

SUMMARY
Peptides have great potential to combat antibiotic resistance. While many platforms can screen peptides for their ability to bind to target cells, there are virtually no platforms that directly assess the functionality of peptides. This limitation is exacerbated when identifying antimicrobial peptides because the phenotype, death, selects against itself and has caused a scientific bottleneck that confines research to a few naturally occurring classes of antimicrobial peptides. We have used this seeming dissonance to develop Surface Localized Antimicrobial displaY (SLAY); a platform that allows screening of unlimited numbers of peptides of any length, composition, and structure in a single tube for antimicrobial activity. Using SLAY, we screened ~800,000 random peptide sequences for antimicrobial function and identified thousands of active sequences dramatically increasing the number of known antimicrobial sequences. SLAY hits present with different potential mechanisms of peptide action and access to areas of antimicrobial physicochemical space beyond what nature has evolved.
Keywords: Antibiotic resistance, bacteria, high-throughput screening, drug discovery, infectious diseases
Graphical Abstract
Identification of antimicrobial peptides with diverse compositions expands the range of efficacious bactericidal agents.
INTRODUCTION
Antibiotic resistant bacteria are projected to kill 30 million people by 2050(O’Neill, 2016). As emphasized by recent World Health Organization reports, antibiotics to treat Gram-negative bacterial infections are needed most(WHO, 2017). The path from antibiotic discovery to clinical therapy has a high attrition rate, with the last new class of antibiotics to combat Gram-negative bacteria being discovered over 40 years ago(Clatworthy et al., 2007; Payne et al., 2007). Most antibiotic screening methods have not evolved far from the innovation of Waksman’s approach developed in the 1930s, and are no longer able to quickly identify new lead compounds(Lewis, 2013; Woodruff, 2014). Necessitated by the lack of new leads and sources for natural products, companies are attempting to resurrect previously unsuccessful drug candidates(Lewis, 2013). Reliable and robust antibiotic discovery platforms are urgently needed to discover new leads against new microbial targets in our arms race against resistance.
There has been a resurgence of interest in developing antimicrobial peptides to supplement our antibiotic arsenal, generate new scaffolds for antibiotic design, and expand our knowledge of antimicrobial action(Hancock and Sahl, 2006; Kang et al., 2014; Zasloff, 2002). However, we lack simple, biologically relevant, means to screen comprehensive peptide libraries and discover peptides with antimicrobial activity for development. This has limited antimicrobial peptide research to the few unique classes that have evolved in nature, with the majority of studies focusing on a single dominant class of naturally occurring cationic antimicrobial peptides (CAMPs)(Bahar and Ren, 2013; Hadley and Hancock, 2010). CAMPs are characterized by strong cationic charge and amphipathic properties with broad-spectrum activity and pore forming mechanisms of action(Peschel and Sahl, 2006). While CAMPs are successful in terms of their broad activity against pathogens in vitro, they have not been successful therapeutically(Fox, 2013). Natural antimicrobials peptides beyond CAMPs have sequences of diverse length, chemistry, and structure acting on a wide range of molecular targets(Bahar and Ren, 2013; Fosgerau and Hoffmann, 2015). This underscores that no single peptide sequence has evolved as singularly effective against all pathogens in all settings(Bahar and Ren, 2013; Gould and Bal, 2013).
With the near infinite possibilities of combinatorial sequence space, and our limited understanding of peptide chemistry with antimicrobial activity, it is nearly impossible to predict bioactive sequences de novo (Fjell et al., 2011; Neme et al., 2017) and necessitates the development of functional approaches for peptide exploration if we hope to capitalize on their therapeutic potential. Many technologies, like phage display, allow screening or selecting for peptides that bind a molecule or cell but do not provide a means to directly assess the functionality and antimicrobial relevance of the peptides or their interaction. Antimicrobial peptide screening through these approaches is further confounded since an antimicrobial interaction eliminates the target bacteria and prevents recovery of the active peptide. Alternative molecular approaches express peptides intracellularly to identify sequences with antimicrobial activity. Unfortunately, peptides identified through these approaches often fail to show activity in synthetic form because they cannot pass through the cell membrane to reach their target. Current chemical synthesis approaches that do allow functional peptide screening are limited to a few thousand short, linear, sequences at a time, and require combinatorial chemistry and robotics for scale-up, which is beyond the reach of most research programs(Hilpert et al., 2005; Hilpert et al., 2007). While marking an important advance in peptide screening, this capacity has not facilitated antimicrobial peptide exploration beyond naturally available templates leaving the majority of potentially therapeutically valuable peptide chemical space undiscovered.
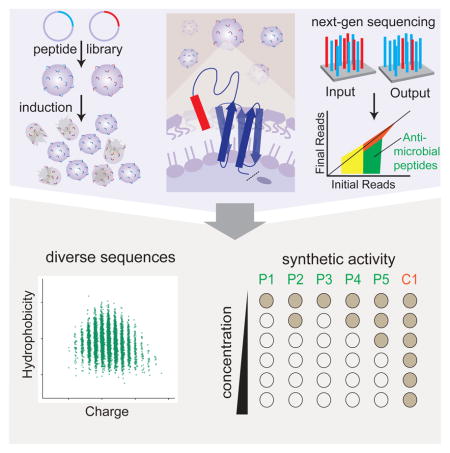
To overcome these roadblocks, we present Surface Localized Antimicrobial displaY (SLAY), a high-throughput screening platform to rapidly identify lead antimicrobial peptides to combat multi-drug resistant Gram-negative bacteria. SLAY drives bacteria to express and self-test peptides of any size, structure, or sequence complexity for antimicrobial activity through a physiologically and therapeutically meaningful interface and provides readout of the interactions via high-throughput DNA sequencing. Using SLAY we quickly screened a library of approximately 800,000 20-mer peptides for antimicrobial activity and identified 7,968 fully synthetic sequences covering an unprecedented range of peptide physicochemical space. Selected peptides with properties far removed from CAMPs showed activity against multi-drug resistant bacteria, different potential mechanisms of action, and low eukaryotic toxicity. SLAY offers a unique approach to peptide discovery and aims to revolutionize our understanding of antimicrobial peptide chemistry that can serve to supplement our antibiotic arsenal, generate antibiotic scaffolds, and expand our knowledge of potential antimicrobial targets to combat the spread of antibiotic-resistant bacteria.
RESULTS
Development of Surface Localized Antimicrobial display (SLAY)
During infection treatment, drugs first interact with a bacterium at its cell surface and then migrate to their target. To recapitulate this scenario during screening, SLAY localizes peptides on the Gram-negative bacterial cell surface as part of a fusion protein consisting of: (1) a murein lipoprotein (lpp) signal sequence that directs proteins for export from the cytoplasm and is subsequently cleaved, (2) five transmembrane domains (residues 46-159) of the OmpA membrane protein for outer membrane localization(Georgiou et al., 1996), (3) a flexible tether that allows spatial freedom(Li et al., 2011), and (4) a C-terminal peptide. We engineered the tether to extend up to 180 Å from its fusion to OmpA, enabling the C-terminal peptide flexibility to interact with the growth environment, the outer membrane, and periplasmic components. With the fluid nature of periplasmic space ranging anywhere from 106 to 253 Å, peptides have the potential to penetrate as far as the cytoplasmic membrane(Graham et al., 1991) (Fig. 1A).
Figure 1. SLAY platform demonstrated in gram-negative bacteria.

(A) Diagram of surface display system. Antimicrobial peptide surface display system composed of (1) Lpp signal sequence, (2) OmpA (46–159) transmembrane protein, (3) flexible tether, (4) C-terminal peptide. The Lpp signal sequence is shown for clarity, but is removed prior to insertion into the outer membrane. (B) Optical density plot over a period of 6 hours of a control peptide, tandem influenza hemagglutinin peptide 2xHA (top), and an antimicrobial peptide, cecropin P1 (bottom) expressed in the surface display system induced with 0 mM, 0.1 mM, and 1 mM IPTG. (C) Surface display expression of cecropin P1 as in (B) reported as colony forming units (cfu/mL) over time. (D) Expression of cecropin P1 at 0.1 mM IPTG in the parent strain W3110 (blue) CAMP resistant W3110 strain WD101 (purple), and eptA deletion in WD101 (red). (E) The surface display is amenable to disulfide-forming peptides. Expression of protegrin 1 (top) and defensin HNP-1 (middle), and a defensin cysteine mutant (bottom) plotted as optical density versus time in the E. coli strain W3110. (F) The surface display system functions across many Gram-negative species such as Acinetobacter baumannii and Pseudomonas aeruginosa. Each strain is displaying protegrin 1 at 0 mM, 0.1 mM and 1 mM IPTG. Plotted are recorded as optical density over 6 hours. (G) Neighboring cells are unaffected by surface expression of antimicrobial peptides. White and blue cells with empty plasmid and cecropin P1 respectively. Input cultures (left) were collected, serial diluted, and spotted before induction of 1 mM IPTG. Cells were induced at a total starting OD 600nm of 0.01. After 3 hours of surface expression, cells were collected, serial diluted, and spotted (right). All growth curves were performed in triplicate. Data are represented as mean ± SEM.
Cecropin P1 is a well-studied CAMP that acts by binding and disrupting the structure of the bacterial outer membrane(Gazit et al., 1995). As a test case, cecropin P1 was cloned as the C-terminal peptide and the construct was expressed in wild-type E. coli K-12 strain W3110. A tandem influenza hemagglutinin peptide (2xHA) was cloned as a C-terminal peptide control. We induced expression with increasing concentrations of IPTG and monitored optical density as an initial measure of cell growth and viability. The cultures expressing the control 2xHA peptide grew similarly at all IPTG concentrations (Fig. 1B). The cultures expressing cecropin P1 showed an induction-dependent decrease in optical density (Fig. 1B). We measured colony-forming units (CFUs) for cecropin P1 cultures and found a correlative decrease in viable cells following induction (Fig. 1C). Cytosolic expression of cecropin P1 alone did not affect W3110 growth or viability (Supplementary Fig. 1).
The length of the flexible tether strongly influenced cecropin P1-dependent growth effects. In addition to the full-length tether (2X), we also cloned cecropin P1 with a half-length tether (1X) and no tether (0X). Induction of each construct at 0.1mM IPTG showed that cecropin P1 displayed with the full 2X tether length had the strongest activity (Supplementary Fig. 2).
Displayed Peptides Mimic Native Interactions
To further demonstrate bacterium-relevant physiological interactions recapitulated through our approach, we introduced the 2X tether cecropin P1 construct in E. coli strain WD101(Trent et al., 2001). WD101 is a derivate of strain W3110 and carries a mutation that decreases its overall surface charge through the addition of amine-containing residues to lipopolysaccharide (LPS) and makes it resistant to CAMPs like cecropin P1. Consistent with the ability our engineered system to recapitulate natural interactions, WD101 was more resistant to antimicrobial activity of surface expressed cecropin P1 compared to the parent CAMP sensitive strain W3110 (Fig. 1d). Furthermore, deletion of the eptA gene, which is required for the LPS modification conferring CAMP resistance, sensitized WD101 to surface displayed cecropin P1(Herrera et al., 2010).
The action of peptides displayed by our platform is also sensitive to relevant environmental conditions. CAMP activity is decreased by the addition of magnesium ions that fortify bacterial cell surfaces and are also sensitive to trypsin degradation due the large numbers of arginine and lysine residues they contain. Addition of up to 2 mM magnesium to the growth medium greatly reduced the antimicrobial action of surface-displayed cecropin P1, and addition of trypsin to the culture medium greatly lessens cecropin P1-induced growth effects (Supplementary Fig. 3).
SLAY Allows Functional Display of Cyclic Peptides in a Broad Range of Gram-Negative Bacteria
In addition to cecropin P1, antimicrobial peptides dermaseptin, protegrin 1, and defensin HNP-1 showed strong antimicrobial activity against W3110 in our system (Fig. 1E, Supplementary Fig. 4). Defensin HNP-1 and protegrin 1 were particularly interesting since they require disulfide bonds for activity. We reconstructed defensin HNP-1 without disulfides and demonstrated that its activity was dramatically reduced, in agreement with biochemical studies(Varkey and Nagaraj, 2005). This indicates that our system supports the formation of cyclic, disulfide bond-dependent antimicrobial peptides.
To ensure the application of our system in a wide range of gram-negative bacteria, we engineered expression and replication of ubiquitous OmpA surface localization on a broad RSF1010 origin-based plasmid. Without any change to our system, we demonstrated that it was transferable and functional in a broad range of gram-negative bacteria, including ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Psuedomonas aeruginosa, and Enterobacter species) pathogens, like Acinetobacter baumannii and Pseudomonas aeruginosa (Fig. 1F).
Batch Screening of a Defined Peptide Library with SLAY
While the full-length tether (2X) is required for the spatial freedom of a peptide to interact with its host bacterium, it is short enough to prevent an antimicrobial peptide from exerting an effect on neighbor cells in culture. We co-cultured W3110 surface expressing cecropin P1, which shows potent CAMP activity, with W3110 containing only the empty plasmid pMMB67EH. When co-cultured in a 1:1 ratio and induced, cecropin P1 only affects the viability of cells expressing it (Fig. 1G). Thus, multiple peptides can be assayed for activity in a single tube.
Our screening workflow for SLAY is shown in Fig. 2. Peptides are cloned into our surface display system and transformed into a Gram-negative strain of interest. Peptide surface expression is then induced by IPTG. Bacteria expressing bactericidal or bacteriostatic peptides will decrease in abundance during the induction period. One PCR reaction generates Illumina next-generation sequencing samples for sequencing from plasmid libraries pre- and post-induction. In silico translation and comparison identifies each peptide in the library and its abundance pre- and post-induction to identify potential antimicrobial hits.
Figure 2. SLAY workflow.

Batch screening of peptides using our surface display system can be achieved by first constructing a random library using random PCR primers that flank the peptide region (i), followed by collection of transformants, plasmid isolation, and subsequent transformation into a bacterial strain of interest. Next, the library is grown in culture and induced (ii). Peptides with antimicrobial activity (colored red) will drop out of the population (iii). Next-generation sequencing of the initial input at time zero and output (iv) at a pre-defined number of hours provides a read out of sequencing counts (v). From this information, top hits can be identified and tested. Further libraries can be constructed based on the identified top hits and the process can be repeated. A more detailed explanation of our workflow can be found in the methods section.
To validate SLAY, a small library of three antimicrobial peptides and two control peptides (Table 1) were transformed into E. coli then pooled, induced, and harvested at 0 (input), 2, 3, and 4 hours. Following next-gen library construction and sequencing, reads were normalized to the input counts (Fig. 3). Log2 fold values, reported in Table 1, indicate the degree to which the peptides were removed from the population. Control peptides 2xHA and defensin HNP-1 cysteine mutant showed a near neutral log2 fold change over the time course examined. Meanwhile, the remaining antimicrobial peptides show a log2 fold change of -1 or lower indicating they were removed from the population over the time course. From these data, we would conclude that protegrin 1, cecropin P1, and defensin HNP-1 have effective antimicrobial activity against our E. coli strain with protegrin 1 exhibiting the strongest activity. Indeed, minimal bactericidal concentration (MBC) assays using synthesized peptides showed correlative bactericidal activity with log2 fold values, with MBCs of <0.125 μM, 1 μM and 8 μM measured for protegrin 1, cecropin P1 and defensin HNP-1, respectively (Table 1).
Table 1.
Defined library statistics and MBCs.
| Peptide | Sequence | log2 fold | p-value | MBC (μM) |
|---|---|---|---|---|
| Protegrin 1 | RGGRLCYCRRRFCVCVGR | −3.19 | <1E-143 | <0.125 |
| Cecropin P1 | SWLSKTAKKLENSAKKRISEGIAIAIQGGPR | −1.74 | 8.54E-27 | 1 |
| Defensin HNP-1 | ACYCRIPACIAGERRYGTCIYQGRLWAFCC | −1.04 | 3.27E-20 | 8 |
| DefensinHNP-1ΔC | AYRIPAIAGERRYGTIYQGRLWAF | −0.09 | 0.09 | N/A |
| 2xHA | YPYDVPDYAAYPYDVPDYAA | −0.01 | 0.63 | N/A |
Figure 3. SLAY platform demonstrated with a small, defined library.

A defined set of 5 peptides were cloned and pooled into a small library. The library was tested as described in Fig. 2 and methods over a period of 4 hours with plasmids isolation at 0, 2, 3 and 4 hour time points in duplicate. Reads were normalized to the input counts and plotted as a function of time.
SLAY Identifies Antimicrobial Sequences from a Massive Random Pool
The breadth of peptide chemical space with antimicrobial and potential therapeutic value is likely much larger than current screening approaches allow us to assess (Fjell et al., 2011; Neme et al., 2017). To test this hypothesis, we applied SLAY to screen a massive and unbiased peptide library of fully random sequences for antimicrobial activity. We constructed a library of approximately 800,000 peptides in E. coli W3110 using NNB codons to produce random peptide sequences with a target length of 20 amino acids. We sequenced this library and generated a sequence logo based on all peptides in the library (Supplementary Figure 5). We also generated a sequence logo based on a similarly computationally generated random 20-mer peptide library (Supplementary Figure 5). The sequence logo generated in both analysis was nearly identical indicating our ~800,000 peptide library did indeed contain a random assortment of sequences.
Library samples were collected pre-induction and post four-hour induction with 0.1mM IPTG in duplicate. Sequencing read counts are listed in Supplementary Table 1. Peptides were taken through two triage stages to identify hits with a high likelihood of true activity. Peptides were first sorted by their log2 reduction values. Peptides with a significant decrease of at least log2 fold −1 were considered to be depleted from the input library and to have potential antimicrobial activity. Next, we removed peptides that had less than or equal to 50 reads in each replicate. While somewhat arbitrary, samples with fewer reads will be more affected by machine errors than those with larger read counts so removing these decreases the overall noise in our analysis. As we anticipated from screening a random library the vast majority (98.3%) of sequences showed no depletion following induction indicating they had no antimicrobial activity (Fig. 4A). However, due to the massive throughput of SLAY, the 1.7% of the peptide library that did show depletion and potential antimicrobial activity represents 7,968 peptides. This single screen nearly doubled the number of unique antimicrobial peptides reported in publicly available databases(Hammami et al., 2009; Novkovic et al., 2012; Piotto et al., 2012; Waghu et al., 2016; Wang et al., 2016; Zhao et al., 2013b).
Figure 4. Computational analysis of the random peptide library screen results.

(A) Mean normalized input and output counts of total peptide library. Peptides considered active with lfcMLE < or = −1 are plotted in green. Peptides with lfcMLE > −1 were considered inactive are plotted in orange. Peptides removed from further analysis contained initial reads in either replicate of less than or equal to 50 and are plotted in yellow. (B) Screened peptides are plotted according to their hydrophobicity and charge properties. Active peptides are colored in green and inactive peptides are colored in orange. Ellipses represent a 95% confidence interval assuming a t-distribution. (C) A charge vs hydrophobicity plot comparing SLAY active peptides and known active peptides. Known antimicrobial peptides complied from six available online databases are colored in black, active peptides from our screen are colored green. Ellipses represent a 95% confidence interval assuming a t-distribution. (D) Plot of amino acid frequencies of known, active and inactive peptides from our screen. The error bars represent the SEM (standard error of the mean) and the asterisks correspond to Bonferroni adjusted p-values (*, **, and *** denote p-value <0.05, <0.01, and <0.001 respectively) derived from Tukey’s range test performed in conjunction with an ANOVA.
SLAY Reveals Untapped Chemical Diversity of Antimicrobial Peptides
Natural antimicrobial peptides are dominated by cationic and amphipathic composition. To begin to explore the range of hits identified by SLAY in the context of currently known antimicrobial chemistry, we plotted each active and inactive peptide from our screen by their charge and hydrophobicity. On average, the chemical composition of the library was centered near neutral charge and neutral hydrophobicity (Fig. 4B). Remarkably, we observed no bias in these parameters between inactive and active sequences from our library with the bulk of both peptide populations centered near neutral charge and neutral hydrophobicity (Fig 4B). Active sequences did not show a propensity towards any specific charge or hydrophobic character. This lack of selection is in sharp contrast to the bulk of naturally occurring antimicrobial peptides in current databases, which are dominated by positive charge and hydrophobic character (Fig 4C). Comparing amino acid frequency further highlights these observations (Fig. 4D). When examining our library we observed little enrichment of any specific amino acid in active vs. inactive sequences. Meanwhile positively charged lysine was found at a much higher frequency in known antimicrobial peptides compared to active sequences from our screen. Hydrophobic residues including alanine, isoleucine, leucine, valine, were also more frequent in known antimicrobial peptides compared to the active sequences from our screen. These results indicate that antimicrobial peptide sequence and chemical space extends far beyond what is known and has evolved in nature, and can be functionally explored through SLAY.
To further explore the composition of active sequences identified in our screen we performed a clustering analysis based on amino acid side chain properties to identify subclasses of peptide sequences that may be present in our hits. To facilitate our analysis, we simplified the amino acid sequence such that all the amino acids were grouped into the broad categories of polar positive, polar negative, polar uncharged, aromatic, nonpolar aliphatic, and cysteine. Supporting the breadth of antimicrobial sequences uncovered, we found large sequence differences between peptides, as measured by Levenshtein edit distances (min = 2, median = 13, max = 20). Using hierarchical clustering we sub-divided the peptides into 81 clusters with group sizes ranging from 8 to 259 peptides and a median of 68 peptides. (Supplementary Figure 6). We performed multiple sequence alignments on the simplified sequences of each of the 81 clusters to look for potential signature motifs. In general, no strong motif could be identified for any cluster although some clusters did have a simplified consensus sequence with an apparent hydrophobic domain in addition to variable domains that may facilitate membrane interactions. From the variance in cluster sizes and in the simplified consensus sequences, outlined in Supplemental Dataset 1, it is evident that the peptides discovered in this screen are extremely diverse and represent a vast potential for research into unexplored antimicrobial peptides. These results further support that active antimicrobial sequences exist in a much wider range of peptide chemical space than previously recognized that extends far beyond what has evolved in nature.
SLAY Hits are Active in Synthetic Form
To validate our hits, we selected 22 peptides based on chemical composition, predicted aqueous solubility (Pepcalc.com), and clustering diversity for chemical synthesis and antimicrobial activity testing. This included two cationic peptides, P1 and P2, that we selected to show SLAY can identify antimicrobial sequences reminiscent of naturally occurring CAMPs. In contrast, the remaining peptides (P3–P18) were selected for opposing characteristics—low hydrophobicity and neutral to negative charge. We chose these sequences to test if SLAY could identify peptide chemistry not typically associated with antimicrobial activity. One control peptide (C1) that had a neutral log2 fold reduction in our screen was used. These peptide sequences that were synthesized and tested for antimicrobial activity are listed in Table 2.
Table 2.
Fold change (log2) and antimicrobial (MBC) activities of peptides from SLAY.
| Peptide | Sequence | Groupa | Charge | Hydrophobicity | log2 fold | p-value | MBC (μM)b | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| W3110 | Ab5075 | PA14 | NDM E. coli | |||||||
| Cecropin P1 | SWLSKTAKKLENSAKKRISEGIAIAIQGGPR | - | 5.000 | −0.590 | - | - | ≤2 | ≤2 | ≤2 | ≤2 |
| P1 | RLVRILVSKRPVAIKPYFRL | 29 | 5.997 | 0.325 | −1.98 | <1.3e-5 | 2 | ≤2 | ≤2 | ≤2 |
| P2 | TTSIRRRYQVSLIRRHRGKR | 67 | 8.088 | −1.490 | −3.33 | <5.2e-21 | 16 | ≤2 | ≤2 | 4 |
| P3 cyclicc | TCRTNRPCFYDLDLNVCRCS | 26 | 0.750 | −0.381 | −4.60 | <2.85e-27 | ≤2 | 4 | ≤2 | 4 |
| P4 | SNGDGTLDAGSTCAPFYARA | 49 | −1.064 | −0.290 | −1.63 | <8.7e-11 | 2 | 16 | 4 | 4 |
| P5 cyclicd | YYNPLPHDCGRDNNTDICSR | 46 | −1.04 | −1.31 | −2.88 | <8.4 e-34 | 4 | 8 | ≤2 | ≤2 |
| P6 | LSVDKRPVLHPEHIYGHNHY | 73 | 0.362 | −0.910 | −3.63 | <1.2e-51 | 4 | 32 | 8 | >128 |
| P7 | IHRDQQHESFLDARPEPGLTE | 2 | −2.814 | −1.348 | −2.09 | <1.4e-9 | 4 | 16 | 4 | 4 |
| P8 | TIDFGVRNINQSNLVYDTER | 33 | −1.000 | −0.670 | −2.76 | <5.1e-44 | 8 | 16 | 8 | >128 |
| P9 | PCNPDHDYRPFGNFRIAFTT | 60 | 0.027 | −0.845 | −2.59 | <2.5e-15 | 8 | 16 | 8 | 8 |
| P10 | TRDTNDLISSRTAAPSMV | 60 | −0.001 | −0.422 | −10.7 | <2.0e-66 | 16 | 64 | 8 | 32 |
| P11 | LPLPSCSSHGGDADNTSQRN | 8 | −0.972 | −1.060 | −4.66 | <8.5e-59 | 16 | 128 | 16 | 32 |
| P12 | PNDPDSPCVYRMPNARGCSI | 75 | −0.126 | −0.765 | −3.80 | <2.3e-27 | 16 | 128 | 16 | 16 |
| P13 | YDLSDSNCLPANRDKRYYVI | 79 | −0.066 | −0.845 | −1.10 | <1.6e-12 | 16 | >128 | 16 | >128 |
| P14 | SMLAYVDKNDHINPPHSPRS | 10 | 0.180 | −1.055 | −5.49 | <8.9e-60 | 32 | 128 | 32 | 64 |
| P15 | DATPHAALFFTVKDHTAGDN | 69 | −1.819 | −0.380 | −2.83 | <8.6e-11 | 32 | 64 | 32 | 64 |
| P16 | SDDAQRCYPHNRTPFTYTYI | 39 | 0.025 | −1.230 | −2.18 | <2.8e-07 | 32 | >128 | 16 | 32 |
| P17 | EPCSPKNNYHDLFYRT | 22 | 0.027 | −1.488 | −1.80 | <6.2e-6 | 128 | >128 | 128 | >128 |
| P18 | CNPLNGADRRTDSFPRFTVI | 51 | 0.937 | −0.545 | −1.11 | <5.2e-8 | 128 | >128 | 64 | >128 |
| P3 | TCRTNRPCFYDLDLNVCRCS | 26 | 0.750 | −0.400 | −4.60 | <2.85e-27 | >128 | 128 | >128 | >128 |
| P5 | YYNPLPHDCGRDNNTDICSR | 46 | −1.035 | −1.380 | −2.88 | <8.4 e-34 | >128 | >128 | 128 | >128 |
| C1 | PDRAIDTYRTSPVADQRYNA | - | −0.002 | −1.245 | 0.09 | 0.999 | >128 | >128 | >128 | >128 |
Peptide group numbers were determined based on clustering as described in the methods.
Minimal bactericidal concentrations (MBC) were determined as the lowest concentration of peptide that results in at least 99.9% killing of the initial inoculums. Data are representative of three independent experiments.
P3 peptide was synthesized into cyclic formation by two disulfide bonds at C2–C19 and C8–C17.
P5 peptide was synthesized into cyclic formation by one disulfide bond at C9–C18. Disulfide bonds in P3 and P5 are represented by black bars.
We tested antimicrobial activity against our host strain used in the screen (E. coli W3110) and three multi-drug resistant strains: Acinetobacter baumannii (Ab 5075), Pseudomonas aeruginosa (PA14), and E. coli conferring New-Delhi metallo-beta-lactamase (NDM) resistance. Antimicrobial peptide activity is highly sensitive to medium conditions(Friedrich et al., 1999; Giacometti et al., 2000; Schwab et al., 1999). We first performed minimal inhibitory concentration (MIC) assays using Mueller-Hinton medium. Cationic peptides, P1 and P2 showed robust activity, like that of our standard CAMP cecropin P1 (Supplementary Table 2). Peptides P3–P22 did not show activity in this medium (data not shown). We next assayed antimicrobial activity using a simple and defined Tris based medium. Since the bacteria did not grow robustly in this medium we assayed the minimal bactericidal concentration (MBC) of each peptide. In this medium, cationic peptides P1 and P2 had potent antibacterial activity, with minimal bactericidal concentration (MBC) values of less than 2μM for P1 for all bacteria tested. Peptides P3–P18 had activity against the strain W3110 except for P3 and P5. Both peptides P3 and P5 contained cysteine residues suggesting possible cyclic formation is needed for activity. P5 contains two cysteine residues within its sequence, while P3 contains four. We had P5 synthesized as a cyclic peptide with a disulfide bond and retested its activity. This cyclic analog of P5 exhibited much higher antimicrobial activity, with MBC changing from >128μM to ≤2–8μM. Similarly, we tested a cyclic configuration of P3 with disulfides C2–C19 and C8–C17 and its antimicrobial activity increased from MBC of 128μM to ≤2–4μM. This further reiterates that SLAY can screen and select for cyclic peptides. Peptides P19–P22 as well as the control peptide C1 did not show activity in any medium we tested (Table 2 and data not shown). Thus, 18 of 22 (~80%) sequences identified by SLAY as active showed antimicrobial activity in at least one medium indicating a high true-positive rate. Select peptides were assayed in two additional media (Supplementary Table 3).
Cationic-hydrophobic peptide P1 showed universal activity, which is commonly associated with non-specific CAMP activity. Interestingly, P2, which is cationic but non-hydrophobic, showed a larger range of activity. Furthermore, many of our atypical, non-cationic, non-hydrophobic peptides (P3–P18) showed varying ranges in activity across the four Gram-negative bacteria tested. For example, P6, P8, P13, and P16 showed antimicrobial action against some strains while having no activity (>128 μM) against others. This suggests many of our peptides may act through a more targeted mechanism.
SLAY Hits Present with Different Potential Mechanisms of Peptide Action
Traditional antimicrobial peptides are considered to act non-specifically through membrane disruption with extremely rapid killing. In addition to the chemical landscape SLAY provides access to, we hypothesized that peptides identified by SLAY might also act through different mechanisms of peptide action.
Defining the target(s) and mechanism(s) of antibiotic action is challenging and is still debated for many clinically used antibiotics(Dwyer et al., 2015; Miller et al., 2016; Trimble et al., 2016; Zipperer et al., 2016). To begin to explore the mechanism of action of peptides identified by SLAY we compared their pore-forming activity and killing kinetics to the traditional CAMP cecropin P1. Peptide-dependent membrane damage is commonly assayed with propidium iodide (PI), which penetrates cells with compromised membranes to stain nucleic acids (Belloc et al., 1994; Darzynkiewicz et al., 1997). The effect of peptides on E. coli was probed by incubating peptide-treated cells with PI followed by flow cytometry analysis to determine peptide-induced membrane damage as previously described(Zhang et al., 2016) (Fig. 5A). Treatment of E. coli with cecropin P1, a known poreforming peptide, resulted in 33.7% of the population staining PI positive, indicating membrane damage. Cationic peptides P1 and P2 identified by SLAY exhibited even stronger membrane damage compared to cecropin P1, with 85.3% and 71.6% PI-positive cells respectively. Remarkably, peptides P3–P18 identified in our screen that contained atypically antimicrobial amino acid compositions compared to known CAMPs, did not cause cell fluorescence over 4%, with majority under 1%. This indicates that peptides P3–P18 identified through SLAY do not damage bacterial membranes, suggesting they kill bacteria via alternative mechanism(s) of action.
Figure 5. Mechanism of action of select peptides.

(A) The membrane damage of E. coli treated by peptides, as measured by an increase in fluorescence intensity of PI. E. coli was treated with 25μM peptide. Controls were processed without peptides. (B) Time-kill analysis of selected active peptides from our screen and cecropin P1. (C) Hemolytic activity of selected peptides at 50 μM.
We further probed the mechanism of SLAY peptides with time-dependent killing assays. While membrane-targeting CAMPs kill rapidly, antimicrobials targeting specific cellular processes tend to elicit their effect over a long period of time(Yang et al., 2006). We selected our top five non-cationic peptides, P3 cyclic, P4, P5 cyclic, P6, and P7 for testing. We assayed all peptides at 4X MBC. In our time-kill assay, cecropin P1 killed >99.9% of bacteria in less than 30 minutes (Fig. 5B). In contrast, our selected peptides acted over a longer time period. Peptides P3, P4, and P5 acted over 12 hours, while P6 and P7 acted over 18 hours. Additionally, development of resistance was not observed in W3110 during continuous serial passaging in the presence of subinhibitory concentration of the cationic peptide P1, while resistance could be generated against anionic P7 (Supplementary Figure 7). Combined with their non-pore forming action, these results suggest that peptides identified through SLAY may represent non-pore forming and diverse mechanism of action.
Hemolysis is a known off-target effect of CAMPs, with peptides such as protegrin-1 showing marked hemolysis at therapeutically relevant concentrations(Edwards et al., 2016). The hemolytic activities of the peptides against human red blood cells were determined as an indication of their toxicity towards mammalian cells. The hemolytic activities of all peptides are summarized in Figure 5C. PBS was used as a negative control and 1% triton was used as a positive control for 100% lysis. None of the peptides P3–P18 identified in our screen exhibited notable hemolytic activity, with all well under 20% hemolysis. However, maximal tolerated dose testing in CD-1 mice with cationic peptide P1 and anionic peptide P7 revealed marked differences in toxic effects. Cationic peptide P1 showed toxic effects at 25mg/kg (seizure-like activity) and caused immediate mortality at 35 mg/kg. This agrees with established literature that cationic peptides frequently have toxic effects (LeBeau et al., 2009). On the other hand, the anionic peptide P7 did not show any toxic effects up to the maximum dose of 50mg/kg.
DISCUSSION
SLAY presents a unique approach that challenges current drug discovery paradigms by replacing robotics, synthetic chemistry, and individual well reactions with molecular and computational techniques in a simple cell-based system for immediate biological relevance. Our screen of ~800,000 unique sequences revealed the untapped potential of peptide chemical space with antimicrobial activity. As anticipated from a random peptide screen, the vast majority of sequences screened (98.3%) showed no activity. However, with the efficient throughput of SLAY the 1.7% of active sequences still represents several thousand potential unique hits. Synthesis and testing of selected hits indicates a high true-positive rate for SLAY with ~80% of sequences tested having antimicrobial activity in synthetic form. Furthermore, since SLAY mounts peptides directly at the bacterial cell surface, it effectively increases their local concentration near potential targets. This may facilitate discovery of peptides with initially weak target interactions or poor medium solubility that can then be developed into more potent analogs.
We showed that SLAY can identify peptides similar to naturally occurring CAMPs and that these peptides have the expected pore forming activity and mammalian toxicity. Importantly we show that SLAY can identify peptide chemistry not typically associated with antimicrobial activity. For this purpose we tested diverse sequences with hydrophilic character and neutral-to-negative charge, and showed they could still kill several types of bacteria. The lack of detectable membrane activity among these peptides suggests they act through different mechanisms of action yet to be explored. The cell envelope of Gram-negative bacteria has many potential targets including essential protein complexes like the Bam, Lpt, and Lol systems(Lorenz et al., 2016; Mori et al., 2012; Srinivas et al., 2010). Alternatively, peptides discovered through SLAY may target and sequester essential metabolites as was suggested for the mechanism of action of teixobactin(Ling et al., 2015). Natural antimicrobial peptides evolved in the context of a complex immune system and were likely selected for more than their antimicrobial activity. Indeed, many naturally occurring CAMPs have been shown to have immune modulatory activity. Thus, while nature has provided predominantly one scaffold and target for antimicrobial peptide chemistry, our results with SLAY highlight the diversity of untapped antimicrobial peptide chemical space that can be explored for therapeutic value.
The power of SLAY lies in the high-throughput molecular foundation of the platform and opens the door for countless iterations. Our pipeline allows for progression from library construction through sequencing-based identification of antimicrobial leads that can then be validated synthetically and tested for in vivo effects. With this framework, peptide libraries of any size and composition can be easily screened in a broad range of Gram-negative bacteria, facilitating a wide range of uses. Analysis of our screen indicated no strong compositional bias between active and inactive peptide sequences. Thus, any area of antimicrobial peptide space can be explored by biasing the sequence composition of the initial library. In addition to composition we demonstrated that SLAY can be used to explore structure with easy identification of cyclic peptides, which have many positive pharmacokinetic properties. Once a lead sequence is identified, SLAY can be used to explore its sequence-function relationship by generating and testing a library of sequence derivatives. Furthermore, screens can be performed under any condition, such as in serum or in the presence of proteases to study the effects of these environmental changes on sequence activity. The unprecedented sequence-activity relationship data of functional and non-functional antimicrobial sequences gained through these screens will facilitate rational development of therapeutic peptides and the ability to broadly understand peptide chemical space with effects on bacterial physiology.
As with all screening procedures, SLAY can generate false-positive hits. False-positives could arise from peptide-fusion proteins that are toxic to the cell because they cause deleterious protein aggregates, stall translation, block essential secretion systems, or inhibit other essential functions during secretion to the cell surface. Some peptides may utilize the tether in their activity on the cell surface and would arise as false positives when synthesized and tested without the tether. Peptides identified by SLAY may not be soluble in synthetic form, limiting their potential use. Thus, it is important to validate hits as synthetic peptides.
Other than polymixins, antimicrobial peptides have not made a clinical impact for treating Gram-negative bacteria. However, clinical testing of peptides has been dominated by CAMPs, likely since they are the most commonly found form of antimicrobial peptide(Brunetti et al., 2017; Eckert, 2011; Fox, 2013; Kang et al., 2017). All lead compounds require development on their path to the clinic. Many features of antimicrobial peptides can be engineered to increase performance including salt tolerance, protease stability, and activity in serum(Carmona et al., 2013; Deslouches et al., 2005; Friedrich et al., 1999; Furman et al., 2015; Kim et al., 2014; Mai et al., 2011; Shin et al., 2015), but it has been challenging to engineer toxicity out of CAMPs. Peptide P7, showed no toxicity in mice suggesting non-cationic peptides identified through SLAY may be able to surmount the toxicity obstacle. These lead sequences will require optimization to improve the robustness of their activity, and future studies will seek to apply the excellent engineering principles developed for CAMPs to improve the activity and pharmacokinetics of well tolerated peptides like P7 and hopefully provide strong leads for preclinical development.
Bacteria have gained resistance to every antibiotic clinically used. There is no doubt they would gain resistance to any antimicrobial peptide discovered. However, the facile implementation of SLAY allows for continual iteration of peptide screens to identify leads as resistance arises. Thus, SLAY allows us to continuously spin the wheel and identify additional sets of antimicrobial peptides to thwart the inevitable rise of resistance. Conventional antibiotics that drive the problem of resistance are broad spectrum. While powerful, these drugs cannot distinguish between a target pathogen and a harmless commensal, and this collateral damage can further spur the development of antibiotic resistance. By screening the same peptide library in multiple Gram-negative bacteria, SLAY could allow for the identification of targeted peptides to eliminate only invading pathogens and reduce off-target consequences. SLAY allows us to enter previously unexplored chemical space for the first time and will facilitate the discovery of antibiotic scaffolds poised for further development.
STAR METHODS
Contact for Reagents and Resource Sharing
Requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Bryan W. Davies (bwdavies@austin.utexas.edu).
Experimental Model and Subject Details
Bacteria
Bacterial strains used in this study are listed in the Key Resources Table. All strains were grown aerobically at 37°C in Luria Bertani (LB) broth/aga r. The antibiotics carbenicillin 75 μg/mL (for E. coli and A. baumannii strains) or 150 μg/mL (for P. aeruginosa) were added for plasmid selection as needed.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| Escherichia coli W3110 Wild type, F−, λ− | Trent et al., 2001 | N/A |
| Escherichia coli W3110, pmrAC, polymyxinR | Trent et al., 2001 | N/A |
| Escherichia coli WD101ΔarnT:CmR | Herrera et al., 2010 | ST01 |
| Escherichia coli WD101ΔeptA:CmR | Herrera et al., 2010 | ST02 |
| Acinetobacter baumannii strain 17978 | ATCC | ATCC 17978 |
| Acinetobacter baumannii strain 5075 | Jacobs et al., 2014 | N/A |
| Pseudomonas aeruginosa strain PA14 | Liberati et al., 2006 | N/A |
| E. coli BAA-2452 | ATCC | ATCC BAA-2452 |
| E. coli W3110 carrying pSD03 | This study | SD01 |
| E. coli W3110 carrying pSD04 | This study | SD02 |
| E. coli WD101 carrying pSD05 | This study | SD03 |
| E. coli W3110 carrying pSD05 | This study | SD04 |
| WD101ΔarnT:CmR carrying pSD05 | This study | SD05 |
| WD101ΔeptA:CmR carrying pSD05 | This study | SD06 |
| E. coli W3110 carrying pSD06 | This study | SD07 |
| E. coli W3110 carrying pSD07 | This study | SD08 |
| A. baumannii 17978 carrying pSD07 | This study | SD09 |
| P. aeruginosa PA14 carrying pSD07 | This study | SD10 |
| E. coli W3110 carrying pSD08 | This study | SD11 |
| E. coli W3110 carrying pSD09 | This study | SD12 |
| E. coli W3110 carrying pMMB67EH | This study | SD13 |
| E. coli W3110 carrying pSD10 | This study | SD14 |
| E. coli W3110 carrying pSD11 | This study | SD15 |
| E. coli C2987 chemically competent cells | NEB | Cat# C2987I |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cecropin P1 | Sigma | Cat# C7927 |
| Denfensin HNP-1 | Sigma | Cat# D2043 |
| Protegrin | Anaspec | Cat# AS-64819-05 |
| Peptide P1 | Genscript | N/A |
| Peptide P2 | Genscript | N/A |
| Peptide P3 cyclicc | Genscript | N/A |
| Peptide P4 | Genscript | N/A |
| Peptide P5 cyclicd | Genscript | N/A |
| Peptide P6 | Genscript | N/A |
| Peptide P7 | Genscript | N/A |
| Peptide P8 | Genscript | N/A |
| Peptide P9 | Genscript | N/A |
| Peptide P10 | Genscript | N/A |
| Peptide P11 | Genscript | N/A |
| Peptide P12 | Genscript | N/A |
| Peptide P13 | Genscript | N/A |
| Peptide P14 | Genscript | N/A |
| Peptide P15 | Genscript | N/A |
| Peptide P16 | Genscript | N/A |
| Peptide P17 | Genscript | N/A |
| Peptide P18 | Genscript | N/A |
| Peptide P3 | Genscript | N/A |
| Peptide P5 | Genscript | N/A |
| Control Peptide C1 | Genscript | N/A |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Raw and analyzed sequencing data | This study | GEO: GSE94531 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: Envigo product 030 ICR (CD-1) outbred mice | Charles River Laboratories | Strain code: 022 |
| Oligonucleotides | ||
| Peptide library oligonucleotides | IDT | Table S4 |
| Recombinant DNA | ||
| pMMB67EH ampR | ATCC | pMMB67EH |
| pMMB67EH with lpp ompA | This study | N/A |
| pMMB67EH with lpp ompA 2x tether | This study | N/A |
| pMMB67EH with lpp ompA cecropin P1 | This study | N/A |
| pMMB67EH with lpp ompA 1x tether cecropin P1 | This study | N/A |
| pMMB67EH with lpp ompA 2x tether cecropin P1 | This study | N/A |
| pMMB67EH with lpp ompA 2x tether 2x HA | This study | N/A |
| pMMB67EH with lpp ompA 2x tether protegrin 1 | This study | N/A |
| pMMB67EH with lpp ompA 2x tether defensin HNP-1 | This study | N/A |
| pMMB67EH with lpp ompA 2x tether defensin HNP-1ΔC | This study | N/A |
| pMMB67EH with 2x tether cecropin P1 | This study | N/A |
| pMMB67EH with lpp ompA 2x tether dermaseptin | This study | N/A |
| Software and Algorithms | ||
| GraphPad Prism 5 for Mac OS X | GraphPad Software, Inc | https://www.graphpad.com/scientificsoftware/prism, RRID:SCR_002798 |
| Flexbar | Dodt et al., 2012 | http://sourceforge.net/projects/flexbar, RRID:SCR_013001 |
| Ustacks | Catchen et al., 2013; Catchen et al., 2011 | N/A |
| Biopython | Cock et al., 2009 | http://biopython.org, RRID:SCR_007173 |
| DESeq2 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq.html, RRID:SCR_000154 |
| R/Bioconductor | Huber et al., 2015 | https://www.bioconductor.org, RRID:SCR_006442 |
| FlowJo | Treestar, USA | http://www.flowjo.com, RRID:SCR_008520 |
| Other | ||
Mice
All procedures involving animals were performed in accordance with Charles River Laboratories’ protocols and SOPs and were approved by the Institution’s Animal Care and Use Committee. Mice studies were carried out with 5 week old male CD-1 mice from Envigo (product 030 ICR (CD-1) outbred mice) and tested at Charles River Laboratories. Mice were allowed to acclimate to the test facility for at least 2 days prior to study start. Mice were given a single intravenous bolus dose of peptide at 5, 10, 25, 35 and 50 mg/kg. Dosing was done in triplicate. All animals were weighed prior to and at dosing and observed twice daily through day 7.
Method Details
Bacterial growth curves with surface displayed peptides
Strains were grown overnight at 37°C. The following day cultures were inoculated and grown to log phase. The cultures were then back diluted to OD 600nm 0.01. IPTG was added to the cultures where appropriate. Data points were collected every 20 mins over a 6 hour period using a SpectraMax Plus384 absorbance microplate reader with SOFTmax Pro v6.2.2 software.
Mixed-culture assay with empty plasmid and Cecropin P1
Strains were grown overnight at 37°C. The following day cultures were inoculated and grown to log phase. The cultures were then back diluted to OD 600nm 0.01 in 5 mLs of LB containing 75 μg/mL carbenicillin. Separate cultures containing E. coli containing the empty plasmid and E. coli containing the plasmid to display cecropin P1 were mixed at a 1:1 ratio with a final OD 600nm of 0.01. Surface expression was induced with 1 mM IPTG. Cultures were serial diluted and spotted on plates containing carbenicillin 75 μg/mL with 80 μg/mL X-gal at 0 and 3 hours.
Peptide library construction
The surface display system was constructed on the broad host plasmid pMMB67EH. Random peptide sequences were generated using NNB codons in a 60-base nucleotide segment to produce 20 amino acid long peptides. Random sequences were cloned into the KpnI and SalI sites using primers with homology to the tether sequence on the reverse primer. The library was then transformed into C2987 competent cells (NEB) in batch and plated. Roughly 800,000 colonies were plated and pooled. Cells were harvested and aliquoted into glycerol stocks. Plasmid DNA was isolated from the library and re-transformed into the E. coli W3110 strain at 3 to 5 times coverage. Colonies were collected and frozen.
Screening and Sequencing the Defined and Random Peptide Library
An aliquot of the frozen library was thawed and added to 10ml of LB supplemented with carbenicillin 75 μg/ml for growth, shaking at 37°C for about 1 hour. The culture was then back diluted into 5ml LB with carbenicillin 75 μg/ml to OD 600nm 0.01 supplemented with 0.1mM IPTG. The remaining culture was collected as the “Input” sample. Induced cultures were allowed to grow, shaking at 37°C. Cells were harvested after 2, 3, and 4 hours for the defined peptide library and harvest after 4 hours for the random peptide library. Plasmids were isolated from each culture using the Zyppy Plasmid Miniprep kit from Zymo Research Corp. (Irvine, CA). Samples were collected in duplicate. Plasmid concentrations were measured using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). Primers with homologous regions to the plasmid were used to amplify and attach adaptors for sequencing (See Table S4). Briefly, 10ng plasmid DNA and 1ul of 10μM primer mix [2ul of forward primer and 2ul of reverse primer diluted with 16ul dH2O] were added to a 2x master mix of Phusion high-fidelity polymerase (NEB) in a total volume of 50ul. Four reactions per sample were run for a total of 12 cycles. The reactions were pooled and cleaned using Zymo DNA Clean and Concentrator (Irvine, CA). The complete libraries were further gel purified and extracted using the Zymoclean Gel DNA Recovery kit (Irvine, CA). The defined peptide library was sequenced using Illumina Mi-seq. The random peptide library was sequenced using Illumina Hi-seq supplemented with Phi-X. DNA was sequenced at The University of Texas Genomic Sequencing and Analysis Facility.
Read Trimming and Counting
We used flexbar(Dodt et al., 2012) to trim the reads of excess sequence. To do this, we searched for a known sequence as part of the peptide display sequence, “CTCCAGCTGCGGGTATC,” and then retained 73 nucleotides downstream of this sequence. This allowed us to retain the test peptide sequence, as well as a preceding ‘GG’ motif and ending stop codon for each read. Next, we used ustacks (from the stacks computational pipeline(Catchen et al., 2013; Catchen et al., 2011)) to consolidate reads that originated from a particular nucleotide sequence. This allowed us to collect reads for each tested peptide as well as reads with <2 nucleotide mismatches. We translated the nucleotide sequences for each nucleotide sequence into an amino acid sequence with a custom python script using the Biopython(Cock et al., 2009) library, and then summed the total reads for each unique peptide. To reduce false positives, we retained only peptides that started with the expected ‘GG’ motif and were present in both input libraries. Hiseq analysis was also performed from ustacks without flexbar followed by sequence trimming.
Differential Abundance Analysis
A file containing each peptide sequence and the total number of reads in each library was then used as input for the DESeq2(Love et al., 2014) R/Bioconductor(Huber et al., 2015) package. DESeq2 is a commonly used R/Bioconductor package for count based differential testing with next generation sequencing data. We used a standard DESeq2 workflow, which includes read count normalization, peptide dispersion estimates, and Wald tests for significance of differential abundance. For each peptide, we compared the total abundance (normalized reads) in the induced (IPTG) libraries to the abundance in the input libraries, resulting in a log2 FoldChange (log2FC) and p-value for each peptide. P-values were adjusted (padj) for multiple testing using Benjamini-Hochberg correction as part of the standard DESeq2 workflow detailed in the package manual (available here: https://www.bioconductor.org/packages/devel/bioc/vignettes/DESeq2/inst/doc/DESeq2.pdf). Peptides with lfcMLE < or = −1 were considered active (7,968 peptides). Peptides with lfcMLE > −1 were considered inactive. Peptides were considered removed if the initial reads in either replicate of less than or equal to 50. Generally, peptides that were selected for further experimental validation had a log2FC < −1 and padj < .05.
Calculating Peptide Properties
After calculating the change in abundance for each peptide, we computed expected properties for each peptide sequence using a custom R script. After removing the preceding ‘GG’ motif and trailing stop codon, we used the “Peptides” R package (https://cran.r-project.org/package=Peptides) to calculate charge and hydrophobicity for the remaining 20 amino acids. Specific methods used to calculate charge(Nelson and Cox, 2004) and hydrophobicity(Kyte and Doolittle, 1982) are detailed in the Peptides package documentation. We compared calculated properties (hydrophobicity and charge) of our screened peptides with those of 8685 known antimicrobial peptides from available online databases (Hammami et al., 2009; Novkovic et al., 2012; Piotto et al., 2012; Waghu et al., 2016; Wang et al., 2016; Zhao et al., 2013b).
Generation of Logo Plots
Logo plots were generated using R package “RWeblogo”. RWebLogo: plotting custom sequence logos. R package version 1.0.3. https://CRAN.R-project.org/package=RWebLogo), a programmatic interface to make sequence logos(Crooks et al., 2004; Schneider and Stephens, 1990). Briefly, sequence logos were generated from either the entire set of possible killing peptides (7,968 sequences), 10,000 randomly sampled sequences of the total library, or an amino acid translation of 10,000 randomly generated nucleotide sequences of a repeated “NNB” motif. All logos are plotted in units of probability.
Generation of Amino Acid Frequencies
Individual amino acid frequencies were determined for each peptide (simply # of amino acids/length of peptide). Then average frequencies were calculated per group, which is what is graphed. The error bars represent the SEM (standard error of the mean) and the asterisks correspond to Bonferroni adjusted p-values (*, **, and *** denote p-value <0.05, <0.01, and <0.001 respectively) derived from Tukey’s range test performed in conjunction with an ANOVA.
Clustering Analysis
The clustering analysis was conducted on the 7,968 peptides with at least a −1 log2 fold depletion from the antimicrobial peptide screen. We then screened these peptides for those that were 15 amino acids or longer and continued with the clustering the resulting 6,565 peptides that fit into this group. This was done to limit inaccurate clustering that could result from biochemically unrepresentative edit distances due to large differences in peptide length. With these 6,565 peptides, we then simplified their amino acid sequence such that all the amino acids were grouped into the broad categories of polar positive (Arg, His, Lys), polar negative (Asp, Glu), polar uncharged (Ser, Thr, Asn, Gln, Pro), aromatic (Phe, Tyr, Trp), nonpolar aliphatic (Ala, Val, Ile, Leu, Met, Gly), and cysteine amino acids. Cysteine was left as its own group do to its unique ability to form disulfide bonds. This simplification of peptide sequences was used due to the incredible diversity of the hits which hindered clustering of non-simplified peptide sequences. We then acquired a Levenshtein distance for every pairing of peptides in this list of 6,565 simplified peptide sequences. These distances were then used as the edit distance inputs for a complete-linkage hierarchal clustering analysis utilizing R’s hclust command. The resulting clustering dendrogram was then arbitrarily sub-divided into 81 groups representing different groups of similar peptides identified in this analysis. To check to see if the cutoff for subgroups was reasonable and whether any patterns could be identified in the groupings multiple sequence alignments for each group’s simplified sequence was generated using the R package msa. The multiple sequence alignment used was Clustal W with default settings. The consensus sequence for a group is made up of amino acids with presence in at least 50% of the sequences for a given position.
Antimicrobial Activity Assays
Minimum bactericidal concentration (MBC) assays were adapted previous methods (Mah, 2014; Qaiyumi, 2007). Briefly, strains were grown overnight on an LB agar plate at 37°C. A small number of bacteria was scraped from the plate and added to LB and grown to log phase. Cells were collected, washed twice and suspended in a 2X concentrations of assay medium at a density of 1 × 10^6 CFU/mL. 50ul of bacteria were added to each well in a polypropylene 96-well plate (Corning Inc., Lowell, MA, USA). Unless otherwise stated the assay, medium was 10mM Tris (pH7.4) + 25mM NaCl. Where indicated 10mM Tris (pH7.4) + 25mM NaCl + 0.05% glucose or 1% Tryptone Broth assay medium was used. Peptides with >90% purity were synthesized by Genscript (GenScript USA Inc., NJ). Synthesized peptides were diluted to 256μM and serial diluted for a total volume of 100ul of each dilution. Then, 50ul of each peptide solution was added to 50ul of cells. Peptides were diluted in 0.2% BSA, 0.01% acetic acid solution for Tris medium assays and water for Tryptone medium assays. Plates were parafilmed and incubated at 37°C overnight. After 20 hours, each well was spotted onto LB agar to assess cell viability. MBCs were determined where cells had a 3-log reduction in growth.
Minimal inhibitory concentration (MIC) assays were adapted previous methods (Wiegand et al., 2008). Briefly, strains were grown overnight on an LB agar plate at 37°C. A small number of bacteria was scraped from the plate and added to Mueller-Hinton growth media and grown to log phase. Cells were diluted to 1 × 10^6 CFU/mL and 50ul were added to each well in a polypropylene 96-well plate. Synthesized peptides were diluted into 0.2% BSA, 0.01% acetic acid solution to 64μM and serial diluted for a total volume of 100ul of each dilution. Then, 50ul of each peptide solution was added to 50ul of cells. Plates were parafilmed and incubated at 37°C overnight. The MIC was determined by OD600nm where cell density was 0.
Detection of Peptide-induced Membrane Permeability
Bacterial cell membrane damage and pore formation induced by the peptides was examined by detection of propidium iodide (PI) influx(Zhang et al., 2016). The bacteria were cultured at 37°C to mid-log phase and then diluted to OD600 0.1 in 10mM Tris (pH 7.4), 25mM NaCl. Synthesized peptides, at a concentration of 25μM, were added to a 500ul bacterial suspension and incubated for 30 min. Bacteria were collected and resuspended in buffer. PI solution was added to a final concentration of 2ug/ml. The fluorescence signal in treated cells was determined by flow cytometry (BD Accuri) and further analyzed with FlowJo (Treestar, USA).
Analysis of Hemolytic Activity
Hemolytic assays were performed as described previously(Zhao et al., 2013a). Briefly, 50 μM solutions of synthesized peptides were prepared by mixing the peptides by inversion in 10 mM PBS at pH 7.4 for a total volume of 0.5 mL. A human red blood cell solution was made by washing 0.4 mL of the red blood cells twice with 7 mL of PBS by centrifugation at 2500 rpm for 10 minutes. The precipitates were then resuspended in 4 mL of PBS. Hemolytic activity of the peptides was measured by first mixing by inversion the 0.5 mL peptide solutions with 0.4 mL of the human red blood cell solution. The mixtures were placed in a 37°C water bath for 1 h. A negative control of 0.5 mL PBS plus 0.4 mL human red blood cell solution and a positive control of 1% (w/v) Triton X-100 plus 0.4 mL of human red blood cell solution were also incubated in the water bath. After one hour, the samples were centrifuged at 2500 rpm for 10 minutes. The absorbance of the supernatant was measured at 540 nm. The percent hemolysis was calculated using the following equation.
Time-course Antimicrobial Assay
Kinetics assays were set up identically to the MBC assay with the following exceptions. A total volume of 200ul was added to 96-well plates in triplicate. At time points of 30 min, 1, 3, 6, 9, 12, 18, and 24 hours, 15ul of sample was removed from each well. Aliquots were serial diluted and plated to assess viability.
Resistance Development Assay
Initial MBC values for peptide P1 and P7 used in this study are reported in Table 2. E. coli W3110 suspensions were inoculated and assayed at 0.125x to 8x-MBC as described for MBC assays above. After incubation, bacteria were plated for MBC and an aliquot from each well was grown in MH medium at 37°C. Bacteria from the highest concentration below the determined MBC (1/2x-MBC) were used to repeat the MBC, adjusting the concentrations for any observed increase in resistance.
Maximal Tolerated Dose Assay
The MTD assay was carried out with 5 week old male CD-1 mice (Charles River Laboratories Inc, MA, USA). Peptides were soluble to 5 mg/mL, which allowed dosing up to 50 mg/kg. Mice were given a single intravenous bolus dose of peptide at 5, 10, 25, 35 and 50 mg/kg. Dosing was done in triplicate. All animals were weighed prior to and at dosing and observed twice daily through Day 7.
Quantification and Statistical Analysis
Experimental Replicates
Replicates are described in the Results and Figure legends. All growth curves were analyzed using GraphPad Prism 5 Software. Standard error of the mean (SEM) from triplicate samples is shown as error bars. Libraries were assayed in duplicate. All MICs and MBCs shown in Tables were determined from at least three biological replicates.
Data and Software Availability
The sequence data have been deposited with the NCBI’s High-Throughput Sequencing Omnibus under Accession Number GSE94531.
Supplementary Material
Cecropin P1 is expressed intracellularly with the 2x tether (without lpp-ompA for localization to the cell surface) at 0 mM, 0.01 mM, 0.1 mM and 1 mM IPTG. Growth curves were performed in triplicate over 6 hours. Data are represented as mean ± SEM.
Cecropin P1 is displayed with no tether, 1x tether and a 2x at 0 mM and 0.1 mM IPTG. Growth curves were performed in triplicate over 6 hours. Data are represented as mean ± SEM.
Cecropin P1 is displayed with increasing concentrations of magnesium and trypsin. Controls were cecropin P1 expressed with no added magnesium or trypsin enzyme. Growth curves were performed in triplicate over 6 hours. Data are represented as mean ± SEM.
Dermaseptin S4L7K is displayed on the surface. IPTG concentrations tested were 0mM, 0.01mM, 0.1mM and 1mM. Growth curves were performed in triplicate over 6 hours. Data are represented as mean ± SEM.
Sequence logo comparison between a randomly generated 20 amino acid library, our library, and the top hits generated from our library. Sequence logos were generated from either the entire set of possible killing peptides (7,968 sequences), 10,000 randomly sampled sequences of the total library, or an amino acid translation of 10,000 randomly generated nucleotide sequences of a repeated “NNB” motif. All logos are plotted in units of probability.
The resulting clustering dendrogram was divided into 81 subgroups representing different groups of similar peptides identified in this analysis.
Serial passage of E. coli W3110 with 15 days of sub-inhibitory concentrations of peptides P1 and P7. Passages were done in triplicate. Resistance is not observed with cationic P1 peptide. One replicate of the anionic P7 peptide developed 64-fold resistance over the 15-day period.
Table S1. Summary of sequencing read counts for sample libraries, Related to STAR Methods
Table S2. MICs of synthesized SLAY peptides and Cecropin P1, Related to Table 2
Table S3. Selected peptides assayed in 1% tryptone broth and TNG media, Related to Table 2
Table S4. Oligonucleotide sequences used in this study, Related to STAR Methods
HIGHLIGHTS.
Development of a high-throughput platform for discovery of antimicrobial peptides
Screening 800,000 peptides uncovered thousands of synthetic antimicrobial sequences
Lead peptides exhibit potent antimicrobial activity and distinctive mechanisms
Lead hit antimicrobial physicochemistry extend far beyond what nature has evolved
Acknowledgments
This work was supported by NIH Grant AI125337 to BWD, Sanofi iAward to BWD, Welch Foundation Grant F-1870 to B.W.D and DARPA HR0011-15-C-0095 to B.W.D. NIH Grants AI064184, AI076322 to M.S.T. and Army Research Office grant W911NF-12-1-0390 to M.S.T; and NIH NRSA Fellowship F32 GM116523 to A.T.T. We would like to thank the Genome Sequencing and Analysis Facility (GSAF) for next-generation sequencing, and the Microscopy and Imaging Facility for flow cytometry use at UT Austin. None of the authors of this manuscript have a financial interest related to this work.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization A.T.T. and B.W.D; Methodology A.T.T., B.W.D, S.P.L., C.D.D., G.A.K., A.L.C.; Software S.P.L., C.D.D., G.A.K., C.O.W.; Validation A.T.T., S.P.L., C.D.D., G.A.K., A.L.C., C.O.W., B.W.D.; Formal Analysis A.T.T., S.P.L., C.D.D., G.A.K., A.L.C., C.O.W., B.W.D.; Investigation A.T.T., S.P.L., C.D.D., G.A.K., A.L.C., B.W.D.; Resources C.O.W., M.S.T., B.W.D.; Data Curation A.T.T., S.P.L., C.D.D., G.A.K.; Writing – Original Draft A.T.T., B.W.D.; Writing – Review & Editing A.T.T., S.P.L., C.D.D., G.A.K., A.L.C., C.O.W., M.S.T., B.W.D.; Visualization A.T.T., S.P.L., C.D.D., G.A.K.; Supervision A.T.T., C.O.W., M.S.T., B.W.D.; Funding Acquisition B.W.D., C.O.W., M.S.T.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bahar AA, Ren D. Antimicrobial peptides. Pharmaceuticals (Basel) 2013;6:1543–1575. doi: 10.3390/ph6121543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloc F, Dumain P, Boisseau MR, Jalloustre C, Reiffers J, Bernard P, Lacombe F. A Flow Cytometric Method Using Hoechst-33342 and Propidium Iodide for Simultaneous Cell-Cycle Analysis and Apoptosis Determination in Unfixed Cells. Cytometry. 1994;17:59–65. doi: 10.1002/cyto.990170108. [DOI] [PubMed] [Google Scholar]
- Brunetti J, Falciani C, Bracci L, Pini A. Models of In-Vivo Bacterial Infections for the Development of Antimicrobial Peptide-based Drugs. Curr Top Med Chem. 2017;17:613–619. doi: 10.2174/1568026616666160713143017. [DOI] [PubMed] [Google Scholar]
- Carmona G, Rodriguez A, Juarez D, Corzo G, Villegas E. Improved protease stability of the antimicrobial peptide Pin2 substituted with D-amino acids. Protein J. 2013;32:456–466. doi: 10.1007/s10930-013-9505-2. [DOI] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22:3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: building and genotyping Loci de novo from short-read sequences. G3 (Bethesda) 2011;1:171–182. doi: 10.1534/g3.111.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F. Cytometry in cell necrobiology: Analysis of apoptosis and accidental cell death (necrosis) Cytometry. 1997;27:1–20. [PubMed] [Google Scholar]
- Deslouches B, Islam K, Craigo JK, Paranjape SM, Montelaro RC, Mietzner TA. Activity of the de novo engineered antimicrobial peptide WLBU2 against Pseudomonas aeruginosa in human serum and whole blood: implications for systemic applications. Antimicrob Agents Chemother. 2005;49:3208–3216. doi: 10.1128/AAC.49.8.3208-3216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt M, Roehr JT, Ahmed R, Dieterich C. FLEXBAR-Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms. Biology (Basel) 2012;1:895–905. doi: 10.3390/biology1030895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer DJ, Collins JJ, Walker GC. Unraveling the physiological complexities of antibiotic lethality. Annu Rev Pharmacol Toxicol. 2015;55:313–332. doi: 10.1146/annurev-pharmtox-010814-124712. [DOI] [PubMed] [Google Scholar]
- Eckert R. Road to clinical efficacy: challenges and novel strategies for antimicrobial peptide development. Future Microbiol. 2011;6:635–651. doi: 10.2217/fmb.11.27. [DOI] [PubMed] [Google Scholar]
- Edwards IA, Elliott AG, Kavanagh AM, Zuegg J, Blaskovich MA, Cooper MA. Contribution of Amphipathicity and Hydrophobicity to the Antimicrobial Activity and Cytotoxicity of beta-Hairpin Peptides. ACS Infect Dis. 2016;2:442–450. doi: 10.1021/acsinfecdis.6b00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell CD, Hiss JA, Hancock RE, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov. 2011;11:37–51. doi: 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20:122–128. doi: 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Fox JL. Antimicrobial peptides stage a comeback. Nat Biotechnol. 2013;31:379–382. doi: 10.1038/nbt.2572. [DOI] [PubMed] [Google Scholar]
- Friedrich C, Scott MG, Karunaratne N, Yan H, Hancock RE. Salt-resistant alpha-helical cationic antimicrobial peptides. Antimicrob Agents Chemother. 1999;43:1542–1548. doi: 10.1128/aac.43.7.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL, Chiu M, Hunter MJ. Early engineering approaches to improve peptide developability and manufacturability. AAPS J. 2015;17:111–120. doi: 10.1208/s12248-014-9681-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit E, Boman A, Boman HG, Shai Y. Interaction of the mammalian antibacterial peptide cecropin P1 with phospholipid vesicles. Biochemistry. 1995;34:11479–11488. doi: 10.1021/bi00036a021. [DOI] [PubMed] [Google Scholar]
- Georgiou G, Stephens DL, Stathopoulos C, Poetschke HL, Mendenhall J, Earhart CF. Display of beta-lactamase on the Escherichia coli surface: outer membrane phenotypes conferred by Lpp’-OmpA’-beta-lactamase fusions. Protein engineering. 1996;9:239–247. doi: 10.1093/protein/9.2.239. [DOI] [PubMed] [Google Scholar]
- Giacometti A, Cirioni O, Barchiesi F, Del Prete MS, Fortuna M, Caselli F, Scalise G. In vitro susceptibility tests for cationic peptides: comparison of broth microdilution methods for bacteria that grow aerobically. Antimicrob Agents Chemother. 2000;44:1694–1696. doi: 10.1128/aac.44.6.1694-1696.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould IM, Bal AM. New antibiotic agents in the pipeline and how they can help overcome microbial resistance. Virulence. 2013;4:185–191. doi: 10.4161/viru.22507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LL, Beveridge TJ, Nanninga N. Periplasmic Space and the Concept of the Periplasm. Trends Biochem Sci. 1991;16:328–329. doi: 10.1016/0968-0004(91)90135-i. [DOI] [PubMed] [Google Scholar]
- Hadley EB, Hancock RE. Strategies for the discovery and advancement of novel cationic antimicrobial peptides. Curr Top Med Chem. 2010;10:1872–1881. doi: 10.2174/156802610793176648. [DOI] [PubMed] [Google Scholar]
- Hammami R, Ben Hamida J, Vergoten G, Fliss I. PhytAMP: a database dedicated to antimicrobial plant peptides. Nucleic Acids Res. 2009;37:D963–968. doi: 10.1093/nar/gkn655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- Herrera CM, Hankins JV, Trent MS. Activation of PmrA inhibits LpxT-dependent phosphorylation of lipid A promoting resistance to antimicrobial peptides. Mol Microbiol. 2010;76:1444–1460. doi: 10.1111/j.1365-2958.2010.07150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilpert K, Volkmer-Engert R, Walter T, Hancock RE. High-throughput generation of small antibacterial peptides with improved activity. Nat Biotechnol. 2005;23:1008–1012. doi: 10.1038/nbt1113. [DOI] [PubMed] [Google Scholar]
- Hilpert K, Winkler DF, Hancock RE. Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nat Protoc. 2007;2:1333–1349. doi: 10.1038/nprot.2007.160. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs AC, Thompson MG, Black CC, Kessler JL, Clark LP, McQueary CN, Gancz HY, Corey BW, Moon JK, Si Y, et al. AB5075, a Highly Virulent Isolate of Acinetobacter baumannii, as a Model Strain for the Evaluation of Pathogenesis and Antimicrobial Treatments. MBio. 2014;5:e01076–01014. doi: 10.1128/mBio.01076-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HK, Kim C, Seo CH, Park Y. The therapeutic applications of antimicrobial peptides (AMPs): a patent review. J Microbiol. 2017;55:1–12. doi: 10.1007/s12275-017-6452-1. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Park SJ, Mishig-Ochir T, Lee BJ. Antimicrobial peptides: therapeutic potentials. Expert Rev Anti Infect Ther. 2014;12:1477–1486. doi: 10.1586/14787210.2014.976613. [DOI] [PubMed] [Google Scholar]
- Kim H, Jang JH, Kim SC, Cho JH. De novo generation of short antimicrobial peptides with enhanced stability and cell specificity. J Antimicrob Chemother. 2014;69:121–132. doi: 10.1093/jac/dkt322. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- LeBeau AM, Brennen WN, Aggarwal S, Denmeade SR. Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Mol Cancer Ther. 2009;8:1378–1386. doi: 10.1158/1535-7163.MCT-08-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K. Platforms for antibiotic discovery. Nat Rev Drug Discov. 2013;12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- Li X, Parker S, Deeudom M, Moir JW. Tied down: tethering redox proteins to the outer membrane in Neisseria and other genera. Biochemical Society transactions. 2011;39:1895–1899. doi: 10.1042/BST20110736. [DOI] [PubMed] [Google Scholar]
- Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A. 2006;103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schaberle TF, Hughes DE, Epstein S, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz C, Dougherty TJ, Lory S. Transcriptional Responses of Escherichia coli to a Small-Molecule Inhibitor of LolCDE, an Essential Component of the Lipoprotein Transport Pathway. J Bacteriol. 2016;198:3162–3175. doi: 10.1128/JB.00502-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah TF. Establishing the minimal bactericidal concentration of an antimicrobial agent for planktonic cells (MBC-P) and biofilm cells (MBC-B) J Vis Exp. 2014:e50854. doi: 10.3791/50854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai J, Tian XL, Gallant JW, Merkley N, Biswas Z, Syvitski R, Douglas SE, Ling J, Li YH. A novel target-specific, salt-resistant antimicrobial peptide against the cariogenic pathogen Streptococcus mutans. Antimicrob Agents Chemother. 2011;55:5205–5213. doi: 10.1128/AAC.05175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WR, Bayer AS, Arias CA. Mechanism of Action and Resistance to Daptomycin in Staphylococcus aureus and Enterococci. Cold Spring Harb Perspect Med. 2016:6. doi: 10.1101/cshperspect.a026997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori N, Ishii Y, Tateda K, Kimura S, Kouyama Y, Inoko H, Mitsunaga S, Yamaguchi K, Yoshihara E. A peptide based on homologous sequences of the beta-barrel assembly machinery component BamD potentiates antibiotic susceptibility of Pseudomonas aeruginosa. J Antimicrob Chemother. 2012;67:2173–2181. doi: 10.1093/jac/dks174. [DOI] [PubMed] [Google Scholar]
- Nelson DL, Cox MML. Principles of Biochemistry. 4 2004. [Google Scholar]
- Neme R, Amador C, Yildirim B, McConnell E, Tautz D. Random sequences are an abundant source of bioactive RNAs or peptides. Nature Ecology & Evolution. 2017:1. doi: 10.1038/s41559-017-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novkovic M, Simunic J, Bojovic V, Tossi A, Juretic D. DADP: the database of anuran defense peptides. Bioinformatics. 2012;28:1406–1407. doi: 10.1093/bioinformatics/bts141. [DOI] [PubMed] [Google Scholar]
- O’Neill J. Tackling drug-resistant infections globally: Final report and recommendations. The Review on Antimicrobial Resistance. 2016:84. [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Peschel A, Sahl HG. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat Rev Microbiol. 2006;4:529–536. doi: 10.1038/nrmicro1441. [DOI] [PubMed] [Google Scholar]
- Piotto SP, Sessa L, Concilio S, Iannelli P. YADAMP: yet another database of antimicrobial peptides. Int J Antimicrob Agents. 2012;39:346–351. doi: 10.1016/j.ijantimicag.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Qaiyumi S. Macro- and Microdilution Methods of Antimicrobial Susceptibility Testing. In: Schwalbe R, Steele-Moore L, Goodwin AC, editors. Antimicrobial susceptibility testing protocols. Boca Raton: Crc Press; 2007. p. 79. [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab U, Gilligan P, Jaynes J, Henke D. In vitro activities of designed antimicrobial peptides against multidrug-resistant cystic fibrosis pathogens. Antimicrob Agents Chemother. 1999;43:1435–1440. doi: 10.1128/aac.43.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin A, Lee E, Jeon D, Park YG, Bang JK, Park YS, Shin SY, Kim Y. Peptoid-Substituted Hybrid Antimicrobial Peptide Derived from Papiliocin and Magainin 2 with Enhanced Bacterial Selectivity and Anti-inflammatory Activity. Biochemistry. 2015;54:3921–3931. doi: 10.1021/acs.biochem.5b00392. [DOI] [PubMed] [Google Scholar]
- Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, Van der Meijden B, Bernardini F, Lederer A, Dias RL, et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science. 2010;327:1010–1013. doi: 10.1126/science.1182749. [DOI] [PubMed] [Google Scholar]
- Trent MS, Ribeiro AA, Doerrler WT, Lin S, Cotter RJ, Raetz CR. Accumulation of a polyisoprene-linked amino sugar in polymyxin-resistant Salmonella typhimurium and Escherichia coli: structural characterization and transfer to lipid A in the periplasm. The Journal of biological chemistry. 2001;276:43132–43144. doi: 10.1074/jbc.M106962200. [DOI] [PubMed] [Google Scholar]
- Trimble MJ, Mlynarcik P, Kolar M, Hancock RE. Polymyxin: Alternative Mechanisms of Action and Resistance. Cold Spring Harb Perspect Med. 2016:6. doi: 10.1101/cshperspect.a025288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkey J, Nagaraj R. Antibacterial activity of human neutrophil defensin HNP-1 analogs without cysteines. Antimicrob Agents Chemother. 2005;49:4561–4566. doi: 10.1128/AAC.49.11.4561-4566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waghu FH, Barai RS, Gurung P, Idicula-Thomas S. CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016;44:D1094–1097. doi: 10.1093/nar/gkv1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016;44:D1087–1093. doi: 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics 2017 [Google Scholar]
- Wiegand I, Hilpert K, Hancock RE. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc. 2008;3:163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- Woodruff HB. Selman A. Waksman, winner of the 1952 Nobel Prize for physiology or medicine. Appl Environ Microbiol. 2014;80:2–8. doi: 10.1128/AEM.01143-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ST, Shin SY, Hahm KS, Kim JI. Different modes in antibiotic action of tritrpticin analogs, cathelicidin-derived Trp-rich and Pro/Arg-rich peptides. Biochim Biophys Acta. 2006;1758:1580–1586. doi: 10.1016/j.bbamem.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- Zhang SK, Song JW, Gong F, Li SB, Chang HY, Xie HM, Gao HW, Tan YX, Ji SP. Design of an alpha-helical antimicrobial peptide with improved cell-selective and potent anti-biofilm activity. Sci Rep. 2016;6:27394. doi: 10.1038/srep27394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhao C, Liang G, Zhang M, Zheng J. Engineering antimicrobial peptides with improved antimicrobial and hemolytic activities. J Chem Inf Model. 2013a;53:3280–3296. doi: 10.1021/ci400477e. [DOI] [PubMed] [Google Scholar]
- Zhao X, Wu H, Lu H, Li G, Huang Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS One. 2013b;8:e66557. doi: 10.1371/journal.pone.0066557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipperer A, Konnerth MC, Laux C, Berscheid A, Janek D, Weidenmaier C, Burian M, Schilling NA, Slavetinsky C, Marschal M, et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature. 2016;535:511–516. doi: 10.1038/nature18634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cecropin P1 is expressed intracellularly with the 2x tether (without lpp-ompA for localization to the cell surface) at 0 mM, 0.01 mM, 0.1 mM and 1 mM IPTG. Growth curves were performed in triplicate over 6 hours. Data are represented as mean ± SEM.
Cecropin P1 is displayed with no tether, 1x tether and a 2x at 0 mM and 0.1 mM IPTG. Growth curves were performed in triplicate over 6 hours. Data are represented as mean ± SEM.
Cecropin P1 is displayed with increasing concentrations of magnesium and trypsin. Controls were cecropin P1 expressed with no added magnesium or trypsin enzyme. Growth curves were performed in triplicate over 6 hours. Data are represented as mean ± SEM.
Dermaseptin S4L7K is displayed on the surface. IPTG concentrations tested were 0mM, 0.01mM, 0.1mM and 1mM. Growth curves were performed in triplicate over 6 hours. Data are represented as mean ± SEM.
Sequence logo comparison between a randomly generated 20 amino acid library, our library, and the top hits generated from our library. Sequence logos were generated from either the entire set of possible killing peptides (7,968 sequences), 10,000 randomly sampled sequences of the total library, or an amino acid translation of 10,000 randomly generated nucleotide sequences of a repeated “NNB” motif. All logos are plotted in units of probability.
The resulting clustering dendrogram was divided into 81 subgroups representing different groups of similar peptides identified in this analysis.
Serial passage of E. coli W3110 with 15 days of sub-inhibitory concentrations of peptides P1 and P7. Passages were done in triplicate. Resistance is not observed with cationic P1 peptide. One replicate of the anionic P7 peptide developed 64-fold resistance over the 15-day period.
Table S1. Summary of sequencing read counts for sample libraries, Related to STAR Methods
Table S2. MICs of synthesized SLAY peptides and Cecropin P1, Related to Table 2
Table S3. Selected peptides assayed in 1% tryptone broth and TNG media, Related to Table 2
Table S4. Oligonucleotide sequences used in this study, Related to STAR Methods