

Abstract
Background and Purpose
Translation of non‐clinical markers of delayed ventricular repolarization to clinical prolongation of the QT interval corrected for heart rate (QTc) (a biomarker for torsades de pointes proarrhythmia) remains an issue in drug discovery and regulatory evaluations. We retrospectively analysed 150 drug applications in a US Food and Drug Administration database to determine the utility of established non‐clinical in vitro IKr current human ether‐à‐go‐go‐related gene (hERG), action potential duration (APD) and in vivo (QTc) repolarization assays to detect and predict clinical QTc prolongation.
Experimental Approach
The predictive performance of three non‐clinical assays was compared with clinical thorough QT study outcomes based on free clinical plasma drug concentrations using sensitivity and specificity, receiver operating characteristic (ROC) curves, positive (PPVs) and negative predictive values (NPVs) and likelihood ratios (LRs).
Key Results
Non‐clinical assays demonstrated robust specificity (high true negative rate) but poor sensitivity (low true positive rate) for clinical QTc prolongation at low‐intermediate (1×–30×) clinical exposure multiples. The QTc assay provided the most robust PPVs and NPVs (ability to predict clinical QTc prolongation). ROC curves (overall test accuracy) and LRs (ability to influence post‐test probabilities) demonstrated overall marginal performance for hERG and QTc assays (best at 30× exposures), while the APD assay demonstrated minimal value.
Conclusions and Implications
The predictive value of hERG, APD and QTc assays varies, with drug concentrations strongly affecting translational performance. While useful in guiding preclinical candidates without clinical QT prolongation, hERG and QTc repolarization assays provide greater value compared with the APD assay.
Abbreviations
- (ΔΔ)QTc
baseline and vehicle changes in duration of the QTc interval corrected for heart rate
- APD
action potential duration
- CRC
clinical reference concentration
- FDA
Food and Drug Administration
- hERG
human ether‐à‐go‐go‐related gene
- ICH
International Conference on Harmonization
- IRT
Interdisciplinary Review Team
- LR
likelihood ratio
- NPV
negative predictive value
- PPV
positive predictive value
- QTc
duration of the QT interval corrected for heart rate
- ROC
receiver operating characteristic
- TdP
torsade de pointes
- TQT
thorough QT
Introduction
Drug‐induced delayed ventricular repolarization, manifested as prolongation of the QT interval on the ECG, remains a serious safety issue in drug discovery and development because of its link to a rare but potentially lethal arrhythmia, known as torsade de pointes (TdP, Kannankeril et al., 2010). The need to detect and prevent this proarrhythmic liability has led to the development and adoption of multiple non‐clinical and clinical approaches during drug discovery and development to assess delayed repolarization, a surrogate marker of proarrhythmic risk.
Regarding non‐clinical evaluations, it is well appreciated that inhibition of the delayed rectifier current I Kr that flows through an ion channel encoded by human ether‐à‐go‐go‐related gene (hERG, also known as KCNH2, Kv11.1) can delay ventricular repolarization and prolong the QT interval. This current plays a prominent role in defining ventricular repolarization (Rampe and Brown, 2013), and because of the channel structure and drug‐binding characteristics, is frequently used early in drug discovery to screen novel chemical entities prior to testing in vivo (Pollard et al., 2010; Valentin et al., 2010). However, it is also appreciated that ventricular repolarization represents the integrated response of several cardiac currents (O'Hara et al., 2011) and that drugs can reduce many different cardiac currents to generate responses that are not easily predicted based on hERG block alone (Kramer et al., 2013). Another recognized in vitro experimental approach involves monitoring repolarization, measured as prolongation of the action potential duration (APD), with ventricular tissues and cells from various species (Lawrence et al., 2008). It is also possible to directly evaluate QT interval prolongation in larger mammalian species in vivo (Leishman et al., 2012). The non‐clinical International Conference on Harmonization (ICH) S7B guideline (Anonymous, 2005b) identified the I Kr/hERG current assay and the in vivo evaluation of duration of the QT interval corrected for heart rate (QTc) interval as key components to be considered in regulatory submissions for assessing the potential of a drug to delay ventricular repolarization.
Clinically, the ICH E14 guidance (Anonymous, 2005a) describes a rigorous approach to assess the potential of a drug to delay ventricular repolarization as defined in so‐called thorough QT (TQT) studies. Arguably, TQT studies have become the present ‘gold standard’ surrogate marker for detecting drug‐induced delayed repolarization. Despite widespread acceptance of many non‐clinical approaches used to detect or predict clinical QTc prolongation, only a few studies have characterized the utility and performance of these non‐clinical approaches. Such studies are limited in that they have studied small numbers of drugs (Wallis, 2010) or included only one of the three widely used non‐clinical approaches (Gintant, 2011; Ewart et al., 2014) compared with clinical findings.
Since implementation of the ICH S7B and E14 guidance documents in 2005, numerous drug dossiers submitted to the Food and Drug Administration (FDA) for regulatory review in support of Investigational New Drug and New Drug Applications have typically included non‐clinical repolarization studies and clinical TQT study results. This extensive regulatory data set (which included non‐clinical repolarization and clinical TQT study results) provided an opportunity to rigorously compare non‐clinical and clinical assessments of delayed repolarization. Towards this goal, the Health and Environmental Sciences Institute Proarrhythmia Working Group collaborated closely with the FDA as part of a public–private–government consortium described previously (Trepakova et al., 2009).
Diagnostic and prognostic performance of these three non‐clinical assays was characterized based on sensitivity and specificity, concordance, positive (PPVs) and negative predictive values (NPVs), receiver operating characteristic (ROC) curves and likelihood ratios (LRs), all of which are widely used in biomarker research.
Methods
Database description
The database consisted of 150 Investigational New Drug and New Drug Applications submitted between 1 March 2006 and 7 July 2012. Inclusion criteria included at least one non‐clinical assessment of repolarization, and either clinical TQT study results or clinical QT prolongation in either non‐TQT studies or FDA QT Interdisciplinary Review Team (IRT) review summaries. QT‐prolonging drugs without TQT studies were included in the database as assay sensitivity was demonstrated by positive clinical findings. Biological products or combination drugs were excluded from this analysis. Out of the 150 drugs in the database, 13.3%, of drugs came from the FDA neurology division, 12.0% from antivirals, 11.3% from metabolic and endocrine and 10.7% from cardiovascular and renal. The remaining divisions represent less than 10% each and are shown in the Supporting Information. Additional characteristics of the database along with specific details regarding each assay are provided in the Supporting Information Table S2.
The following information was collected from each submission: FDA application number, approval status and division, clinical QT correction methodology, drug plasma protein binding in relevant species, study doses, study design, study initiation dates and study results. When multiple studies were reported for the same non‐clinical assay (i.e. preliminary vs. definitive good laboratory practice hERG assays), only good laboratory practice study results were included. Metabolite data were not included for any of the compounds in this analysis. Drug identities were blinded, and data were anonymized to preserve proprietary information to consortium colleagues not employed by the FDA. This allowed researchers to access the data, enabling an unbiased comparison of matched concentration–response effects from using composite graphs (plotting non‐clinical and clinical data) and associated concordance tables while remaining blinded to drug identities.
This retrospective study evaluated in vitro and in vivo non‐clinical experimental data previously submitted to the FDA to support regulatory approvals from 2006 to 2012, originated from different laboratories across many years. Some data may not adhere to current standards set out in the Declaration of Transparency and Scientific Rigour of the BJP but are considered to represent accepted best practices at the time of submissions.
Comparing assay results based on free drug concentrations
Results from non‐clinical studies and clinical TQT study findings were compared based on the clinical reference concentration (CRC), defined as the highest mean free Cmax achieved for TQT‐negative drugs (−TQT studies) or the lowest mean free Cmax showing QT prolongation (+TQT studies). Thus, non‐clinical concentration–response curves were constructed based on exposure multiples relative to the CRC. Nominal drug concentrations were used to characterize hERG and APD assay results (conducted in the absence of plasma proteins). When only a range of concentrations was provided without specific test concentrations (e.g. 1–100 μM), minimum and maximum test concentrations were used. For in vivo QTc and clinical TQT studies, free (unbound) maximum drug concentrations in plasma, or plasma concentrations near time at which plasma drug concentration is maximal were used. When Cmax measurements were not available from in vivo QTc assays, estimations of Cmax were obtained from pharmacokinetic or toxicokinetic studies in the same species using the same or similar dose levels. If doses differed between the in vivo QTc and pharmacokinetic/toxicokinetic studies, Cmax was estimated by assuming a linear dose–pharmacokinetic relationship. Free drug plasma concentrations from in vivo QTc and clinical TQT studies were based on measured plasma concentrations and adjusted for plasma protein binding in the relevant species (assuming a linear relationship between binding values for non‐clinical species and human studies). An arithmetic mean was used when a range of plasma protein binding values was provided by the sponsor.
Dichotomous endpoints for assays and concordance assessments
For the hERG assay, a positive response was defined as concentrations ≥IC50 value for I Kr block (half maximal inhibitory concentration). While it has been suggested that IC10 or IC20 values for I Kr block may provide a more sensitive measure for predicting QTc effects (Wallis, 2010), these were not used as they are subject to high variability and were not typically reported by sponsors.
For the APD assay, the primary endpoint was the absolute change in APD at 90% repolarization from baseline control values, and a positive response was defined by the sponsor's claim of a significant drug‐related effect. If a drug's response was expressed as ‘no effect’ instead of a numerical value, the response was marked as 0%.
The maximum, baseline‐adjusted and vehicle‐adjusted (ΔΔ)QTc (ms) was used as the primary response endpoint for human TQT studies and maximum, baseline‐adjusted (Δ)QTc (ms) for in vivo QTc studies (Anonymous, 2005a; Darpo, 2010; Stockbridge et al., 2012; Darpo, 2015). A positive response in the in vivo QTc assay was defined by the sponsor's claim of a significant drug‐related effect. For clinical TQT studies, a positive response (+TQT study) was defined using ICH E14 criteria. Specifically, a positive effect was one where either the upper 95% confidence interval or two‐sided 90% of mean (ΔΔ)QTc ≥ 10 ms (Anonymous, 2005a; Garnett et al., 2012; Stockbridge et al., 2012). The FDA QT‐IRT analysed TQT study results and determined the most appropriate correction methodology based on statistical outcomes. The FDA QT‐IRT most often used the Fridericia correction method for QTc values, whereas sponsors used Fridericia, individual or sponsor‐specific correction methods. In some cases, exposure–response modelling was used to interpret equivocal findings (Garnett et al., 2008).
For each drug, a standardized composite exposure–response template was constructed to display study results based on multiples of the free CRC (Figure 1). All assay results were plotted with SEMs (non‐clinical) or confidence intervals (clinical) applied to responses where appropriate. This template supported construction of a summary concordance table (Figure 1 lower panel) Concordance was evaluated by comparing positive or negative results in non‐clinical studies to clinical TQT study outcomes at CRC multiples ranging from 0.1× to 1000×. For example, if the TQT study result was positive at the 1× CRC, a positive hERG study finding between 10× and 30× multiple was considered concordant at multiples of 30× and higher and discordant (false negative) at concentration multiples of 10× and less (Figure 1). As a second example, if a TQT study did not show QTc prolongation at the CRC but a significant increase in QTc was reported at the highest dose of the in vivo QTc assay (i.e. at 30× of the CRC), then the in vivo QTc assay was considered concordant (true negative) at concentration multiples of 10× and less and discordant (false positive) at concentration multiples of 30× and above.
Figure 1.
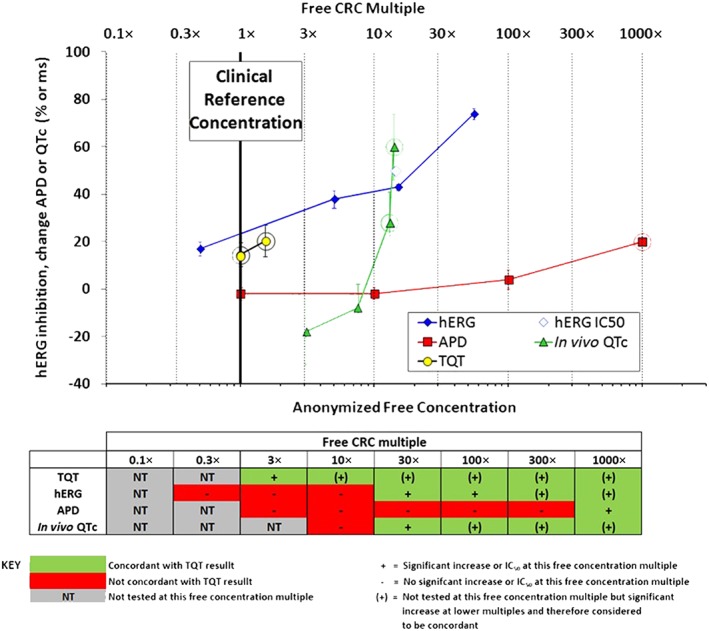
Example of the format for evaluating assay performance. Upper panel: for each drug, concentration–response data from the non‐clinical assays and clinical TQT study were plotted as means ± SEM (non‐clinical data) or confidence intervals (clinical data). The CRC (bold vertical line) was defined from the TQT study as either the highest free (unbound) maximum drug concentration (Cmax,free) tested for TQT‐negative drugs or the lowest Cmax,free showing QT prolongation for TQT‐positive drugs. The CRC was set as the 1× multiple upon which all comparisons were made. Dotted vertical lines indicate multiples from the CRC. Rings around non‐clinical data results indicate concentration multiples at which positive effects occurred. Rings around TQT study results indicate TQT‐positive findings as defined by the FDA IRT. Lower panel: concordance was evaluated by comparing positive or negative results in non‐clinical studies with the TQT study outcome based on free CRC multiples. The green colour code in the table signifies concordance with TQT study results, red indicates discordance and grey (NT) indicates concentrations below the CRC that were not tested.
Evaluating sensitivity, specificity, concordance, positive and negative predictive values and likelihood ratios
The sensitivity, specificity and overall concordance of hERG, APD and in vivo QTc assays were calculated across CRC multiples. Sensitivity represents the proportion of drugs testing positive in the TQT that were correctly identified in the non‐clinical assays. Specificity represents the proportion of drugs testing negative in the TQT that were correctly identified in the non‐clinical assays (Altman and Bland, 1994a; Loong, 2003). The concordance defines how well model results agree with clinical outcomes. Equations used for calculating all performance parameters are summarized in the Supporting Information.
Predictive values are useful in describing the ability to prospectively describe a clinical result based on non‐clinical findings. PPVs and NPVs were calculated at multiples of the CRC to express the probability that a non‐clinical model correctly predicted the outcome of a clinical TQT study. As PPV and NPV are dependent on the prevalence of true positive results (Altman and Bland, 1994b; Loong, 2003), their values were adjusted based on the proportion of positive clinical TQT studies in the complete data set (29%, see below).
LRs were also calculated at multiples of the CRC. LRs characterize the ability of an assay to improve the probability of a prediction or diagnosis, thus providing a meaningful influence on test accuracy. LRs are independent of the prevalence of the measured event, a particularly important benefit for low‐incidence events.
Receiver operating characteristic curve analysis and Youden's J statistic
To assess the predictive accuracy of the non‐clinical assays, ROC curves were generated by plotting sensitivity against (1‐specificity) at the different CRC exposure multiples tested (Hanley and McNeil, 1982). Quantitative measures of ROC curve were assessed as the AUC calculated using SAS statistical software (SAS, 2008). The accuracy of a diagnostic test is defined as excellent (0.90–1.0), good (0.80–0.90), fair (0.70–0.80), poor (0.60–0.70) or fail (0.50–0.60) (Vaidya et al., 2010). Youden's J statistic was calculated to evaluate the cut‐off point that optimizes sensitivity and specificity as the maximum value (sensitivity + 1‐specificity) at different exposure multiples (Schisterman et al., 2005).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Concentration ranges tested in non‐clinical assays
There was a wide exposure range for non‐clinical studies relative to clinical exposures, ranging from less than 0.001× to more than 1 000 000× the free CRC. In general, concentrations tested in in vitro assays tended to be higher than the concentration achieved in TQT studies and covered an exposure range up to 1000× the CRC. In contrast, in vivo QTc assays were generally conducted at lower exposure multiples compared with in vitro assays and covered exposures up to 100× the CRC (Figure 2). In a small number of studies (5, 0 and 17 for hERG, APD and in vivo QTc, respectively), the highest non‐clinical concentration explored was below the CRC.
Figure 2.
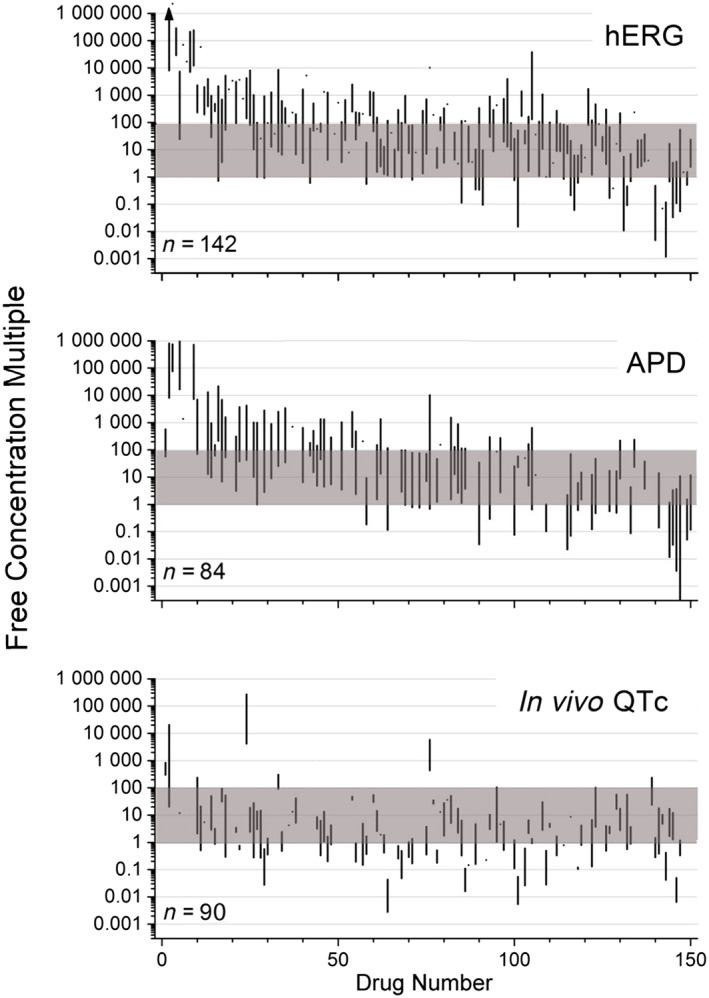
A comparison of nominal free drug concentrations tested in non‐clinical (hERG, APD and in vivo QTc) assays compared with CRCs obtained in TQT studies. Ranges of non‐clinical concentrations (represented as vertical lines) are plotted based on multiples of CRC values for each drug. The shaded area represents 1×–100× multiples of clinical values. Drugs are arranged (left to right) based on increasing clinical exposures in TQT studies. The n‐size shown in each panel represents the number of drugs evaluated in that assay (from a total of 150 drugs evaluated). The arrow (hERG assay, upper left) indicates a maximum value of 10 000 000 attained for one drug.
Sensitivity, specificity and concordance analysis
Sensitivity, specificity and concordance values for the hERG, APD and in vivo QTc assays are illustrated in Figure 3. Assay sensitivity was low (0–0.33) at lower multiples of the free CRC (1×–10×). Sensitivity increased (to 0.67–0.80) with higher exposure multiples for the hERG and in vivo QTc assays, while remaining consistently low (≤0.53) for the APD assay. In contrast, specificity of all assays was consistently high across all free CRC multiples, with highest values (0.89–1.0) at up to 30× multiples and slight decreases at higher multiples. The highest concordance of each individual non‐clinical assay occurred at a 30× multiple of the free CRC.
Figure 3.
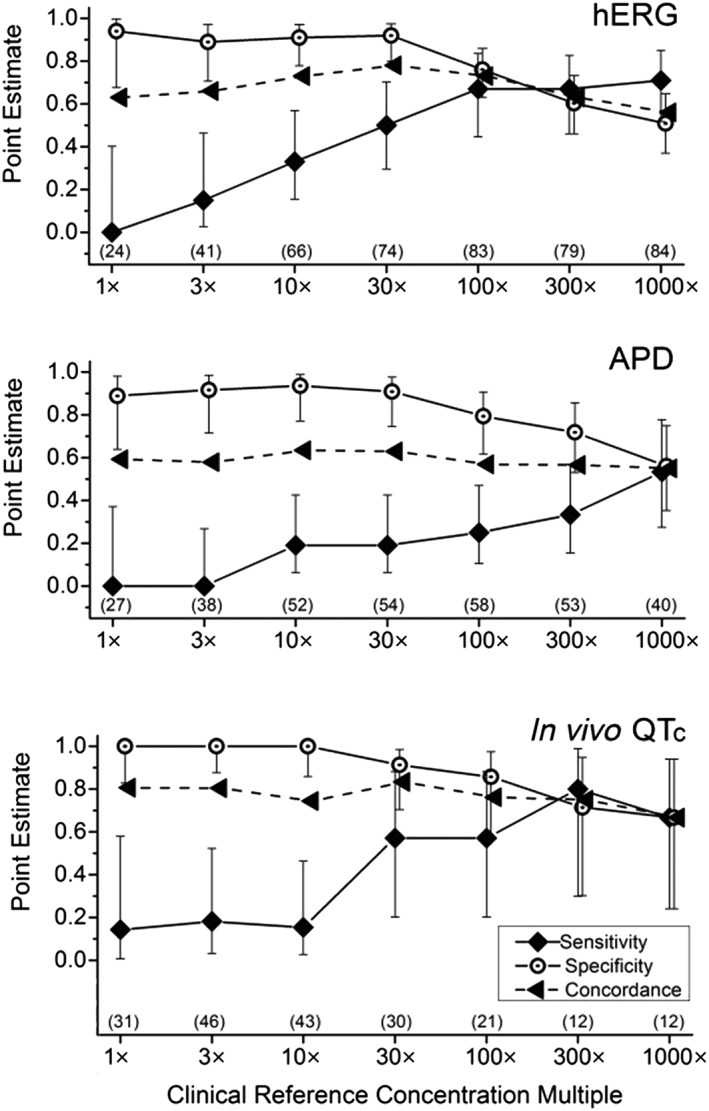
Sensitivity, specificity and concordance for each non‐clinical assay compared with clinical TQT studies. Findings for each non‐clinical are plotted across multiples of CRCs. Each assay demonstrated high specificity across lower (1×–30×) multiples of clinical exposures. In contrast, sensitivity was low for each assay across similar multiples, with values increasing at higher multiples for the hERG and in vivo QTc assay and less so for the APD assay. Values represent mean ± 95% confidence intervals for each performance parameter. Sample sizes at each concentration multiple are shown in parentheses.
Predictivity and optimal cut‐off points
Figure 4 compares the overall diagnostic accuracy for each preclinical assay using ROC curves across free CRC multiples. AUC values were greatest for the in vivo QTc assay (AUC = 0.75), lesser for the hERG assay (0.69) and minimal for the APD assay (0.55). Using Youden's index (J statistic), the optimum threshold for correctly categorizing TQT study results was achieved with 30×–100× and 30×–300× exposure multiples for the hERG and in vivo QTc assays, respectively, with corresponding J values for these thresholds of 0.42–0.44 and 0.48–0.51 respectively (see the Supporting Information Table S2). AUC and Youden's index values for the APD assay were low at all free CRC multiples consistent with poor assay performance.
Figure 4.
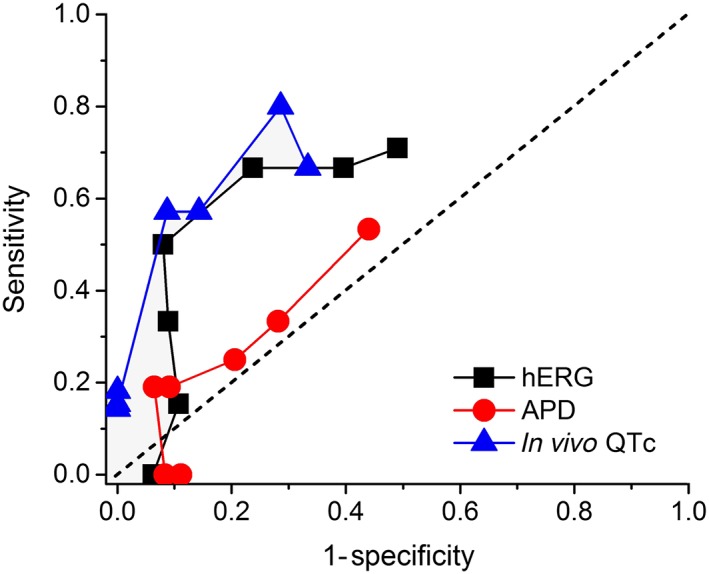
ROC curves for non‐clinical assays. The hERG and in vivo QTc assays provided the best overall performance for detecting TQT prolonging drugs (AUC values of 0.69 and 0.75), while the APD assay provided minimal overall utility (AUC = 0.55). Free CRC multiples ranging from 1× to 1000× (half log unit increments) are plotted for each assay, with 1× being the lowest point on the curves. Results were derived from data set summarized in Figure 3, which indicates sample sizes. The line of identity (dashed diagonal line) represents no discriminatory value.
Likelihood ratios
Figure 5 compares LRs for each repolarization assay at multiples of the CRC drug exposure. The greatest LR+ values were obtained for the hERG (6.25) and in vivo QTc (6.57) assays at the 30× exposure multiple, suggesting that drugs that elicit clinical QT prolongation are 6 times more likely to demonstrate hERG block or in vivo QTc prolongation (compared with drugs that do not elicit QT prolongation) at that exposure multiple. An LR+ of this magnitude suggests a positive hERG or in vivo QTc result causes a moderate increase on the post‐test probability of clinical QT prolongation. Lower LR+ values were obtained at higher or lower exposure multiples, declining to values near 1 at higher multiples, thus providing minimal influence on post‐test probabilities at supratherapeutic concentrations. In contrast, LR+ values for the APD assay were low across all exposure multiples, consistent with minimal utility in predicting clinical QTc prolongation (maximum LR+ of 2.95 at 10× multiple). This finding was further supported by the 95% confidence intervals that spanned the value of 1 at all exposure multiples for the APD assay.
Figure 5.
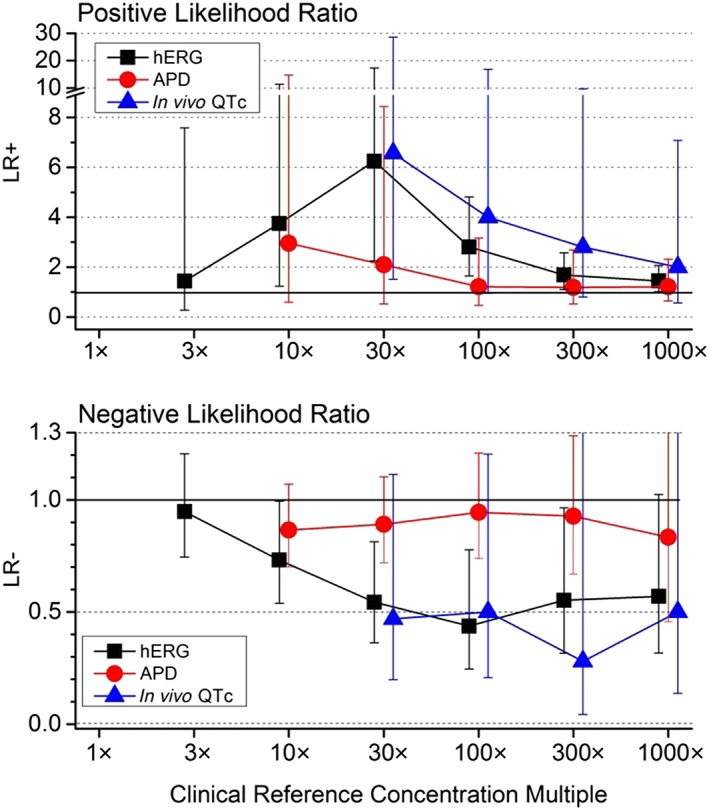
Positive (upper panel) and negative LRs (lower panel) compared for the three non‐clinical assays across CRC multiples. A value of 1 (dashed lines) represents no influence on post‐test probabilities for predicting clinical QTc prolongation and served as a threshold value for plotting data at low concentration multiples. The absent data points at lower exposure multiples for in vivo QTc assays represent indeterminate values due to zero values in the denominator. Error bars represent 95% confidence intervals. Results were derived from data set summarized in Figure 3, which lists corresponding group sizes at each multiple for each assay.
Meaningful LR− values were obtained for the hERG and in vivo QTc assays at exposure multiples >30×, while values for the APD assay were consistently near 1 thus failing to provide useful information. For the hERG assay, 95% confidence intervals for negative LRs did not include 1 (no utility) over the 10×–300× range of exposure multiples. However, the 95% confidence intervals for the APD and in vivo QTc assays overlapped a value of 1, consistent with minimum utility for LR− values with these assays. Thus, positive hERG and positive in vivo QTc assay results influence the likelihood of a positive clinical TQT study, a negative hERG assay influences the likelihood of a negative TQT study, whereas a negative in vivo QTc assay result has minimal influence. Positive or negative findings from the APD assay have minimal influence on predicting clinical QT effects.
Negative and positive predictive values analysis
NPV values were consistent and comparable for all three non‐clinical assays and moderately predictive (range 68–90%) across the full range of exposure multiples (Figure 6A), proving useful in predicting negative clinical TQT outcomes. In contrast, distinct differences in PPV values for the three assays were evident across CRC multiples (Figure 6B). The in vivo QTc assay proved superior in predicting clinical QTc prolongation at lower exposure multiples, with values declining below 73% as exposures multiples increased beyond 30×. PPV values for the hERG assay were minimal at lower exposure multiples (ranging from 0 to 62% at exposure multiples increased from 1× to 10×), increasing to a maximal 72% at the 30× multiples (a value approximately equal to the in vivo QTc assay), before declining at higher multiples. The APD assay had the lowest PPV values over the full range of multiple exposures, attaining a maximal value of 55% at a 10× exposure multiple. Thus, while negative findings in all three assays provided comparable value predicting no clinical QTc prolongation over a wide range of exposure multiples, only positive findings in the vivo QTc assay proved valuable in predicting clinical QTc prolongation over reasonable exposure multiples ranging from 1× to 30× CRC.
Figure 6.
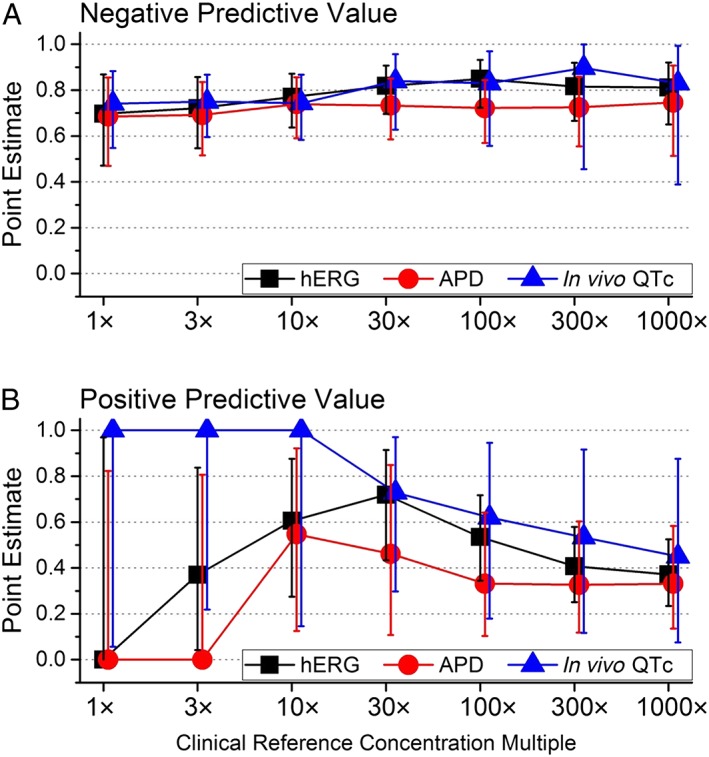
A comparison of assay performance based on NPVs and PPVs across exposure multiples. Panel A: NPVs were comparable and consistent for all assays, predicting the absence of clinical QTc prolongation (NPV values ranging from 70 to 90%). Panel B: PPVs were high for the in vivo QTc assay (values ranging from 0.73 to 1.00 across 1×–30× exposure multiples), consistent with the ability to predict clinical QTc prolongation. In contrast, values for hERG and APD assay were low across similar exposure multiples. Values represent mean +/−95% confidence intervals. The n‐sizes for each group are provided in Figure 3.
Discussion
This study assessed the ability of three widely used non‐clinical cardiac repolarization assays (the in vitro hERG current and APD repolarization assays, as well as in vivo QTc assay) to provide information relevant to on clinical QT prolongation measured in rigorous TQT studies for a unique anonymized proprietary database of 150 drug candidates (43 TQT positive drugs and 107 TQT negative drugs) submitted to the FDA. These results demonstrate that the utility of each assay is dependent on the diagnostic or prognostic question addressed and drug concentrations tested relative to clinical exposures (see Table 1 below).
Table 1.
Performance of three non‐clinical repolarization assays in translation to clinical QTc prolongation
| Performance parameter | hERG | APD | In vivo QTc | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1×–10× | 30×–100× | 300×–1000× | 1×–10× | 30×–100× | 300×–1000× | 1×–10× | 30×–100× | 300×–1000× | |
| Sensitivity | Low | Mod | Mod | Low | Low | Mod | Low | Mod | Mod |
| Specificity | High | High | Mod | High | High | Mod | High | High | Mod |
| Concordance | Mod | High | Mod | Mod | Mod | Mod | High | High | Mod |
| PPV | Low | Mod | Mod | Low | Mod | Low | High | Mod | Mod |
| NPV | Mod | Mod | Mod | Mod | Mod | Mod | Mod | Mod | Mod |
| Low = 0.0–0.3, Mod = 0.3–0.7 and High = 0.7–1.0 | |||||||||
| LR+ | Low | Mod | Low | Low | Low | Low | Low | Mod | Low |
| LR− | Low | Mod | Mod | Low | Low | Low | Low | Mod | Mod |
| For LR+: Low = 1–3, Mod = 3–10 and High >10 | For LR−: Low = 1.0–0.3, Mod = 0.3–0.1 and High <0.1 | ||||||||
| ROC overall AUC | Mod | Low | Mod | ||||||
| Low = 0.5–0.7, Mod = 0.7–0.85 and High = 0.85–1.0 | |||||||||
The relative utility of each assay at different exposure multiples was categorized as low, mod and high defined based on the range of possible values for each performance metric. Exposure multiples (1×–10×, 30×–100× and 300×–1000×) refer to multiples of the CRC achieved in TQT studies. Mod, moderate.
In this study, sensitivity and specificity describe the diagnostic ability of non‐clinical assays to accurately discriminate between TQT study outcomes. Each non‐clinical assay demonstrated robust specificity, but poor sensitivity, at low exposure multiples (<30× free exposure multiples), with sensitivity increasing at higher exposure multiples. High specificity reflects a low false‐positive rate, which is useful for ‘ruling in’ clinical QTc prolongation. However, the low sensitivity observed over the same range of exposure multiples is consistent with a high false‐negative rate and provides less confidence of ‘ruling out’ clinical QTc prolongation. The increased sensitivity, and somewhat reduced specificity, at higher exposures seen for the hERG assay is not unexpected as block of the I Kr current is more likely at supratherapeutic exposures for a channel described as ‘promiscuous’ based on numerous drug studies (Stansfeld et al., 2006).
Based on the European Centre for the Validation of Alternative Methods criteria for defining the robustness of a model (Genschow et al., 2002), the concordance of the hERG and in vivo QTc assays would be characterized as sufficient (>65%) and good (>75%) respectively. In contrast, concordance for the APD assay would be characterized as insufficient (<65%). Earlier studies (although limited in scope and typically using strongly positive or negative QT prolonging drugs) have confirmed overall good concordance between in vivo QTc assay results with clinical outcomes using conscious telemeterized dogs, mini pigs or monkeys (Ando et al., 2005, Kano et al., 2005, Miyazaki et al., 2005, Sasaki et al., 2005, Toyoshima et al., 2005, Hanson et al., 2006, Sivarajah et al., 2010, Leishman et al., 2012, Chain et al., 2013; Parkinson et al., 2013).
The in vivo QTc assay had specificity values comparable with previous results from Wallis (2010) and Ewart et al. (2014) (1.0, 0.86 and 0.98, respectively) at clinically relevant exposures, consistent with a low false‐positive rate with this assay. Similar to results from Ewart et al. (2014), assay sensitivity in the present study was low at therapeutic exposures (0.14), with both results substantially lower than reported by Wallis (0.83; Wallis, 2010). As in the present study, the data set from Ewart et al. (2014) was composed of a relatively large and arguably diverse data set from many different companies using different methods and interpretations of an effect, whereas the smaller data set from Wallis (2010) was from a single company using consistent methods and criteria for an effect. Further, most clinical QT‐positive compounds reported by Wallis (2010) were acknowledged to be hERG‐blocking agents, which was not the case for either Ewart et al. (2014) or the present study. The overall good concordance of in vivo QTc assays with TQT studies is not surprising due to electrophysiological similarities of hearts of larger mammals and humans and a common assay endpoint (QTc), reinforcing the importance of this non‐clinical in vivo assay in drug development.
The overall diagnostic power of an assay is captured by ROC curves and characterized by AUC values. In general, AUC values >0.9 represent excellent accuracy, and values between 0.6 and 0.7 represent rather poor accuracies; values between 0.7 and 0.9 represent fair to good accuracies and are considered useful for some purposes and may still provide optimal criteria for decision making (Swets, 1988). In the present study, AUC values were greatest for the in vivo QTc assay (0.75) and hERG assay (0.69) and lowest for the APD assay (0.55). In comparison, AUC values for the high sensitivity, point of care, cardiac troponin assays used to detect myocardial infarction range from 0.87 to 0.96 (Palamalai et al., 2013), and AUC values for the preclinical renal injury biomarker Kim‐1 for different nephrotoxicants range from 0.88 to 0.91 (Vaidya et al., 2010). The relatively low overall AUC values for the hERG (0.69) and in vivo QTc (0.75) assays do not support their use as sole assays for predicting clinical QTc prolongation. However, their high specificity values at low to moderate exposure multiples (depicted graphically by the vertical configuration of the left portions of their ROC curves) demonstrate utility for ruling in clinical QTc prolongation over this range of exposures.
PPVs and NPVs are prognostic measures that describe the probability that a condition is present (or absent) when an assay result is positive (or negative). While NPV values for all assays were essentially constant (0.7–0.9) across all clinical exposure multiples, clear differences in PPV values were apparent at low exposure multiples, with the in vivo QTc assay provided robust PPVs (PPV = 1) at low CRC multiples (1×–10×). In contrast to the in vivo QTc assay, the hERG and APD assay provided minimal value to predicted clinical QTc prolongation low (1×–3×) exposure multiples.
It should be noted that PPVs and NPVs are strongly dependent on the true prevalence and that PPV values will be farther from one with lower prevalence despite high sensitivity and specificity. However, the prevalence for clinical QTc prolongation in this data set (29%) was not excessively low and somewhat surprising given the well‐established industry practice of early screening with in vitro assays (Cavero, 2009). This prevalence rate may be due to some compounds entering clinical development prior to the implementation of ICH S7B guideline, uncertainty in translating potency of I Kr/hERG block to clinical QTc prolongation and the exquisite sensitivity of TQT studies to detect small QTc changes (less than 10 ms relative to baseline values of 400 ms).
Positive and negative LRs define the influence of positive and negative assay results on post‐test probabilities. Values above 10 and below 0.1 are considered to provide strong evidence to rule in or rule out diagnoses (Deeks and Altman, 2004), while values near 1 provide minimal to moderate influence on post‐test probabilities (Jaeschke et al., 1994). The hERG and in vivo QTc assays provided the highest LR+ ratios (values 6.25 and 6.57, respectively) at 30× CRC exposure multiples. Based on this result, a positive test result in the in vivo QTc assay within 30× CRC (LR+ = 6.57) would significantly change the pre‐test probability of a positive TQT outcome from 0.29 to a post‐test probability of 0.73 (calculated using the 0.29 prevalence of clinical QTc prolongation). Lower LRs (at exposures above or below 30×) result in less influence on post‐test probabilities. Multi‐tier contingency tables also demonstrated a moderate influence on positive LRs for the hERG assay on post‐test probabilities for 1×–30× exposure multiples (Gintant, 2011).
In contrast to hERG and in vivo QTc assays, low LRs for the APD assay suggest minimal overall influence on post‐test probabilities. Similar results for APD assays have been reported with smaller datasets using canine Purkinje fibres (Hanson et al., 2006; Wallis, 2010) and guinea pig papillary muscles (Hayashi et al., 2005; Hashimoto, 2008). This poor performance may reflect significant heterogeneity in drug responses based on different protocols and models used (see Limitations of assays below). Various in vitro cardiac APD models employing additional indices of repolarization (including triangulation of the APD, instability of the time course and duration of repolarization and APD alternans with rapid stimulation) (Hondeghem and Hoffmann, 2003, Champeroux et al., 2005, Redfern and Valentin, 2011) have proven useful in identifying drugs linked to proarrhythmia. By defining integrated electrophysiological responses, APD studies can identify the net effect of many drugs that unexpectedly affect several cardiac ionic currents (Martin et al., 2004; Kramer et al., 2013). The utility of repolarization assays using standardized protocols and human stem cell‐derived cardiomyocytes to assess proarrhythmic risk is currently being evaluated as part of the Comprehensive in vitro Proarrhythmia Assay paradigm to improve preclinical cardiac safety testing (Sager et al., 2014; Gintant et al., 2016).
Alignment of repolarization assays in drug discovery and development
It is important to identify and eliminate, wherever possible, safety hazards early during the drug discovery phases, e.g. during lead optimization (Bowes et al., 2012), to ensure the best compounds move forward. Arguably, it is preferable to employ assays with high specificity (low false‐positive detection rates) when selecting lead candidates, thereby allowing downstream efficacy studies along with safety screening efforts to proceed. While each non‐clinical assay demonstrated high specificity (low false‐positive rate) at low to moderate exposure multiples, the hERG assay is arguably the better early diagnostic test to employ based on practical considerations (including higher throughput and minimal compound requirements). However, the overall limited performance of this assay (ROC curves and LRs) is likely to reflect the inability of the single ionic current (I Kr) to predict drug effects on repolarization involving many other ionic currents. Once a candidate has progressed further into non‐clinical development, an assay with high PPV should be used as part of an integrated strategy balancing target‐based efficacy with safety assessments. The in vivo QTc assay is well suited for this task, providing moderate utility based on sensitivity and LR. This assay typically also evaluates other valuable functional cardiovascular endpoints, such as heart rate and BP, along with pharmacokinetic data, clinical endpoints (electrolytes) and potential effects of metabolites from telemeterized animals (Hoffmann and Warner, 2006; Leishman et al., 2012) to support further progression of candidates.
Relation of clinical findings to product labelling and proarrhythmic risk
The prevalence of TQT positive drugs in this data set was 29% (43/150 drugs) with a mean (ΔΔ)QTc effect ranging from −2.9 to 38 ms. The data can be stratified based on the magnitude of effect size as follows: 17 drugs (mean <10 ms), 19 drugs (mean ≥10 and <20 ms) and 7 drugs (mean ≥20 ms). While no relationship between the magnitude of QTc prolongation in TQT studies and drug approval rates has been identified, the severity of safety warnings (warnings and precautions, contradictions and/or boxed warnings) on product labelling is related to QTc prolongation (Anonymous, 2011; Park et al., 2013), demonstrating that drugs associated with QTc prolongation can be approved based on significant benefit and unmet medical need, such as oncology drugs, relative to delayed repolarization and TdP risk.
Limitations of assays
This study used results from rigorous clinical TQT studies as the ‘gold standard’ to compare with non‐clinical findings submitted for regulatory consideration (and thus reflecting ‘real‐world’ scenarios). QTc prolongation is well recognized as a biomarker for TdP proarrhythmia risk (Salvi et al., 2010; France and Pasqua, 2015), and a link between delayed ventricular repolarization and TdP risk is well established for both congenital and drug‐induced (or acquired) long QT syndromes. However, it is also well established that QTc prolongation is an imperfect surrogate marker of proarrhythmic risk (Anonymous, 2005a). Questions remain regarding how proarrhythmic risk is influenced by the extent of QTc prolongation, morphological changes in the QT interval and many ionic mechanisms (beside hERG current) that may mediate and influence delayed ventricular repolarization.
A number of specific limitations regarding the data set and methods of analysis used should be considered. One important limitation were gaps noted in the data set; not all drugs were tested in each assay or over the same concentration multiples. Indeed, the range of test concentrations or free exposures achieved often did not reach 1×–100× multiples of the clinical values, with exposures for many in vivo studies not even attaining a 1× clinical multiple. Also, deficiencies resulting from smaller numbers of drugs populating different categories within contingency tables were noted despite the large number of drugs (n = 150) evaluated. Further, possible effects of active metabolites (which may differ in non‐clinical vs. clinical studies) will not be captured in hERG or APD/repolarization assays.
It should be recognized that the IC50 values used to describe potency of hERG current block represent a simplified assessment of drug effects and that the influence of experimental conditions, such as temperature, Hill coefficients used in defining potency and more detailed characterization, i.e., kinetics of block and drug trapping, were typically not considered (and often not available). Such information might have enhanced the performance of the hERG assay. It should also be recognized that this data set from investigational new drug/new drug application submissions to the FDA was very diverse, with numerous differences in the non‐clinical studies used by sponsors. For example, differences in the contributions of repolarizing currents across species (guinea pig, rabbit and dog) and between tissue types (Purkinje, papillary muscles and whole perfused hearts), along with differences in protocols and uncertainty of drug concentrations (nominal vs. measured bath concentration) all likely contributed to the response heterogeneity and overall poor performance of the APD repolarization assay.
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). While less heterogeneity might be expected in the in vivo QT assays, differences in animal species, study design (anaesthetized vs. conscious animal) and QT correction methods for heart rate changes all collectively contributed to response heterogeneity with this assay. We also relied on the sponsors' determination of positive versus negative responses for APD and in vivo QTc assays, which is likely to vary with experimental designs and between sponsors. Thus, while the present study provides an overall global assessment of the utility of popular non‐clinical repolarization assays, it may not reflect the diagnostic or predictive capacity of a specific assay employing consistent methods.
Overall, there was also paucity of information regarding the quality of the non‐clinical studies, such as positive controls, or possible influence of sex in most submitted assays. This highlights the need for greater standardization of experimental designs and analysis methods to minimize assay variability and the importance of model characterization with appropriate calibrating reference substances. All studies utilized healthy tissues or animals without structural heart disease or altered repolarization that does not reflect increased proarrhythmic risks in some patients. While such factors affect the ability to translate findings of delayed repolarization and proarrhythmia to at risk populations, it does not affect the ability to alter delayed repolarization in healthy human subjects in typical TQT studies.
Conclusions
In conclusion, a comparison of the diagnostic and prognostic performance of three widely used non‐clinical assays with QTc prolongation assessed with well‐controlled clinical TQT study results for 150 drug candidates demonstrated good specificity (low false‐positive rate) but relatively poor sensitivity (high false‐negative rate) at low multiples of clinical concentrations. Moderate overall diagnostic utility for the hERG and in vivo QTc (but not APD) assays was demonstrated from ROC curves. Assay performance for the hERG and in vivo QTc assays was marginal to good based on LRs and predictive values, with the APD assay providing the least utility for assessing the potential for clinical delaying repolarization. The performance of all assays was strongly influenced by drug concentrations tested (placed in the context of clinical exposures). The hERG and in vivo QT repolarization assays provide greater value compared with the APD assay in guiding the selection of drug candidates devoid of clinical QTc prolongation.
Author contributions
The contributions of each of the authors are as follows: E.P., J.W. and J.K. for manuscript drafting and editing, data set compilation and curation and data analysis; G.A.G. for manuscript drafting and editing, data analysis and data set review; D.B. and D.K. for data set compilation and curation and data analysis; S.D.P. and J.B.P. for manuscript editing, data set review and project management; M.S. and T.W. for manuscript drafting and editing, data set review and data analysis; and J.‐P.V. for study design, manuscript drafting and editing, data set review and data analysis.
Conflict of interest
The authors declare no conflicts of interest. The opinions presented here are those of the authors. No official support or endorsement by the US Food and Drug Administration is intended or should be inferred.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Definitions of Outcomes Criteria, Positive and Negative Findings for Non‐clinical and Clinical Assays.
Table S2 Dichotomous Contingency Table and Basis for Assay Characterizations.
Table S3 Characteristics of the 150 Drug Anonymized Dataset.
Table S4 Characterization of ROC curves.
Table S5 Therapeutic Areas of Drugs in Dataset.
Acknowledgements
The authors wish to express their gratitude to the HESI Cardiac Safety Committee Proarrhythmia Working Group members for supporting the efforts of this collaborative project. Additionally, the authors would like to specifically thank the following individuals for their contributions to the data set analysis and review of the manuscript: Louis Cantilena, William Link, Monica Fiszman, Peter Hoffmann, Hugo Vargas, Jean‐Michel Guillon, Alan Bass and Derek Leishman.
US FDA Center for Drug Development and Research, Oak Ridge Institute for Science fellowships programme supported Dr E.P. from 15 August 2011 to 10 August 2013. All other work was provided in kind by the ILSI HESI Cardiac Safety Committee Proarrhythmia Working Group, which is supported by sponsorships from member companies. HESI's scientific initiatives are primarily supported by the in‐kind contributions (from public and private sector participants) of time, expertise and experimental effort. These contributions are supplemented by direct funding (that primarily supports programme infrastructure and management) provided primarily by HESI's corporate sponsors. The following authors are employed in the pharmaceutical industry (at the time of manuscript submission): G.A.G., M.S., T.W. and J.‐P.V.
Park, E. , Gintant, G. A. , Bi, D. , Kozeli, D. , Pettit, S. D. , Pierson, J. B. , Skinner, M. , Willard, J. , Wisialowski, T. , Koerner, J. , and Valentin, J.‐P. (2018) Can non‐clinical repolarization assays predict the results of clinical thorough QT studies? Results from a research consortium. British Journal of Pharmacology, 175: 606–617. doi: 10.1111/bph.14101.
References
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017). The Concise Guide to Pharmacology 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman DG, Bland JM (1994a). Diagnostic tests 2: predictive values. Br Med J 309: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman DG, Bland JM (1994b). Diagnostic tests 1: sensitivity and specificity. Br Med J 308: 1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Hombo T, Kanno A, Ikeda H, Imaizumi M, Shimizu N et al (2005). QT PRODACT: in vivo QT assay with a conscious monkey for assessment of the potential for drug‐induced QT interval prolongation. J Pharmacol Sci 99 : 487–500. [DOI] [PubMed] [Google Scholar]
- Anonymous (2005a). International Conference on Harmonisation; guidance on E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs. Fed Regist 70: 61134–61135 Available at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf [PubMed] [Google Scholar]
- Anonymous (2005b). International Conference on Harmonisation; guidance on S7B Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals; availability. Fed Regist 70: 61133–61134 Available at: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7B/Step4/S7B_Guideline.pdf [PubMed] [Google Scholar]
- Anonymous (2011) Guidance for industry: warnings and precautions, contraindication, boxed warning sections of labeling for human prescription drug and biology products – content and format. US FDA October 2011. Available at: http://www.fda.gov/downloads/Drugs/Guidances/ucm075096.pdf
- Bowes J, Brown AJ, Hamon J, Jarolimek W, Sridhar A, Waldron G et al (2012). Reducing safety‐related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov 11: 909–922. [DOI] [PubMed] [Google Scholar]
- Cavero I (2009). Exploratory safety pharmacology: a new safety paradigm to de‐risk drug candidates prior to selection for regulatory science investigations. Expert Opin Drug Saf 8: 627–647. [DOI] [PubMed] [Google Scholar]
- Chain AS, Dubois VF, Danhof M, Sturkenboom MC, Della Pasqua O (2013). Cardiovascular Safety Project Team, TI Pharma PKPD Platform. Identifying the translational gap in the evaluation of drug‐induced QTc interval prolongation. Br J Clin Pharmacol 76: 708–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champeroux P, Viaud K, El Amrani AI, Fowler JS, Martel E, Le Guennec JY et al (2005). Prediction of the risk of torsade de pointes using the model of isolated canine Purkinje fibres. Br J Pharmacol 144: 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darpo B (2010). The thorough QT/QTc study 4 years after the implementation of the ICH E14 guidance. Br J Pharmacol 159: 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darpo B (2015). Clinical ECG assessment. Handb Exp Pharmacol 229: 435–468. [DOI] [PubMed] [Google Scholar]
- Deeks JJ, Altman DG (2004). Diagnostic tests 4: likelihood ratios. Br Med J 329: 168–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewart L, Aylott M, Deurinck M, Engwall M, Gallacher DJ, Geys H et al (2014). The concordance between nonclinical and phase I clinical cardiovascular assessment from a cross‐company data sharing initiative. Toxicol Sci 142: 427–435. [DOI] [PubMed] [Google Scholar]
- France NP, Pasqua D (2015). The role of concentration–effect relationships in the assessment of QTc interval prolongation. Br J Clin Pharmacol 79: 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett CE, Beasley N, Bhattaram VA, Jadhav PR, Madabushi R et al (2008). Concentration–QT relationships play a key role in the evaluation of proarrhythmic risk during regulatory review. J Clin Pharmacol 48: 13–18. [DOI] [PubMed] [Google Scholar]
- Garnett CE, Zhu H, Malik M, Fossa AA, Zhang J, Badilini F et al (2012). Methodologies to characterize the QT/corrected QT interval in the presence of drug‐induced heart rate changes or other autonomic effects. Am Heart J 163: 912–930. [DOI] [PubMed] [Google Scholar]
- Genschow E, Spielmann H, Scholz G, Seiler A, Brown N, Piersma et al (2002). The ECVAM international validation study on in vitro embryotoxicity tests: results of the definitive phase and evaluation of prediction models. European Centre for the Validation of Alternative Methods. Altern Lab Anim 30: 151–176. [DOI] [PubMed] [Google Scholar]
- Gintant G (2011). An evaluation of hERG current assay performance: translating preclinical safety studies to clinical QT prolongation. Pharmacol Ther 129: 109–119. [DOI] [PubMed] [Google Scholar]
- Gintant G, Sager PT, Stockbridge N (2016). Evolution of strategies to improve preclinical cardiac safety testing. Nat Rev Drug Discov 15: 457–471. [DOI] [PubMed] [Google Scholar]
- Hanley JA, McNeil BJ (1982). The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143: 29–36. [DOI] [PubMed] [Google Scholar]
- Hanson LA, Bass AS, Gintant G, Mittelstadt S, Rampe D, Thomas K (2006). ILSI‐HESI cardiovascular safety subcommittee initiative: evaluation of three non‐clinical models of QT prolongation. J Pharmacol Toxicol Methods 54: 116–129. [DOI] [PubMed] [Google Scholar]
- Hashimoto K (2008). Torsades de pointes liability inter‐model comparisons: the experience of the QT PRODACT initiative. Pharmacol Ther 119: 195–198. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Kii Y, Tabo M, Fukuda H, Itoh T, Shimosato T et al (2005). QT PRODACT: a multi‐site study of in vitro action potential assays on 21 compounds in isolated guinea‐pig papillary muscles. J Pharmacol Sci 99: 423–437. [DOI] [PubMed] [Google Scholar]
- Hoffmann P, Warner B (2006). Are hERG channel inhibition and QT interval prolongation all there is in drug‐induced torsadogenesis? A review of emerging trends. J Pharmacol Toxicol Methods 53: 87–105. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Hoffmann P (2003). Blinded test in isolated female rabbit heart reliably identifies action potential duration prolongation and proarrhythmic drugs: importance of triangulation, reverse use dependence, and instability. J Cardiovasc Pharmacol 41: 14–24. [DOI] [PubMed] [Google Scholar]
- Jaeschke R, Guyatt GH, Sackett DL (1994). Users' guides to the medical literature. III. How to use an article about a diagnostic test. B. What are the results and will they help me in caring for my patients? The Evidence‐Based Medicine Working Group. JAMA 271: 703–707. [DOI] [PubMed] [Google Scholar]
- Kannankeril P, Roden DM, Darbar D (2010). Drug‐induced long QT syndrome. Pharmacol Rev 62: 760–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Toyoshi T, Iwasaki S, Kato M, Shimizu M, Ota T (2005). QT PRODACT: usability of miniature pigs in safety pharmacology studies: assessment for drug‐induced QT interval prolongation. J Pharmacol Sci 99: 501–511. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer J, Obejero‐Paz CA, Myatt G, Kuryshev YA, Bruening‐Wright A, Verducci JS et al (2013). MICE models: superior to the HERG model in predicting torsade de pointes. Sci Rep 3: 2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CL, Pollard CE, Hammond TG, Valentin JP (2008). In vitro models of proarrhythmia. Br J Pharmacol 154: 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leishman DJ, Beck TW, Dybdal N, Gallacher DJ, Guth BD, Holbrook M et al (2012). Best practice in the conduct of key nonclinical cardiovascular assessments in drug development: current recommendations from the Safety Pharmacology Society. J Pharmacol Toxicol Methods 65: 93–101. [DOI] [PubMed] [Google Scholar]
- Loong TW (2003). Understanding sensitivity and specificity with the right side of the brain. Br Med J 327: 716–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RL, McDermott JS, Salmen HJ, Palmatier J, Cox BF, Gintant GA (2004). The utility of hERG and repolarization assays in evaluating delayed cardiac repolarization: influence of multi‐channel block. J Cardiovasc Pharmacol 43: 369–379. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki H, Watanabe H, Kitayama T, Nishida M, Nishi Y, Sekiya K et al (2005). QT PRODACT: sensitivity and specificity of the canine telemetry assay for detecting drug‐induced QT interval prolongation. J Pharmacol Sci 99: 523–529. [DOI] [PubMed] [Google Scholar]
- O'Hara T, Virág L, Varró A, Rudy Y (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7: e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamalai V, Murakami MM, Apple FS (2013). Diagnostic performance of four point of care cardiac troponin I assays to rule in and rule out acute myocardial infarction. Clin Biochem 46: 1631–1635. [DOI] [PubMed] [Google Scholar]
- Park E, Willard J, Bi D, Fiszman M, Kozeli D, Koerner J (2013). The impact of drug‐related QT prolongation on FDA regulatory decisions. Int J Cardiol 168: 4975–4976. [DOI] [PubMed] [Google Scholar]
- Parkinson J, Visser SA, Jarvis P, Pollard C, Valentin JP, Yates JW et al (2013). Translational pharmacokinetic-pharmacodynamic modeling of QTc effects in dog and human. J Pharmacol Toxicol Methods 68: 357–366. [DOI] [PubMed] [Google Scholar]
- Pollard CE, Abi Gerges N, Bridgland‐Taylor MH, Easter A, Hammond TG, Valentin JP (2010). An introduction to QT interval prolongation and non‐clinical approaches to assessing and reducing risk. Br J Pharmacol 159: 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampe D, Brown AM (2013). A history of the role of the hERG channel in cardiac risk assessment. J Pharmacol Toxicol Methods 68: 13–22. [DOI] [PubMed] [Google Scholar]
- Redfern WS, Valentin JP (2011). Trends in safety pharmacology: posters presented at the annual meetings of the Safety Pharmacology Society 2001–2010. J Pharmacol Toxicol Methods 64: 102–110. [DOI] [PubMed] [Google Scholar]
- Sager PT, Gintant G, Turner JR, Pettit S, Stockbridge N (2014). Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J 167: 292–300. Epub 2013 Dec 2. [DOI] [PubMed] [Google Scholar]
- Salvi V, Karnad DR, Panicker GK, Kothari S (2010). Update on the evaluation of a new drug for effects on cardiac repolarization in humans: issues in early drug development. Br J Pharmacol 159: 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAS (2008). SAS 9.2 Cary, NC, USA.
- Sasaki H, Shimizu N, Suganami H, Yamamoto K (2005). QT PRODACT: inter‐facility variability in electrocardiographic and hemodynamic parameters in conscious dogs and monkeys. J Pharmacol Sci 99: 513–522. [DOI] [PubMed] [Google Scholar]
- Schisterman EF, Perkins NJ, Liu A, Bondell H (2005). Optimal cut‐point and its corresponding Youden index to discriminate individuals using pooled blood samples. Epidemiology 16: 73–81. [DOI] [PubMed] [Google Scholar]
- Sivarajah A, Collins S, Sutton MR, Regan N, West H, Holbrook M et al (2010). Cardiovascular safety assessments in the conscious telemetered dog: utilisation of super‐intervals to enhance statistical power. J Pharmacol Toxicol Methods 62: 12–19. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld PJ, Sutcliffe MJ, Mitcheson JS (2006). Molecular mechanisms for drug interactions with hERG that cause long QT syndrome. Expert Opin Drug Metab Toxicol 2: 81–94. [DOI] [PubMed] [Google Scholar]
- Stockbridge N, Zhang J, Garnett C, Malik M (2012). Practice and challenges of thorough QT studies. J Electrocardiol 45: 582–587. [DOI] [PubMed] [Google Scholar]
- Swets JA (1988). Measuring the accuracy of diagnostic systems. Science 240: 1285–1293. [DOI] [PubMed] [Google Scholar]
- Toyoshima S, Kanno A, Kitayama T, Sekiya K, Nakai K, Haruna M et al (2005). QT PRODACT: in vivo QT assay in the conscious dog for assessing the potential for QT interval prolongation by human pharmaceuticals. J Pharmacol Sci 99: 459–471. [DOI] [PubMed] [Google Scholar]
- Trepakova ES, Koerner J, Pettit SD, Valentin JP (2009). A HESI consortium approach to assess the human predictive value of non‐clinical repolarization assays. J Pharmacol Toxicol Methods 60: 45–50. [DOI] [PubMed] [Google Scholar]
- Vaidya VS, Ozer JS, Dieterle F, Collings FB, Ramirez V, Troth S et al (2010). Kidney injury molecule‐1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol 28: 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin JP, Pollard C, Lainee P, Hammond T (2010). Value of non‐clinical cardiac repolarization assays in supporting the discovery and development of safer medicines. Br J Pharmacol 159 (1): 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis RM (2010). Integrated risk assessment and predictive value to humans of non‐clinical repolarization assays. Br J Pharmacol 159: 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Definitions of Outcomes Criteria, Positive and Negative Findings for Non‐clinical and Clinical Assays.
Table S2 Dichotomous Contingency Table and Basis for Assay Characterizations.
Table S3 Characteristics of the 150 Drug Anonymized Dataset.
Table S4 Characterization of ROC curves.
Table S5 Therapeutic Areas of Drugs in Dataset.