,3 subtype‐selective agonist, compared to lorazepam and placebo in healthy volunteers. J Psychopharmacol (Oxford, England) 21: 374–383. [DOI] [PubMed] [Google Scholar]
- de Haas SL, de Visser SJ, van der Post JP, Schoemaker RC, van Dyck K, Murphy MG et al (2008). Pharmacodynamic and pharmacokinetic effects of MK‐0343, a GABAA α2,3 subtype selective agonist, compared to lorazepam and placebo in healthy male volunteers. J Psychopharmacol (Oxford, England) 22: 24–32. [DOI] [PubMed] [Google Scholar]
- de Haas SL, Franson KL, Schmitt JA, Cohen AF, Fau JB, Dubruc C et al (2009). The pharmacokinetic and pharmacodynamic effects of SL65.1498, a GABA‐A α2,3 selective agonist, in comparison with lorazepam in healthy volunteers. J Psychopharmacol (Oxford, England) 23: 625–632. [DOI] [PubMed] [Google Scholar]
- de Haas SL, Schoemaker RC, van Gerven JM, Hoever P, Cohen AF, Dingemanse J (2010). Pharmacokinetics, pharmacodynamics and the pharmacokinetic/pharmacodynamic relationship of zolpidem in healthy subjects. J Psychopharmacol (Oxford, England) 24: 1619–1629. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981). Improved patch‐clamp techniques for high‐resolution current recording from cells and cell‐free membrane patches. Pflugers Arch 391: 85–100. [DOI] [PubMed] [Google Scholar]
- Ivarsson M, Paterson LM, Hutson PH (2005). Antidepressants and REM sleep in Wistar–Kyoto and Sprague–Dawley rats. Eur J Pharmacol 522: 63–71. [DOI] [PubMed] [Google Scholar]
- Kantor S, Jakus R, Bodizs R, Halasz P, Bagdy G (2002). Acute and long‐term effects of the 5‐HT2 receptor antagonist ritanserin on EEG power spectra, motor activity, and sleep: changes at the light–dark phase shift. Brain Res 943: 105–111. [DOI] [PubMed] [Google Scholar]
- Kenward MG, Roger JH (2010). The use of baseline covariates in crossover studies. Biostatistics (Oxford, England) 11 (1): 1–7. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, Ahmadi S et al (2008). Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature 451: 330–334. [DOI] [PubMed] [Google Scholar]
- Kopp C, Rudolph U, Keist R, Tobler I (2003). Diazepam‐induced changes on sleep and the EEG spectrum in mice: role of the α3‐GABAA receptor subtype. Eur J Neurosci 17: 2226–2230. [DOI] [PubMed] [Google Scholar]
- Kopp C, Rudolph U, Low K, Tobler I (2004). Modulation of rhythmic brain activity by diazepam: GABAA receptor subtype and state specificity. Proc Natl Acad Sci U S A 101: 3674–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen NA, Bartenstein PA, Lammertsma AA et al (1995). Benzodiazepine receptor quantification in vivo in humans using [11C]flumazenil and PET: application of the steady‐state principle. J Cereb Blood Flow Metab 15 (1): 152–165. [DOI] [PubMed] [Google Scholar]
- Licata SC, Platt DM, Cook JM, Van Linn ML, Rowlett JK (2009). Contribution of α1 subunit‐containing γ‐aminobutyric acidA (GABAA) receptors to motor‐impairing effects of benzodiazepines in squirrel monkeys. Psychopharmacology (Berl) 203: 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingford‐Hughes A, Wilson SJ, Feeney A, Grasby PG, Nutt DJ (2005). A proof‐of‐concept study using [11C]flumazenil PET to demonstrate that pagoclone is a partial agonist. Psychopharmacology (Berl) 180: 789–791. [DOI] [PubMed] [Google Scholar]
- MacDonald RL, Twyman RE (1992). Kinetic properties and regulation of GABAA receptor channels. Ion Channels 3: 315–343. [DOI] [PubMed] [Google Scholar]
- Mandema JW, Tuk B, van Steveninck AL, Breimer DD, Cohen AF, Danhof M (1992). Pharmacokinetic–pharmacodynamic modeling of the central nervous system effects of midazolam and its main metabolite α‐hydroxymidazolam in healthy volunteers. Clin Pharmacol Ther 51: 715–728. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR et al (2000). Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nat Neurosci 3: 587–592. [DOI] [PubMed] [Google Scholar]
- McKernan RM, Whiting PJ (1996). Which GABAA‐receptor subtypes really occur in the brain? Trends Neurosci 19: 139–143. [DOI] [PubMed] [Google Scholar]
- Miranda JA, Stanley P, Gore K, Turner J, Dias R, Rees H (2014). A preclinical physiological assay to test modulation of knee joint pain in the spinal cord: effects of oxycodone and naproxen. PLoS One 9: e106108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan P, Van Der Graaf PH, Arrowsmith J, Feltner DE, Drummond KS, Wegner CD et al (2012). Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving phase II survival. Drug Discov Today 17: 419–424. [DOI] [PubMed] [Google Scholar]
- Nickolls S, Mace H, Fish R, Edye M, Gurrell R, Ivarsson M et al (2011). A comparison of the α2/3/5 selective positive allosteric modulators L‐838,417 and TPA023 in preclinical models of inflammatory and neuropathic pain. Adv Pharm Sci 2011: 608912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoto K, Owen RM, Pryde DC, Watson CAL, Takeuchi M (2014) Imidazopyridazine derivatives as GABAA receptor modulators. PCT. Int. Appl. WO2014091368.
- Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, Fritschy JM et al (1999). Benzodiazepine actions mediated by specific γ‐aminobutyric acidA receptor subtypes. Nature 401: 796–800. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Knoflach F (2011). Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat Rev Drug Discov 10: 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steveninck AL, Gieschke R, Schoemaker RC, Roncari G, Tuk B, Pieters MS et al (1996). Pharmacokinetic and pharmacodynamic interactions of bretazenil and diazepam with alcohol. Br J Clin Pharmacol 41: 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steveninck AL, Schoemaker HC, Pieters MS, Kroon R, Breimer DD, Cohen AF (1991). A comparison of the sensitivities of adaptive tracking, eye movement analysis and visual analog lines to the effects of incremental doses of temazepam in healthy volunteers. Clin Pharmacol Ther 50: 172–180. [DOI] [PubMed] [Google Scholar]
- van Steveninck AL, van Berckel BN, Schoemaker RC, Breimer DD, van Gerven JM, Cohen AF (1999). The sensitivity of pharmacodynamic tests for the central nervous system effects of drugs on the effects of sleep deprivation. J Psychopharmacol (Oxford, England) 13: 10–17. [DOI] [PubMed] [Google Scholar]
- van Steveninck AL, Verver S, Schoemaker HC, Pieters MS, Kroon R, Breimer DD et al (1992). Effects of temazepam on saccadic eye movements: concentration–effect relationships in individual volunteers. Clin Pharmacol Ther 52: 402–408. [DOI] [PubMed] [Google Scholar]
- Sullivan SK, Petroski RE, Verge G, Gross RS, Foster AC, Grigoriadis DE (2004). Characterization of the interaction of indiplon, a novel pyrazolopyrimidine sedative‐hypnotic, with the GABAA receptor. J Pharmacol Exp Ther 311: 537–546. [DOI] [PubMed] [Google Scholar]
- Tzourio‐Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N et al (2002). Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single‐subject brain. Neuroimage 15: 273–289. [DOI] [PubMed] [Google Scholar]
- Van Laere K, Bormans G, Sanabria‐Bohorquez SM, de Groot T, Dupont P, De Lepeleire I et al (2008). In vivo characterization and dynamic receptor occupancy imaging of TPA023B, an α2/α3/α5 subtype selective γ‐aminobutyric acid – a partial agonist. Biol Psychiatry 64: 153–161. [DOI] [PubMed] [Google Scholar]
- de Visser SJ, van der Post JP, de Waal PP, Cornet F, Cohen AF, van Gerven JM (2003). Biomarkers for the effects of benzodiazepines in healthy volunteers. Br J Clin Pharmacol 55: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T, Waldron G, Addley N, Knight T, Staveley L, Barrow M et al (2009). Translation of in vitro CB1 agonist potency to in vivo CNS effects, the challenge in designing selective CB2 agonists for pain. Soc Neurosci Abstr . [Google Scholar]
- Zuiker RG, Chen X, Osterberg O, Mirza NR, Muglia P, de Kam M et al (2016). NS11821, a partial subtype‐selective GABAA agonist, elicits selective effects on the central nervous system in randomized controlled trial with healthy subjects. J Psychopharmacol (Oxford, England) 30: 253–262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Trial Design of SAD.
Figure S2 Disposition of subjects for the SAD and PET clinical studies.
Table S1 Demographic Characteristics of SAD Study (Cohorts 1–4).
Table S2 Incidence of Treatment‐Emergent Adverse Events, All Causality (Treatment‐Related) [Limited to AEs reported in 2 or more subjects].
Table S3 Functional activity of clinical comparators.
Table S4 In vitro binding affinity comparisons of clinical candidates.