

Abstract
Reactive oxygen species (ROS) are generated by virally-infected cells however the physiological significance of ROS generated under these conditions is unclear. Here we show that inflammation and cell death induced by exposure of mice or cells to sources of ROS is not altered in the absence of canonical ROS-sensing pathways or known cell death pathways. ROS-induced cell death signaling involves interaction between the cellular ROS sensor and antioxidant factor KEAP1, the phosphatase PGAM5 and the proapoptotic factor AIFM1. Pgam5−/− mice show exacerbated lung inflammation and proinflammatory cytokines in an ozone exposure model. Similarly, challenge with influenza A virus leads to increased virus infiltration, lymphocytic bronchiolitis and reduced survival of Pgam5−/− mice. This pathway, which we term ‘oxeiptosis’, is a ROS-sensitive, caspase independent, non-inflammatory cell death pathway and is important to protect against inflammation induced by ROS or ROS-generating agents such as viral pathogens.
Introduction
Reactive nitrogen and oxygen species (RNS and ROS) such as ·O2 and ·OH are generated as natural byproducts of normal oxygen metabolism. ROS play an important role in cell signaling by regulating cell proliferation and survival 1,2. However, exposure to pollutants, increased protein folding load, elevated fatty acid oxidation and energy metabolism can result in pathological accumulation of ROS in the endoplasmic reticulum, peroxisomes and mitochondria 3,4. Since oxidation by ROS can cause irreversible conformational changes on proteins and lipids as well as DNA mutations 5, several enzymatic and non-enzymatic mechanisms have evolved to protect cells from detrimental accumulation of ROS.
The main intracellular sensor monitoring ROS is KEAP1 6. Under physiological conditions, KEAP1 ubiquitinates and degrades the transcription factor NRF2, a positive regulator of cytoprotective gene expression. Increased intracellular ROS oxidizes KEAP1, resulting in its inability to degrade NRF2 the latter then accumulates, translocates to the nucleus and induces expression of antioxidant factors such as NQO1, HOX1 and TXN 7–10. Prolonged or high oxidative stress, however, does not induce sufficient expression of cytoprotective proteins and can result in cell damage and/or cell death 11.
Exogenous stimuli such as UV light, heat or inflammatory cytokines increase intracellular ROS that contributes to the severity of pathological disorders 12,13. Furthermore, infection with viruses can increase intracellular ROS levels due to perturbation of cellular metabolism 14,15. It has been proposed that in cases of persistent virus infection, ROS contributes to organ damage and exacerbates disease progression 16. Some viruses have evolved mechanisms to avoid detrimental ROS generation to facilitate viral proliferation. For instance, dengue virus (DENV) 17, human herpes virus 8 (KSHV)18and encephalomyocarditis virus (EMCV) 14 evolved distinct mechanisms to modulate ROS-mediated cell death pathways. Conversely, other viruses, such as Marburg virus (MarV), specifically perturb stress responses by promoting cytoprotective programs 19,20.
ROS-induced cell death can induce caspase-dependent apoptosis, inflammasome-driven pyroptosis, as well as caspase-independent cell death pathways including necroptosis, ferroptosis and autophagic cell death 21–25. Despite the apparent importance of ROS in physiological and pathological processes, relatively little is known about their sensors and downstream signaling pathways that lead to cell death in response to oxidative stress. We demonstrate here a ROS-induced form of apoptosis-like cell death which we term ‘oxeiptosis’. This novel form of cell death is independent of all previously characterized cell death pathways and plays an important role in limiting harmful ROS-associated inflammation.
Results
Ozone exposure induces inflammasome-independent inflammation in vivo
To investigate the involvement of known pathways in ROS-induced cell death we used an in vivo ozone exposure model (Fig. 1a). Single exposure to ozone induced ROS accumulation in murine airway cells in vivo and led to severe inflammation, evidenced by increased abundance of proteins, lymphocytes and cytokines in bronchoalveolar lavage (BAL) of treated mice (Fig. 1a, Supplementary Fig. 1a, b) 26. In this in vivo model, ozone-driven inflammation was not significantly changed in mice that lacked key components of the inflammasome pathway such as NACHT, LRR and PYD domains-containing protein 3 (Nlrp3−/−), caspases 1 and 11 (Casp1/11−/−) or the adaptor protein apoptosis-associated speck-like protein containing a CARD (Asc−/−) (Fig. 1a). Furthermore, caspases 3 and -7, required for apoptosis, were not activated after single exposure to ozone 27,28. Thus, in this experimental model it is not clear which pathways are involved in ozone-mediated injury, inflammation or cell death.
Figure 1. Inflammasome-, caspase- and necroptosis-independent but KEAP1-dependent cell death after ROS stimulation.
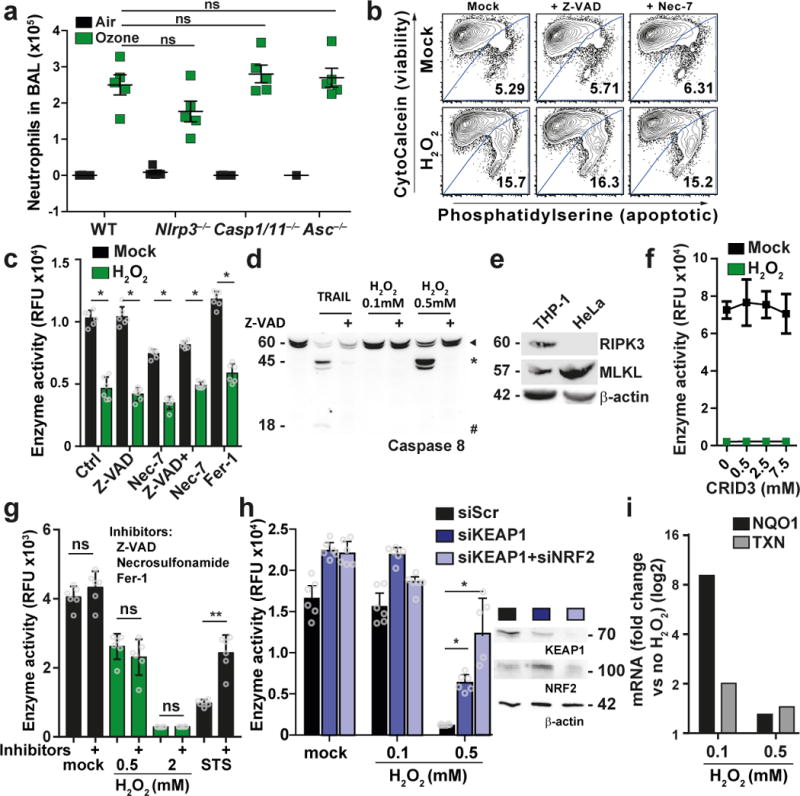
(a) Mouse model to analyze ROS effect in vivo. C57BL/6 mice (wt) or Nlrp3−/−, Casp1/11−/− and Asc−/− mice were exposed to 1 ppm of ozone for 1h, BAL was analyzed by cytospin measurements 24h later. Graph shows number of neutrophils of five individual mice. ns: non significant, Mann Whitney test, two-tailed. Graph shows mean +/- SEM. (b) Flow cytometry analysis for viability of mouse embryo fibroblasts (MEFs) pretreated with Z-VAD (20 µM) or Nec-7 (10 µM) for 1h, followed by 20h H2O2 treatment. (c) HeLa cells were left untreated or treated as indicated (Ferrostatin-1 (Fer-1, 1 µM)) for 1h, followed by 21h exposure to 0.5 mM H2O2. Cell viability was determined by resazurin conversion assay. The plot shows the mean ± S.D. of six individual treatments. (d) Caspase 8 cleavage in cells which were left untreated or treated with 20 µM Z-VAD for 1 h, followed by 10h treatment with 0.1 mM H2O2, 0.5 mM H2O2 or 300 ng/mL TRAIL. ► full-length caspase 8, * cleaved caspase 8, # p18 fragment of caspase 8. (e) Immunoblot analysis of THP-I and HeLa cell lysates for expression of RIPK3 and MLKL. (f) as (c) but cells were pretreated with CRID3. (g) as (c) but cells were pretreated with a combination of Z-VAD, 5 µM Necrosulfonamide and Fer-1 followed by stimulation with 0.5 mM H2O2 or 1 mM staurosporine (STS). (h) as (c) but HeLa were transfected for 48h with siRNAs as indicated followed by H2O2. Immunoblot of indicated proteins at the time of treatment. (i) Expression of NRF2 regulated target genes after 21h of H2O2 treatment. (c), (g), (h) * p-value < 0.001, ** p-value < 0.0001; 2way ANOVA. One representative experiment of at least two (h) or three (b), (c), (d), (e), (h) with similar results is shown.
Caspase-independent and RIPK3-independent ROS-induced cell death
To investigate the pathways involved in ROS-mediated cell death in more detail, we studied cell survival in response to the ROS inducer hydrogen peroxide (H2O2). Mouse embryonic fibroblasts (MEFs) exposed to H2O2 showed reduced cell viability (Fig. 1b). Induction of cell death was not changed by pre-treatment with the caspase inhibitor Z-VAD or the necroptosis inhibitor Nec-7 (Fig. 1b). Furthermore, we tested the morphology of MEFs upon exposure to H2O2 by electron microscopy. Upon H2O2 treatment cells showed apoptotic membrane blebbing, which was also present in Z‐VAD treated H2O2 exposed cells (Supplementary Fig. 1c). To confirm this data in different cell lines we tested H2O2 sensitivity of HeLa and Jurkat T-cells. As expected, H2O2 treatment induced cell death as measured by resazurin reduction assay, ATP abundance assay measured by CellTiter-Glo®, MTT or LDH release assay (Fig. 1c, Supplementary Fig. 1d, e, f). Z-VAD was not able to rescue cell viability (Fig. 1c, Supplementary Fig. 1f), despite successful inhibition of caspase 8 cleavage (Fig. 1d), suggesting a caspase-independent process. HeLa cells are naturally deficient in RIPK3 (Fig. 1e), a central component of the necroptosis pathway. Moreover, in line with published data 29, pretreating cells with Nec-7 or combination of Nec-7 with Z-VAD did not rescue from H2O2-induced cell death (Fig. 1c). The ferroptosis inhibitor Fer-1 did as well not rescue cells from H2O2 triggered cell death (Fig. 1c, Supplementary Fig. 1h, i). Furthermore, concentrations of H2O2 used did not lead to accumulation of LC3-A, an indicator of autophagic cell death (Supplementary Fig. 1g), nor did the chemical inhibition of inflammasome by CRID3 show an effect (Fig. 1f). Combining inhibitors for apoptosis, necroptosis and ferroptosis did not influence H2O2 induced toxicity (Fig. 1g). Collectively, our data suggested that H2O2‐induced cell death can be mediated by a pathway that is independent of caspases, ferroptosis, pyroptosis, autophagy or necroptosis.
The ROS sensor KEAP1 is required for H2O2-induced cell death
A protein that directly links ROS sensing to cell death is currently not known. However, a well-studied sensor of ROS is KEAP1. KEAP1 contains C-terminal cysteine residues that are oxidized by ROS and mediate a conformational change of the protein, resulting in the release of NRF2 to regulate expression of cytoprotective genes in order to counterbalance oxidative stress 30. We aimed to establish cellular systems that are differentially sensitive to ROS and therefore depleted KEAP1 and NRF2 in HeLa cells to test survival upon treatment with H2O2. As expected, cells depleted for KEAP1 led to upregulation of cytoprotective genes compared to controls (Supplementary Fig. 1j) and significantly decreased cell death after treatment with H2O2 (Fig. 1h). Co-depletion of KEAP1 and NRF2, which was expected to increase sensitivity towards H2O2, also rescued cells from H2O2-induced cell death (Fig. 1h), despite no detectable upregulation of cytoprotective genes (Supplementary Fig. 1j). Moreover, expression of NRF2-dependent genes was increased at low concentrations of H2O2, while toxic concentrations of H2O2 did not increase gene expression (Fig. 1i). Collectively we concluded that NRF2 transcriptional activity is dispensable for cytoprotection after KEAP1 depletion, suggesting that KEAP1 is a concentration-dependent ROS sensor that plays an active role in a ROS-dependent cell death pathway.
KEAP1 utilizes PGAM5 to mediate ROS-induced cell death
We next sought to identify potential interaction partners of KEAP1 that could convey signals leading to cell death. Using affinity purification followed by tandem mass spectrometry (AP-LC-MS/MS) we identified 32 proteins significantly enriched in KEAP1 purifications as compared to an unrelated control protein (Fig. 2a, Supplementary Table 1). These 32 proteins included 14 known binding partners of KEAP1 (Fig. 2a, brown dots), including NRF2. Only one identified protein has been previously associated with cell death pathways, namely PGAM5 (Fig. 2a, green dot). PGAM5 is a convergence point of multiple cell death pathways and has been shown to be involved in ROS-induced cell death 29. PGAM5 bears an N-terminal mitochondrial localization signal (MLS) and a transmembrane (TM) domain. Furthermore, PGAM5 interacts with KEAP1 and tethers it to the mitochondrial membrane 31. We validated association of KEAP1 and PGAM5 by co-immunoprecipitation of StrepII-HA-tagged (SII-HA) KEAP1 with endogenous PGAM5 (Fig. 2b) as well as SII-HA-tagged PGAM5 with endogenous KEAP1 (Fig. 2c) and confirmed that amino acids 69 to 89 of PGAM5 are critical for its interaction with KEAP1 (Supplementary Fig. 2a) 31. KEAP1 releases NRF2 after exposure to ROS and thereby allows NRF2 accumulation. This prompted us to test whether the interaction between KEAP1 and PGAM5 may be similarly regulated by ROS. Indeed, binding of PGAM5 to KEAP1 was reduced in the presence of 0.5 mM H2O2, while non-toxic amounts of H2O2, which regulate NRF2-dependent gene expression (Fig. 1h, i), did not affect the interaction between KEAP1 and PGAM5 (Fig. 2c). KEAP1 and PGAM5 co-localized with the mitochondrial marker COX IV under steady state conditions (Supplementary Fig. 2b) 32. In the presence of 0.5 mM H2O2, only KEAP1 but not PGAM5 lost co-localization with COX IV (Supplementary Fig. 2b), further supporting a ROS-sensitive interaction between KEAP1 and PGAM5.
Figure 2. KEAP1 binds PGAM5, which is required to induce ROS-dependent cell death.
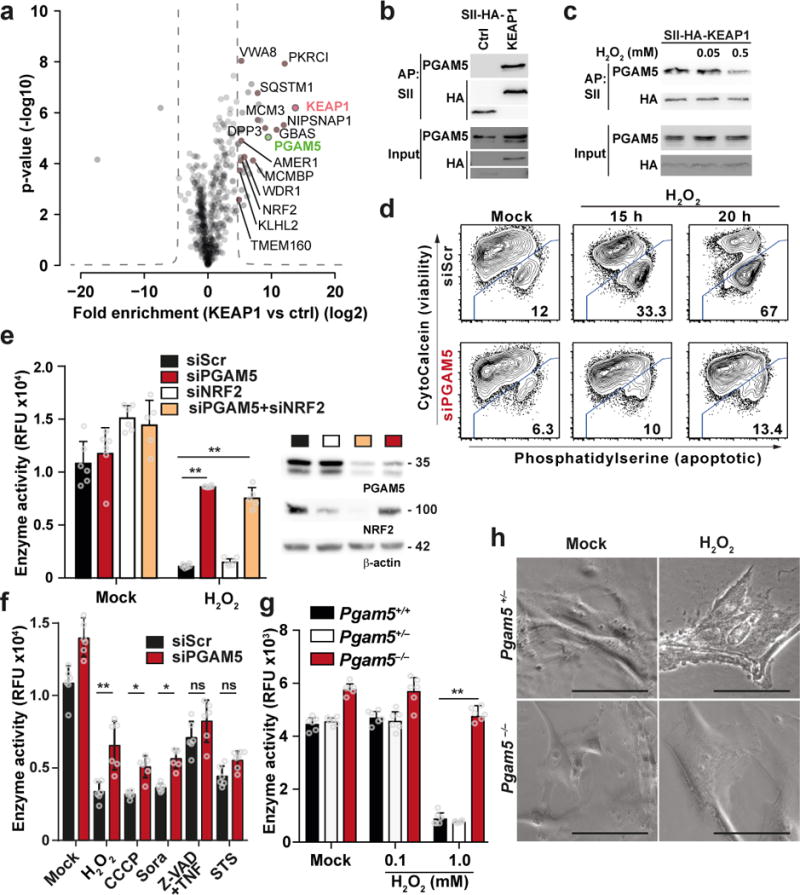
(a) Identification of KEAP1 binding partners. SII-Affinty purification (AP) of SII-HA-tagged KEAP1 and THYN1 as control (ctrl) followed by LC-MS/MS analysis. Volcano plots show the average degree of enrichment of label-free quantitation (LFQ) values by KEAP1 over control (x-axis) and transformed p-values (two-tailed t-test; y‐axis) for each identified protein. The hyperbolic curve separates significantly enriched proteins (FDR < 0.00001, S0=100) background. Red: KEAP1, brown: known KEAP1 interactors (based on Biogrid), green dot: protein associated with cell death. Four independent APs were performed for each bait. (b) Immunoblot of precipitates after AP with SII-HA-KEAP1 or SII-HA-THYN1 (ctrl). (c) SII-HA-KEAP1 expressing HeLa Flp-In cells were treated with the indicated amounts of H2O2 for 8h followed by SII-AP and Immunoblotting. (d) Flow cytometry analysis for viability of HeLa cells transfected with the indicated siRNAs for 48h, followed by treatment with 0.5 mM of H2O2 for the indicated time. (e) HeLa cells were treated with the indicated siRNAs for 48h and stimulated with H2O2 for 20h. The graph shows mean resazurin conversion activity ± S.D. of six measurements. Knockdown efficiency was confirmed by Immunoblotting against indicated proteins 48h after siRNA transfection (right panel). (f) as (e) but stimulation with H2O2, 20 µM CCCP, 20 µM Sorafenib (Sora), 20 µM Z-VAD + 20 µM TNF and 1 µM STS,, respectively. (g) as (e) but MEFs with indicated genotype were stimulated as indicated. (h) Light microscopic images of Pgam5+/+ and Pgam5−/− MEFs after 20h treatment with 1 mM H2O2. Scalebar: 100 µM. (e)-(g) ** p-value < 0.001, * p-value < 0.05, ns: non-significant, 2way ANOVA. One representative experiment of three (b)-(f) (h), four (g) or six (e) is shown.
To functionally assess whether PGAM5 is involved in ROS-mediated cell death we used siRNA-mediated knockdown of PGAM5 and applied a flow cytometry-based cell survival assay after treatment with H2O2. In control cells, H2O2 treatment led to 33% and 67% of dying cells after 15h and 20h, respectively (Fig. 2d). PGAM5 depletion rescued the majority of cells and only 10% and 13% showed signs for cell death at 15h and 20h after treatment (Fig. 2d). Similarly, reduction enzyme activity was severely decreased (<10%) in control cells treated with H2O2, while cells lacking PGAM5 showed more than 73% activity (Fig. 2e, Supplementary Fig. 2c, d). Co-depletion of PGAM5 and NRF2 also rescued cells from H2O2-induced cell death (Fig. 2e). While depletion of PGAM5 rescued from H2O2-induced cell death, it did not affect induction of apoptosis by staurosporin, necroptosis by Z-VAD+TNF and mildly affected ferroptosis induced by sorafenib and autophagic cell death triggered by carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (Fig. 2f). Inhibition of caspases in PGAM5 depleted cells did not further increase cell viability, further excluding apoptosis in this experimental setting (Supplementary Fig. 2e). These data collectively suggested that PGAM5 depletion in HeLa cells did not have a general role, but was specific for H2O2-induced cell death. We reproduced these data in a genetically clean system: Pgam5+/+ and Pgam5+/− MEFs (Supplementary Fig. 2f) treated with H2O2 showed severe signs of cytotoxicity (Fig. 2g). Morphologically these cells showed membrane blebbing (Fig. 2h), classical for apoptotic cell death. However, Pgam5−/− MEFs tolerated treatment with high-dose H2O2 (Fig. 2g) and were morphologically similar to untreated MEFs (Fig. 2h). Sorafenib decreased cell viability similarly in Pgam5+/− and Pgam5−/− MEFs (Supplementary Fig. 2g). We concluded that PGAM5 is specifically involved in a H2O2-induced signaling cascade that is unrelated to other known cell death pathways.
PGAM5-dependent cell death induction utilizes AIFM1
PGAM5 has previously been proposed to be of central importance to transmit cell death signals 29, but its molecular partners are not known. AP-LC-MS/MS using PGAM5 as bait identified 8 high confidence interactors (Fig. 3a, Supplementary Table 2). As expected, PGAM5 associated with KEAP1 (Fig. 3a, brown dot). Furthermore, PGAM5 co-precipitated proteins of the TIM complex (TIMM50, TIMM8A and TIMM13) which facilitates the transport of proteins to the inner mitochondrial membrane 33. Only HCLS1-associated protein X-1 (HAX1) and AIFM1 were previously associated with cell death pathways (Fig. 3a, green dots). Intriguingly, AIFM1 has previously been implicated in caspase-independent cell death 34. We validated the PGAM5-AIFM1 interaction by co-precipitation followed by immunoblotting (Fig. 3b). Domain mutants of AIFM1 indicated that it binds to PGAM5 through N-terminal domains (amino acids (aa)2-103), which contain the MLS and TM domain, while C-terminal sequences (aa 480-613) were not involved in PGAM5 binding (Supplementary Fig. 3a).To test whether AIFM1 serves as a downstream target of PGAM5, we depleted HeLa cells for AIFM1 or PGAM5 and monitored for cell survival after stimulation with H2O2 or the mitochondrial ROS-inducer tBHQ. As expected, control cells were highly sensitive to H2O2 and tBHQ treatment (Fig. 3c, Supplementary Fig. 3b). Notably, HeLa and SKN-BE2 cells depleted for AIFM1 or PGAM5 exhibited superior survival rates after H2O2 and tBHQ treatment (Fig. 3c, Supplementary Fig. 3b, c). Combination of KEAP1, PGAM5 and AIFM1 depletion did not significantly increase viability in response to H2O2 treatment (Supplementary Fig. 3d), suggesting that all three proteins are operating in the same pathway. AIFM1 has been described to be central for the oxidoreductase pathway 35. However, mitochondrial respiration of HeLa cells left untreated or treated with different doses of H2O2 was indistinguishable (Supplementary Fig. 3e), suggesting that death-inducing amounts of intracellular ROS are independent of the oxidoreductase function of AIFM1.
Figure 3. PGAM5 interacts with AIFM1.
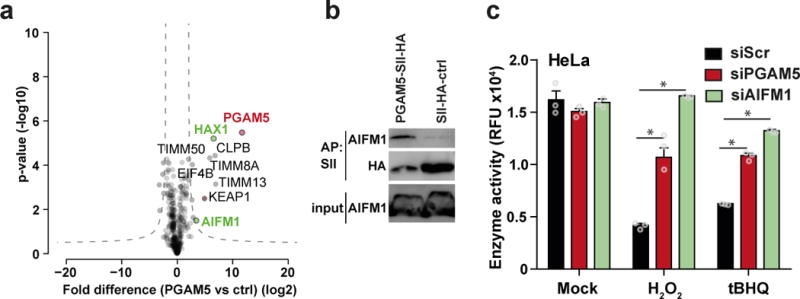
(a) Identification of PGAM5 binding partners. Proteins enriched with SII-HA-PGAM5 and THYN1 (ctrl) from HeLa FlpIn cells were analyzed by LC-MS/MS. Volcano plots show the average degrees of enrichment by PGAM5 over control (ratio of label-free quantitation (LFQ) protein intensities; x-axis) and p-values (two-tailed t-test; y‐axis) for each protein. Significantly enriched proteins (FDR 0.001, S0=0.2) are separated from background proteins by a hyperbolic curve (dotted line). Red: PGAM5, green: proteins known to be involved in cell death. Four independent affinity purifications were performed for both baits. (b) Binding of endogenous AIFM1 to PGAM5. Immunoblot analysis of SII-HA-PGAM5 or SII-HA-ctrl precipitates and input lysate. One representative experiment of eight is shown. (c) Viability of HeLa cells treated with siRNA against PGAM5, AIFM1 and siScr after 21h of H2O2 or tert-Butylhydroquinone (tBHQ) treatment. Cell viability was tested by resazurin conversion assay. The plot shows the mean ± S.D. of three individual treatments. One representative experiment of four is shown. * p-value < 0.001, 2way ANOVA.
PGAM5 dephosphorylates AIFM1 on S116
We next investigated the functional interaction between PGAM5 and AIFM1. Intriguingly, binding of AIFM1 to PGAM5 was reduced when cells were treated with H2O2 (Fig. 4a). Binding could be rescued by the ROS scavenger N-acetylcysteine (NAC) (Fig. 4a), suggesting that presence of ROS can modulate the interaction between PGAM5 and AIFM1. PGAM5 was previously reported to possess serine/threonine-protein phosphatase activity 29, and several studies have shown that processed substrates often lose interactions with their modifying enzymes 31. Notably, binding between PGAM5 and AIFM1 was also reduced in the absence of phosphatase inhibitors (Fig. 4b). We mutated known phosphorylated residues in AIFM1 N-terminal region (phopsphoyites.org) to alanine and tested for their ability to co-precipitate with endogenous PGAM5. Mutation of AIFM1 serine 116 to alanine (S116A) impaired binding to PGAM5, whereas other residues did not affect PGAM5 association (Fig. 4c). Sequence alignment analysis revealed a remarkable conservation of AIFM1 S116 and surrounding amino acid residues within mammals (Fig. 4d, Supplementary Fig. 4a). Of particular interest in this region are two glutamic acid residues located three and five amino acids C-terminally of S116 which contribute negative charges (Fig. 4d, Supplementary Fig. 4a). Such contribution of negative charges has been reported to support binding of substrates to the positively charged phosphatase active site of PGAM5 (Supplementary Fig. 4c) 36. A custom-raised antibody against pS116 of AIFM1 indicated that AIFM1 S116 is phosphorylated under steady state conditions (Fig. 4e, Supplementary Fig. 4b). Strikingly, treatment of cells with H2O2 led to pS116 dephosphorylation (Fig. 4e).
Figure 4. PGAM5 dephosphorylates AIFM1 S116.
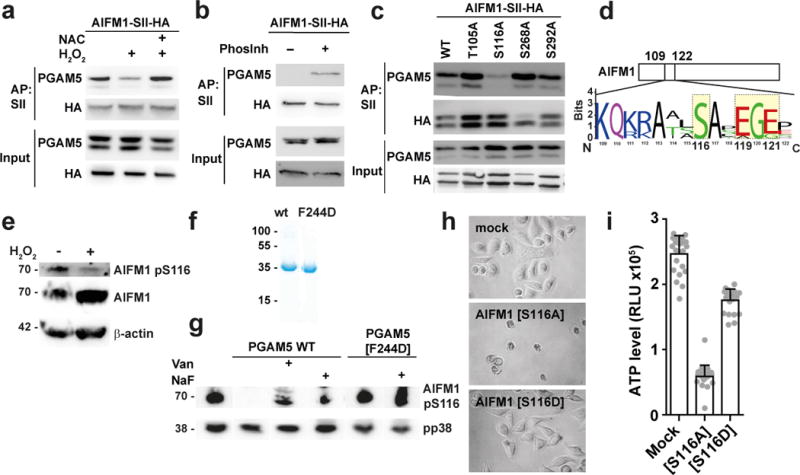
(a) AIFM1-SII-HA Hela FlpIn cells were treated for 1h with 2 mM N-Acetylcysteine (NAC) followed by 10h with 0.5 mM H2O2. Immunoblot analysis of precipitates after SII-AP. (b) PGAM5 precipitated with SII-HA-AIFM1 in the presence or absence of phosphatase inhibitors (Phosinh) followed by Immunoblotting. (c) Co-precipitation of PGAM5 to the indicated AIFM1-SII-HA mutant proteins expressed in HEK293R1 AIFM1 knockout cells. Immunoblot analysis of precipitates after AP with SII-beads. (d) Sequence logo of AIFM1 S116 and surrounding amino acids in 13 mammalian species. (e) Immunoblot analysis of Jurkat cells that were left untreated or were treated with 0.5 mM H2O2 for 2h. (f) Coomassie gel of purified recombinant proteins. (g) HeLa cell lysates blotted onto nitrocellulose membrane was incubated with 11 µg of recombinant PGAM5 wt or PGAM5[F244D] mutant protein for 30 min at 30°C and subjected to Immunoblot analysis for the indicated proteins. One representative experiment of three is shown. (h, i) Viability of HEK293 AIFM1 knockout cells 12h after infection with the indicated lentivirus. (h) Representative images of cells transduced with the indicated constructs. (i) Cell viability was determined by Cell-Titer-Glo viability assay. The plot shows the mean ± S.D. of four individual treatments consisting of six replicates each. One representative experiment of three (b), (e), (f), four (c) or five (h) with similar restuls.
We generated recombinant PGAM5 and a phosphatase inactive PGAM5[F244D] dimerization mutant (Fig. 4f, Supplementary Fig. 4d–f, h) in order to test the phosphatase activity ofPGAM5. PGAM5Δ2-28 was able to release phosphates from phospho-serine but not when phospho-threonine pseudosubstrate-peptides (Supplementary Fig. 4i) while PGAM5Δ2-28[F244D] was inactive (Supplementary Fig. 4i). Dephosphorylation of pseudosubstrate could be inhibited by the phosphatase inhibitor sodium ortho-vanadate (Van) (Supplementary Fig. 4i). We proceeded using membrane-bound HeLa cell lysate as substrate for recombinant PGAM5. Treatment of membranes with recombinant proteins followed by staining for phosphorylated AIFM1 showed that incubation with PGAM5Δ2-28 led to AIFM1 pS116 dephosphorylation (Fig. 4g). This process could be inhibited with Van and sodium fluoride (Fig. 4g). The PGAM5Δ2-28[F244D] mutant protein did not exhibit dephosphorylation activity on AIFM1 pS116 (Fig. 4g). The effect of recombinant PGAM5 was specific for AIFM1 pS116 since p38 was not dephosphorylated in this assay.
To assess the consequence of AIFM1 S116 dephosphorylation on cell survival, we reconstituted AIFM1 CRISPR/Cas9 knockout HEK293 cells with expression plasmids for AIFM1, phosphomutant AIFM1[S116A] and phosphomimetic AIFM1[S116D] and monitored for cell survival. Expression of AIFM1[S116A] but not AIFM1[S116D] was sufficient to induce cell death, evidenced by cell rounding up and reduced intracellular ATP levels (Fig. 4h, i).
These data show that AIFM1 pS116 is dephosphorylated during oxidative stress and that PGAM5 has the ability to mediate this dephosphorylation. Furthermore, it highlights AIFM1 S116 dephosphorylation as a hallmark of this cell death pathway. We name this pathway “oxeiptosis” as it deals with a cell death pathway (“-ptosis”) that can be triggered by oxidative stress.
Pgam5−/− mice show severe inflammation in response to ozone treatment
In order to gain information on the role of PGAM5 in vivo we tested the response of Pgam5+/+ and Pgam5−/− mice to the ROS inducer ozone. We exposed control mice (Pgam5+/+) and Pgam5−/− mice to ozone and tested inflammation parameters 4h and 24h later. Ozone treatment led to increased protein abundance in BAL of Pgam5−/− mice as compared to controls (Fig. 5a, Supplementary Fig. 5a). Specifically, we could see a significant increase in myeloperoxidase (MPO), an inflammation-specific marker generated by myeloid cells (Fig. 5a). This increase in inflammation markers was corroborated by increased total cell numbers in the BAL of ozone-treated Pgam5−/− compared to control animals, most strikingly being a highly significant increase in neutrophils in Pgam5−/− mice as compared to controls (Fig. 5b, Supplementary Fig. 5b). Compared to Pgam5+/+ ozone-treated Pgam5−/− mice showed significantly increased levels of IL-6, CXCL1, CXCL2 and CCL2 (Fig. 5c). Collectively, these data indicate that Pgam5 deficiency in mice leads to increased inflammation when exposed to a ROS inducer. This suggests that PGAM5 is required to dampen inflammatory responses and that absence of PGAM5 in vivo leads to compensatory inflammatory responses.
Figure 5. Pgam5−/− mice show severe inflammation in response to ozone exposure.
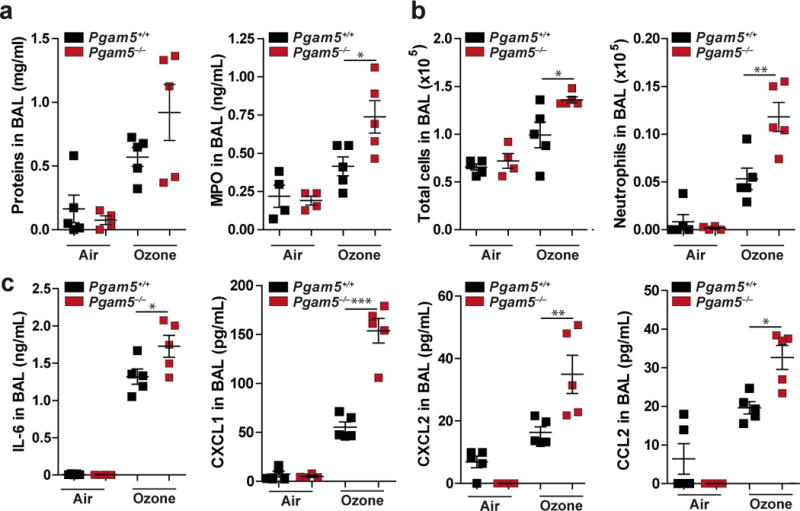
Pgam5−/− and wild-type (wt) mice were treated 1 ppm ozone for 1h and sacrificed after 4h recovery time. (a) Determination of proteins and MPO in BAL. (b) Total cells and neutrophils in BAL. (c) Indicated cytokines were measured by Luminex analysis in BAL of mice. * p-value < 0.05, ** p-value < 0.05, *** p-value < 0.0001 by one-way ANOVA and Bonferroni post t-test. Graphs show individual mice (n=5) (dots), mean and SEM. One representative experiment of two with similar results is shown.
Oxeiptosis is targeted by viral ORFs
Virus infection commonly triggers generation of intracellular ROS 18, which could lead to cell death. As a countermeasure, many viruses regulate cell death pathways in order to optimize their own replication and spread. Mining an AP-MS dataset that identified cellular binding partners of innate immune regulators 37 for interactors of KEAP1, PGAM5 or AIFM1, revealed four proteins from distantly related viruses that interacted with any of these three proteins (Fig. 6a). VP24 of MarV bound KEAP1 19,38, measles virus VΔC and KSHV K3 protein associated with PGAM5 (Supplementary Fig. 6–d, Supplementary Table 3a) and the non-structural protein small (NSs) of La Crosse virus (LaCV) bound AIFM1 (Supplementary Fig. 6e, f). In additional interaction studies of the non-structural protein 2 (NS2) of respiratory syncytial virus (RSV) we identified the PGAM5 interaction site (amino acids 2-55) of AIFM1 as a cellular target (Supplementary Fig. 6g–i, Supplementary Table 4). We proceeded to functionally test the interaction of K3 (KSHV) and PGAM5. Binding of PGAM5 to K3 required amino acids 29-69 in PGAM5 (Supplementary Fig. 6d), a region that has previously been identified to be important for full PGAM5 functionality 36. Precipitates of PGAM5 from cells co-expressing K3 showed reduced phosphatase activity (Fig. 6b), confirming a functional interaction between PGAM5 and K3. Collectively, these data indicate that distantly related viruses share their ability to bind KEAP1, PGAM5 or AIFM1, suggesting evolutionary pressure to target this pathway.
Figure 6. Oxeiptosis is activated by virus infection and targeted by viral proteins.
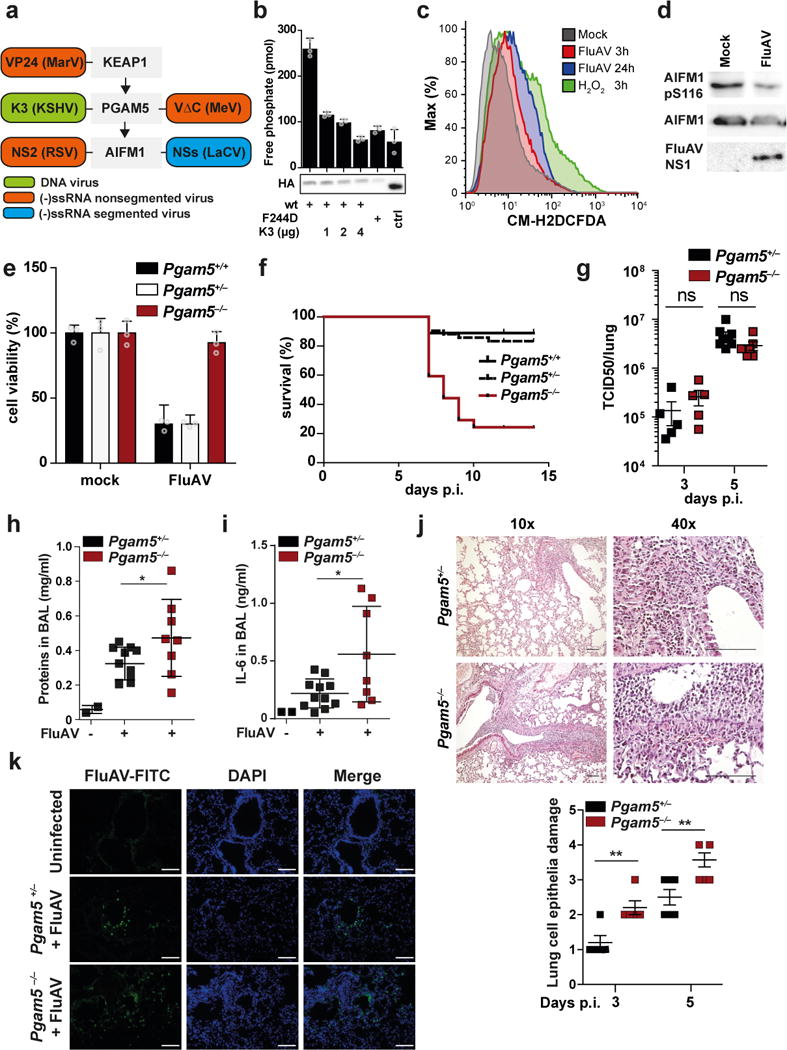
(a) Schematic representation of viral proteins identified in 48 and Supplementary Fig. 6 to bind KEAP1, PGAM5 or AIFM1. (b) HEK293 cells were co-transfected with SII-HA-PGAM5 (wt) or SII-HA-PGAM5[F244D] and Ren-K3 (KSHV) as indicated. SII-tagged proteins were precipitated and phosphatase activity was evaluated using a phospho-serine containing peptide as pseudosubstrate. Immunoblot shows expression of SII-HA-tagged proteins in cell lysates. Shown are mean of triplicate measurements +/- SD. (c) Flow cytometry analysis of mitochondrial ROS in HeLa cells treated with 0.5 mM H2O2 for 3h or infected with FluAV MOI 3 for 3h and 24h. Graphs show mean +/- SD of three replicates. (d) Immunoblot of Jurkat cells infected with FluAV for 24h. (e) Viability of MEFs after infection with FluAV (MOI 3) for 21h tested by resazurin viability assay. The plots shows the mean ± S.D. of three individual treatments. (f) – (k) Mice of the indicated genotype were infected with FluAV. (f) Survival of littermate Pgam5+/+ (n=9), Pgam5+/− (n=42) and Pgam5−/− (n=20) mice monitored for 14 days and euthanized when they lost >25% of their initial body weight. (g) Virus titer in lungs of mice 3 and 5 days after infection. (h) Total protein and (i) IL6 in BAL after 5 days. (j) Top panel: representative micrographs (H&E stain) of lungs five days after infection. Bottom panel: scoring of lung damage in 3 and 5 day infected animals. (g)-(j) dots indicate individual mice, shown is mean +/- SEM. (k) Immunofluorescence analysis of lung tissuefrom mice infected for five days. (j), (k) Scale bar is 100 µM. * p-value < 0.05, ** p-value < 0.02, ns: non-significant, two sided t-test. Graphs show representative experiments of two (h), (i), three (b), (c), (k), four (j), five (d), eight (e).
PGAM5 regulates virus-induced inflammation in vivo
We used influenza A virus infection as model to test the involvement of oxeiptosis in virus infections. HeLa cells infected with influenza A virus (FluAV; strain SC35M) showed accumulation of intracellular ROS (Fig. 6c) and expression of cytoprotective genes (Supplementary Fig. 6j). Furthermore, FluAV infection led to reduced AIFM1 pS116 as compared to uninfected control cells (Fig. 6d), indicating activation of oxeiptosis. In line with this notion, HeLa cells depleted for PGAM5 showed significantly less FluAV-induced cytotoxicity as compared to control knockdowns (Supplementary Fig. 6k). Similarly, compared to Pgam5+/+ and Pgam5+/− control cells, FluAV infection of Pgam5−/− MEFs resulted in superior survival rates (Fig. 6e), pointing towards a role of Pgam5 in virus-induced cell death in vitro. To assess a function of Pgam5 in vivo, we infected Pgam5+/+, Pgam5+/− and Pgam5−/− mice with 1,500 pfu of FluAV. This infection regime only caused 10% mortality in Pgam5+/+ and Pgam5+/− mice (Fig. 6f). In contrast, Pgam5−/− mice showed a markedly increased mortality rate since 78% of animals succumbed to FluAV infection (Fig. 6f). Surprisingly, virus titers and virus mRNA levels in lungs on days 3 and 5 after infection were indistinguishable in Pgam5+/− and Pgam5−/− animals (Fig. 6g, Supplementary Fig. 6l), suggesting that Pgam5 does not directly affect virus replication and that the increased susceptibility to FluAV infection is not due to exacerbated virus load. However, in line with the ozone exposure experiments (Fig. 5c), lungs showed significantly increased amounts of Il6 mRNA in the absence of Pgam5 (Supplementary Fig. 6m). Similarly, BAL fluid of infected Pgam5−/− mice contained increased levels of protein (Fig. 6h) and significantly more inflammatory cytokines such as IL6, CXCL1 and CCL2 (Fig. 6i, Supplementary Fig. 6n). These experiments suggested exacerbated inflammatory processes during FluAV infection in the absence of Pgam5. Indeed, histological analysis of lungs of infected mice showed severely increased lung pathology, with significantly more destruction of the epithelial layer in the bronchioles and increased infiltration of lymphocytes in Pgam5−/− mice as compared to Pgam5+/− mice (Fig. 6j). However, immunohistochemistry for FluAV protein distribution in lungs of infected mice showed much deeper infiltration of FluAV in Pgam5−/− mice as compared to Pgam5+/− controls (Fig. 6k). From these experiments we concluded that Pgam5 is required to modulate inflammatory responses in vivo and that lack of Pgam5 in the context of FluAV infection results in acute necrotic intrabronchial and peribronchial inflammation, which allows deeper infiltration of viruses.
Discussion
Cells have to make critical decisions as how to respond to physiological and pathological insults. Infections, inflammatory cytokines and other environmental cues can raise ROS levels to detrimental concentrations that contribute to pathological disease manifestation 16,39,40. However, a dedicated sensor of ROS that is linked to cell death or mediates inflammatory responses has not been described. Here we show that the well-described ROS sensor KEAP1 can induce a cell death pathway that is signaling through PGAM5 and AIFM1.
Interestingly, KEAP1 bears 27 cysteine residues in its C-terminus, which can be modified in a stimulus-dependent manner 6. These cysteine residues may not only allow for sensing of the presence or absence of ROS but may also be able to quantify ROS levels and induce concentration-specific responses. Such a system may allow a single protein to integrate quantitative information in order to regulate diverse cellular signaling pathways and to respond to environmental needs in an appropriate manner. This model is supported by the dissociation of KEAP1 and PGAM5 that was only observed in the presence of high H2O2 concentration, while concentrations that activate KEAP1-dependent NRF2 activation did not perturb this interactions. This likely explains the dual character of KEAP1 to regulate NRF2-dependent protection at low levels of ROS, while mediating the induction of cell death through release of PGAM5 at high levels of oxidative stress.
PGAM5 has been proposed to function on a convergence point of multiple caspase-independent cell death pathways 29. Notably, cell death stimuli reported to signal through PGAM5, such as TNF, H2O2, t-butyl hydroxide (TBH) and A23187, have all been shown to generate intracellular ROS 41,42, which may explain the ability of PGAM5 to integrate many diverse cell death signals.
Signaling downstream of PGAM5 which results in cell death remained enigmatic. By AP-LC-MS/MS analysis we identified AIFM1 as a PGAM5 binding partner. AIFM1 is residing inside mitochondria and as such should be spatially dissociated from PGAM5. However, KEAP1 has been reported to be tethered to the outer membrane of mitochondria through PGAM5 engagement 32. Conversely, the same mechanism could mediate retention of PGAM5 at the outer mitochondrial membrane in order to spatially dissociate it from its phosphatase target AIFM1. Interestingly, PGAM5 bears a CX9C (C229 to C239) motif, which can be used by the Mia40 complex to import proteins into mitochondria 43. In line with an activity in mitochondria, PGAM5 physically interacts with members of the TIMM complex (TIMM8/-13), which is known to be involved in regulating protein import into mitochondria 33. Release of PGAM5 by KEAP1 during oxidative stress may be part of an activation process that allows internalization of PGAM5 into the mitochondria in order to interact with its target AIFM1. We identified the highly conserved Serine 116 residue in AIFM1 as target of PGAM5 phosphatase activity during treatment with high amounts of H2O2, by using recombinant PGAM5 and after virus infection. Functionally, expression of AIFM1 S116A is sufficient to induce cell death in AIFM1-deficient cells.
Based on these data we propose the existence of a caspase-independent cell death pathway, which we name oxeiptosis. This pathway encompasses KEAP1-PGAM5-AIFM1 and is activated by detrimental levels of ROS. Dephosphorylated S116 on AIFM1 serves as a marker and is at the same time a key regulator for the activity of the oxeiptosis pathway.
An increase of oxidative stress has been linked to necroptosis, apoptosis, ferroptosis, autophagy and pyroptosis 44,45. These pathways are operating in parallel and integrate additional signals. The engagement of these pathways is also cell-type dependent since some of the individual pathways are limited to certain cell types. We propose that oxeiptosis operates in parallel to other cell death pathways and leads to a non-inflammatory, caspase-independent, apoptosis-like cell death phenotype. It is operational in a wide range of cells with the possible exception of macrophages since PGAM5 deficiency has been reported to be dispensable for ROS-dependent cell death in these cells 46. In line with an apoptosis-like anti-inflammatory activity of PGAM5 in vitro, Pgam5−/− mice showed increased inflammatory parameters in BAL after exposure to ozone and infection with FluAV.
ROS is produced during numerous diseases including allergies, autoimmunity, allograft rejection, cancer, and infection with pathogens, which suggests that oxeiptosis could be activated in many different pathological situations 47. Notably, it has been shown that treatment of tumors with ROS-inducing agents such as BZL101 induces cell death in an AIFM1-dependent manner 48. Keap1 is often mutated in lung, gall bladder, and head and neck cancers, and Keap1 gene expression is silenced by promoter hypermethylation in various cell lines derived from lung and prostate cancer 49, which could lead to dysfunction of the oxeiptosis pathway and could thereby promote survival of transformed cells. Similarly, evolutionary distinct viruses interfere with KEAP1, PGAM5 and AIFM1, respectively. This indicates that modulation of oxeiptosis is involved in antiviral immunity. Indeed, we could show that PGAM5 plays an important role during FluAV infection and Pgam5 deficiency in mice leads to increased inflammatory responses. Recent reports suggest activation of Ripk3-dependent necroptosis by FluAV and Ripk3−/− mice show decreased inflammatory parameters in lungs of infected animals 50. It is likely that the pro-inflammatory phenotype in FluAV infected Pgam5−/− mice is mediated by compensatory activation of Ripk3-dependent necroptosis.
An important complication of treating virus-derived tumors could be the activity of viral proteins, such as the K3 protein of KSHV, which directly affects functionality of PGAM5.
Our study identified oxeiptosis, a ROS-induced cell death pathway that leads to a caspase-independent non-inflammatory cell death and encompasses KEAP1, PGAM5 and AIFM1. Similar to viral interference with KEAP1, PGAM5 or AIFM1, we propose that targeting oxeiptosis through therapeutic intervention may constitute a means to modulate progression of diseases that involve oxidative stress.
Online Methods
Plasmids
Expression constructs were generated by PCR amplification of plasmids from ImaGenes cDNA Library (MPI core Facility) and a cDNA library obtained from HeLa cells followed by Gateway cloning (Invitrogen) into the plasmids pcDNA3-Ren-GW, pTO-SII-HA-GW and pLenti6-V5-GW 1,2. Mutations and truncations were introduced by PCR, respectively. pI.18_3xFlag_NSs (LaCV) and pI.18_3xFlag_DMx_1xFlag were kindly provided by Friedemann Weber (Giessen). Sequences were verified by Sanger sequencing.
Cells, reagents and viruses
HeLa S3 (CCL-2.2) and Vero E6 cells (CRL-1586) were purchased from ATCC. THP-I cells (300356) were purchased from CLS. Jurkat cells were a gift from Felix Meissner (MPI of Biochemistry, Munich). SKN-BE2 cells were kindly provided by Rüdiger Klein (MPI of Neurobiology, Munich). HEK293 cells were a gift from Andrew Bowie. HeLa FlpIn cells (a gift from Andrea Musacchio, MPI of Cell Biology, Dresden) stably expressing SII-HA-tagged human PGAM5, AIFM1, KEAP1, K3, K5, NS2 and THYN1 under control of the CMV promoter were generated by hygromycin selection. Mouse embryonic fibroblasts (MEFs) were isolated from 13.5 day old embryos from heterozygous breeding pairs.
Cell lines were maintained in DMEM (PAA Laboratories) or RPMI (PAA Laboratories) (for THP-I and Jurkat cells) containing 10% fetal calf serum (GE Healthcare) and antibiotics (100 U/ml penicillin, 100 µg/ml streptomycin). Streptavidin-agarose beads were obtained from Novagen. Hydrogen peroxide, M2-Flag beads, Malachite green, N‐Acetylcysteine, Necrostatin-7, Resazurin, sodium fluoride, tert-Butylhydroquinone (tBHQ) and Thiazolyl Blue Tetrazolium Bromide (MTT) were from Sigma, Necrosulfomamide was from Merck, Polyethylenimine linear MW 25.000 from Polysciences, ortho-Vanadate from Alfa Aesar. Pan Caspase Inhibitor Z-VAD-FMK from R&D Systems. Primary antibodies used in this study were as follows: PGAM5 (Sigma: HPA036978), Renilla-tag (Millipore: MAB4410), and AIFM1, Caspase-8, KEAP1, MLKL, NRF2, pp38, RIPK3, COX IV (Cell Signaling: 5318, 9746, 8047, 14993, 12721, 4511, 13526, 11967), AIFM1 pS116 (ECM Bioscience: AP5501). Antibody against FluAV NS1 was a gift from Georg Kochs (Freiburg). Antibodies against actin and His-tag (Santa Cruz: sc-47778, sc-8036), Flag-M2-tag and HA-tag (Sigma: A8592, H6533), and secondary antibodies detecting mouse or rabbit IgG (Jackson ImmunoResearch, Dako) were horseradish peroxidase (HRP)-coupled. CM-H2DCFDA, 4′,6-diamidino-2-phenylindole (DAPI) and secondary antibodies used for immunofluorescence were purchased from Invitrogen. Apoptosis/Necrosis assay Detection Kit was purchased from Abcam. CellTiter-Glo assay kit and CytoTox 96® Non-Radioactive Cytotoxicity Assay (LDH-assay) were purchased from Promega. Recombinant influenza virus (strain SC35M wt) was described previously 3. Reagents for electron microscopy were the following Sodium Cacodylate 0.2M (Electron Microscopy Sciences, cat.#11653), 2% Osmium Tetroxide (Electron Microscopy Sciences, cat.#19152), low Viscosity Embedding Media Spurr’s Kit (Electron Microscopy Sciences, cat.# E14300).
Cloning and expression of recombinant proteins and phosphatase assay
N-terminal His-tagged PGAM5 (pNIC28-Bsa4-PGAM5(Δ2-28)) was a kindly provided by Apirat Chaikuad and Stefan Knapp (Oxford) 4. For solubility reasons the recombinant protein carried a deletion from amino acid 2-28. Mutation F244D in PGAM5 was introduced by site directed mutagenesis. Sequences of all cloning primers are available on request. Expression of recombinant proteins was induced over night at 18°C in E. coli strain Rosetta(DE3) using 0.5 mM IPTG (Thermo). Cells were lysed in lysis buffer (50 mM Tris-HCl pH 8.5, 500 mM NaCl, 10% glycerol, 40 mM imidazole, 1 mM DTT and protease inhibitor cocktail (EDTA-free, cOmplete; Roche)) using an Emulsiflex-C3 homogenizer and cleared lysate was used for protein purification using a HisTrap HP column (GE Healthcare: 17-5247-01) and further purified by gel filtration (mobile phase: 30 mM Tris-HCl pH 8.5, 300 mM NaCl, 10% glycerol, 1 mM DTT). Identity of recombinant PGAM5Δ2-28 wt and PGAM5Δ2-28[F244D] was confirmed by mass spectrometry. Far ultraviolet (UV) circular dichroism (CD) spectra of wild type and a mutant version of PGAM5 were recorded on a Jasco J-810 CD-Photometer at room temperature in 20 mM sodium phosphate buffer pH 7.4 and 50 mM NaF. For each sample and the buffer (baseline), four scans were recorded and averaged. The averaged baseline spectrum was subtracted from the averaged sample spectra and the resulting spectra were smoothed using an FFT filter (as part of the software package). Measurements were only made down to wavelengths where the instrument dynode voltage indicated the detector was still in its linear range (190 nm). Spectra are shown as the mean residue elipticity. Secondary structure compositions were estimated using the CONTINLL program 5.
To determine phosphatase activity in vitro of affinity purified proteins, beads with bound proteins were resuspended in ddH2O and incubated in phosphatase buffer (50 mM imidazole pH 7.2, 0.2 mM EGTA, 0.02 % β-mercaptoethanol, 0.1 mg/ml BSA) and 50 µM phosphorylated peptide (peptide sequences: RRA(pT)VA and AAL(pS)ASE) for 20 minutes at 30°C. The reaction was stopped by addition of Malachite Green Reagent as described before 6. After 15 minutes incubation at room temperature absorbance at 630 nm was measured using an Infinite 200 PRO series micro plate reader (Tecan). To determine phosphatase activity in silico recombinant proteins were used incubated in phosphatase buffer as described above. To detect dephosphorylation of AIFM1 HeLa cell lysate was separated by SDS page and transferred onto a nitrocellulose membrane. The membranes were incubated with recombinant PGAM5Δ2-28wt and PGAM5Δ2-28[F244D] in phosphatase buffer for 60 min at 30°C, washed, blocked and stained with primary antibodies.
Cell viability assays, flow cytometry and Immunofluorescence
To test cell viability we used three different assays. We measured ATP levels by CellTiter-Glo assay according to manufacturer’s protocol. We measured released LDH activity by LDH-assay according to manufacturer’s protocol. We measured succinate dehydrogenase mitochondrial activity by MTT assay. Briefly, 0.5 mg/mL MTT were added to the cells and incubated for 3 h at 37°C, reaction was stopped by aspirating the medium and solving the crystals in 1:1 mix of DMSO:ethanol for 15 min shaking at room temperature, followed by absorbance (570 nm) measurement using an Infinite 200 PRO series micro plate reader (Tecan). We measured general enzyme reduction activity by resazurin-based cell viability assay. 50 µg/mL resazurin was added to each well of a 96-well plate and incubated for 30 min at 37°C, followed by fluorescence (535/590 nm) measurement using an Infinite 200 PRO series micro plate reader (Tecan). Flow cytometry based cell death assays were performed using the apoptosis/necrosis assay according to the manufacturer’s instructions. To measure intracellular ROS, cells were incubated for 30 min with 1 μM CM-H2DCFDA in the dark and fixed using 4 % paraformaldehyde. Flow cytometry analyses were done using a FACS Calibur (BD) or MACSQuant analyzer (Miltenyi). For immunofluorescence, HeLa cells were grown on coverslips and fixed with 4% (w/v) paraformaldehyde for 15 min, permeabilize with 0.1% (v/v) Triton X-100 for 5 min and washed three times with blocking buffer (1× PBS containing 0.1% fetal calf serum (w/v)). Immunofluorescence analysis was performed as described previously 2. Confocal imaging was performed using a LSM780 confocal laser scanning microscope (ZEISS, Jena, Germany) equipped with a Plan-APO 63x/NA1.46 oil immersion objective (ZEISS).
Real-time RT-PCR
RNA was reverse transcribed with PrimeScript™ RT Master Mix (TAKARA) and quantified by real-time RT-PCR using the QuantiFast SYBR Green RT-PCR Kit (Qiagen) and a CFX96 Touch Real-Time PCR Detection System (BioRad). Each cycle consisted of 10 sec at 95°C and 30 sec at 60°C, followed by melting curve analysis. Primer sequences were as follows: huGAPDH (5′-GATTCCACCCATGGCAAATTC-3′ and 5′-AGCATCGCCCCACTTGATT-3′), hTBP (5′-GTTCTGAATAGGCTGTGGGG-3′ and 5′-ACAACAGCCTGCCACCTTAC-3′), KEAP1 (5′-GCTGATGAGGGTCACCAGTT-3′ and 5′-CCAACTTCGCTGAGCAGATT-3′), NRF2 (5′- GCTCATACTCTTTCCGTCGC-3′ and 5′- ATCATGATGGACTTGGAGCTG-3′), NQO1 (5′-GCATAGAGGTCCGACTCCAC-3′ and 5′-GGACTGCACCAGAGCCAT-3′), TXN (5′-AATGTTGGCATGCATTTGAC-3′ and 5′-CCTTGCAAAATGATCAAGCC-3′), mIL6 (5′-TAGTCCTTCCTACCCCAATTTCC-3′ and 5′-TTGGTCCTTAGCCACTCCTTC-3′), NS1 (5′-GACCAGGCGATCATGGATAA-3′ and 5′-TGCTTCTCCAAGCGAATCTC-3′).
Affinity purification and quantitative LC-MS/MS, bioinformatics analysis
For affinity purification, cell lysates were prepared by lysing cells for 5 min on ice in TAP lysis buffer (50 mM Tris pH 7.5, 100 mM NaCl, 5% (v/v) glycerol, 0.2 % (v/v) Nonidet-P40, 1.5 mM MgCl2 and protease inhibitor cocktail (EDTA-free, cOmplete; Roche)) or Cell Signaling IP buffer (20 mM TrisHCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1 % Triton and protease inhibitor cocktail (EDTA-free, cOmplete; Roche)). Where indicated, phosphatase inhibitor cocktail (PhosSTOP; Roche) or sodium fluoride (NaF) and sodium orthovanatade (Van) were added. For affinity-purification with HA-SII-tagged proteins, streptavidin affinity resin was incubated with cell lysate in either TAP lysis buffer or Cell Signaling IP buffer for 60 min at 4°C on a rotary wheel. For affinity-purification with Flag-M2-tagged proteins, Flag-M2 affinity resin was incubated with cell lysate and processed as above. Beads were washed four times with TAP lysis buffer, followed by two times with TAP wash buffer [lacking 0.2 % (v/v) Nonidet-P40] or five times with Cell Signaling IP buffer, boiled in 2x Cell Signaling SDS buffer for 5 min at 95°C and subjected to SDS-PAGE and Immunoblot analysis. For LUMIER experiments cell lysates (1:10 diluted in TAP wash buffer) or beads were resuspended in TAP wash buffer and transferred in 4x 20 µL aliquots to white well plate (Nunc) and mixed with 2x Renilla reagent (100 mM K3PO4, 500 mM NaCl, 1 mM EDTA, 25 mM Thiourea, 30 µM Coelenterazine) and luminescence was measured using an Infinite 200 PRO series micro plate reader (Tecan).
To detect and quantify proteins bound to HA-SII-tagged bait proteins by affinity purification and mass spectrometry, samples were prepared as described above. After the final four washes in TAP lysis buffer, samples were in addition washed four times with TAP wash buffer to remove residual detergent. Four independent affinity purifications were performed for each bait. Sample preparations and LC-MS/MS analysis was performed as described previously 2. Briefly, samples were sequentially digested with LysC (Wako Chemicals USA) and Trypsin (Promega), acidified with 0.1% TFA, desalted with C18 stage tips and analyzed by liquid chromatography coupled to mass spectrometry on an Orbitrap XL platform (Thermo Fisher Scientific).
For analysis of interaction proteomics data, mass spectrometry raw files were processed with MaxQuant version 1.3.0.5 or 1.4.0.6 7 using the built-in Andromeda engine to search against human proteome (UniprotKB, release 2012_06) containing forward and reverse sequences. In MaxQuant the label-free quantitation (LFQ) algorithm 8 and Match Between Runs option were used as described previously 2. Only proteins identified on the basis of at least two peptides and a minimum of three quantitation events in at least one experimental group were considered. LFQ protein intensity values were log-transformed and missing values filled by imputation. Specific enrichment was determined by multiple equal variance t-tests with permutation-based false discovery rate (FDR) statistics, performing 250 permutations. FDR thresholds and S0 parameters were empirically set to separate background from specifically enriched proteins. Data was analysed using Perseus version 1.5.2.11. Results were plotted using R (www.R-project.org). Sequence logo was generated using WebLogo 9.
Mitochondrial respiration
Hela cells were mock treated or treated with either 0.1 mM or 0.5 mM H2O2 for 5 h. After 5 h media was changed to seahorse assay media (DMEM D5030 with 10mM Pyruvate, 2mM Glutamine) without H2O2 treatment. After 1 h read out in SeaHorse Analyzer was performed with injections of 1.5 µM OliogA (ATP coupler) after 24 min, 1 µM CCCP (Electron transport chain accelerator) after 45 min and 4 µM Antimycin A and 2 µM Rotenone (mitochondrial inhibitors) after 69 min. Results were normalized by total DNA using CyQuant.
Electron microscopy
MEFs were treated as indicated, than washed once and fixed with glutaraldehyde. Fixed cells were washed with 0.2 M Sodium Cacodylate, osmicated with 1% OSO4 for 40 min, washed twice and subsequently dehydrated in Ethanol and infiltrated with Low Viscosity Embedding Media Spurr’s Kit (Electron Microscopy Sciences, cat.# E14300) according to the manual. After polymerization (overnight at 70-80°C) ultrathin serial sections (thickness 60nm) were counterstained with 0.5% Uranyl acetate and 3% Lead citrate. Image were acquired with JEOL JEM-1230 transmission electron microscope (magnification 5000-30000x), with 80kV, Gatan Orius SC1000 digital Camera and software Gatan DigitalMicrograph™.
In vivo experiments
Heterozygous Pgam5 knockout mice (Pgam5+/−) C57BL/6N background were obtained from the European Mouse Mutant Archive (EMMA). Mice were bred at the MPI of Biochemistry animal facility under license number 55.2-54-2532.116.2015. Primer sequences for genotyping were as follow L3f_6764 5′-AGGCTGGATCACTATAAGGC-3′ and L3r_6765 5′‐CTGGAGACATTGTGACCATC-3′. Mice deficient in Asc−/− (C57BL/6J) Nlrp3−/− (C57BL/6J) and Casp1/11−/− (SV129 background) or wild-type (wt, C57BL/6J) were housed and bred in animal facility at Transgenose Institute (TAAM-CNRS, Orléans, France, UPS 44 under agreement D-45-234-6, 2014) under SPF conditions in a temperature controlled environment with free access to food and water. All animal experiments have been performed according to animal welfare regulations and have been approved by the responsible authorities (University Freiburg, Germany, G-12/46, CNRS Orleans, France, CLE CCO 2012-047). For ozone treatment experiments mice were exposed to ozone (1 ppm) for 1h in a plexiglas chamber (EMB 104, EMMS). An ozonisator (Ozoniser S 500mg, Sander) was used to generate ozone and ozone levels were controlled by an ozone sensor (ATI 2-wire transmitter, Analytical Technology®). Mice were euthanatized 4h or 24h after ozone exposure by progressive CO2 inhalation. BAL was collected and cardiac perfusion was performed with ISOTON II (Acid free balanced electrolyte solution Beckman Coulter, Krefeld, Germany) before removal of lungs for further analyses. BAL was isolated by four consecutive saline lavages (500 µL/lavage, NaCl 0.9%) via a cannula introduced into trachea. After centrifugation (2000 rpm for 10 min at 4 °C) the supernatants were removed and stored at -20 °C for ELISA analysis and cell pellets were analyzed by cytopsin (Thermo Scientific, Waltham, USA) followed by Diff-Quick coloration (Merz & Dade A.G., Dudingen, Switzerland). Differential cell counts were performed with at least 400 cells. For FluAV infection experiments, mice were anesthetized with ketamine (100 µg per gram body weight) and xylazine (5 µg per gram body weight) before infection by the intranasal route with 1.5×103 pfu of SC35M per animal in a 40 µL volume. Mice were weight monitored for 14 days and euthanized when they lost >25% of their initial body weight.
Measurement of infection, cell death and inflammatory parameters in vivo
Mouse MPO was determined by ELISA (R&D systems, Abingdon, UK), cytokines were measured by Multiplex Immunoassay (ProcartaPlex, eBioscience) and total protein levels in BAL were analyzed using the Bio-Rad DC Protein Assay according to the manufacturer’s instructions.
For histological analyses left lobes of lungs were fixed in 4% buffered formaldehyde and embedded in paraffin. After tissue sections (3 µm), cuts were stained with hematoxylin and eosin. Epithelial damage and inflammatory cell infiltration were scored for severity of epithelial injury and inflammation (0 to 5) 10. Lungs were stained with anti-FluAV antiserum, ApopTag Fluorescein In Situ Apoptosis Detection Kit (Merck) and DAPI.
RNAi-mediated knockdown
Duplex siRNAs (1nmol of siRNA per 1×106 cells) were transfected using the Neon Transfection System (Invitrogen) according to the manufacturer´s instructions for HeLa and SKN-BE2 cells. siRNAs were either purchased from Qiagen, Dharmacon or synthesized by the Core Facility at the MPI of Biochemistry. siRNA target sequences were as follows: human PGAM5 [#1: 5′-CCCGCCCGTGTCTCATTGGAA-3′; #2: 5′-TCCAAGCTGGACCACTACAAA-3′; #3: 5′-CTCGGCCGTGGCGGTAGGGAA-3′; #4: 5′-CGCTAGTGACAGCCCAATAT-3′; #5: 5′-GGAGAAACCAATATAGAATT-3′], human AIFM1 [#1: 5′-GAACATCTTTAACCGAATG-3′; #2: 5′-GCATGAAGATCTCAATGAA-3′; #3: 5′-CAAGGAAGATCATTAAGGA-3′; #4: 5′-GGTAGAAACTGACCACATA-3″; #5: 5′-GATCAAGTCCTTTGTG-3″; #6: 5′-CCTGAGAGGAGGTCGA-3″; #7: 5′-GGAATTGGCAAACCCAC-3″; #8: 5′-GGAGGTCGAATGGGTA-3′], human KEAP1 [#1: 5′-GGACAAACCGCCTTAATTC-3′; #2: 5′-CAGCAGAACTGTACCTGTT-3′; #3: 5′-GGGCGTGGCTGTCCTCAAT-3′; #4: 5′-CGAATGATCACAGCAATGA-3′], human NFE2L2 [#1: 5′-TAAAGTGGCTGCTGAGAAT-3′; #2: 5′-GAGTTACAGTGTCTTAATA-3′; #3: 5′-TGGAGTAAGTCGAGAAGTA-3′; #4: 5′-CACCTTATATCTCGAAGTT-3′] and scrambled [5′- AAGGTAATTGCGCGTGCAACT-3].
Supplementary Material
Acknowledgments
We want to acknowledge the innate immunity laboratory for critical discussions and suggestions, A. Mann for technical assistance, the MPI-B core facility for technical assistance with protein purification, analysis and imaging. We further thank M. Wilson and ECM bioscience for raising the AIFM1 pS116 antibody, K. Mayr, I. Paron and G. Sowa for maintaining mass spectrometers, S. Kaphengst and the MPI-B animal facility for breeding mice. This work was supported by the Max-Planck Free Floater program to A.P., the German research foundation (PI 1084/2 and TRR179) to A.P. and (SFB 1160, project 13) to P.S., an ERC starting grant (ERC-StG iVIP, 311339) to A.P. Infect-Era and the German Federal Ministry of Education and Research (ERASE) to A.P. and United States National Institutes of Health grants R01 AI107056 and 1R41AI127009 (Moir) to D.W.L and G.K.A.
Footnotes
Author Contributions
C.H., Ch.M., Cl.M., D.A.H., D.S., F.L.P., J.W., M.B. and P.S. conducted the experiments, C.B. performed structural modelling, C.H., D.A.H. and P.H. were analyzing data. D.W.L and G.K.A contributed reagents. C.H., F.P., D.W.L, G.K.A., B.R., P.S. and A.P. designed the experiments and wrote the paper.
Competing Financial Interests Statement
The authors declare no competing financial interests.
References
- 1.Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2001;11:173–186. doi: 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura H, Nakamura K, Yodoi J. Redox regulation of cellular activation. Annual review of immunology. 1997;15:351–369. doi: 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- 3.Sandalio LM, Rodriguez-Serrano M, Romero-Puertas MC, del Rio Luis A. Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Sub-cellular biochemistry. 2013;69:231–255. doi: 10.1007/978-94-007-6889-5_13. [DOI] [PubMed] [Google Scholar]
- 4.Rosca MG, et al. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes. 2012;61:2074–2083. doi: 10.2337/db11-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu JM, Lin PH, Yao Q, Chen C. Chemical and molecular mechanisms of antioxidants: experimental approaches and model systems. Journal of cellular and molecular medicine. 2010;14:840–860. doi: 10.1111/j.1582-4934.2009.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes to cells : devoted to molecular & cellular mechanisms. 2011;16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 7.Song JJ, et al. Role of glutaredoxin in metabolic oxidative stress. Glutaredoxin as a sensor of oxidative stress mediated by H2O2. The Journal of biological chemistry. 2002;277:46566–46575. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- 8.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Molecular and cellular biology. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochemical pharmacology. 2013;85:705–717. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 10.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free radical biology & medicine. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free radical biology & medicine. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang D, et al. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Experimental eye research. 2007;85:462–472. doi: 10.1016/j.exer.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hroudova J, Singh N, Fisar Z. Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer’s disease. BioMed research international. 2014;2014:175062. doi: 10.1155/2014/175062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ano Y, et al. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neuroscience letters. 2010;469:39–43. doi: 10.1016/j.neulet.2009.11.040. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Boehning DF, Qian T, Popov VL, Weinman SA. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:2474–2485. doi: 10.1096/fj.06-7345com. [DOI] [PubMed] [Google Scholar]
- 16.Bhattacharya A, et al. Superoxide Dismutase 1 Protects Hepatocytes from Type I Interferon-Driven Oxidative Damage. Immunity. 2015;43:974–986. doi: 10.1016/j.immuni.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olagnier D, et al. Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus-infected dendritic cells. PLoS pathogens. 2014;10:e1004566. doi: 10.1371/journal.ppat.1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bottero V, Chakraborty S, Chandran B. Reactive oxygen species are induced by Kaposi’s sarcoma-associated herpesvirus early during primary infection of endothelial cells to promote virus entry. Journal of virology. 2013;87:1733–1749. doi: 10.1128/JVI.02958-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards MR, et al. The Marburg virus VP24 protein interacts with Keap1 to activate the cytoprotective antioxidant response pathway. Cell reports. 2014;6:1017–1025. doi: 10.1016/j.celrep.2014.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson B, et al. Dimerization Controls Marburg Virus VP24-dependent Modulation of Host Antioxidative Stress Responses. Journal of molecular biology. 2016;428:3483–3494. doi: 10.1016/j.jmb.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amri F, Ghouili I, Amri M, Carrier A, Masmoudi-Kouki O. Neuroglobin protects astroglial cells from hydrogen peroxide-induced oxidative stress and apoptotic cell death. Journal of neurochemistry. 2016 doi: 10.1111/jnc.13876. [DOI] [PubMed] [Google Scholar]
- 22.Kim JY, Park JH. ROS-dependent caspase-9 activation in hypoxic cell death. FEBS letters. 2003;549:94–98. doi: 10.1016/s0014-5793(03)00795-6. [DOI] [PubMed] [Google Scholar]
- 23.Radogna F, et al. Cell type-dependent ROS and mitophagy response leads to apoptosis or necroptosis in neuroblastoma. Oncogene. 2016;35:3839–3853. doi: 10.1038/onc.2015.455. [DOI] [PubMed] [Google Scholar]
- 24.Ravindran J, Gupta N, Agrawal M, Bala Bhaskar AS, Lakshmana Rao PV. Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis : an international journal on programmed cell death. 2011;16:145–161. doi: 10.1007/s10495-010-0554-0. [DOI] [PubMed] [Google Scholar]
- 25.Shen C, et al. Aldehyde dehydrogenase 2 deficiency negates chronic low-to-moderate alcohol consumption-induced cardioprotecion possibly via ROS-dependent apoptosis and RIP1/RIP3/MLKL-mediated necroptosis. Biochimica et biophysica acta. 2016 doi: 10.1016/j.bbadis.2016.11.016. [DOI] [PubMed] [Google Scholar]
- 26.Wiegman CH, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. The Journal of allergy and clinical immunology. 2015;136:769–780. doi: 10.1016/j.jaci.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Triantaphyllopoulos K, et al. A model of chronic inflammation and pulmonary emphysema after multiple ozone exposures in mice. American journal of physiology. Lung cellular and molecular physiology. 2011;300:L691–700. doi: 10.1152/ajplung.00252.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kosmider B, Loader JE, Murphy RC, Mason RJ. Apoptosis induced by ozone and oxysterols in human alveolar epithelial cells. Free radical biology & medicine. 2010;48:1513–1524. doi: 10.1016/j.freeradbiomed.2010.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi A, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Molecular and cellular biology. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lo SC, Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. The Journal of biological chemistry. 2006;281:37893–37903. doi: 10.1074/jbc.M606539200. [DOI] [PubMed] [Google Scholar]
- 32.Lo SC, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Experimental cell research. 2008;314:1789–1803. doi: 10.1016/j.yexcr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ceh-Pavia E, Spiller MP, Lu H. Folding and biogenesis of mitochondrial small Tim proteins. International journal of molecular sciences. 2013;14:16685–16705. doi: 10.3390/ijms140816685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Susin SA, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 35.Joza N, et al. AIF: not just an apoptosis-inducing factor. Annals of the New York Academy of Sciences. 2009;1171:2–11. doi: 10.1111/j.1749-6632.2009.04681.x. [DOI] [PubMed] [Google Scholar]
- 36.Wilkins JM, McConnell C, Tipton PA, Hannink M. A conserved motif mediates both multimer formation and allosteric activation of phosphoglycerate mutase 5. The Journal of biological chemistry. 2014;289:25137–25148. doi: 10.1074/jbc.M114.565549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pichlmair A, et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature. 2012;487:486–490. doi: 10.1038/nature11289. [DOI] [PubMed] [Google Scholar]
- 38.Page A, et al. Marburgvirus hijacks nrf2-dependent pathway by targeting nrf2-negative regulator keap1. Cell reports. 2014;6:1026–1036. doi: 10.1016/j.celrep.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 39.Medvedev R, Ploen D, Hildt E. HCV and Oxidative Stress: Implications for HCV Life Cycle and HCV-Associated Pathogenesis. Oxidative medicine and cellular longevity. 2016;2016:9012580. doi: 10.1155/2016/9012580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren JH, et al. Protective Role of Sirtuin3 (SIRT3) in Oxidative Stress Mediated by Hepatitis B Virus X Protein Expression. PloS one. 2016;11:e0150961. doi: 10.1371/journal.pone.0150961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kajitani N, et al. Mechanism of A23187-induced apoptosis in HL-60 cells: dependency on mitochondrial permeability transition but not on NADPH oxidase. Bioscience, biotechnology, and biochemistry. 2007;71:2701–2711. doi: 10.1271/bbb.70304. [DOI] [PubMed] [Google Scholar]
- 42.Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS Crosstalk in Inflammation. Trends in cell biology. 2016;26:249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 43.Chacinska A, et al. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. The EMBO journal. 2004;23:3735–3746. doi: 10.1038/sj.emboj.7600389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shindo R, Kakehashi H, Okumura K, Kumagai Y, Nakano H. Critical contribution of oxidative stress to TNFalpha-induced necroptosis downstream of RIPK1 activation. Biochemical and biophysical research communications. 2013;436:212–216. doi: 10.1016/j.bbrc.2013.05.075. [DOI] [PubMed] [Google Scholar]
- 45.Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moriwaki K, et al. The Mitochondrial Phosphatase PGAM5 Is Dispensable for Necroptosis but Promotes Inflammasome Activation in Macrophages. Journal of immunology (Baltimore, Md.: 1950) 2016;196:407–415. doi: 10.4049/jimmunol.1501662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer research. 1991;51:794–798. [PubMed] [Google Scholar]
- 48.Marconett CN, et al. BZL101, a phytochemical extract from the Scutellaria barbata plant, disrupts proliferation of human breast and prostate cancer cells through distinct mechanisms dependent on the cancer cell phenotype. Cancer biology & therapy. 2010;10:397–405. doi: 10.4161/cbt.10.4.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang R, et al. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochemical and biophysical research communications. 2008;373:151–154. doi: 10.1016/j.bbrc.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 50.Nogusa S, et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell host & microbe. 2016;20:13–24. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1.Pichlmair A, et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature. 2012;487:486–490. doi: 10.1038/nature11289. [DOI] [PubMed] [Google Scholar]
- 2.Habjan M, et al. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS pathogens. 2013;9:e1003663. doi: 10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabriel G, et al. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18590–18595. doi: 10.1073/pnas.0507415102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaikuad A, Alfano I, Picaud S, Filippakopoulos P, Barr A, von Delft F, Arrowsmith CH, Edwards AM, Weigelt J, Bountra C, Takeda K, Ichijo H, Knapp S. 3MXO, Crystal structure oh human phosphoglycerate mutase family member 5 (PGAM5) PDB database. 2010 [Google Scholar]
- 5.Provencher SW, Glockner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 6.Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS Crosstalk in Inflammation. Trends in cell biology. 2016;26:249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 8.Cox J, et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Molecular & cellular proteomics : MCP. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome research. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Besnard AG, et al. Dual Role of IL-22 in allergic airway inflammation and its cross-talk with IL-17A. American journal of respiratory and critical care medicine. 2011;183:1153–1163. doi: 10.1164/rccm.201008-1383OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.