

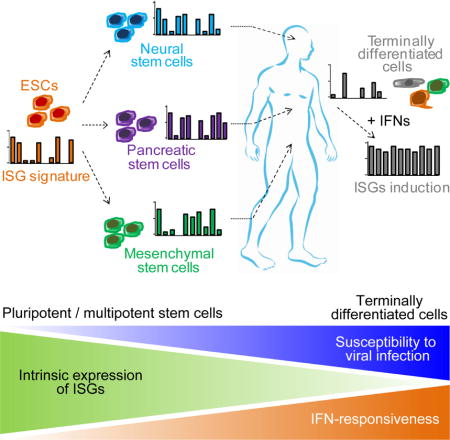
SUMMARY
Stem cells are highly resistant to viral infection compared to their differentiated progeny; however, the mechanism is mysterious. Here we analyzed gene expression in mammalian stem cells and cells at various stages of differentiation. We find that, conserved across species, stem cells express a subset of genes previously classified as interferon (IFN) stimulated genes (ISGs), but that expression is intrinsic, as stem cells are refractory to interferon. This intrinsic ISG expression varies in a cell type-specific manner and many ISGs decrease upon differentiation, at which time cells become IFN-responsive, allowing induction of a broad spectrum of ISGs by IFN signaling. Importantly, we show that intrinsically expressed ISGs protect stem cells against viral infection. We demonstrate the in vivo importance of intrinsic ISG expression for protecting stem cells and their differentiation potential during viral infection. These findings have intriguing implications for understanding stem cell biology and the evolution of pathogen resistance.
eTOC
Intrinsic expression of interferon stimulating genes makes stem cells resistant to infections, preserving their pool throughout the organism’s lifespan
INTRODUCTION
Over four decades ago researchers made a curious observation that pluripotent, but not differentiated teratocarcinoma cells were resistant to murine polyomavirus infection (Swartzendruber and Lehman, 1975). Since then, others have made similar observations with a diverse array of viruses and tissue stem cells, suggesting that resistance to viral infection is a general property of pluri/multipotent cells (Belzile et al., 2014; Gonczol et al., 1984; Shen et al., 1999; Villa et al., 2016; Weichold et al., 1998; Wolf and Goff, 2009). Myxoma virus, for example, a therapeutic oncolytic poxvirus, infects differentiated human monocytes, B cells, and natural killer cells, but does not infect the hematopoietic stem cells (HSCs) from which those cells derive (Villa et al., 2016). Still, the mechanism by which these progenitor cells resist viral infection remains unexplained.
In most cells, interferon (IFN) response is a major first line of defense against viral infection. Viral infection triggers production of IFNs which then bind to ubiquitously expressed receptors on nearby cells and induce a powerful transcriptional program comprising hundreds of antiviral IFN-stimulated genes (ISGs) (Schneider et al., 2014). Unlike most cells, however, pluripotent embryonic stem cells (ESCs) and embryonic carcinoma cells do not produce type I IFN in response to viral infection or treatment with poly(I:C) (Burke et al., 1978), and they respond weakly to exogenous IFN (Hong and Carmichael, 2013). This suggests that pluripotent cells do not rely on canonical IFN signaling for antiviral protection.
Consistent with this, it was recently reported that mouse ESCs use an IFN-independent RNA interference-based mechanism for antiviral defense (Maillard et al., 2013). Nevertheless, a protein-based defense via ISGs may still be important. We were previously intrigued to find that two classic ISGs (IFITM1 and IFI30) are highly expressed in hESCs, and that their expression progressively decrease across differentiation of hESC into hepatocyte-like cells (HLCs) (Wu et al., 2012). This suggests that ISG expression may relate to pluripotency.
In this study, we used a variety of viruses and stem cells of different lineages and differentiation potential to better define the role that these intrinsically expressed ISGs play in defending pluri/multipotent cells from viral infection. We found that like hESCs, induced pluripotent stem cells (iPSCs), germ layer cells, and tissue stem cells all intrinsically express canonical ISGs. Importantly, different types of stem cell display distinct ISG signatures and many of these ISGs decrease as cells terminally differentiate. Further, by knocking out several of these ISGs, we show that intrinsically expressed ISGs are indeed important for protecting stem cells from viral infection. Finally, we used a chimeric mouse model to demonstrate the importance of protecting human stem cells from viral infection. Without the expression of critical ISGs, stem cells are susceptible to infection and fail to regenerate tissues after viral challenge.
RESULTS
Intrinsic expression of ISGs in pluripotent and multipotent human stem cells
We first sought to more thoroughly define how pluripotency correlates with resistance to viral infection. To do this, we challenged pluripotent hESC, multipotent endoderm cells, and differentiated HLCs with a diverse panel of viruses, including positive-strand RNA viruses from the flavivirus family including dengue virus (DENV), yellow fever virus (YFV) and Zika virus (ZIKV), as well as several negative-strand RNA viruses including vesicular stomatitis virus (VSV), Newcastle disease virus (NDV), and respiratory syncytial virus (RSV). We found that while differentiated HLCs were highly permissive to infection, multipotent endoderm cells were only partially permissive, and pluripotent hESCs were highly resistant (Figure S1A).
Of the viruses we tested, it is well known that DENV and VSV have broad tissue tropism (Anderson, 2003; Finkelshtein et al., 2013), suggesting that their entry might depend on ubiquitously expressed factors. Their inability to infect pluripotent and early progenitor cells therefore suggests these cell types may express antiviral factors. To test this, we performed RNA-Seq transcriptome analyses on two ESC and two iPSC lines. We found an overlapping set of ISGs that was highly expressed by all lines (Figures 1A–B and S1B). This is consistent with a previous study showing that IFITM1 is highly expressed by hESC (Grow et al., 2015). Western blot analyses of representative ISGs including EIF3L, IFITM1, IFITM3, and BST2 confirmed high baseline expression in the stem cells, similar to the levels achieved in HLCs after IFN-β stimulation (Figure 1C). Meanwhile, many other ISGs such as APOBEC3G, MX1/2, OAS1-3, RSAD2, and members of the IFIT family were not expressed (Figure 1B). Nevertheless, representatives of these unexpressed ISGs were readily induced in HLCs by IFN-β (Figure 1C).
Figure 1. Intrinsic expression of ISGs in pluripotent and multipotent human stem cells.
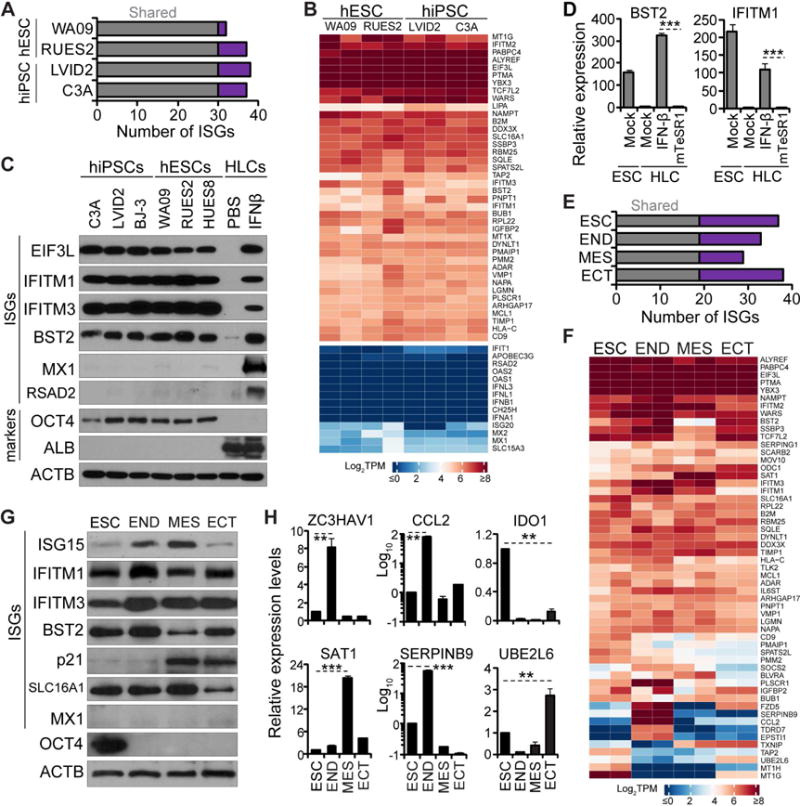
(A, E) Bar diagram illustrating the number of highly expressed ISGs unique to each hESC/iPSC line (purple) and shared by all lines (gray) (A), unique to each cell type and shared by all cell types (E)
(B, F) Heatmap of z-score TPM of highly expressed ISGs in hES/iPSC lines (B), in hESCs and derived germ layers (F). Additional ISGs and IFN genes with low expression values were selected for comparison (lower block in B).
(C) Western blot analyses of ISGs and markers in the indicated hES/iPSCs at baseline and in hESC-derived HLCs treated with IFN-β or PBS.
(D) Analyses of representative ISGs for hESCs at baseline and for HLCs after treatment with IFN-β or conditioned mTeSR1 by qRT-PCR. Expression levels relative to mock HLC were for normalization.
(G–H) Analyses of ISGs and markers in hESCs and germ layers by western blot (G) and qRT-PCR (H). ISG expression in hESC at baseline was for normalization.
qRT-PCR data shown as the means ± standard deviations (SD) from 3 independent experiments. Throughout this study, Student’s t test was used and asterisks indicate statistically significant difference (*, p<0.05, **, p<0.01; ***, p<0.001).
See also Tables S1 and S2 and Figure S1.
These results raised the possibility that expression of a subset of ISGs is an intrinsic property of ESC/iPSCs. It is also possible, however, that a component of the stem cell medium (mTeSR1) directly induced ISGs and/or that ESC/iPSCs do in fact produce and respond to IFN leading to ISG expression. We tested these possibilities by culturing HLCs in the conditioned mTeSR1 but found that select ISGs were not induced (Figure 1D). Moreover, even in the presence of effective concentrations of IFN-neutralizing antibody, ISG expression was stable (Figure S1C). These results indicate that ISG expression is not due to IFN expression and are consistent with a previous study showing that IFN response is severely attenuated in ESC/iPSCs (Hong and Carmichael, 2013).
Like pluripotent ESCs, multipotent endoderm cells were also largely resistant to viral infection (Figure S1A). This suggested that they too might express ISGs. In order to measure ISG expression in each of the three multipotent germ layers, we differentiated hESCs into endoderm (END), mesoderm (MES), and ectoderm (ECT) (Figure S1D). Using RNA-Seq, we found that like hESCs, all three germ layers showed high expression of many ISGs (Figure 1E–F). Many of the highly expressed ISGs, such as BST2, DDX3X, EIF3L and IFITM family members showed comparable protein levels across the three germ layers and the parental ESCs. Other ISGs, however, showed germ layer-specific expression patterns. SERPINB9, CCL2 and PLSCR1, for example, were highly expressed in END; SAT1 and TIMP1 were expressed in MES; and UBE2L6 and TXNIP were expressed in ECT (Figure 1F–H). Overall, we found that different germ layers have distinct ISG signatures, and like ESCs, each cell type only highly expresses a subfraction of the known ISGs.
Distinct ISG expression patterns in different tissue stem cells
We next examined whether expression of ISGs was apparent in tissue stem cells. We began by differentiating hESCs into three types of tissue stem cells derived from the three germ layers: pancreatic stem cells (PSCs), mesenchymal stem cells (MSCs), and neural stem cells (NSCs) (Figure 2A). Additional markers were confirmed by qRT-PCR (Figure S2A) and multipotency was confirmed by their ability to further terminally differentiate (Figure S2B).
Figure 2. Distinct ISG expression patterns in different tissue stem cells.
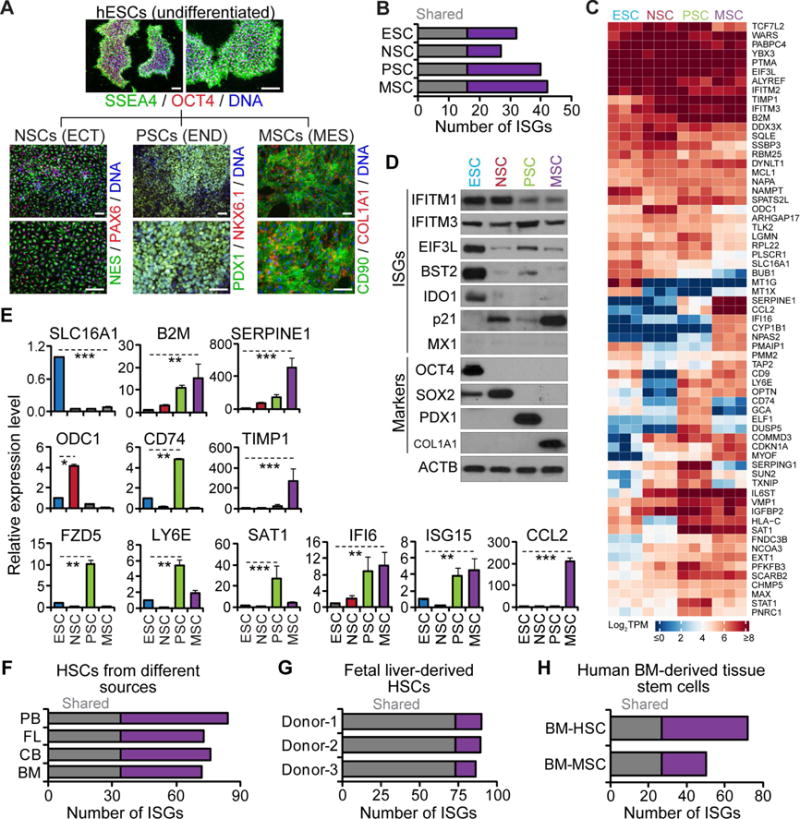
(A) Representative images of hESCs and derived tissue stem cells. Scale bars: 100 μm.
(B) Bar diagram illustrating the number of highly expressed ISGs unique to each cell type and common to all cell types.
(C) Heatmap of z-score TPM of ISGs highly expressed in hESCs and derived tissue stem cells.
(D–E) Analyses of ISGs and markers in hESCs and tissue stem cells by western blot (D) and qRT-PCR (E), shown as the means ± SD from 3 independent experiments.
(F–H) Bar diagram illustrating the number of highly expressed ISGs unique to each source and shared by all sources for primary HSCs (F), unique to individual donor and shared by all donors for FL-derived HSCs (G), unique to each cell type and shared by both cell types for BM-derived MSCs and HSCs (H).
See also Tables S3 and Figure S2.
We then performed RNA-Seq to assess gene expression in the three tissue stem cells. We found that each tissue stem cell type expressed a number of ISGs at high levels, and their expression pattern differed from those of the germ layers and ESCs (Figures 2B–E and S2C–D). ESCs, for example, expressed higher levels of IDO1, SLC16A1, and BST2 than tissue stem cells. Likewise, several ISGs including CDKN1A and COMMD3 were highly expressed in tissue stem cells but not in ESCs. Further, between each tissue stem cell types, each had a distinguishable ISG signature. CD74, SERPING1 and FZD5, for example, were highly expressed in PSCs, whereas SERPINE1 and CCL2 were highly expressed in MSCs.
To confirm these observations in vivo, we extended our analyses to primary tissue stem cells. We isolated hematopoietic stem cells (HSCs) from four different sources: bone marrow (BM), peripheral blood (PB), umbilical cord blood (CB) and fetal liver (FL). We found that primary HSCs expressed numerous ISGs at high levels and their ISG signatures were consistent across donors, but varied among different tissue sites (Figures 2F–G and S2E). Analyses of a published data set for MSCs isolated from placenta (PL) and BM yielded similar observations (Roson-Burgo et al., 2014) (Figure S2F). Again, intercellular comparison suggested diverging ISG expression patterns between primary HSCs and MSCs (Figures 2H and S2G–I), corroborating what we observed in hESC-derived stem cells.
ISG expression changes during terminal differentiation of hESCs
We observed that differentiation of hESCs into HLCs was accompanied by a progressive loss of antiviral resistance (Figure S1A). This raised the possibility that decreased antiviral resistance during differentiation may associate with changes in ISG expression patterns. To test this, we differentiated hESCs towards different tissue types and monitored ISG expression during stages of differentiation.
We began by differentiating hESCs towards pancreatic β-like cells (Im-β). Through the process, we confirmed differentiation by measuring the expression of lineage-specific markers by RNA-Seq, IFA, and qRT-PCR (Figures 3A and S3A-B). Interestingly, many ISGs highly expressed in hESCs decreased in terminally differentiated cells (Figure 3B). We confirmed the expression changes of these ISGs by western blot and qRT-PCR (Figure 3C–D). Due to dramatic differences in gene expression patterns between cells during differentiation, for qRT-PCR, expression levels were normalized to a panel of housekeeping genes (Eisenberg and Levanon, 2013), all of which were stably expressed (Figure S3C). Interestingly, we also found that cells at each stage also displayed high expression of a subgroup of ISGs (Figure S3J). We next performed an analogous set of experiments by differentiating hESCs into HLCs and obtained similar results (Figure S4A–E).
Figure 3. ISG expression changes during terminal differentiation of hESCs.
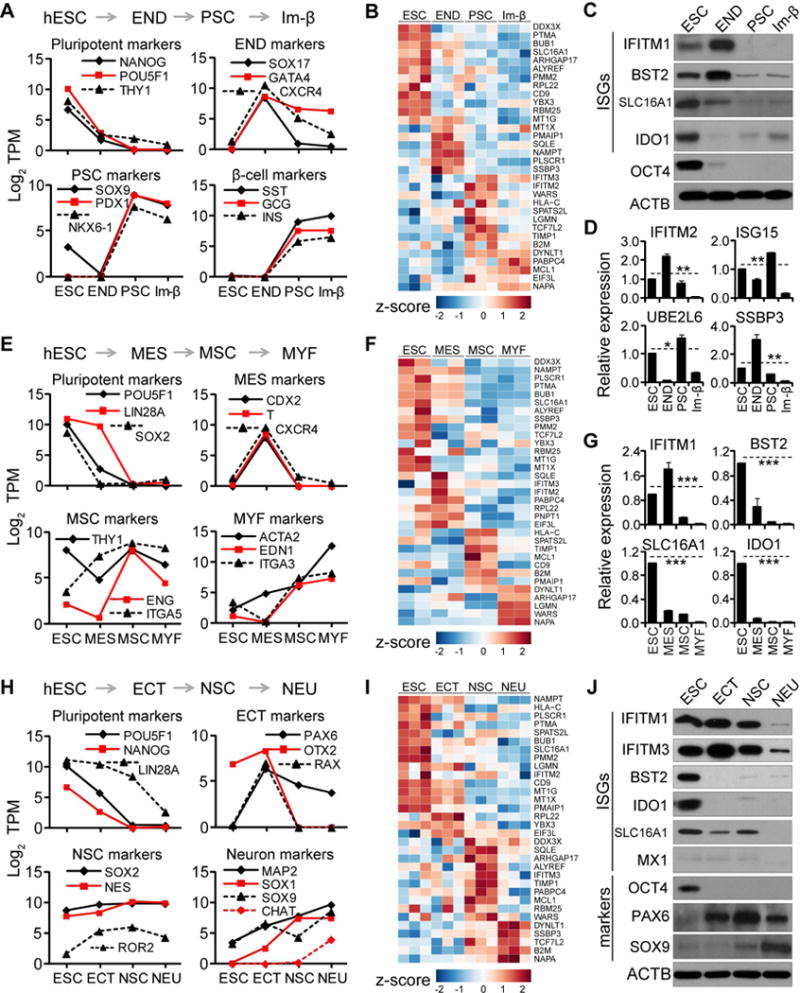
(A, E, and H) Analyses of markers at specified stages along terminal differentiation of hESCs, shown as average log2 TPM values from 2 to 3 independent experiments. (A) Stages shown for β-cell differentiation: ESC, END, pancreatic stem (PSC) and immature β-like cell (Im-β); (E) For myofibroblast differentiation: ESC, MES, MSC, and myofibroblast (MYF); (H) for neuronal differentiation: ESC, ECT, NSC, and neuron (NEU).
(B, F, and I) Heatmap of z-score TPM of ISGs highly expressed in hESC along β-cell (B), myofibroblast (F), and neuronal differentiation (I).
(C–D, G, and J) Analyses of ISGs and markers along differentiation of hESCs to β-cell by western blot (C) and qRT-PCR (D), to myofibroblast by qRT-PCR (G), to neuron by western blot (J). qRT-PCR data shown as the means ± SD from 3 independent experiments.
See also Figures S3 and S4.
Both pancreatic β-like cell and HLC are within the endoderm lineage. We next tested whether a similar dynamic pattern of ISG expression occurs when hESCs are differentiated into the other two lineages – mesoderm and ectoderm. Indeed, we found that many ISGs highly expressed in hESCs decreased along the mesoderm lineage when hESCs were differentiated into either myofibroblasts (MYF) (Figures 3E–G and S3D–F) or macrophage-like cells (MLC) (Figure S4F–I), and along the ectoderm lineage when hESCs were differentiated into neuron-like cells (NEU) (Figures 3H–J and S3G–I). Similarly, cells at each differentiation stage also displayed high expression of a subgroup of ISGs (Figure S3K–L).
ISGs expression changes during development of human tissue stem cells in vivo
To exclude the possibility that the changes observed in ISG expression were an artifact of cell culture manipulations, we extended our analysis to primary cells isolated from human fetal liver.
The fetal liver is enriched in both HSCs and their derivatives including cells from morphologically distinct and successive developmental stages: proerythroblasts (Pro), early basophilic erythroblasts (E-Baso), late basophilic erythroblasts (L-Baso), polychromatic erythroblasts (Poly) and orthochromatic erythroblasts (Ortho). We isolated these cell populations using our recently described method (Hu et al., 2013) and further confirmed their identity using RNA-Seq and Giemsa staining (Figure 4A–B). As expected, we found that HSC-specific transcription factors, including GFI1, SPI1, GATA4, were highly expressed in HSCs. Likewise, the expression of erythrocyte-specific markers GYPA, SLC4A1, and HBG1 progressively increased during differentiation to the Ortho stage (Figure 4C).
Figure 4. ISGs expression changes during in vivo differentiation of human tissue stem cells.
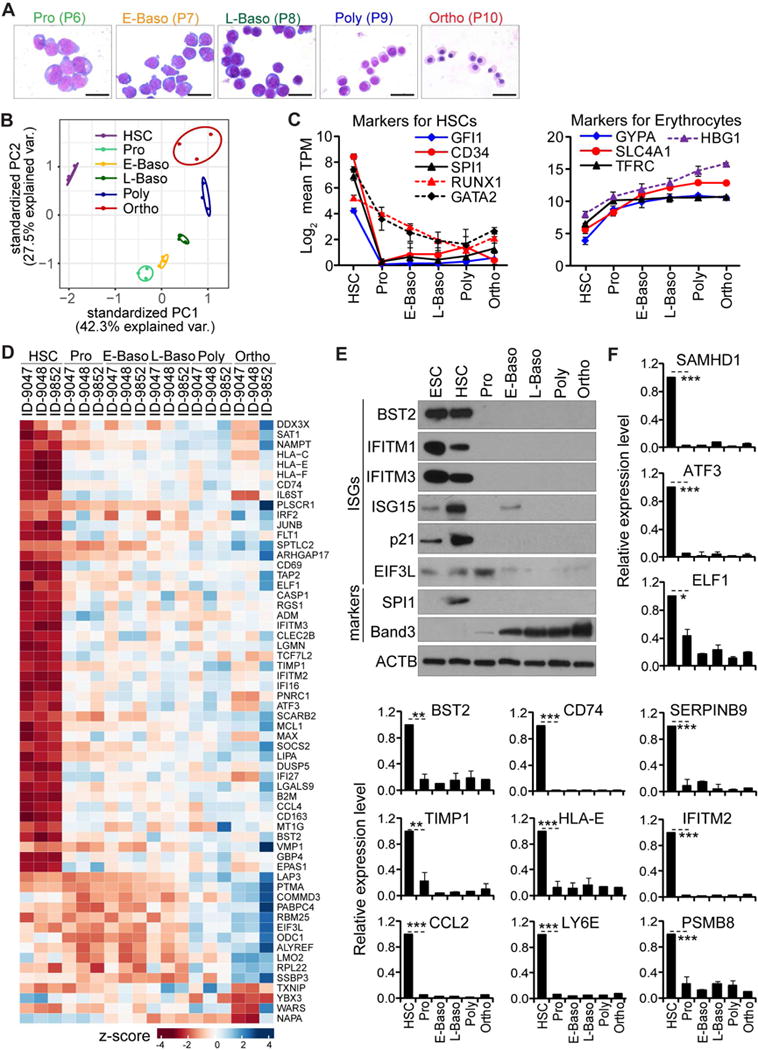
(A) Representative images of erythroblasts on Giemsa stained cytospins from the five sorted cell populations.
(B) PCA of expressed genes in the indicated cell populations.
(C) Analyses of HSC and erythrocyte markers at the specified stages, shown as average log2 TPM ± SD from 3 biological replicates.
(D) Heatmap of z-score TPM of highly expressed ISGs in HSCs during in vivo erythropoiesis.
(E–F) Analyses of ISGs and markers in HSCs and differentiated populations by western blot (E) and qRT-PCR (F), shown as the means ± SD from 3 biological replicates.
Similar to in vitro observations, expression of ISGs displayed dynamic changes: many ISGs were highly expressed in HSCs in vivo, but not in further differentiated cells; some remained stable across differentiation, while very few were expressed at relatively higher levels at later stages (Figure 4D). For example, most of the ISGs highly expressed in HSCs, such as IFITM3, SAMHD1, LY6E, and CCL2 were expressed at lower levels in Pro cells, as further confirmed by western blot and qRT-PCR (Figure 4E–F). In contrast to ISGs, housekeeping gene expression was stable in all six cell populations (data not shown). These results agree with our findings in cell culture and show that stem cells maintain high expression levels a subgroup of ISGs in vivo and that these genes display changing expression patterns as the stem cells differentiate.
Conserved ISG expression in stem cells across species
We then investigated whether ISG expression in stem cells is conserved across species. To test this, we analyzed published transcriptomic data obtained from mouse ESC (mESC) lines derived from different mouse strains (Klattenhoff et al., 2013; Li et al., 2014; Wamstad et al., 2012) for expression levels of a set of known mouse ISGs (Table S2). Like hESCs, we found that mESCs intrinsically express a subset of ISGs at high levels (Figure S5B). Similar intrinsic expression of ISGs was also observed in iPSCs of chimpanzee (Marchetto et al., 2013) (Figure S5A). Interestingly, while many of the ISGs highly expressed in hESCs differed from those highly expressed in pluripotent stem cells from mouse and chimpanzee, some ISGs such as BST2, MOV10, and members of the IFITM family, were highly expressed in ESCs from all three species (Figure S5C). Also, like hESCs, many ISGs highly expressed in mESCs appear to decrease upon terminal differentiation (Wamstad et al., 2012) (Figure S5D–E). These data suggest that intrinsic ISG expression is a conserved property of mammalian stem cells.
The elevated expression of distinct ISG subsets in each stem cell lineage rather than the full ISG repertoire suggests distinct transcriptional regulatory mechanisms. To understand the differential regulation of ISGs in stem cells and during differentiation, we then analyzed published ChIP-Seq data from terminal differentiation of mESCs (Wamstad et al., 2012). We found that, in mESCs, H3K4me3 and H3K27ac modifications (associated with active promoters and/or enhancers) were enriched at the promoters and/or enhancers of highly expressed ISGs (e.g. IFITM3 and IGFBP2), but not unexpressed ISGs (e.g. MX1 and OAS1) (Figure S5F–G). Similar observations were also made from hESCs (Figure S5H). During terminal differentiation, dynamic changes of these epigenetic modifications correlated with ISG expression (Figure S5F). These analyses suggest that chromatin modifications may be involved in the differential regulation of ISGs.
Antiviral activity of ISGs in undifferentiated human ESCs
We next tested whether ISGs expressed in ESCs confer resistance to viral infection. For this, we chose to knock out IFITM genes because these ISGs have well-known antiviral activity (Bailey et al., 2014) and were highly expressed in most of the stem cells we analyzed.
The IFITM gene family includes, among others, IFITM1, IFITM2, and IFITM3, all of which share high sequence homology. As a result, CRISPR/Cas9 gene editing that targeted the IFITM3 locus yielded double (IFITM2 and IFITM3) and triple (IFITM1, IFITM2, and IFITM3) knockout (KO) hESCs, designated as KO23 and KO123, respectively (Figure 5A). Loss of these genes did not alter the expression of pluripotent markers nor did it affect cell growth rates. Most importantly, the IFITM KO hESCs retained the capacity to differentiate into the three germ layers (Figure S6A–C). These results are consistent with observations made in Ifitm knockout mice, which are viable and developmentally normal (Lange et al., 2008).
Figure 5. Antiviral activity of ISGs in human stem cells.
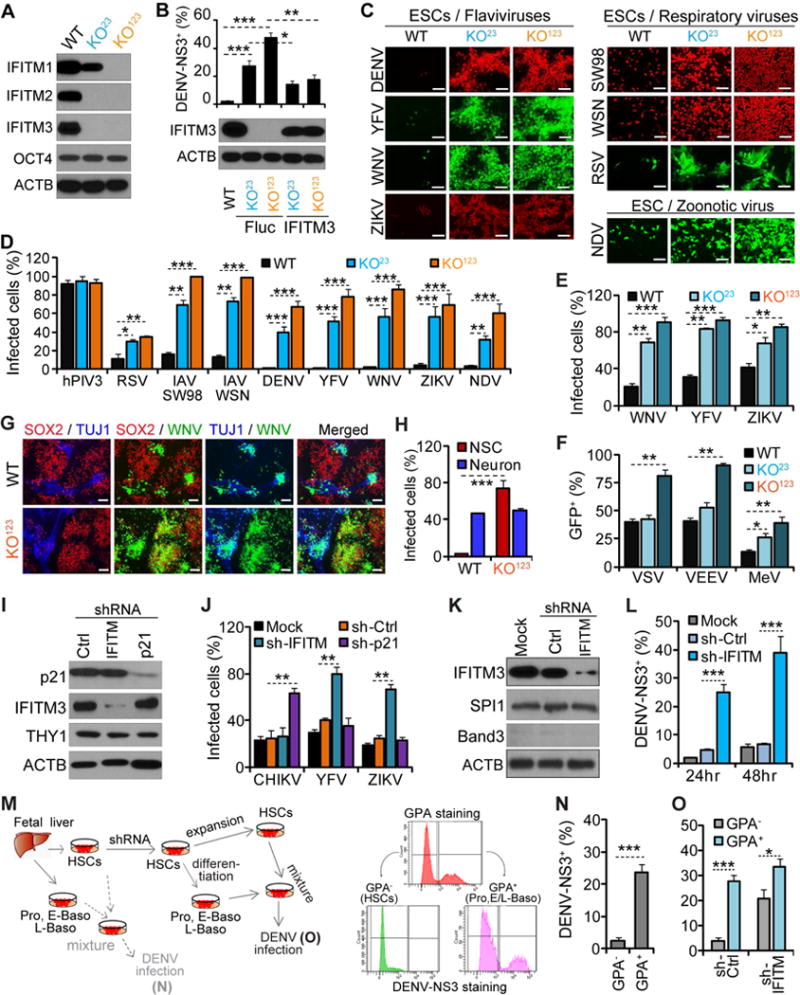
(A) Analyses of IFITM family members by western blot in WT and two KO ESC clones.
(B) WT or KO23 and KO123 ESCs reconstituted with IFITM3 or Fluc were infected with DENV (MOI =1.0). Upper panel: the percentages of infected cells at 48 hr post infection (p.i.). Lower panel: western blot analysis of IFITM3.
(C–D) Fluorescence microscopy (C) or flow cytometry (D) showing infection of WT, KO23, and KO123 ESCs by different viruses. Scale bars: 100 μm.
(E–F) Flow cytometry showing infection of WT and IFITM KO NSCs by the indicated viruses.
(G–H) hESC-derived co-cultures were infected with GFP-expressing WNV. (G): NSC marker SOX2 (red); neuronal marker TUJ1 (blue). Scale bars: 100 μm; (H): flow cytometry showing the percentages of infected cells.
(I–J) Analyses of primary MSCs transduced with indicated shRNAs by western blot (I) or by flow cytometry (J) after infection with flaviviruses or CHIKV.
(K–L) Analyses of FL-derived HSCs mock treated or transduced with the indicated shRNAs by western blot (K) or by flow cytometry (L) after infection with DENV.
(M–O) Left: schematic of infection of FL-derived hematopoietic cells with DENV; Right: flow cytometry gating strategy for GPA and DENV infection; (N–O) DENV infection.
Shown in (B, D, E–F, J, L, N–O) are the mean percentages of infected cells ± SD from 3 independent experiments.
See also Figure S6.
To determine if these ISGs were critical for ESC resistance to viral infection, we exposed wild-type (WT) and IFITM KO hESC clones to DENV. Unlike WT hESCs, we found that both the double and triple IFITM KO clones were now highly permissive to DENV infection (Figure 5B–C). Next, to address concerns of off-target effects, we reconstituted the IFITM KO clones with IFITM3 and found that this protein alone was sufficient to restore partial resistance to DENV infection (Figure 5B).
We then extended these observations to a panel of human viral pathogens. We analyzed four respiratory viruses [influenza A virus (IAV) strains A/WSN/33 (WSN) and A/swine/Texas/98 (SW98), RSV, and human parainfluenza virus 3 (hPIV3)], three additional flaviviruses [YFV, West Nile virus (WNV), and ZIKV], and one zoonotic virus (NDV). Again, except for hPIV3 (Figure S6D), we found that the IFITM KO hESCs were strikingly more permissive to viral infection than WT cells (Figure 5C–D). Of the viruses tested, the flaviviruses were the most affected. WT ESCs were almost completely resistant to infection by DENV, WNV, and YFV, and to a lesser extent by ZIKV. On the other hand, 65–85% of the IFITM triple KO cells labeled positive for viral antigens (Figure 5D). We further confirmed these results in a second hESC line, WA09, by using shRNAs to knockdown IFITM proteins expression (Figure S6E–G). Together, these results provide proof-of-principle that ISGs expressed in pluripotent stem cells can indeed be functionally antiviral.
Antiviral activity of ISGs in primary and hESC-derived multipotent tissue stem cells
We then extended our analyses to tissue stem cells, analyzing the effects of ISGs on viral infection in NSCs. As seen for hESCs, marker expression, cell growth rates, and the capacity to differentiate into neuronal cells were not affected by the loss of IFITM genes (Figure S6H–I).
We then challenged WT and IFITM KO NSCs with WNV, which is naturally neurotropic. Unlike WT hESCs, WT NSCs were susceptible to WNV infection; however, infection was markedly increased in IFITM KO NSCs (Figure 5E). ZIKV is also neurotropic – it has been linked to microcephaly, serious neurological complications, and it was recently demonstrated to infect NSCs (Cao-Lormeau et al., 2016; Tang et al., 2016). Again, triple IFITM KO NSCs were significantly more susceptible to ZIKV infection. We made similar observations when we infected WT and IFITM KO NSCs with YFV-17D, a live-attenuated vaccine strain, which in rare cases can cause encephalitis (McMahon et al., 2007) (Figure 5E).
In addition to the flaviviruses, we tested several neuroinvasive viruses, including VSV, Venezuelan equine encephalitis virus (VEEV), and measles (MeV). Unlike flaviviruses where both double and triple IFITM KO were more susceptible to infection, for VSV and VEEV, only the triple IFITM KO cells showed increased susceptibility (Figures 5F and S6J). This suggests that VSV and VEEV are most sensitive to inhibition by IFITM1, with IFITM2 and IFITM3 having less antiviral activity.
Next we developed a co-culture system comprising NSCs and neurons to more dramatically visualize the antiviral capacity of ISGs in tissue stem cells. We established this co-culture system by culturing hESC-derived NSCs under conditions where NSCs give rise to additional NSCs – through self-renewal – and to differentiated neurons (Figure 5G). Using this system, we generated WT-derived and IFITM KO-derived co-cultures and then infected them with WNV. In the WT co-cultures, infection occurred primarily in differentiated neuronal cells, where approximately 50% of the neuronal cells were infected, while less than 1% of the WT NSCs were infected. However, infection of the IFITM KO co-cultures was dramatically different. While KO neuronal cells were similarly susceptible to infection as WT cells, the IFITM KO NSCs were now highly susceptible to WNV infection, with ~70% becoming infected (Figure 5H). This suggests that IFITM proteins are critically important for protecting NSCs from infection with WNV. Similar results were obtained when cultures were infected with YFV-17D (Figure S6K–L).
Finally, we extended this analysis to primary human tissue stem cells. Silencing p21/CDKN1A, one of the most highly expressed ISGs in MSCs, specifically increased their susceptibility to chikungunya virus (CHIKV, Figure 5I–J), which mainly targets MSC-derived tissue (e.g. muscles and joints) in both humans and mice (Couderc et al., 2008). Infection by YFV or ZIKV in MSCs was not affected by p21/CDKN1A silencing whereas knockdown of IFITM3 in MSCs increased their permissiveness (Figure 5J). However, neither p21/CDKN1A nor IFITM3 was critical for the self-renewal and terminal differentiation of MSCs (Figure S6M–N). In human FL-derived HSCs, knockdown of IFITM3 increased permissiveness to DENV (Figure 5K–L), a virus that infects a variety of differentiated hematopoietic cells, including macrophages and megakaryocytes (Theofilopoulos et al., 1976). Interestingly, in co-culture experiments with HSCs and their differentiated progeny, significantly more differentiated erythroid cells (Pro, E-Baso, and L-Baso) were infected by DENV than HSCs (Figure 5M–N). However, this differential permissiveness between HSCs and differentiated erythroid cells declined and became less significant after knockdown of IFITM3, since knockdown of this ISG resulted in enhanced DENV infection of the HSCs (Figure 5O).
Stage-specific antiviral programs during stem cell differentiation
While stem cells are refractory to IFN stimulation, terminally differentiated cells are highly responsive (Burke et al., 1978). To understand how these differences affect the antiviral state of the cells, we monitored DENV infection in hESCs and hESC-derived HLCs, with and without IFN pre-stimulation, in both WT and IFITM KO cells. Consistent with our previous results, WT ESCs were completely resistant to DENV infection, while IFITM KO ESCs were highly permissive, irrespective of whether they had been pre-treated with IFN (Figure 6A). As seen previously, HLCs were highly permissive to DENV infection. In HLCs, pre-treatment with IFN-β strongly inhibited the infection in both WT and IFITM KO cells, likely through induction of ISGs such as MX1, IFITM3 (in WT cells) and others (Figure 6A–B). Thus, in the absence of IFITM proteins, induction of other ISGs (e.g. IFI6) can provide protection against DENV (Figure 6C). The failure of IFN pre-treatment to reduce DENV infection in the IFITM KO ESCs is not likely due to an inability of IFN-β to induce IFITM proteins, but rather a general lack of response to IFN, as we did not observe robust induction of any ISG in these cells (Figure 6A–B).
Figure 6. Stage-specific antiviral program during stem cells differentiation.
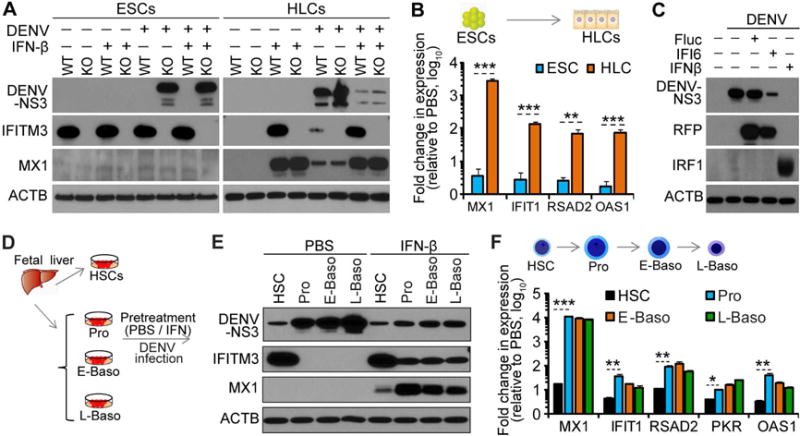
(A) WT and KO ESCs (KO123), and their derived HLCs were pre-treated with PBS or IFN-β before being exposed to DENV (MOI=1.0). Cells were analyzed by western blot at 48 hr p.i.
(B) WT and KO ESCs (KO123), and their derived HLCs were treated with PBS or IFN-β and analyzed by qRT-PCR.
(C) IFITM KO (KO123) HLCs reconstituted with IFI6 or Fluc, or pre-treated with IFN-β, were infected with DENV (MOI =1.0). At 48 hr p.i., DENV infection and ISG expression were analyzed by western blot (for RFP).
(D–E) Schematic of DENV infection (MOI=3.0) of hematopoietic cells. Cells were treated with IFN-β or PBS for 12 hr prior to DENV exposure and analyzed by western blot at 48 hr p.i.
(F) Analyses of ISGs by qRT-PCR for cells treated with IFN-β or PBS for 12 hr.
Shown in (B and F) are the mean fold changes ± SD from 3 independent experiments.
We also compared antiviral protection of IFN in FL-derived primary HSCs and three erythroid progeny (Pro, E-baso, and L-Baso) (Figure 6D). Without pre-treatment with IFN-β, differentiated progeny were more permissive to DENV infection, compared to HSCs from which they derive. Pre-treatment with IFN-β, however, strongly inhibited the infection in the three differentiation intermediates, but not in HSCs (Figure 6E). Again, differential protection is likely due to different responses to IFN-β stimulation, as demonstrated by strong induction of ISGs such as MX1, IFITM3 and others in the three progeny, but not in HSCs. This was further supported by the finding that induction of other ISGs, such as IFIT1, RSAD2, PKR and OAS1 was significantly less in HSCs than in differentiated progeny following IFN-β treatment (Figure 6F).
Taken together, these results demonstrated the existence of two distinct, cell stage-specific antiviral mechanisms. Stem cells, including ESCs and various tissue stem cells, show an attenuated IFN response but instead constitutively maintain elevated levels of select ISGs to function as prophylactic mediators of an antiviral protection. In contrast, terminally differentiated cells respond to IFN, leading to ISG induction and broad viral resistance.
ISGs protect stem cells from viral infection during development
Finally, to further validate our findings in a physiological context, we tested whether ISGs could protect primary stem cells from viral infections and preserve their differentiation potential ex vivo and in vivo. To this end, we used shRNAs to knock down IFITM genes in human fetal liver (FL)-derived primary HSCs (Figure 7A–B). We then exposed control (CTRL) and IFITM knockdown (KD) primary HSCs to IAV or DENV and tested their ability to form colonies and to differentiate into erythrocytes or macrophages. We found that, with pre-exposure to virus, KD HSCs produced fewer colonies and fewer differentiated erythrocytes and macrophages compared to control HSCs (Figures 7C–D and S7A–B). However, uninfected control and KD HSCs behaved similarly in these assays (Figures S7A–E).
Figure 7. ISGs protect stem cells from viral infection during development.
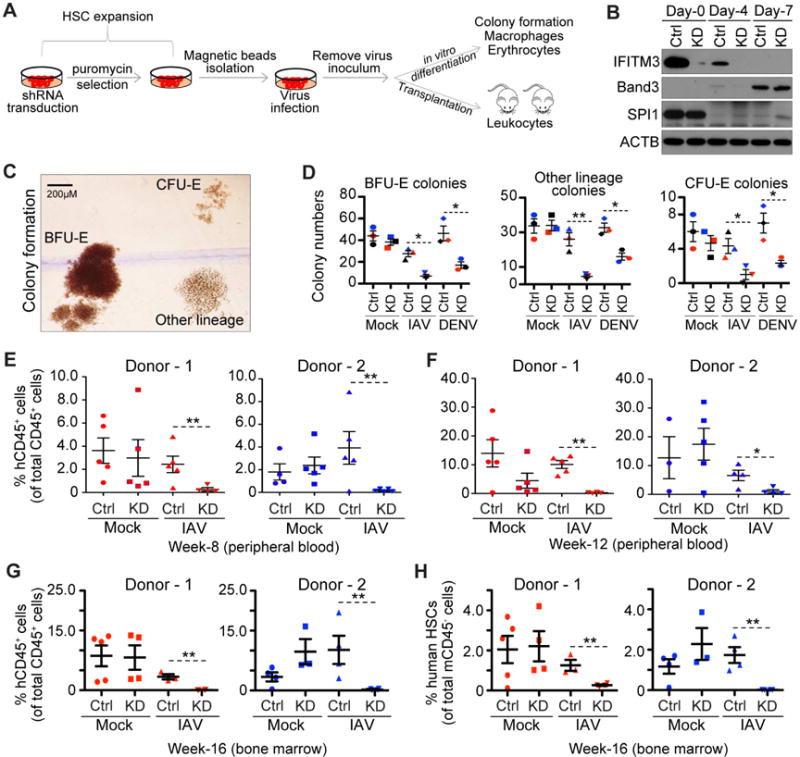
(A) Schematic of ex vivo transduction, expansion, virus infection and downstream assays for HSCs.
(B) Analyses of ISGs and markers in HSCs transduced with control (CTRL) or IFITM shRNA (KD) during erythropoiesis by western blot.
(C) Colony morphology: BFU-E: erythroid burst-forming units; CFU-E: hematopoietic colony forming unit. Scale bars: 200 μm.
(D) Colony formation assay: the number of colonies in each condition is plotted; colored symbols correspond to different donors. Error bars: 1 SD.
(E–F) Percentages of peripheral human CD45+ cells determined by flow cytometry at 8 weeks (E) and 12 weeks (F). Bars are the means ± SD from n=5 mice per group.
(G–H) Percentages of human CD45+ cells (G) and HSCs (H) in BM determined by flow cytometry at 16 weeks post transplantation. Each symbol represents the value obtained for an individual mouse. Bars are the means ± SD from n=5 mice per group.
See also Figure S7.
We next transplanted primary human FL-derived control and KD HSCs into immunodeficient mice to test whether these results could be recapitulated in vivo. Similar to the ex vivo results described above, we found that both control and KD HSCs differentiated with comparable efficiency in vivo, and both could reconstitute hematopoietic cells in peripheral blood (Figure 7E). Further, pre-exposure to IAV severely reduced the ability of KD HSCs to yield hematopoietic cells in periphery, whereas control HSCs were minimally affected. This difference was not due to delayed reconstitution kinetics in KD HSCs, since reconstitution remained low even as late at 12 weeks post-transplantation (Figure 7F). We also observed fewer human CD45+ cells (mainly B cells and myeloid cells, Figure S7F) in the femoral bone marrow of mice transplanted with KD HSCs pre-exposed to IAV (Figure 7G).
To further probe the basis of these defects, we analyzed both human (CD34+/CD38−) and mouse (Sca-1+/c-KIT+) HSC populations in the bone marrow of recipient mice (Figure S7G). All groups had a similar overall frequency of mouse HSCs (Figure S7H); however, there were fewer human HSCs in mice transplanted with KD HSCs pre-exposed to IAV (Figure 7H). This suggests that the defects in hematopoietic cell reconstitution we observed in peripheral blood were likely due to a reduction in the number of human HSCs in bone marrow. Together these results show that by protecting stem cells from viral infection, ISGs can facilitate hematopoietic repopulation by HSCs.
DISCUSSION
Our study demonstrates, rather surprisingly, that mammalian stem cells intrinsically express a subset of ISGs at high levels, which differ by stem cell type. We show that several of these ISGs protect stem cells from viral infection, and that knocking out the IFITM family ISGs renders stem cells susceptible to infection by a variety of viruses.
In this study, we focused attention on ISGs given their known roles in innate antiviral immunity. As discussed below, we believe there is strong biological justification for protecting stem cells from the deleterious effects of pathogen infection. However, it is highly likely that other genes, not designated as ISGs and yet to be discovered, also confer intrinsic pathogen resistance in stem cells. For example, mouse Zfp809 (Wolf and Goff, 2009) and Zfp819 (Tan et al., 2013), two known antiviral genes against endogenous retroviruses, are intrinsic and not regulated by IFN.
Why should stem cells be intrinsically protected?
As the operational units for growth and regeneration, stem cells are essential for tissue maintenance and repair (Weissman, 2000). To carry out their functions, they must communicate with their surroundings and be protected from damage and loss. Their ability to communicate with their surroundings and respond to extracellular cues is facilitated by their microenvironments – stem cell niches – which are often in close proximity to the blood supply (Doetsch et al., 1999; Kiel et al., 2005). Some stem cells even circulate in peripheral blood (Kuznetsov et al., 2001; Saiura et al., 2001). But access to the blood comes with increased risks of exposure to pathogens.
Several recent findings highlight the fact that stem cells are often at risk of viral infection. In vivo studies, for example, have shown that some retroviruses and herpesviruses infect HSCs in bone marrow (Carter et al., 2010; Feuer et al., 1996; Mendelson et al., 1996). Similarly, recent studies showed that ZIKV infects human (Tang et al., 2016) and mouse NSCs (Li et al., 2016).
Loss of stem cells from viral infection can have severe consequences. For example, infection of early human embryos by rubella virus can destroy a fetus in utero or cause severe congenital defects (Naeye and Blanc, 1965). HIV or CMV infection of HSCs in some cases may serve as a latent reservoir for long-term virus dissemination and can lead to a reduced hematopoiesis (Carter et al., 2010; Sindre et al., 1996). Our transplantation experiments showed that removing the intrinsic protection of HSCs by IFITM KD prior to exposure to IAV can severely diminish hematopoiesis in vivo.
Why do stem cells exhibit intrinsic ISG expression rather than IFN inducible expression?
Given that stem cells are rare yet critical for tissue maintenance and repair, they may express ISGs intrinsically to preempt viral infection. If so, this may explain why many types of stem cells specifically express ISGs such as CDKN1A and members of IFITM family that act on early steps of viral life cycle and block viruses before they can establish infection (Schoggins et al., 2011). We found that through constitutive ISG expression hESCs maintain a persistent and efficient antiviral status, rendering them highly resistant to infection by a panel of viruses. Such antiviral potency is even stronger than that induced by IFN stimulation in differentiated cells.
However, while this may explain why stem cells intrinsically express ISGs, it does not explain why they are refractory to IFN and fail to induce the full spectrum of ISGs when IFN is present. There are several reasons why stem cells may need to be refractory to IFN. For one, IFN has well-known anti-proliferative activity mediated by a subset of ISGs, which would be incompatible with a stem cell’s role in self renewal, tissue regeneration and repair (Borden et al., 1982). Of note, we did not detect expression of ISGs such as IFIT1, CH25H, or members of the OAS family, which have well-known anti-proliferative activity (de Veer et al., 2001), in any of the stem cell ISG signatures tested. Further, we found that IFN had strong anti-proliferative effects on differentiated hematopoietic cells, but not on HSCs (data not shown). This resistance to IFN-mediated anti-proliferative responses could be important for maintaining a functional stem cell niche during both acute and chronic infection.
IFN treatment can also sensitize cells to apoptosis upon subsequent viral infection. It could be deleterious for these pro-apoptotic genes to be expressed in stem cells. In line with this speculation, none of the stem cells we analyzed highly expressed ISGs with known pro-apoptotic activity (e.g. ISG12, TRAIL, and TNFSF10) (de Veer et al., 2001).
Finally, IFN has been shown to stimulate differentiation (Hertzog et al., 1994), suggesting that stem cells may dampen their IFN response to maintain pluri/multipotency. A recent study showed that under normal physiological conditions, a low level of IFN stimulates very low differentiation of dormant HSCs (Baldridge et al., 2010). However, sustained IFN-mediated HSC differentiation during chronic infection could lead to impaired stem cell function. Disruption of such fine-tuning has been implicated in the pathophysiology of bone marrow suppression in patients with infections such as tuberculosis and HIV/AIDS (Castella et al., 1985; Singh et al., 2001).
Why is ISG expression inducible rather than intrinsic in terminally differentiated cells?
Innate immune signaling pathways can be dismantled by viruses. There are numerous examples of viral proteins that degrade, inhibit or sequester different components of the IFN induction and signaling pathways (García-Sastre, 2017). Such counter-mechanisms can determine the species and tissue tropism of viruses and the outcome of infection (Douam et al., 2015). Constitutive expression of the full spectrum of antiviral ISGs in differentiated cells, which often reside at the portals of pathogen encounters, would subject this important multipronged host defense system to constant pathogen surveillance and the possible selection of resistant, disease causing variants.
In the context of an infection, a mix of intrinsic and inducible innate antiviral defenses could have multiple benefits. First, the pathogen detection and innate response capabilities of differentiated cells is not uniform. Heterogeneity of transcriptional responses has been observed during inducible IFN stimulation and ascribed to stochastic variation in what otherwise appear to be identical differentiated cells (Shalek et al., 2014). Such variation may be contributed by a relatively small fraction of virus-susceptible cells that initiate the response and eventually disseminate an alarm signal through the population via paracrine responses to IFN. These “sentinels” could be important for the host to sense and respond to initial viral infection. Inducible ISG expression in the majority of differentiated cells would allow the host to unleash the full spectrum of ISGs to dampen local virus infection and spread. We found that in the IFITM KO HLCs, IFN was still able to exert an efficient antiviral state against DENV infection. This protection occurs through induction of other ISGs that have redundant and/or complementary anti-DENV activity to that of the IFITMs. Unlike in stem cells, a low level of direct virus-induced killing of initially infected cells or in cell primed by IFN should be tolerable in differentiated cells provided that stem cells are protected and capable of replenishing this compartment.
Interestingly, during ex vivo differentiation of hESCs, we found that cells at terminal stage also displayed relatively high expression of some ISGs. However, this high ISG expression phenotype was significantly less pronounced during in vivo differentiation of HSCs. Recently, some studies have shown high expression of certain ISGs in terminally differentiated primary cells (Allen et al., 2017; Laguette et al., 2011). For example, we found that hESC-derived macrophages intrinsically expressed SAMHD1, a well-known antiretroviral restriction factor, which is consistent with previous study showing SAMHD1 expression in other myeloid lineage cells (Laguette et al., 2011). Such constitutive expression of ISGs in differentiated cells may provide additional protection to cells of critical importance to the host.
Evolutionary perspectives on selective expression of ISGs
ISGs highly expressed in human stem cells are similar among different cell lines/donors. Similarly, different mouse ESC lines express a shared list of murine ISGs. An intriguing question centers on how such intrinsic programs have evolved. Both humans and mice have been exposed to viral infections for millions of years. Viruses, as obligatory intracellular parasites, pose a persistent threat to all living beings, generating strong positive selection for effective protective mechanisms, including physical barriers (Bomsel and Alfsen, 2003), as well as intrinsic, innate and adaptive immunity (Marques and Carthew, 2007). Yet, given the evolutionary time scale, such sculpting is difficult to demonstrate. We speculate that the overlapping but distinct intrinsic ISG signatures in different stem cell types, reflect the outcome of millions of year of host-pathogen encounters and selection (Figure S2G–I). However, we are handicapped by our narrow cross-sectional view, not knowing the actual pathogens that have exerted these selective pressures and how they relate to those we are aware of today.
Some recent examples provide a fascinating glimpse of how intrinsic resistance mechanisms can unfold in the context of infection. In mice resolving an influenza virus infection, surviving lung tissue-resident memory CD8+ T cells were found to selectively maintain expression of IFITM3, a potent ISG against influenza virus (Wakim et al., 2013). Interestingly, a human polymorphism leading to decreased IFITM3 expression in CD8+ T cells is associated with a decrease in this important effector subset and more severe influenza (Allen et al., 2017). This suggests possible evolutionary selection for this antiviral effector but also the ability of pathogen encounters to elicit intrinsic antiviral resistance in long-lived mammalian tissue stem cell-like cells. Grow et al. recently found that rec, an early viral transcript from HERVK endogenous retroviruses is expressed and sufficient to increase expression of IFITM1 protein in hESCs, raising the possibility that endogenous retroviruses may also be involved in the shaping intrinsic stem cell immunity (Grow et al., 2015).
Our preliminary epigenetic analyses revealed that chromatin modifications and differential TF binding are involved in the regulation of intrinsic ISGs expression in stem cells. Our analysis also suggests that the regulatory mechanisms responsible for the expression of highly expressed ISGs are multifactorial and complex: different regulatory factors may be responsible for the expression of different ISGs in different cell types. Notably, future studies should aim at elucidating how such transcriptional networks regulate differential expression of ISGs in different types of stem cells. These studies will have important implications for our understanding of stem cell biology and the evolution of the vertebrate pathogen defense response.
STAR ★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charles M. Rice (ricec@rockefeller.edu). Some materials may require execution of a simple UBMTA.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
NOD.Cg-Rag1 IL2rgtmlWjl/Sz (NRG) mice were obtained from the Jackson Laboratory. NRG mice were bred and maintained at the Comparative Bioscience Center of the Rockefeller University. For transplantation of primary human HSCs, male and female mice around four-week old were randomly assigned to experimental groups. All procedures involving mice were in accord with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by the Rockefeller University Institutional Animal Care and Use Committees (protocol 12536).
Cell lines
Human ESC and iPSC lines
WA09 (human; sex: female; NIHhESC-10-0062), RUES2 (human; sex: female; NIHhESC-09-0013), and HUES8 (human; sex: male; NIHhESC-09-0021), iPSC.C3A (human; sex: male), iPSC.LVID2 (human; sex: male), and iPSC.BJ-3 (human; sex: male) were cultured on growth factor reduced Matrigel according to the manufacturer’s recommendations in feeder-independent mTeSR1 medium. Cultures were replenished with fresh medium every day. Cells were passaged every four to six days as clumps using ReLeSR. For all the experiments in this study, hES/iPSCs were used between passage 30 and 40.
Other cell lines
293T cells (human; sex: female) were maintained in DMEM supplemented with 10% FBS and 1% NEAA. Primary MSCs (human; sex: male; Lonza pt-2501) were maintained in Lonza MSC basal medium supplemented with MSCGM™ Singlequots™ according to the manufacturer’s instructions.
METHOD DETAILS
Stem cell differentiation
Differentiation into three germ layers
Ectoderm: hESCs were differentiated into neuroectoderm cells using the dual SMAD inhibition method. Briefly, hESCs were dissociated into single cells using Accutase and plated onto Matrigel-coated plates with ROCK inhibitor (Y-27632, final concentration 10 μM) to reach approximately 90% confluence on the next day. Differentiation was induced with SRM (DMEM/F12, 15% knockout serum replacement (KOSR), and 1% GlutaMax) supplemented with 10 μM SB431542 and 200nM Noggin for five to seven days.
Mesoderm: Differentiation of hESCs into mesoderm was performed as described in the literature (Lian et al., 2010) with some modifications. Briefly, hESCs were dissociated into single cells using Accutase and plated onto Matrigel-coated plates with ROCK inhibitor (Y-27632) to reach approximately 30% confluence on the next day. Differentiation was initiated with RPMI/B-27 (RPMI/1640, 2% B-27 (minus insulin), 0.5% GlutaMax, and 0.5% non-essential amino acid) supplemented with 10 μM CHIR-98014 for four days, with daily medium replenishment.
Endoderm: hESCs were dissociated into single cells using Accutase and plated onto Matrigel-coated plates with ROCK inhibitor (Y-27632) to reach approximately 90% confluence. The next day, differentiation was induced with RPMI/B-27 (RPMI/1640, 2% B-27, 0.5% GlutaMax, and 0.5% non-essential amino acid) supplemented with 100ng/ml Activin-A and 3 μM CHIR99021 for one day and then with 0.25ng/ml BMP4, 5ng/ml bFGF and 100ng/ml Activin-A for three more days. Alternatively, very consistent and reproducible endoderm could also be generated using the STEMdiff Definitive Endoderm Kit following the manufacturer’s instructions.
Differentiation into tissue stem cells
Neural stem cells (NSCs): Confluent ectoderm cells were passaged and maintained in the induction medium containing the dual SMAD inhibitors for one additional week. Derived NSCs were then maintained in neural progenitor medium (DMEM/F12, 2% StemPro Neural Supplement, and 1% GlutaMax) supplemented with 20ng/ml bFGF and 20ng/ml EGF. Alternatively, NSCs were derived from hESCs using an embryonic body (EB) protocol (Kim et al., 2006). NSC cultures were replenished with fresh medium every two days.
Mesenchymal stem cells (MSCs): mesoderm cells were passaged and maintained at a low density in MSC enrichment medium. Briefly, cells were cultured on gelatin coated plates with basal medium (knockout-DMEM, 10% KOSR, 1% sodium pyruvate, 1% Insulin-Transferrin-Selenium-X (ITS-X), and 0.1% β-mercaptoethanol) supplemented with 5ng/ml bFGF, 10ng/ml PDGF-AB, and 10ng/ml EGF. Differentiating cells were passaged at a 1:4 split for several times (usually around 4–5) to enrich MSCs. Alternatively, CD105+CD24− MSCs were sorted by FACS and expanded according to conditions described above. MSC cultures were replenished with fresh medium every two days.
Pancreatic stem cells (PSCs): Differentiation of hESCs into PSCs was performed as described (Rezania et al., 2014). Briefly, endoderm cells were cultured in BLAR medium (custom formulated as described) supplemented with 0.5% BSA, 10mM glucose, 0.25mM ascorbic acid, and 50ng/ml FGF7 for one day and with additional supplement (2% BSA, 0.25 μM SANT-1, 1 μM retinoid acid, 100nM LDN193189, and 0.5% ITS-X) for two more days. Cells were then exposed to the same medium with reduced FGF7 (2ng/ml) for three days. Finally, cells were cultured in the last stage differentiation medium (BLAR medium with 20mM glucose, 2% BSA, 0.25 μM SANT-1, 0.05 μM retinoid acid, 100nM LDN193189, and 0.5% ITS-X, 1 μM T3, 10 μM ALK5 inhibitor II, 10 μM zinc sulfate and 10 μg/ml heparin) for four days.
Hematopoietic stem cells (HSCs): hESCs were differentiated into hematopoietic stem cells using the Spin-EB method. Briefly, 3000 cells were seeded into U-bottom 96-well non-tissue culture plates in 50 μl of SFM (IMDM/Ham’s F12 (1:1), 5mg/ml BSA, 1× insulin-transferrin-selenium, 1× synthetic lipids, 50 μg/ml of ascorbic acid and 2mM GlutaMax) supplemented with 10ng/ml BMP4, 10ng/ml bFGF, and 10 μM ROCK inhibitor (Y-27632) for two days. SFM supplemented with 10ng/ml BMP4, 10ng/ml bFGF, and 20ng/ml VEGF was added to cultures (50 μl/well) every three days. On day 8, half of the culture medium was removed and fresh SFM medium containing 10ng/ml bFGF, 10ng/ml VEGF, and 50ng/ml SCF was added every three days until day 14 when cells were collected for purification of HSCs using the EasySep human CD34 positive selection kit according to the manufacturer’s instructions.
Terminal differentiation of tissue stem cells
Hepatocyte-like cells (HLCs): Endoderm cells were dissociated into single cell suspensions using Accutase and plated onto Matrigel-coated plates with ROCK inhibitor in RPMI/B-27 supplemented with 20ng/ml BMP4 and 10ng/ml bFGF. Medium was replaced daily for five days. Cells were then exposed to RPMI/B-27 containing 20ng/ml HGF for five days. HLCs were further matured in Lonza hepatocyte culture medium containing ascorbic acid, BSA-FAF, hydrocortisone, transferrin, insulin, GA-1000 and 20ng/ml OSM for an additional one to two weeks. HLCs cultures were replenished with fresh medium every two days.
Immature pancreatic β-like cells (Im-β): Further differentiation of PSCs into insulin+ β-like cells was performed as described (Zhang et al., 2009) with some modifications. Briefly, confluent PSCs were dissociated into single cells using Accutase and plated onto Matrigel-coated plates with ROCK inhibitor (Y-27632) in last stage differentiation medium described above. Once the culture reached confluence, cells were further maintained in maturation medium (DMEM/F12, 0.2% BSA, 0.5×N2, 0.5×B-27, and 1% GlutaMax) supplemented with 1% ITS, 10ng/ml bFGF, 10mM nicotinamide, 50ng/ml Exendin-4, and 10ng/ml BMP4 for seven to nine days.
Myofibroblast-like cells (MYF): Confluent MSCs was dissociated into single cells using Accutase and plated onto cell culture plates with ROCK inhibitor in the differentiation medium (DMEM/F12, 2% FBS, and 1% GlutaMax) supplemented with 1 μM retinoic acid, 30ng/ml VEGF, and 10ng/ml PDGF-AB for two weeks. Cells were further cultured in maturation medium (DMEM/F12, 5% FBS, and 1% GlutaMax) supplemented with the above factors for one week. MYF cultures were replenished with fresh medium every two days.
Adipocyte-like cells (Adipocyte): Further differentiation of MSCs into adipocytes was based on the Lonza adipogenic differentiation system with some modifications. Briefly, hESC-derived MSCs were dissociated and plated onto cell culture plate at relatively low density in MSCGM medium (Lonza). Medium was replaced every three days until the cultures reached super confluence. Cells were then treated with three to four cycles of induction/maintenance according to the manufacturer’s instructions, followed by one-week treatment with maintenance medium.
Macrophage-like cells (MLCs): Briefly, purified HSCs were expanded in the expansion medium (StemSpan SFEM II supplemented with 10% CD34+ expansion supplement) for three days and then transferred to Matrigel-coated plates in the macrophage differentiation medium (IMDM, 10% FBS, 20ng/ml M-CSF, and 10ng/ml IL-1β). Medium was replenished every three days. Macrophage-like cells (MLCs) were collected at one week post differentiation. For functional characterization, MLCs were stimulated with lipopolysaccharides (LPS, 5 μg/ml) for 6 hr and analyzed for induction of major cytokines and chemokines by qRT-PCR.
Neuron-like cells (Neuron): Briefly, hESC-derived NSCs were dissociated into single cells using Accutase and plated onto poly-L-ornithine/laminin-coated cell culture plate in the neuronal differentiation medium (BrainPhys basal medium, 2% SM1 neuronal supplement, 1% N2 supplement-A, 200nM ascorbic acid, 1mM dibutyryl cAMP) supplemented with 20ng/ml BDNF and 20ng/ml GDNF. A half-medium change was performed every two to three days for three to four weeks.
Astrocyte-like cells: For differentiation of NSCs into astrocyte-like cells, hESC-derived NSCs were dissociated into single cell suspensions using Accutase and plated onto matrigel-coated cell culture plate in astrocyte differentiation medium (DMEM, 1% N-2 supplement, 2 mM GlutaMax-1 supplement, 1% FBS). Medium change was performed every three to four days for three to four weeks.
Human ESC-derived NSCs/Neurons coculture system: NSCs were derived from hESCs using an EB protocol (Kim et al., 2006) and neural rosettes were isolated using a commercial selection reagent and cultured on Matrigel-coated plates at a low density in neural induction medium for three to five days. Cells were then treated with several cycles of induction/differentiation medium for one to two weeks until neuronal morphology became apparent around rosette structures. Cultures were replenished with fresh medium every two days.
Isolation of hematopoietic cells
Deidentified fetal livers (16–24 weeks gestation) were procured through Advanced Bioscience Resources (Alameda, CA). Livers received on ice were washed with hepatocyte wash buffer (HWB) consisting of Williams' E Medium (WEM) plus 10mM HEPES, 50 μg/ml gentamicin, 100U/ml penicillin, and μ100 g/ml streptomycin. Tissue was minced then resuspended in 20–40ml warm digestion buffer consisting of Hanks Balanced Salt Solution plus 40mM HEPES, 3.26mM CaCl2, 2U/ml DNase I Grade II (Roche), and 0.2% Collagenase type IV. Tissue was digested for 30 minutes at 37°C, then diluted 1:1 w ith HWB and gently pushed through 70 μm cell-strainers. The suspension was centrifuged at 100×g for 3 minutes and the supernatant containing hematopoietic cells was transferred to a new 50ml tube and further centrifuged at 400×g for 5 min. The cell pellet was washed twice by resuspension in HWB. Cells were then resuspended in PBS containing 1% FBS and 1mM EDTA and divided into two fractions for subsequent isolations. CD34+ cells were isolated from one fraction by using the EasySep human CD34 positive selection kit.
The other fraction was further diluted in appropriate volume (80 μl/10 million cells) of CD45 selection buffer (PBS, 0.5% BSA and 2mM EDTA) with CD45 microbeads (Miltenyi Biotec,20 μl/10 million cells) and incubated at 4°C for 15 min . CD45− cells were isolated by immunomagnetic negative selection according to the manufacturer’s instructions. FACS-based purification of erythroblasts of different developmental stages was performed as described previously(Hu et al., 2013). Briefly, CD45− cells were first incubated in blocking buffer (PBS, 0.5% BSA and 0.4% human AB serum) and 1×106 cells/100 μl were stained by the addition of 0.2ng PE anti-GPA, 1000ng FITC anti-Band3, and 100ng APC anti-α4 integrin antibodies. After incubation at room temperature for 15 min in the dark, the cells were washed once with washing buffer (PBS and 0.5% BSA), and were suspended at a concentration of 1.5×107/ml for sorting on a MoFlo high-speed cell sorter (Beckman). The specific antibodies used for FACS are indicated in Key Resource Table.
For subsequent analyses such as RNA-Seq, qRT-PCR and western blot, hematopoietic cells were immediately pelleted, washed with DPBS, and lysed accordingly. For RNA extraction, pellets were lysed directly in the RLT buffer from RNeasy Mini Kit (Qiagen); for western blot, pellets were lysed in 2× SDS-loading buffer (100mM Tris-Cl (pH 6.8), 4% SDS, 0,2% bromophenol blue, 20% glycerol, and 200mM β-mercaptoethanol), followed by boiling at 100°C for 10 min.
Isolation of CD34+ cells from other sources
Briefly, cells were diluted with an equal volume of buffer (PBS and 0.5% BSA) and were subsequently separated on a Ficoll density gradient at 400×g for 30 min at room temperature. The mononuclear cells were collected and washed twice with washing buffer (PBS and 0.5% BSA) by sequential centrifugation at 300×g and 200×g for 10 min. The cells were incubated with CD34 microbeads for positive selection using the Magnetic-activated cell sorting system (Miltenyi Biotec) according to the manufacturer’s instructions. Bone marrow derived CD34+ cells were isolated from a single deidentified donors, while peripheral and cord blood derived CD34+ cells were isolated from pooled mixtures of 2 to 4 deidentified donors. For subsequent experiments such as RNA-seq, qRT-PCR and western blot, purified cells were handled similarly as described for fetal liver-derived cells.
RNA-Seq analysis
Approximately 100ng of total RNA were used as input. Libraries were prepared according to Illumina's instructions for the TruSeq Stranded mRNA LT Sample Prep Kit and sequenced on an Illumina HiSeq with a read length of 50 nucleotides (single end configuration).
Libraries were sequenced to generate approximately 13 to 20 million reads per sample. Following quality assessment with FASTQC (Version 0.11.5), gene and transcript expression levels (TPM and estimated counts) were quantified using the Salmon pseudo-alignment tool (v0.8.2) with Ensembl v88 reference transcriptome annotation. Gene level expression values were assembled in the R statistical framework using the Tximport package (v0.99.2). Genes with TPM=0 across all samples were removed. For each sample, the distribution of gene expression (Log2 TPM) values was plotted and expression thresholds were established as the local minimum partitioning the bimodal distributions: genes above this threshold were classified as “expressed” in a given sample. For each sample, “highly expressed” genes were defined as the top 20% of these expressed genes ranked by TPM values. Interferon-stimulated genes (ISGs, Table S2) within this set were defined as “highly expressed ISGs” for that sample. Heatmaps were plotted with the pheatmap tool (v1.0.8); genes with maximal TPM values in each cell type (relative to other cell types) are grouped together for visualization.
For PCA visualization, estimated read counts were normalized and variance stabilized by regularized log transformation with the rlog() function from the DESeq2 package (v1.14.1, available through Bioconductor). When present, donor or preparation specific effects were corrected using the remove Batch Effect() function in limma. For heatmaps in Figures 1B, 1F, 2C and S5A, shown are log2 scaled TPM values for ISGs highly expressed (within top 20% of all expressed genes) by any cell types compared. In Figures 1B and S5A, additional ISGs and IFN genes with low expression values were manually selected for comparison. For the heatmaps shown in Figures 3B, 3F, 3I and S4C, shown are z-score scaled TPM values for ISGs highly expressed in hESCs along ex vivo differentiation of hESCs. For the heatmap shown in Figure 4D, shown are z-score scaled TPM values for ISGs highly expressed in HSCs along in vivo differentiation of human primary HSCs. For the heatmap shown in Figure S5D, shown are z-score scaled TPM values for ISGs highly expressed in mouse ESCs along ex vivo differentiation towards cardiomyocytes. z-score scaled heatmaps were used to show general relative abundance patterns of ISGs along ex vivo differentiation of stem cells, therefore “blue” color does not necessarily indicate absent of the gene of interest in the given cell type.
Generation of IFITM-knockout hESCs
Two guide RNAs targeting IFITM3 exon 1 were transfected into HUES8-iCas9 cells, which upon treatment with 2 μg/ml doxycycline (Dox) inducibly expressed CRISPR-Cas9. HUES8-iCas9 cells were treated with Dox for 48 hr prior to and for 24 hr post transfection. Transfected cells were then seeded for limiting dilution cloning and grown on mouse embryonic fibroblast cells in cloning medium (DMEM/F12, 20% KOSR, 1% GlutaMax, 1% NEAA, 0.1% β-mercaptoethanol, and 10ng/ml bFGF). Medium was replenished daily. Clones were expanded and screened by western blot analysis and homozygous IFITM knockout clones were chosen for further characterization. Pluripotency was evaluated by checking expression of pluripotent markers (e.g. OCT4 and SSEA4) and differentiation capability into three germ layers, using the conditions described above. Proliferation of modified hESCs and their derivatives (hESC-derived NSCs, MSCs) was measured by using CellTiter-Glo Assay Kit according to manufacturer’s instructions.
Transduction of lentiviral particles
shRNAs targeting the gene of interest were cloned into the pLKO.1-puro vector using standard techniques. Lentiviral particles were generated as reported previously (Zhang and Stefanovic, 2017) with some modifications. Briefly, packaging plasmids were transfected into 293T cells using Lipofectamine 2000 according to the manufacturer’s directions, medium was changed to DMEM supplement with 3% ESC-qualified FBS (Life Technologies) at 6 hr post transfection. Medium containing the lentiviral particles was harvested at 48 hr post transfection and filtered through 0.2 μm filter. shRNA targeting sites are shown in Key Resource Table.
For generation of shRNA-mediated knockdown in hESCs and primary MSCs, lentiviral particles were further concentrated using Lenti-X Concentrator according to the manufacturer’s directions. For transduction of hESCs, cells were dissociated into single cells using Accutase and plated onto Matrigel-coated plates with ROCK inhibitor (Y-27632) to a confluency of approximately 20 – 30%. Cells were transduced twice in mTeSR1 medium with concentrated lentiviral particles and puromycin (1 μg/ml) was added to cultures at 48 hr post transduction. For transduction of primary MSCs, cells were seeded at 20% confluency and puromycin (1 μg/ml) was used for selection at 48 hr post transduction.
For lentiviral transduction of primary hematopoietic cells, lentiviral particles were further concentrated by ultracentrifugation: briefly, filtered supernatant containing the lentiviral particles was ultracentrifuged at 70,000×g for 2 hr at room temperature with the brake off. Pellets were resuspended in sterile PBS (concentrated by 1000:1). Purified CD34+ cells were first recovered overnight in the expansion medium before lentiviral transduction. Briefly, 0.5×106 cells were spun down and resuspended in 0.5ml of transduction medium (IMDM, 10% FBS, 3IU/ml heparin, and 8 μg/ml polybrene) with concentrated lentiviral particles (20 to 40 μl virus stock containing approximately 2×107 transduction units, MOI=40). Cells and viral particles were mixed by pipetting up and down several times before transfer to one well of a 24-well plate (flat bottomed non-tissue culture treated). The plate was spinoculated at 1,400×g for 2 hr at room temperature prior to overnight incubation in transduction medium. Cells were washed and resuspended in expansion medium on the next day. Puromycin (1 μg/ml) was added to the cultures at 36 hr post transduction for three days. At one week post transduction, CD34+ cells were further purified using the EasySep human CD34+ positive selection kit and used for subsequent experiments, including virus infection, ex vivo differentiation, colony formation assay, and transplantation into immunodeficient mice.
For lentiviral transduction of hESCs-derived HLCs, lentiviral particles (ISG overexpression) were concentrated using Lenti-X Concentrator as described above. Differentiated HLCs were transduced in Lonza hepatocyte culture medium with concentrated viruses and spinoculated at 1,400×g for 2 hr at room temperature. Viral inoculum was then removed at 4 hr post spinoculation and HLCs were washed with basal medium twice.
Virus infection
Viruses. The construction, characterization and generation of viral stocks for the following viruses have been previously described: recombinant clones were obtained for the (+) RNA viruses CHIKV-GFP (derived from pCHIKV-LR 5′GFP) (Tsetsarkin et al., 2006), VEEV-GFP (derived from pTC83-GFP infectious clone) (Schoggins et al., 2011), YFV-Venus (derived from YF17D-5′C25Venus2AUbi), DENV (derived from IC30P-A infectious clone of strain 16681), and WNV-GFP (derived from pBELO-WNV-GFP-RZ ic) (Schoggins et al., 2011). ZIKV (Puerto Rican strain PRVABC59) was from obtained from the CDC and passaged three times on Vero cells and twice on Huh-7.5 cells. Infectious virus stocks were obtained for the (−) RNA viruses VSV-GFP (based on strain Indiana) (Dalton and Rose, 2001), IAV strain A/WSN/33 (H1N1) and A/swine/Texas/98 (H3N2), RSV-GFP (based on stain A2) (Biacchesi et al., 2004), MeV-GFP (MVvac2-GFP, based on the vaccine strain, Edmonston lineage measles virus) (del Valle et al., 2007), hPIV3-GFP (based on strain JS) (Zhang et al., 2005), and NDV-GFP (based on strain Hitchner B1) (Park et al., 2003). All infections were performed by incubation of virus inoculum with cells for 2 to 4 hr before the cells were washed and changed to the medium appropriate for the specific cell type and differentiation stage. Viral infection was performed using MOI=1.0 (as determined of the cell type used to titrate each virus), unless stated otherwise in the figure legend.
After infection, cells were fixed and stained as follows: infections with two stains of IAV were fixed at 9 hr p.i. and stained with anti-NP antibody (red); infections with GFP-expressing RSV, NDV, MeV, YFV, and WNV were fixed at 48 hr p.i. (green); infections with GFP-expressing CHIKV, VSV, and VEEV were fixed at 16 hr p.i. (green); infections with GFP-expressing hPIV3 was fixed at 24 hr p.i. (green); infection with DENV was fixed at 48 hr p.i. and stained with NS3 antibody (red); and infection with ZIKV was fixed at 72 hr p.i. and stained with anti-flavivirus E (4G2) antibody (red).
For IFN-β mediated inhibition of DENV infection in hESC and ESC-derived HLCs, cells were pretreated with IFN-β (200IU/ml) for 8 to12 hr before being exposed to DENV. IFN-β was kept in culture medium during virus infection. At 48 hr post infection, cells were harvested for western blot using 2 × SDS-loading buffers as described before, or fixed with 4% paraformaldehyde and stained with anti-NS3 antibody (anti-dengue nonstructural protein 3, serotype-2).
Virus infection of hematopoietic cells
CD34+ cells (hematopoietic stem cells) were maintained in expansion medium described above and differentiated progenies (including Pro, Early and Late-Baso cells) were cultured in stage-two medium. For DENV infection, cells were exposed to virus overnight before the inoculum was removed by centrifugation. For testing differential permissiveness to DENV infection, the mixed cells (including HSC, Pro, Early and Late-Baso) were maintained in stage-one medium and exposed to virus overnight before the inoculum was removed by centrifugation. At 48 hr post infection, cells were first stained with anti-CD34 and/or anti-GPA/CD235a antibodies, followed by fixation and staining with anti-NS3 antibody or were directly collected for western blot analysis.
For the experiments shown in Figure 6N, HSCs and three differentiated progeny cell types (Pro, E-Baso, and L-Baso) were isolated from one donor by FACS, mixed together, and exposed to DENV. For the experiments shown in Figure 6O, HSCs were isolated from one donor, and transduced with control shRNA or shRNA targeting IFITM3. After puromycin selection, cells were divided into two portions: one for ex vivo expansion and the other for ex vivo terminal differentiation into Pro, E-Baso, and L-Baso. Expanded HSCs were mixed with the differentiated cells and were infected with DENV. For both N and O, infection was terminated at 48 hr p.i. and cells were stained with erythrocyte-specific marker glycophorin A (GPA) and NS3 antibodies.
IFN mediated inhibition of virus infection in hematopoietic cells was performed by pre-treating cells with IFN-β (200U/ml) for 12 to 16 hr before subsequent exposure to virus. IFN-β was kept in the culture medium during virus infection. Cells were collected at the indicated time points post infection for western blot or flow cytometry analysis. For ISG induction by IFN stimulation, hematopoietic cells were treated with IFN-β (100U/ml) for 6 hr before being collected for qRT-PCR analysis.
Transplantation of primary human HSCs
After a one-week ex vivo expansion, control and shRNA-transduced human CD34+ cells were further purified using the EasySep human CD34 positive selection kit. For infection with IAV (A/swine/Texas/98) or DENV (strain 16681), cells were exposed to viruses for 3 hr in expansion medium, then washed twice with IMDM. Viable cells were then counted and resuspended in PBS. An equal number of viable cells from each condition were used in the subsequent experiments: for ex vivo differentiation, cells were seeded at 3×105/ml and 2×105/ml for erythrocytes and macrophages, respectively; for transplantation, each mouse was given 1×105 cells intrasplenically.
Ex vivo differentiation of HSCs towards erythrocytes was performed using the “3 phase method” as described (Hu et al., 2013). Ex vivo differentiation of HSCs to macrophages was performed as follows: cells were cultured in macrophage differentiation medium (IMDM, 2% human plasma, 3% human AB serum, 200 mg/ml Holo human transferrin, 3 IU/ml heparin, and 10 mg/ml insulin) supplemented with 10ng/ml SCF, 1ng/ml IL-3, 50ng/ml Flt3, and 50ng/ml M-CSF for one to two weeks. For colony formation assay, briefly, cells from each condition were diluted to a density of 200/ml of MethoCult H4434 classic medium for BFU-E colony assay in 37°C incubator. The different colonies were defined according to previously described criteria (Hu et al., 2013) and counted on day 14 post seeding. During ex vivo differentiation, samples were collected at the indicated time points to monitor expression of surface markers using flow cytometry. Proliferation was examined by counting cells every day during the first week and is plotted as fold change after normalization to the seeding number on the first day.
Mouse (NOD.Cg-Rag1 IL2rgtmlWjl/Sz (NRG)) was used for HSCs transplantation. At weeks 8 and 12 post transplantation, peripheral blood samples were obtained from recipients and subset reconstitution of human leukocytes was evaluated by flow cytometry. At week 16, mice were sacrificed and single cell suspensions of bone marrow were prepared by flushing the bone marrow cells out of the femurs into PBS supplemented with 10% FBS using a syringe and 22-gauge needle. Human and mouse hematopoietic cells were analyzed by flow cytometry as follows: human B cells were stained with anti-CD19 antibody, and human myeloid cells were stained with anti-CD33 antibody; murine cells were stained with antibodies to CD45, c-KIT, and Sca-1. Human HSCs were defined as the CD34+CD38− population and mouse HSCs were defined as the c-KIT+Sca-1+ population. Antibodies used for flow cytometry are listed in Key Resource Table.
Immunofluorescence analysis
Cells were fixed in 4% para-formaldehyde in phosphate-buffered saline (PBS) at room temperature for 10 min and blocked with PBTG (PBS containing 10% normal goat serum, 1% bovine serum albumin (BSA), 0.1% Triton-X100) at room temperature for 2 to 3 hr. Cells were incubated with primary antibodies (diluted in PBTG) at 4°C overnight or 2 hr at room temperature. Isotype mouse or rabbit IgGs were used as negative controls. After four washes with PBS, Alexa-350, 488, or 594 conjugated secondary antibodies (1:500 – 1:1000 diluted in PBTG) were added and incubated in the dark at room temperature for 1 hr, followed by four washes with PBS. For lipid droplets staining in hESC-derived adipocytes, cells grown on coverslips were fixed with 10% formalin for 30 min, and washed with 60% isopropanol for 10 min at room temperature. Cells were then stained with 0.18% freshly made oil-red in PBS for 5 min at room temperature and washed thoroughly with double-distilled water four times. Nuclei were stained with DAPI for 2 min at room temperature. Primary antibodies used for Immunofluorescence analysis are listed in Key Resource Table.
Quantitative real-time RT-PCR (qRT-PCR)
Total RNA was isolated from cell lysates using the RNAeasy Mini Kit (Qiagen) followed by reverse transcription using Superscript III First-Strand Synthesis System. Gene expression was quantified using the LightCycler SYBR Green I Master Mix on a LightCycler 480 Instrument (Roche Life Science) with gene-specific primers shown in Key Resource Table. PCR conditions were as follows: initial denaturation step at 50°C for 2 min and 95°C for 10 min, then 45 cycles of 95°C for 15 sec, 56°C for 15 sec, and 72°C for 2 0 sec; followed by a melting step of 95°C for 10s, 65°C for 10s and a 0.07°C/s decrease from 95°C ; and finally a cooling step of 50°C for 5s. PCR product specificity was confirmed by a melting-curve analysis.
Western blot
Cell lysates were separated by 10% or 4% – 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by transfer onto polyvinylidene fluoride membrane. Comparisons between different groups/cells were made by analyzing equal amounts of loaded protein from lysates, as determined by BCA protein assay. GAPDH or ACTB was used as loading control, as indicated in the figures and figures legends. Primary antibodies used for western blot analysis are listed in Key Resource Table.
IFN neutralization
Human ESCs were cultured in mTeSR1 medium supplemented with anti-human IFNAR2 antibody or isotype control antibody for 48 hr before being collected for qRT-PCR. Antibodies were replenished daily with medium changes. For neutralization assays on HLCs, cells were pretreated with antibodies for 6 hr prior to exposure to IFN-β (100U/ml) and collected for qRT-PCR at 6 hr post IFN-β treatment. mTeSR1 medium that had been exposed to hESCs for 24 hr was collected as conditioned mTeSR1, which was used to examine possible ISG induction in HLCs. Primary antibodies used for neutralization are listed in Key Resource Table.
QUANTIFICATION AND STATISTICAL ANALYSIS
For RT-qPCR, fold changes in mRNA expression for cellular markers and ISGs were determined using the ΔΔCt method relative to the values in control samples as indicated in figure legends, after normalization to housekeeping genes (Eisenberg and Levanon, 2013). Results are presented as means ± standard deviation (SD), unless stated otherwise. Comparisons between groups/cells were made using the two-tailed Student’s t test, unless stated otherwise. Statistical significance was denoted with * (p<0.05), ** (p<0.01), and *** (p<0.001) in the figures and figures legends. Statistical analysis was performed in Graph Pad PRISM 5.
DATA AND SOFTWARE AVAILABILITY
All RNA-Seq data have been deposited to the Sequence Read Archive (SRA) under accession number GSE97987.
Gene expression data for human HSCs and MSCs are included in Table S3; gene expression data for mouse ESCs and Chimpanzee iPSCs are included in Table S4. Processed mouse epigenetic data from (Wamstad et al., 2012) are included in Table S4.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-AFP | Sigma-Aldrich | Cat# A8452 RRID:AB_258392 |
| Mouse monoclonal anti-ALB | Cedarlane | Cat# CL2513A RRID:AB_10086438 |
| Mouse monoclonal anti-CD90/THY1 | Stemcell Technologies | Cat# 60045 |
| Rabbit polyclonal anti-COL1A1 | Rockland | Cat# 600-401-103-0.1 RRID:AB_2074625 |
| Rabbit polyclonal anti-DNEV NS3 | GeneTex | Cat# GTX124252 RRID:AB_11171668 |
| Mouse polyclonal anti- Flavivirus E | This paper | N/A |
| Mouse monoclonal anti-IAV NP | EMDmillipore | Cat# MAB8251 RRID:AB_95293 |
| Rabbit monoclonal anti-FoxA2 | Cell Signaling Technology | Cat# 8186P RRID:AB_10891055 |
| Rabbit polyclonal anti-GATA2 | GeneTex | Cat# GTX113441 RRID:AB_10617761 |
| Rabbit monoclonal anti-GATA4 | Cell Signaling Technology | Cat# 36966S |
| Rabbit monoclonal anti-HNF4α | Cell Signaling Technology | Cat# 3113S RRID:AB_2295208 |
| Rat monoclonal anti-INS | DSHB | Cat# GN-ID4 RRID: AB_2255626 |
| Rabbit polyclonal anti-MAP2 | Cell Signaling Technology | Cat# 4542S RRID:AB_10693782 |
| Mouse monoclonal anti-NES | R&D Systems | Cat# MAB1259 RRID:AB_2251304 |
| Rat monoclonal anti-NKX6.1 | DSHB | Cat# F55A10 RRID: AB_532378 |
| Rabbit monoclonal anti-OCT4 | Cell Signaling Technology | Cat# 2890S RRID:AB_2167725 |
| Rabbit polyclonal anti-OCT4 | Stemgent | Cat# 09-0023 RRID:AB_2167689 |
| Rabbit polyclonal anti-PAX6 | Covance | Cat# PRB-278P RRID: AB_2565003 |
| Rabbit monoclonal anti-PDX1 | Cell Signaling Technology | Cat# 5679S RRID:AB_10706174 |
| Mouse monoclonal anti-SMAα | Sigma-Aldrich | Cat# A2547 RRID:AB_476701 |
| Mouse monoclonal anti-SOX17 | R&D Systems | Cat# MAB1924 RRID:AB_2195646 |
| Rabbit monoclonal anti-SOX2 | Cell Signaling Technology | Cat# 3579S RRID:AB_2195767 |
| Mouse monoclonal anti-SSEA4 | Stemcell Technologies | Cat# 60062 |
| Rabbit monoclonal anti-T/Brachyury | Cell Signaling Technology | Cat# 81694S |
| Mouse monoclonal anti-TUJ1 | Covance | Cat# MMS-435P RRID: AB_2313773 |
| Rabbit monoclonal anti-SLC4A1/Band3 | This paper | N/A |
| Mouse monoclonal anti-BST2 | SCBT | Cat# sc-390719 |
| Rabbit monoclonal anti-CDKN1A | Cell Signaling Technology | Cat# 2947S RRID:AB_823586 |
| Rabbit polyclonal anti-EIF3L | Bethyl | Cat# A304-753A RRID:AB_2620948 |
| Rabbit polyclonal anti-GAPDH | Abcam | Cat# ab9385 RRID:AB_449791 |
| Rabbit polyclonal anti-IRF1 | SCBT | Cat# sc-50366 RRID:AB_2126309 |
| Rabbit monoclonal anti-IDO1 | Abcam | Cat# ab76157 RRID:AB_1310357 |
| Rabbit polyclonal anti-IFITM1 | GeneTex | Cat# GTX101728 RRID:AB_11176549 |
| Mouse monoclonal anti-IFITM2 | Ptglab | Cat# 66137-1-Ig |
| Rabbit polyclonal anti-IFITM3 | GeneTex | Cat# GTX115407 RRID:AB_11172546 |
| Rabbit polyclonal anti-MX1 | Sigma-Aldrich | Cat# SAB1100069 RRID:AB_10607751 |
| Rabbit monoclonal anti-SF3B1 | Abcam | Cat# ab172634 |
| Rabbit polyclonal anti-SLC16A1 | Bethyl | Cat# A304-358A RRID:AB_2620553 |
| Rabbit polyclonal anti-SOX9 | EMD Millipore | Cat# AB5535 RRID:AB_2239761 |
| Rabbit monoclonal anti-SPI1/Pu.1 | Cell Signaling Technology | Cat# 2258S RRID:AB_10693421 |
| Mouse monoclonal anti-RSAD2 | Peter Creswell (Yale) | N/A |
| PE Mouse anti-human CD34 | BD Biosciences | Cat# 555822 RRID:AB_396151 |
| APC Mouse anti-Human CD235a | BD Biosciences | Cat# 551336 RRID:AB_398499 |
| PE Mouse anti-human CD235a | BD Biosciences | Cat# 555570 RRID:AB_395949 |
| FITC Rabbit anti-human Band3 | This paper | N/A |
| APC Mouse anti-human α4 integrin/CD49d | Miltenyi Biotec | Cat# 130-093-281 RRID:AB_1036218 |
| Alexa Rat 488 anti-mouse CD45 | BD Biosciences | Cat# 103122 RRID:AB_493531 |
| Pacific Orange Mouse anti-human CD45 | Life Technologies | Cat# MHCD4530 RRID:AB_10376143 |
| APC Mouse anti-human CD19 | BD Biosciences | Cat# 555415 RRID:AB_398597 |
| PerCP-Cy5.5 Mouse anti-human CD33 | eBioscience | Cat# 45-0338-42 RRID:AB_10714975 |
| Alexa 488 Mouse anti-human CD38 | BioLegend | Cat#303512 RRID:AB_493088 |
| PE Cy7 Rat anti-mouse c-KIT | Thermofisher | Cat# 25-1171-82 RRID:AB_469644 |
| Pacific Blue Rat anti-mouse Sca-1 | BioLegend | Cat# 108120 RRID:AB_493273 |
| Mouse anti-Human IFNAR2 Antibody | pbl assay science | Cat# 21385-1 RRID:AB_387828 |
| Mouse anti-IgG2a control antibody | BioLegend | Cat# 401503 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor® 594 conjugate | Life Technologies | Cat# A-11012 RRID:AB_2534079 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate | Life Technologies | Cat# A-11008 RRID:AB_143165 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 594 conjugate | Life Technologies | Cat# A-11005 RRID:AB_2534073 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate | Life Technologies | Cat# A-11001 RRID:AB_2534069 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 350 | Life Technologies | Cat# A-11045 RRID:AB_2534100 |
| Viruses | ||
| CHIKV-GFP (based on strain LR2006 OPY1) | Laboratory of Stephen Higgs | Tsetsarkin et al., 2006 |
| VEEV-GFP (based on strain TC83) | Laboratory of Ilya Frolov | Schoggins et al., 2011 |
| YFV-Venus (based on strain 17D) | This laboratory | Schoggins et al., 2011 |
| DENV (based on IC30P-A infectious clone of strain 16681 | This laboratory | Schoggins et al., 2011 |
| WNV-GFP (based on isolate WNV-TX02) | This laboratory | Schoggins et al., 2011 |
| ZIKV (strain PRVABC59) | CDC, Ft. Collins | GenBank: KU501215.1 |
| VSV-GFP (based on strain Indiana) | Laboratory of John Rose | Dalton and Rose, 2001 |
| IAV strain A/WSN/33 (H1N1) and A/swine/Texas/98 (H3N2) | This laboratory | N/A |
| RSV-GFP (based on strain A2) | Laboratory of Peter L. Collins | Biacchesi et al., 2004 |
| MeV-GFP (based on strain Edmonston) | Laboratory of Roberto Cattaneo | del Valle et al., 2007 |
| hPIV-3-GFP (based on strain JS) | Laboratory of Peter L. Collins | Zhang et al., 2005 |
| NDV-GFP (based on strain Hitchner B1) | Laboratory of Christopher F. Basler | Park et al., 2003 |
| Biological Samples | ||
| Human fetal liver tissue | Advanced Bioscience Resources (Alameda, CA) | N/A |
| Human cord blood | New York Blood Center | N/A |
| Human peripheral blood | New York Blood Center | N/A |
| Human bone marrow (normal) | New York–Presbyterian Hospital | N/A |
| Chemicals, Reagents, and Recombinant Proteins | ||
| Animal Free Human BMP-4 | Peprotech | Cat# AF-120-05ET |
| Animal-Free Human BDNF | Peprotech | Cat# AF-450-02 |
| Animal-Free Human EGF | Peprotech | Cat# AF-100-15 |
| Animal-Free Human GDNF | Peprotech | Cat# AF-450-10 |
| Exendin-4 | Sigma-Aldrich | Cat# E7144 CAS: 141758-74-9 |
| FGF-Basic (AA 10-155) | Life Technologies | Cat# PHG0024 |
| Human Flt3 ligand | Peprotech | Cat# 300-19 |
| Human G-CSF | Peprotech | Cat# 300-23 |
| Human HGF | Peprotech | Cat# 100-39 |
| Human Holo-transferrin | Sigma-Aldrich | Cat# T4132 |
| Human IFN-β | Peprotech | Cat# 300-02BC |
| Human IL-1β | Peprotech | Cat# 200-01B |
| Human IL-3 | Peprotech | Cat# 200-03 |
| Human IL-6 | Peprotech | Cat# 200-06 |
| Human insulin | Sigma-Aldrich | Cat# 91077C |
| Human KGF/FGF7 | Peprotech | Cat# 100-19 |
| Human M-CSF | Peprotech | Cat# 300-25 |
| Human Noggin | Peprotech | Cat# 120-10C |
| Human oncostatin M (OSM) | R&D systems | Cat# 295-OM-050 |
| Human PDGF-AB | Peprotech | Cat# 100-00AB |
| Human SCF | Peprotech | Cat# 300-07 |
| Human VEGF | Peprotech | Cat# 100-20 |
| Human/mouse/rat activin A | R&D systems | Cat# 338-AC-010/CF |
| Mouse Wnt-3A | R&D systems | Cat# 1324-WN-002 |
| 2-Mercaptoethanol (55mM) | Life Technologies | Cat# 21985-023 |
| 3,3′,5-Triiodo-L-thyronine sodium salt | Sigma-Aldrich | Cat# T6397 CAS: 55-06-1 |
| ALK5 inhibitor II | Enzo Life Science | Cat# ALX-270-445 CAS: 446859-33-2 |
| B-27 | Life Technologies | Cat# 0080085-SA |
| B-27 (minus insulin) | Life Technologies | Cat# A1895601 |
| CD34+ Expansion Supplement (10X) | Stemcell Technologies | Cat# 02691 |
| CHIR-98014 | selleckchem | Cat# S2745 CAS: 252935-94-7 |
| CHIR99021 | Stemcell Technologies | Cat# 72054 CAS: 252917-06-9 |
| Dibutyryl cAMP | Sigma-Aldrich | Cat# D0627 CAS: 16980-89-5 |
| ESC-qualified FBS | Life Technologies | Cat# 16141061 |
| GlutaMAX Supplement | Life Technologies | Cat# 35050-061 |
| Heparin, sodium salt from porcine intestine | Sigma-Aldrich | Cat# H3149 CAS: 9041-08-1 |
| Human AB serum | Atlanta Biologicals | Cat# s40110H |
| Human PB plasma | Stemcell Technologies | Cat# 70039 |
| Insulin-Transferrin-Selenium (ITS -G) (100X) | Life Technologies | Cat# 41400-045 |
| Insulin-Transferrin-Selenium-X (100X) | Life Technologies | Cat# 51500056 |
| JAK inhibitor | EMD Millipore | Cat# 420099 CAS: 457081-03-7 |
| KnockOut™ Serum Replacement (KOSR) | Life Technologies | Cat# 10828028 |
| L-Ascorbic acid | Sigma-Aldrich | Cat# A4544 CAS: 50-81-7 |
| LDN193189 | Stemgent | Cat# 04-0074 CAS; 1062368-24-4 |
| Lipid Mixture 1, Chemically Defined (100X) | Sigma-Aldrich | Cat# L0288 |
| Lonza MSC medium | Lonza | Cat# PT-3238 & PT-4105 |
| MethoCult H4434 classic medium | Stemcell Technologies | Cat# 04434 |
| N-2 Supplement (100X) | Life Technologies | Cat# 17502-048 |
| N2 supplement-A | Stemcell Technologies | Cat# 05712 |
| Nicotinamide | Sigma-Aldrich | Cat# N0636 CAS: 98-92-0 |
| Non-essential amino acid | Life Technologies | Cat# 11140050 |
| Retinoid acid | Sigma-Aldrich | Cat# R2625 CAS: 302-79-4 |
| ROCK inhibitor (Y-27632) | Stemcell Technologies | Cat# 72308 CAS: 129830-38-2 |
| SB 431542 | Tocris Bioscience | Cat# 1614 CAS: 301836-41-9 |
| SM1 neuronal supplement | Stemcell Technologies | Cat# 05711 |
| StemPro Neural Supplement | Life Technologies | Cat# A10508-01 |
| Accutase | Innovative Cell Technologies | Cat# AT104-500 |
| Basement Membrane Matrix | Corning | Cat# 356230 |
| EDTA solution | Life Technologies | Cat# AM9260G |
| Gentamicin | Life Technologies | Cat# 15750078 CAS: 1405-41-0 |
| Poly-L-ornithine hydrobromide | Sigma-Aldrich | Cat# P3655 CAS: 27378-49-0 |
| ReLeSR | Stemcell Technologies | Cat# 05872 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | Qiagen | Cat# 74014 |
| LightCycler SYBR Green I Master Mix | Roche Life Science | Cat# 04707516001 |
| TruSeq Stranded mRNA LT Sample Prep Kit | Illumina | Cat# RS-122-2101, RS-122-2102 |
| STEMdiff Definitive Endoderm Kit | Stemcell Technologies | Cat# 05110 |
| EasySep human CD34 positive selection kit | Stemcell Technologies | Cat# 18056 |
| CellTiter-Glo Assay Kit | Promega | Cat# G7571 |
| Lenti-X Concentrator | Clontech | Cat# 631231 |
| Superscript III First-Strand Synthesis System | Life Technologies | Cat# 18080051 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE97987 |
| Genome Reference Consortium Human Build 38 | Genome Reference Consortium | N/A |
| Experimental Models: Cell Lines | ||
| Human ESC line (WA09), passage 30 - 40 | WiCell | N/A |
| Human ESC line (RUES2), passage 30 - 40 | Ali Brivanlou (Rockefeller University) | N/A |
| Human ESC line (HUES8), passage 30 - 40 | Danwei Huangfu (MSKCC) | N/A |
| Human iPSC line (C3A), passage 30 - 40 | Stephen Duncan (Medical University of South Carolina) | N/A |
| Human iPSC line (BJ-3), passage 30 - 40 | Fred H. Gage (Salk Institute) | N/A |
| Human iPSC line (LVID2), passage 30 - 40 | This paper | N/A |
| Human: Lenti-X 293Tcells, passage 25 - 30 | Clontech | Cat# 632180; RRID: CVCL_4401 |
| Human Mesenchymal stem cells, passage 2 - 8 | Lonza | Cat# PT-2501 |
| Human fibroblasts CCD-1112Sk, passage 2 - 5 | ATCC | Cat# CRL-2429 |
| Experimental Models: Organisms/Strains | ||
| Immunodeficient mouse | NOD.Cg-Rag1 IL2rgtmlWjl/Sz (NRG) | Jackson Laboratory |
| Oligonucleotides | ||
| IFITM3 gRNA-1: TTCTTCTCTCCTGTCAACAG | This paper | N/A |
| IFITM3 gRNA-2: CCCAGTAACCCGACCGCCGC | This paper | N/A |
| shRNA-IFITM1: ACCTGTCTACAGTGTCATTCA | This paper | N/A |
| shRNA-IFITM3: CCCAACTATGAGATGCTCAAG | This paper | N/A |
| shRNA-CDKN1A-1: GTCACTGTCTTGTACCCTTGT | This paper | N/A |
| shRNA-CDKN1A-2: AGAGGTTCCTAAGAGTGCTGG | This paper | N/A |
| Primers for RT-qPCR, see Table S1 | This paper | N/A |
| Primers for ChIP-PCR, see Table S1 | This paper | N/A |
| Software and Algorithms | ||
| FASTQC (Version 0.11.5) | https://www.bioinformatics.babraham.ac.uk | N/A |
| Salmon pseudo-alignment tool (v0.8.2) | http://salmon.readthedocs.io/en/latest/salmon.html | N/A |
| Tximport package (v0.99.2) | https://bioconductor.org | N/A |
| DESeq2 package (v1.14.1) | https://bioconductor.org | N/A |
| Limma package (v3.32.5) | http://web.mit.edu | Gordon Smyth and Carolyn de Graaf |
| Prism | https://www.graphpad.com/scientific-software/prism/ | GraphPad Software |
| FlowJo | https://www.flowjo.com | Tree Star; FlowJo |
| Others | ||
| Human and Chimpanzee ISG lists | Schoggins et al., 2011 | See Table S2 |
| Mouse ISG list | https://www.qiagen.com | See Table S2 |
| Gene expression data for human HSCs | This paper | See Table S3 |
| Gene expression data for human MSCs | Roson-Burgo et al., 2014 | See Table S3 |
| Gene expression data for mouse ESC line-1 | Wamstad et al., 2012 | See Table S4 |
| Gene expression data for mouse ESC line-2 | Klattenhoff et al., 2013 | See Table S4 |
| Gene expression data for mouse ESC line-3 | Li et al., 2014 | See Table S4 |
| Epigenetic data for mESCs | Wamstad et al., 2012 | See Table S4 |
| Gene expression data for Chimpanzee iPSC lines | Nuttle et al., 2016 | See Table S4 |
Supplementary Material
Figure S1, related to Figure 1. Intrinsic expression of ISGs in pluripotent and multipotent human stem cells.
(A) Representative images showing increasing permissiveness to virus infection along hepatic differentiation of hESCs. Cells were infected with indicated viruses, fixed at the indicated time points post infection (p.i.) and were visualized directly (green, GFP-expressing viruses) or stained with the indicated antibodies (red: anti-NS3 for DENV and anti-flavivirus E for ZIKV). Nuclei were visualized by DAPI staining (lower panels in each grouping). Scale bars: 100 μm.
(B) Schematic workflow for identifying ISG signature in stem cells. Briefly, for any given stem cell type, we focused on genes with TPM values above a minimum threshold (approximately TPM >1.0, detailed in STAR methods) in all biological replicates, thereby defined as “expressed” genes. The number of expressed genes ranged from 13,000 to 15,000, depending on the cell type and cell stage. Within the highly expressed group (top 20% of genes, ranked by TPMs), we focused on interferon-stimulated genes (ISGs), based on the compiled human and mouse ISG lists (Table S2). ISGs within the highly expressed set were defined as “highly expressed ISGs” for the cells analyzed. Shown are data from hESC line WA09.
(C) Blockade of IFN response by IFNAR2 blocking antibodies in HLCs, but not hESCs. Left: hESCs-derived HLCs were pre-treated with isotype control or IFNAR2 blocking antibodies for 4 hr, followed by stimulation with IFN-β (100U/ml) for 12 hr. The magnitude of representative ISG induction was measured by qRT-PCR. Expression in untreated HLC was utilized for normalization. Mock indicates cells not treated with antibody prior to IFN stimulation. Right: Relative expression of the indicated ISGs in hESCs after being maintained in the presence of 10 μg/ml neutralizing antibody against IFNAR2 for 48 hr by qRT-PCR. Expression of mock cells was utilized for normalization. Shown are the means ± SD from 3 independent experiments.
(D) Representative images at low and high magnification of hESCs and hESC-derived germ layers. Cells were stained with indicated antibodies and analyzed by IFA. Nuclei were stained with DAPI. Scale bars: 100 μm.
(E) Relative expression of cell type-specific markers in hESCs and derived germ layers was measured by qRT-PCR. Expression levels in hESC were utilized for normalization. Shown are the means ± SD from 3 independent experiments. Statistical analyses were calculated between ESCs and tissue stem cells, as indicated by the dotted lines.
Asterisks indicate statistically significant differences (Student’s t test was used throughout this study) (*, p<0.05; **, p<0.01; ***, p<0.001).
Figure S2, related to Figure 2. Distinct ISG expression patterns in different tissue stem cells.
(A) Relative expression of the indicated cell type-specific markers in hESCs and derived tissue stem cells was measured by qRT-PCR. PSC makers: PDX1 and GCG; MSC markers: CD44, CD90, and CD105; NSC markers: SOX2 and PAX6. CD90 and SOX2 are also markers for hESCs. Expression levels in hESC were utilized for normalization. Shown are the means ± SD from 3 independent experiments.
(B) Representative images showing terminal differentiation of hESC-derived tissue stem cells. The left panels represent tissue stem cells and the right panels show terminally differentiated cells. PSC to β-cell differentiation was monitored by insulin expression (INS, red); MSC to myocyte differentiation by smooth muscle actin (SMAα, red) and to adipocyte by the presence of characteristic cytoplasmic lipid droplets (oil-red staining); NSC differentiation to neuron was monitored by expression of tubulin beta 3 class III (TUJ1, red). Nuclei were stained by DAPI. Scale bars: 100 μm.
(C) Relative expression of selected ISGs in hESC and three tissue stem cells was determined by qRT-PCR. Expression levels in hESC were utilized for normalization. Shown are the means ± SD from 3 independent experiments.
(D) Expression of housekeeping genes was measured by qRT-PCR and compared between ESCs and three tissue stem cells. Shown are the means of threshold cycles ± SD from 3 independent experiments.
(E) Bar diagram illustrating the number of highly expressed ISGs unique to individual donor (purple) and common to both donors (gray) in HSCs isolated from bone marrow (BM).
(F) Bar diagram illustrating the number of highly expressed ISGs unique to individual donor (purple) and common to all donors (gray). Top: MSCs were isolated from placenta tissue (PL) of three donors; Bottom: MSCs were isolated from bone marrow (BM) of three donors, as described in (Roson-Burgo et al., 2014)
(G) Expression of selected ISGs (anti-HIV and anti-DENV) in primary tissue stem cells (pHSC and pMSC) and hESC-derived HSCs and MSCs (iHSC and iMSC) was measured by qRT-PCR. Primary HSCs were from fetal livers of three different donors, and MSCs were from adipose tissue of two donors; iMSCs and iHSCs were derived from hESC WA09 line. Expression levels in iMSC were utilized for normalization. Shown are the means ± SD from 2 or 3 biological replicates and 3 independent experiments.
(H) Expression of selected ISGs (anti-CHIKV ISGs) in primary tissue stem cells (pHSC and pMSC) and hESC-derived HSCs and MSCs (iHSC and iMSC) was measured by qRT-PCR. Cells were from the same sources as described in (G). Expression levels in iMSC were utilized for normalization. Shown are the means ± SD from 2 or 3 biological replicates and 3 independent experiments.
(I) Primary HSCs and MSCs transduced with sh-Ctrl or sh-CDKN1A were infected with CHIKV (MOI =1.0). At 16 hr p.i., the cells were harvested for analyses of CHIKV infection and CDKN1A expression. The percentages of CHIKV infection were determined by flow cytometry for GFP (+) cells are shown (upper panel). Data are shown as the means ± SD from 3 independent experiments. Statistically significant differences, relative to infection of sh-Ctrl are indicated. Lower panels show western blot analysis of CDKN1A. ACTB served as a loading control.
Asterisks indicate statistically significant differences (Student’s t test was used) (*, p<0.05; **, p<0.01; ***, p<0.001).
Figure S3, related to Figure 3. ISG expression changes during terminal differentiation of hESCs.
(A) Pancreatic β-cell differentiation from hESCs was monitored by staining at days 0, 5, 14, 20, and 27 during the differentiation process using antibodies that recognized OCT4, SOX17, PDX1, NKX6.1, and INS, as indicated. Representative images at specified stages are shown. Scale bars: 100 μm.
(B) Expression of the indicated stage-specific markers along β-cell differentiation from hESCs was analyzed by qRT-PCR. Shown are the means ± SD from 3 independent experiments.
(C) Expression of the indicated housekeeping genes from cells at the specified stages along β-cell differentiation was analyzed by qRT-PCR. Shown are the means of threshold cycles ± SD from 3 independent experiments.
(D) Top: Cells undergoing differentiation into myofibroblasts were fixed and stained with the indicated antibodies and analyzed by IFA. Nuclei were stained with DAPI and merged images are shown. Scale bars: 100 μm.
Bottom: Expression of the indicated selected stage markers along myofibroblast differentiation was analyzed by qRT-PCR. Shown are the means ± SD from 3 independent experiments.
(E) Expression of the indicated selected ISGs from cells at the specified stages along myofibroblast differentiation was analyzed by qRT-PCR. Shown are the means ± SD from 3 independent experiments.
(F) Expression of the indicated housekeeping genes from cells at the specified stages along myofibroblast differentiation was analyzed by qRT-PCR. Shown are the means of threshold cycles ± SD from 3 independent experiments.
(G) Neuronal differentiation from hESCs was monitored by staining at days 0, 18, 40, and 60 during the differentiation process using antibodies that recognized SSEA-4, SOX2, PAX6, NES, TUJ1 and MAP2, as indicated. Representative images at specified stages are shown. Scale bars: 100 μm.
(H) Expression of the indicated selected ISGs from cells at the specified stages along neuronal differentiation was analyzed by qRT-PCR. Shown are the means ± SD from 3 independent experiments.
(I) Expression of the indicated housekeeping genes from cells at the specified stages along neuronal differentiation was analyzed by qRT-PCR. Shown are the means of threshold cycles ± SD from 3 independent experiments.
(J, K, L) Bar diagram illustrating the number of highly expressed ISGs unique to each stage (light blue) and shared by all lines (dark blue) along ex vivo differentiation of hESC into three lineages.
Asterisks indicate statistically significant differences (Student’s t test was used) (*, p<0.05; **, p<0.01; ***, p<0.001).
Figure S4, related to Figure 3. ISG expression changes during terminal differentiation of hESCs.
(A) HLC differentiation from hESCs was monitored during the differentiation process by staining at days 0, 5, 10, 15, 20, and 25 using antibodies that recognized Oct-4, AFP and ALB followed by Alexa Fluor 594 conjugated secondary antibody (red), or SOX17, FoxA2, and HNF4α followed by Alexa Fluor 488 conjugated secondary antibody (green). Representative images at specified stages are shown. Scale bars: 100 μm.
(B) Relative transcript abundance of pluripotent and differentiation markers at different stages along hepatic differentiation of hESCs as measured by RNA-Seq. Data represent average log2 TPM values for the indicated genes from two independent experiments. Five stages are shown: ESCs, END, hepatic stem cells (HepSC), immature hepatocyte (ImHep), Hepatocyte-like cells (HLC).
(C) Heatmap of RNA-Seq expression pattern (z-score TPM) of ISGs highly expressed in hESCs along ex vivo HLC differentiation of hESCs.
(D-E) Expression analyses of selected ISGs and markers during differentiation of hESCs along the hepatic lineage by western blot using the indicated antibodies (D) and qRT-PCR for the indicated transcripts (E). Shown in (E) are the means ± SD from 3 independent experiments.
(F) hESC-derived macrophage-like cells (MLCs) and primary macrophage isolated from fetal liver were collected by cytospin and stained with Giemsa. Representative images are shown. Scale bars: 100 μm.
(G) Expression of the indicated selected stage markers in cells at the specified stages during MLC differentiation in determined by qRT-PCR. Expression levels in hESC were utilized for normalization. Shown are the means ± SD from 3 independent experiments. The morphological features of the indicated stages are shown schematically at the top.
(H) MLCs were treated for 6 hr with lipopolysaccharides (LPS, 5 μg/ml) or PBS and RNA was isolated for qRT-PCR analysis of selected cytokines. The fold change in the LPS-treated cells relative to the PBS-treated controls for each gene is plotted. Shown are the means ± SD from 3 independent experiments.
(I) Expression of the indicated ISGs from cells at specified stages during differentiation of hESC into MLCs was measured by qRT-PCR. Expression levels in hESC were utilized for normalization. Shown are the means ± SD from 3 independent experiments.
Asterisks indicate statistically significant differences (Student’s t test was used) (*, p<0.05; **, p<0.01; ***, p<0.001).
Figure S5, related to Figure 1. Intrinsic ISG expression in pluripotent and multipotent stem cells from other species.
(A) Heatmap of expression patterns of highly expressed ISGs in four chimpanzee iPSC lines, log2 TPM. Data are from (Marchetto, et al. Nature. 2013).
(B) Bar diagram illustrating the number of highly expressed ISGs unique to each mESC line (purple) and shared by all three lines (shared/gray). Data are from (Wamstad et al., Cell. 2012; Klattenhoff et al., Cell. 2013; Li et al., PLoS One. 2014)
(C) Bar diagram illustrating the number of highly expressed pluripotent cell ISGs unique to each species (purple) or shared by all three species (shared/gray).
(D) Heatmap of gene expression (z-score TPM) for highly expressed ISGs along cardiomyocyte differentiation. Shown are highly expressed ISGs in mouse ESCs. Data are from (Wamstad et al., Cell. 2012)
(E) Relative expression of the indicated ISGs at different stages along mESCs differentiation towards cardiomyocyte. Average log2 TPM values of two independent experiments are shown.
(F) The epigenetic landscape showing dynamic levels of H3K4me3, H3K27ac and H3K2me3 for unexpressed or lowly expressed and highly expressed ISGs along cardiomyocyte differentiation of mESCs. Examples of lowly expressed ISGs (Lowly exp ISGs) include MX1 and OAS1, and examples of highly expressed ISGs (Highly ISGs) are IFITM3 and IGFBP2. ChIP-Seq (H3K4me3, H3K27ac and H3K2me3, y axis represents reads over million unique mapped reads) are shown. The scale for each modification is constant throughout the time course.
(G) Statistical analyses showing association between levels of H3K4me3 or H3K27ac and expression (RPKM) of all ISGs along cardiomyocyte differentiation of mESCs. Each dot represents value for an ISG. Shown are the means ± SD from all the ISGs analyzed in each group. Asterisks indicate statistically significant difference (***, p<0.001). For F-G, data are from published data set (Wamstad et al. 2012) and analyzed as shown in Table S4. Asterisks indicate statistically significant differences (Student’s t test was used) (***, p<0.001).
(H) The epigenetic landscape showing different histone modification markers, RNA-Seq, and DNase-Seq for unexpressed or lowly expressed and highly expressed ISGs in hESCs. Examples of lowly expressed ISGs (Lowly exp ISGs) include APOBEC3A, IFIT1 and RSAD2, and examples of highly expressed ISGs (highly exp ISGs) are IFITM1-3, BST2 and MOV10. ChIP-Seq (H3K4me1, H3K4me2, H3K4me3, H3K9me3, H3K9ac, H3K27me3, H3K27ac, y axis represents reads over million unique mapped reads) are shown. The scale for each modification is among different genes. Data are from the Encyclopedia of DNA Elements (ENCODE) Consortium.
See also Table S4 for epigenetic data.
Figure S6, related to Figure 5. Antiviral activity of ISGs in human stem cells.
(A) Representative images showing expression of pluripotent markers in three hESC clones. Cells were fixed and stained with the indicated antibodies. Scale bars: 100 μm.
(B) WT, KO23, and KO123 clones were differentiated ex vivo into cells representing the three germ layers and were fixed and stained with antibodies for the indicated markers. Nuclei were stained with DAPI. Representative merged images are shown. Scale bars: 100 μm.
(C) Equal numbers of WT, KO23, and KO123 clones were plated and at the indicated time points, viable cell numbers were determined using the CellTiter-Glo kit. Shown are the means of relative luciferase units (RLU) ± SD from 3 independent experiments.
(D) WT, KO23, and KO123 clones were infected with GFP-expressing human parainfluenza virus type-3 (hPIV3, MOI=1.0) and after 24 hr the cells were fixed and nuclei stained with DAPI. Representative images showing infection (green, top) and nuclei (blue, bottom) are shown. Scale bars: 100 μm.
(E) WA09 hESCs were transduced with control (CTRL) shRNA or shRNA targeting IFITM3 (sh-1 and sh-2) and puromycin-selected stable clones were analyzed by western blot for the indicated IFITM family members. GAPDH served as a loading control.
(F) Stable clones derived from (E) were fixed and stained with antibodies against the indicated pluripotent markers. Representative merged images are shown. Scale bars: 100 μm.
(G) Stable clones derived from (E) were infected with the indicated viruses (MOI=1.0) and after 48 hr fixed and analyzed by flow cytometry. DENV infected cells were stained with anti-NS3 antibody prior to analysis, while NDV and YFV were GFP tagged. Shown are the means ± SD from 3 independent experiments. Asterisks indicate statistically significant differences (***, p<0.001).
(H) Equal numbers of WT, KO23, and KO123 NSCs were plated and at the indicated time points, cell viable numbers were determined using the CellTiter-Glo kit. Shown are the means of RLU ± SD from 3 independent experiments.
(I) WT and KO123 NSC were differentiated into neurons or astrocytes; after fixation they were stained with antibodies against TUJ1 or GFAP, respectively. Nuclei were stained with DAPI (blue). Representative merged images are shown. Scale bars: 100 μm.
(J) WT and KO123 NSC were infected with the indicated GFP-expressing neurotropic viruses (MOI=1.0). After infection, cells were fixed and stained as follows: infections with VSV and VEEV were fixed at 12 hr p.i. and infection with MeV was fixed at 48 hr p.i. Representative images of infection (green, top) and nuclei (blue, bottom) are shown. Scale bars: 100 μm.
(K–L) hESC-derived NSCs were cultured under conditions where NSCs gave rise to more NSCs as well as differentiated neurons; cultures were then infected with a GFP-expressing YFV (stain 17D). Cells were fixed at 48 hr p.i. and stained with NSC marker SOX2 (red) and neuronal marker TUJ1 (blue). Representative images are shown in (K). Scale bars: 100 μm. Shown in (L) are the mean percentages of infected cells ± SD in the NSC (SOX2+) and neuronal (TUJ1+) populations from 3 independent experiments are plotted.
(M) Primary MSCs were transduced with the indicated shRNAs [control (CTRL), IFITM or p21/CDKN1A)] and 5 days later fixed and stained with the indicated antibodies against MSC markers (CD90/THY1, COL1A1). Nuclei were stained with DAPI (blue). Representative fluorescence microscopy images are shown. Scale bars: 100 μm.
(N) Primary WT MSCs or MSCs transduced with the indicated shRNAs were differentiated into adipocyte cells and analyzed by qRT-PCR for induction of adipocyte markers. Data were normalized to RPS11 expression. Fold changes from expression levels in parental MSCs are shown as the means ± SD from 3 independent experiments.
Asterisks indicate statistically significant differences (Student’s t test was used) (**, p<0.01; ***, p<0.001).
Figure S7, related to Figure 7. ISGs protect stem cells from viral infection during development.
(A–B) Top: Schematic representation of conditions for ex vivo differentiation of HSCs into erythrocyte and macrophage. Bottom: HSCs transduced to express control (CTRL) or IFITM shRNA (KD) were mock infected or exposed to IAV or DENV for 3 hr and then subjected to ex vivo differentiation into erythrocytes (A) or macrophages (B). At the indicated time points the number of cells were counted and normalized to the number present at day 0. The means ± SD of 3 biological replicates are shown per condition. Different colored symbols correspond to 3 different donors.
(C–E) Erythrocyte differentiation conducted in (A) was monitored at the indicated time points by flow cytometry of the indicated surface markers (CD36, CD34, ITGA4, and Band3). (C) Representative FACS plots from 3 biological replicates. (D–E) The percentages of the indicated cell types. CFU-E: colony forming unit-erythroid, defined as CD36+CD34−; BFU-E: burst-forming units-erythroid, defined as CD36−CD34+.
(F) Representative flow cytometry plots showing the composition of human leukocytes in the bone marrow from the transplanted mice described in Figure 7E–H. CD19, surface marker for human B-cells; CD33, surface marker for human cells of myeloid lineage. Shown are representative plots from mice transplanted with mock infected control HSCs. No detectable human hematopoietic cells were seen in mice transplanted with IAV pre-exposed KD HSCs.
(G) Representative flow cytometry plots showing the gating strategies for human and mouse HSCs isolated from mouse bone marrow. Human HSCs, CD34+CD38−; mouse HSCs: c-KIT+Sca-1+.
(H) Bone marrow harvested at 16 weeks form the mice described in Figure 8E–H was analyzed by flow cytometry for the percentages of murine HSCs. Each symbol represents the value obtained in an individual mouse. Bars represent the means ± SD from n=5 mice per group from two donors.
Asterisks indicate statistically significant differences (Student’s t test was used) (*, p<0.05; **, p<0.01; ***, p<0.001).
Table-S1, related to Figure 1. RT-qPCR primers used.
Primer sequences for all primers used for real-time RT-PCR and primers used for generating shRNAs against ISGs.
Table-S2, related to Figure 1. Human and mouse ISG lists.
Detailed information for human and mouse interferon-stimulated genes (ISGs). Entrez Gene IDs and detailed descriptions included.
Table-S3, related to Figure 2. Gene expression data for human HSCs and MSCs.
Transcriptome analysis of human hematopoietic stem cells isolated from four different sources and human mesenchymal stem cells isolated from two different sources.
Table-S4, related to Figure S5. Gene expression data for non-human primate stem cells.
Transcriptional analysis of three mouse embryonic stem cell lines and four induced pluripotent stem cell line from chimpanzee; comparison between mouse, chimpanzee, and human pluripotent stem cells; and processed epigenetic data used for generating Figure S5 (S5A–B).
HIGHLIGHTS.
Pluri/multipotent stem cells exhibit intrinsic expression of ISGs
Different stem cells express cell type-specific groups of ISGs
Intrinsically expressed ISGs mediate antiviral resistance ex vivo and in vivo
Dynamic expression of ISGs is conserved across species
Acknowledgments
We thank all Rice lab members for helpful comments on the manuscript; A. Brivanlou for RUES2, D. Huangfu for HUES8-iCas9 and S. Duncan for iPS.C3A cells. This work was supported by NIH Grant R01-AI091707 (C.M.R. and B.R.R), by NIH grant U19 AI111825 and the John C. Whitehead Presidential Fellowship (B.R.R.), in part by NIH grant DK100810 and a grant 81530005 from NSF of China (X.A.). Additional funding was provided by the Greenberg Medical Research Institute, the Starr Foundation, and anonymous donors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Concept, X.W. and C.M.R.; Methodology, X.W. and B.R.R; Investigation, X.W., V.L.DT., Y.H., E.B., H.-H.H., Y.W., S.S., T.S., L.A., and C.Q.; Bioinformatic Analysis, X.W., D.S., L.A.VS., and Y. Y.; Writing – Original Draft, X.W.; Writing – Review & Editing, X.W., V.L.DT., E.B., L.A.VS., S.S., L.A., M.L., M.R.M., W.M.S., X.A., B.R.R., and C.M.R.; Visualization, X.W. and E.B.; Supervision, X.A., B.R.R., and C.M.R.
References
- Allen EK, Randolph AG, Bhangale T, Dogra P, Ohlson M, Oshansky CM, Zamora AE, Shannon JP, Finkelstein D, Dressen A, et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat Med. 2017;23:975–983. doi: 10.1038/nm.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R. Manipulation of cell surface macromolecules by flaviviruses. In Adv Virus Res. 2003:229–274. doi: 10.1016/S0065-3527(03)59007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CC, Zhong G, Huang IC, Farzan M. IFITM-Family Proteins: The Cell's First Line of Antiviral Defense. Annu Rev Virol. 2014;1:261–283. doi: 10.1146/annurev-virology-031413-085537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzile JP, Stark TJ, Yeo GW, Spector DH. Human Cytomegalovirus Infection of Human Embryonic Stem Cell-Derived Primitive Neural Stem Cells Is Restricted at Several Steps but Leads to the Persistence of Viral DNA. J Virol. 2014;88:4021–4039. doi: 10.1128/JVI.03492-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biacchesi S, Skiadopoulos MH, Tran KC, Murphy BR, Collins PL, Buchholz UJ. Recovery of human metapneumovirus from cDNA: optimization of growth in vitro and expression of additional genes. Virology. 2004;321:247–259. doi: 10.1016/j.virol.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Bomsel M, Alfsen A. Entry of viruses through the epithelial barrier: pathogenic trickery. Nat Rev Mol Cell Biol. 2003;4:57–68. doi: 10.1038/nrm1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden EC, Hogan TF, Voelkel JG. Comparative Antiproliferative Activity in Vitro of Natural Interferons α and β for Diploid and Transformed Human Cells. Cancer Res. 1982;42:4948–4953. [PubMed] [Google Scholar]
- Burke DC, Graham CF, Lehman JM. Appearance of interferon inducibility and sensitivity during differentiation of murine teratocarcinoma cells in vitro. Cell. 1978;13:243–248. doi: 10.1016/0092-8674(78)90193-9. [DOI] [PubMed] [Google Scholar]
- Cao-Lormeau VM, Blake A, Mons S, Lastère S, Roche C, Vanhomwegen J, Dub T, Baudouin L, Teissier A, Larre P, et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet. 2016;387:1531–1539. doi: 10.1016/S0140-6736(16)00562-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell J, Bixby D, Savona MR, Collins KL. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med. 2010;16:446–451. doi: 10.1038/nm.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castella A, Croxson TS, Mildvan D, Witt DH, Zalusky R. The Bone Marrow in AIDS: A Histologic, Hematologic, and Microbiologic Study. Am J Clin Pathol. 1985;84:425–432. doi: 10.1093/ajcp/84.4.425. [DOI] [PubMed] [Google Scholar]
- Couderc T, Chrétien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F, Touret Y, Barau G, Cayet N, Schuffenecker I, et al. A Mouse Model for Chikungunya: Young Age and Inefficient Type-I Interferon Signaling Are Risk Factors for Severe Disease. PLoS Pathog. 2008;4:e290. doi: 10.1371/journal.ppat.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton KP, Rose JK. Vesicular Stomatitis Virus Glycoprotein Containing the Entire Green Fluorescent Protein on Its Cytoplasmic Domain Is Incorporated Efficiently into Virus Particles. Virology. 2001;279:414–421. doi: 10.1006/viro.2000.0736. [DOI] [PubMed] [Google Scholar]
- de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BRG. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- del Valle JR, Devaux P, Hodge G, Wegner NJ, McChesney MB, Cattaneo R. A Vectored Measles Virus Induces Hepatitis B Surface Antigen Antibodies While Protecting Macaques against Measles Virus Challenge. J Virol. 2007;81:10597–10605. doi: 10.1128/JVI.00923-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular Zone Astrocytes Are Neural Stem Cells in the Adult Mammalian Brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Douam F, Gaska JM, Winder BY, Ding Q, Schaewen MV, Ploss A. Genetic Dissection of the Host Tropism of Human-Tropic Pathogens. Annu Rev Genet. 2015;49:21–45. doi: 10.1146/annurev-genet-112414-054823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29:569–574. doi: 10.1016/j.tig.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Feuer G, Fraser JK, Zack JA, Lee F, Feuer R, Chen IS. Human T-cell leukemia virus infection of human hematopoietic progenitor cells: maintenance of virus infection during differentiation in vitro and in vivo. J Virol. 1996;70:4038–4044. doi: 10.1128/jvi.70.6.4038-4044.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci. 2013;110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sastre A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe. 2017;22:176–184. doi: 10.1016/j.chom.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczol E, Andrews P, Plotkin S. Cytomegalovirus replicates in differentiated but not in undifferentiated human embryonal carcinoma cells. Science. 1984;224:159–161. doi: 10.1126/science.6322309. [DOI] [PubMed] [Google Scholar]
- Grow EJ, Flynn RA, Chavez SL, Bayless NL, Wossidlo M, Wesche DJ, Martin L, Ware CB, Blish CA, Chang HY, et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature. 2015;522:221–225. doi: 10.1038/nature14308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzog P, Hwang S, Kola I. Role of interferons in the regulation of cell proliferation, differentiation, and development. Mol Reprod Dev. 1994;39:226–232. doi: 10.1002/mrd.1080390216. [DOI] [PubMed] [Google Scholar]
- Hong XX, Carmichael GG. Innate Immunity in Pluripotent Human Cells: attenuated response to interferon-β. J Biol Chem. 2013;288:16196–16205. doi: 10.1074/jbc.M112.435461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Liu J, Xue F, Halverson G, Reid M, Guo A, Chen L, Raza A, Galili N, Jaffray J, et al. Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo. Blood. 2013;121:3246–3253. doi: 10.1182/blood-2013-01-476390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz ÖH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Kim BK, Kim SE, Shim JH, Woo DH, Gil JE, Kim SK, Kim JH. Neurogenic effect of vascular endothelial growth factor during germ layer formation of human embryonic stem cells. FEBS Lett. 2006;580:5869–5874. doi: 10.1016/j.febslet.2006.09.053. [DOI] [PubMed] [Google Scholar]
- Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, et al. Braveheart, a Long Noncoding RNA Required for Cardiovascular Lineage Commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov SA, Mankani MH, Gronthos S, Satomura K, Bianco P, Robey PG. Circulating Skeletal Stem Cells. J Cell Biol. 2001;153:1133–1140. doi: 10.1083/jcb.153.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange UC, Adams DJ, Lee C, Barton S, Schneider R, Bradley A, Surani MA. Normal Germ Line Establishment in Mice Carrying a Deletion of the Ifitm/Fragilis Gene Family Cluster. Mol Cell Biol. 2008;28:4688–4696. doi: 10.1128/MCB.00272-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Saucedo-Cuevas L, Regla-Nava Jose A, Chai G, Sheets N, Tang W, Terskikh Alexey V, Shresta S, Gleeson Joseph G. Zika Virus Infects Neural Progenitors in the Adult Mouse Brain and Alters Proliferation. Cell Stem Cell. 2016;19:593–598. doi: 10.1016/j.stem.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Rivera CM, Ishii H, Jin F, Selvaraj S, Lee AY, Dixon JR, Ren B. CRISPR Reveals a Distal Super-Enhancer Required for Sox2 Expression in Mouse Embryonic Stem Cells. PLoS One. 2014;9:e114485. doi: 10.1371/journal.pone.0114485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian Q, Zhang Y, Zhang J, Zhang HK, Wu X, Zhang Y, Lam FFY, Kang S, Xia JC, Lai WH, et al. Functional Mesenchymal Stem Cells Derived From Human Induced Pluripotent Stem Cells Attenuate Limb Ischemia in Mice. Circulation. 2010;121:1113–1123. doi: 10.1161/CIRCULATIONAHA.109.898312. [DOI] [PubMed] [Google Scholar]
- Maillard PV, Ciaudo C, Marchais A, Li Y, Jay F, Ding SW, Voinnet O. Antiviral RNA Interference in Mammalian Cells. Science. 2013;342:235–238. doi: 10.1126/science.1241930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MCN, Narvaiza I, Denli AM, Benner C, Lazzarini TA, Nathanson JL, Paquola ACM, Desai KN, Herai RH, Weitzman MD, et al. Differential L1 regulation in pluripotent stem cells of humans and apes. Nature. 2013;503:525–529. doi: 10.1038/nature12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques JT, Carthew RW. A call to arms: coevolution of animal viruses and host innate immune responses. Trends Genet. 2007;23:359–364. doi: 10.1016/j.tig.2007.04.004. [DOI] [PubMed] [Google Scholar]
- McMahon AW, Eidex RB, Marfin AA, Russell M, Sejvar JJ, Markoff L, Hayes EB, Chen RT, Ball R, Braun MM, et al. Neurologic disease associated with 17D-204 yellow fever vaccination: A report of 15 cases. Vaccine. 2007;25:1727–1734. doi: 10.1016/j.vaccine.2006.11.027. [DOI] [PubMed] [Google Scholar]
- Mendelson M, Monard S, Sissons P, Sinclair J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol. 1996;77:3099–3102. doi: 10.1099/0022-1317-77-12-3099. [DOI] [PubMed] [Google Scholar]
- Naeye RL, Blanc W. Pathogenesis of congenital rubella. JAMA. 1965;194:1277–1283. [PubMed] [Google Scholar]
- Park MS, Shaw ML, Muñoz-Jordan J, Cros JF, Nakaya T, Bouvier N, Palese P, García-Sastre A, Basler CF. Newcastle Disease Virus (NDV)-Based Assay Demonstrates Interferon-Antagonist Activity for the NDV V Protein and the Nipah Virus V, W, and C Proteins. J Virol. 2003;77:1501–1511. doi: 10.1128/JVI.77.2.1501-1511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O'Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32:1121. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Roson-Burgo B, Sanchez-Guijo F, Del Cañizo C, De Las Rivas J. Transcriptomic portrait of human Mesenchymal Stromal/Stem cells isolated from bone marrow and placenta. BMC Genomics. 2014;15:910. doi: 10.1186/1471-2164-15-910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiura A, Sata M, Hirata Y, Nagai R, Makuuchi M. Circulating smooth muscle progenitor cells contribute to atherosclerosis. Nat Med. 2001;7:382–383. doi: 10.1038/86394. [DOI] [PubMed] [Google Scholar]
- Schneider WM, Dittmann Chevillotte M, Rice CM. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, Chen P, Gertner RS, Gaublomme JT, Yosef N, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510:363–369. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Cheng T, Preffer FI, Dombkowski D, Tomasson MH, Golan DE, Yang O, Hofmann W, Sodroski JG, Luster AD, et al. Intrinsic Human Immunodeficiency Virus Type 1 Resistance of Hematopoietic Stem Cells Despite Coreceptor Expression. J Virol. 1999;73:728–737. doi: 10.1128/jvi.73.1.728-737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindre H, Tjoonnfjord G, Rollag H, Ranneberg-Nilsen T, Veiby O, Beck S, Degre M, Hestdal K. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood. 1996;88:4526–4533. [PubMed] [Google Scholar]
- Singh KJ, Ahluwalia G, Sharma SK, Saxena R, Chaudhary VP, Anant M. Significance of haematological manifestations in patients with tuberculosis. J Assoc Physicians India. 2001;49:788, 790–784. [PubMed] [Google Scholar]
- Swartzendruber DE, Lehman JM. Neoplastic differentiation: Interaction of simian virus 40 and polyoma virus with murine teratocarcinoma cells in vitro. J Cell Physio. 1975;85:179–187. doi: 10.1002/jcp.1040850204. [DOI] [PubMed] [Google Scholar]
- Tan X, Xu X, Elkenani M, Smorag L, Zechner U, Nolte J, Engel W, Pantakani DVK. Zfp819, a novel KRAB-zinc finger protein, interacts with KAP1 and functions in genomic integrity maintenance of mouse embryonic stem cells. Stem Cell Res. 2013;11:1045–1059. doi: 10.1016/j.scr.2013.07.006. [DOI] [PubMed] [Google Scholar]
- Tang H, Hammack C, Ogden Sarah C, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee Emily M, et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell. 2016;18:587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN, Brandt WE, Russell PK, Dixon FT. Replication of Dengue-2 Virus in Cultured Human Lymphoblastoid Cells and Subpopulations of Human Peripheral Leukocytes. J Immunol. 1976;117:953–961. [PubMed] [Google Scholar]
- Tsetsarkin K, Higgs S, McGee CE, Lamballerie XD, Charrel RN, Vanlandingham DL. Infectious Clones of Chikungunya Virus (La Réunion Isolate) for Vector Competence Studies. Vector Borne Zoonotic Dis. 2006;6:325–337. doi: 10.1089/vbz.2006.6.325. [DOI] [PubMed] [Google Scholar]
- Villa NY, Bais S, Chan WM, Meacham AM, Wise E, Rahman MM, Moreb JS, Rosenau EH, Wingard JR, McFadden G, et al. Ex vivo virotherapy with myxoma virus does not impair hematopoietic stem and progenitor cells. Cytotherapy. 2016;18:465–480. doi: 10.1016/j.jcyt.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakim LM, Gupta N, Mintern JD, Villadangos JA. Enhanced survival of lung tissue-resident memory CD8+ T cells during infection with influenza virus due to selective expression of IFITM3. Nat Immunol. 2013;14:238–245. doi: 10.1038/ni.2525. [DOI] [PubMed] [Google Scholar]
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, et al. Dynamic and Coordinated Epigenetic Regulation of Developmental Transitions in the Cardiac Lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichold FF, Zella D, Barabitskaja O, Maciejewski JP, Dunn DE, Sloand EM, Young NS. Neither Human Immunodeficiency Virus-1 (HIV-1) nor HIV-2 Infects Most-Primitive Human Hematopoietic Stem Cells as Assessed in Long-Term Bone Marrow Cultures. Blood. 1998;91:907–915. [PubMed] [Google Scholar]
- Weissman IL. Stem Cells. Cell. 2000;100:157–168. doi: 10.1016/s0092-8674(00)81692-x. [DOI] [PubMed] [Google Scholar]
- Wolf D, Goff SP. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature. 2009;458:1201–1204. doi: 10.1038/nature07844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Robotham JM, Lee E, Dalton S, Kneteman NM, Gilbert DM, Tang H. Productive Hepatitis C Virus Infection of Stem Cell-Derived Hepatocytes Reveals a Critical Transition to Viral Permissiveness during Differentiation. PLoS Pathog. 2012;8:e1002617. doi: 10.1371/journal.ppat.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Jiang W, Liu M, Sui X, Yin X, Chen S, Shi Y, Deng H. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res. 2009;19:429–438. doi: 10.1038/cr.2009.28. [DOI] [PubMed] [Google Scholar]
- Zhang L, Bukreyev A, Thompson CI, Watson B, Peeples ME, Collins PL, Pickles RJ. Infection of Ciliated Cells by Human Parainfluenza Virus Type 3 in an In Vitro Model of Human Airway Epithelium. J Virol. 2005;79:1113–1124. doi: 10.1128/JVI.79.2.1113-1124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Stefanovic B. mTORC1 phosphorylates LARP6 to stimulate type I collagen expression. Sci Rep. 2017;7:41173. doi: 10.1038/srep41173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, related to Figure 1. Intrinsic expression of ISGs in pluripotent and multipotent human stem cells.
(A) Representative images showing increasing permissiveness to virus infection along hepatic differentiation of hESCs. Cells were infected with indicated viruses, fixed at the indicated time points post infection (p.i.) and were visualized directly (green, GFP-expressing viruses) or stained with the indicated antibodies (red: anti-NS3 for DENV and anti-flavivirus E for ZIKV). Nuclei were visualized by DAPI staining (lower panels in each grouping). Scale bars: 100 μm.
(B) Schematic workflow for identifying ISG signature in stem cells. Briefly, for any given stem cell type, we focused on genes with TPM values above a minimum threshold (approximately TPM >1.0, detailed in STAR methods) in all biological replicates, thereby defined as “expressed” genes. The number of expressed genes ranged from 13,000 to 15,000, depending on the cell type and cell stage. Within the highly expressed group (top 20% of genes, ranked by TPMs), we focused on interferon-stimulated genes (ISGs), based on the compiled human and mouse ISG lists (Table S2). ISGs within the highly expressed set were defined as “highly expressed ISGs” for the cells analyzed. Shown are data from hESC line WA09.
(C) Blockade of IFN response by IFNAR2 blocking antibodies in HLCs, but not hESCs. Left: hESCs-derived HLCs were pre-treated with isotype control or IFNAR2 blocking antibodies for 4 hr, followed by stimulation with IFN-β (100U/ml) for 12 hr. The magnitude of representative ISG induction was measured by qRT-PCR. Expression in untreated HLC was utilized for normalization. Mock indicates cells not treated with antibody prior to IFN stimulation. Right: Relative expression of the indicated ISGs in hESCs after being maintained in the presence of 10 μg/ml neutralizing antibody against IFNAR2 for 48 hr by qRT-PCR. Expression of mock cells was utilized for normalization. Shown are the means ± SD from 3 independent experiments.
(D) Representative images at low and high magnification of hESCs and hESC-derived germ layers. Cells were stained with indicated antibodies and analyzed by IFA. Nuclei were stained with DAPI. Scale bars: 100 μm.
(E) Relative expression of cell type-specific markers in hESCs and derived germ layers was measured by qRT-PCR. Expression levels in hESC were utilized for normalization. Shown are the means ± SD from 3 independent experiments. Statistical analyses were calculated between ESCs and tissue stem cells, as indicated by the dotted lines.
Asterisks indicate statistically significant differences (Student’s t test was used throughout this study) (*, p<0.05; **, p<0.01; ***, p<0.001).
Figure S2, related to Figure 2. Distinct ISG expression patterns in different tissue stem cells.
(A) Relative expression of the indicated cell type-specific markers in hESCs and derived tissue stem cells was measured by qRT-PCR. PSC makers: PDX1 and GCG; MSC markers: CD44, CD90, and CD105; NSC markers: SOX2 and PAX6. CD90 and SOX2 are also markers for hESCs. Expression levels in hESC were utilized for normalization. Shown are the means ± SD from 3 independent experiments.
(B) Representative images showing terminal differentiation of hESC-derived tissue stem cells. The left panels represent tissue stem cells and the right panels show terminally differentiated cells. PSC to β-cell differentiation was monitored by insulin expression (INS, red); MSC to myocyte differentiation by smooth muscle actin (SMAα, red) and to adipocyte by the presence of characteristic cytoplasmic lipid droplets (oil-red staining); NSC differentiation to neuron was monitored by expression of tubulin beta 3 class III (TUJ1, red). Nuclei were stained by DAPI. Scale bars: 100 μm.
(C) Relative expression of selected ISGs in hESC and three tissue stem cells was determined by qRT-PCR. Expression levels in hESC were utilized for normalization. Shown are the means ± SD from 3 independent experiments.
(D) Expression of housekeeping genes was measured by qRT-PCR and compared between ESCs and three tissue stem cells. Shown are the means of threshold cycles ± SD from 3 independent experiments.
(E) Bar diagram illustrating the number of highly expressed ISGs unique to individual donor (purple) and common to both donors (gray) in HSCs isolated from bone marrow (BM).
(F) Bar diagram illustrating the number of highly expressed ISGs unique to individual donor (purple) and common to all donors (gray). Top: MSCs were isolated from placenta tissue (PL) of three donors; Bottom: MSCs were isolated from bone marrow (BM) of three donors, as described in (Roson-Burgo et al., 2014)
(G) Expression of selected ISGs (anti-HIV and anti-DENV) in primary tissue stem cells (pHSC and pMSC) and hESC-derived HSCs and MSCs (iHSC and iMSC) was measured by qRT-PCR. Primary HSCs were from fetal livers of three different donors, and MSCs were from adipose tissue of two donors; iMSCs and iHSCs were derived from hESC WA09 line. Expression levels in iMSC were utilized for normalization. Shown are the means ± SD from 2 or 3 biological replicates and 3 independent experiments.
(H) Expression of selected ISGs (anti-CHIKV ISGs) in primary tissue stem cells (pHSC and pMSC) and hESC-derived HSCs and MSCs (iHSC and iMSC) was measured by qRT-PCR. Cells were from the same sources as described in (G). Expression levels in iMSC were utilized for normalization. Shown are the means ± SD from 2 or 3 biological replicates and 3 independent experiments.
(I) Primary HSCs and MSCs transduced with sh-Ctrl or sh-CDKN1A were infected with CHIKV (MOI =1.0). At 16 hr p.i., the cells were harvested for analyses of CHIKV infection and CDKN1A expression. The percentages of CHIKV infection were determined by flow cytometry for GFP (+) cells are shown (upper panel). Data are shown as the means ± SD from 3 independent experiments. Statistically significant differences, relative to infection of sh-Ctrl are indicated. Lower panels show western blot analysis of CDKN1A. ACTB served as a loading control.
Asterisks indicate statistically significant differences (Student’s t test was used) (*, p<0.05; **, p<0.01; ***, p<0.001).
Figure S3, related to Figure 3. ISG expression changes during terminal differentiation of hESCs.
(A) Pancreatic β-cell differentiation from hESCs was monitored by staining at days 0, 5, 14, 20, and 27 during the differentiation process using antibodies that recognized OCT4, SOX17, PDX1, NKX6.1, and INS, as indicated. Representative images at specified stages are shown. Scale bars: 100 μm.
(B) Expression of the indicated stage-specific markers along β-cell differentiation from hESCs was analyzed by qRT-PCR. Shown are the means ± SD from 3 independent experiments.
(C) Expression of the indicated housekeeping genes from cells at the specified stages along β-cell differentiation was analyzed by qRT-PCR. Shown are the means of threshold cycles ± SD from 3 independent experiments.
(D) Top: Cells undergoing differentiation into myofibroblasts were fixed and stained with the indicated antibodies and analyzed by IFA. Nuclei were stained with DAPI and merged images are shown. Scale bars: 100 μm.
Bottom: Expression of the indicated selected stage markers along myofibroblast differentiation was analyzed by qRT-PCR. Shown are the means ± SD from 3 independent experiments.
(E) Expression of the indicated selected ISGs from cells at the specified stages along myofibroblast differentiation was analyzed by qRT-PCR. Shown are the means ± SD from 3 independent experiments.
(F) Expression of the indicated housekeeping genes from cells at the specified stages along myofibroblast differentiation was analyzed by qRT-PCR. Shown are the means of threshold cycles ± SD from 3 independent experiments.
(G) Neuronal differentiation from hESCs was monitored by staining at days 0, 18, 40, and 60 during the differentiation process using antibodies that recognized SSEA-4, SOX2, PAX6, NES, TUJ1 and MAP2, as indicated. Representative images at specified stages are shown. Scale bars: 100 μm.
(H) Expression of the indicated selected ISGs from cells at the specified stages along neuronal differentiation was analyzed by qRT-PCR. Shown are the means ± SD from 3 independent experiments.
(I) Expression of the indicated housekeeping genes from cells at the specified stages along neuronal differentiation was analyzed by qRT-PCR. Shown are the means of threshold cycles ± SD from 3 independent experiments.
(J, K, L) Bar diagram illustrating the number of highly expressed ISGs unique to each stage (light blue) and shared by all lines (dark blue) along ex vivo differentiation of hESC into three lineages.
Asterisks indicate statistically significant differences (Student’s t test was used) (*, p<0.05; **, p<0.01; ***, p<0.001).
Figure S4, related to Figure 3. ISG expression changes during terminal differentiation of hESCs.
(A) HLC differentiation from hESCs was monitored during the differentiation process by staining at days 0, 5, 10, 15, 20, and 25 using antibodies that recognized Oct-4, AFP and ALB followed by Alexa Fluor 594 conjugated secondary antibody (red), or SOX17, FoxA2, and HNF4α followed by Alexa Fluor 488 conjugated secondary antibody (green). Representative images at specified stages are shown. Scale bars: 100 μm.
(B) Relative transcript abundance of pluripotent and differentiation markers at different stages along hepatic differentiation of hESCs as measured by RNA-Seq. Data represent average log2 TPM values for the indicated genes from two independent experiments. Five stages are shown: ESCs, END, hepatic stem cells (HepSC), immature hepatocyte (ImHep), Hepatocyte-like cells (HLC).
(C) Heatmap of RNA-Seq expression pattern (z-score TPM) of ISGs highly expressed in hESCs along ex vivo HLC differentiation of hESCs.
(D-E) Expression analyses of selected ISGs and markers during differentiation of hESCs along the hepatic lineage by western blot using the indicated antibodies (D) and qRT-PCR for the indicated transcripts (E). Shown in (E) are the means ± SD from 3 independent experiments.
(F) hESC-derived macrophage-like cells (MLCs) and primary macrophage isolated from fetal liver were collected by cytospin and stained with Giemsa. Representative images are shown. Scale bars: 100 μm.
(G) Expression of the indicated selected stage markers in cells at the specified stages during MLC differentiation in determined by qRT-PCR. Expression levels in hESC were utilized for normalization. Shown are the means ± SD from 3 independent experiments. The morphological features of the indicated stages are shown schematically at the top.
(H) MLCs were treated for 6 hr with lipopolysaccharides (LPS, 5 μg/ml) or PBS and RNA was isolated for qRT-PCR analysis of selected cytokines. The fold change in the LPS-treated cells relative to the PBS-treated controls for each gene is plotted. Shown are the means ± SD from 3 independent experiments.
(I) Expression of the indicated ISGs from cells at specified stages during differentiation of hESC into MLCs was measured by qRT-PCR. Expression levels in hESC were utilized for normalization. Shown are the means ± SD from 3 independent experiments.
Asterisks indicate statistically significant differences (Student’s t test was used) (*, p<0.05; **, p<0.01; ***, p<0.001).
Figure S5, related to Figure 1. Intrinsic ISG expression in pluripotent and multipotent stem cells from other species.
(A) Heatmap of expression patterns of highly expressed ISGs in four chimpanzee iPSC lines, log2 TPM. Data are from (Marchetto, et al. Nature. 2013).
(B) Bar diagram illustrating the number of highly expressed ISGs unique to each mESC line (purple) and shared by all three lines (shared/gray). Data are from (Wamstad et al., Cell. 2012; Klattenhoff et al., Cell. 2013; Li et al., PLoS One. 2014)
(C) Bar diagram illustrating the number of highly expressed pluripotent cell ISGs unique to each species (purple) or shared by all three species (shared/gray).
(D) Heatmap of gene expression (z-score TPM) for highly expressed ISGs along cardiomyocyte differentiation. Shown are highly expressed ISGs in mouse ESCs. Data are from (Wamstad et al., Cell. 2012)
(E) Relative expression of the indicated ISGs at different stages along mESCs differentiation towards cardiomyocyte. Average log2 TPM values of two independent experiments are shown.
(F) The epigenetic landscape showing dynamic levels of H3K4me3, H3K27ac and H3K2me3 for unexpressed or lowly expressed and highly expressed ISGs along cardiomyocyte differentiation of mESCs. Examples of lowly expressed ISGs (Lowly exp ISGs) include MX1 and OAS1, and examples of highly expressed ISGs (Highly ISGs) are IFITM3 and IGFBP2. ChIP-Seq (H3K4me3, H3K27ac and H3K2me3, y axis represents reads over million unique mapped reads) are shown. The scale for each modification is constant throughout the time course.
(G) Statistical analyses showing association between levels of H3K4me3 or H3K27ac and expression (RPKM) of all ISGs along cardiomyocyte differentiation of mESCs. Each dot represents value for an ISG. Shown are the means ± SD from all the ISGs analyzed in each group. Asterisks indicate statistically significant difference (***, p<0.001). For F-G, data are from published data set (Wamstad et al. 2012) and analyzed as shown in Table S4. Asterisks indicate statistically significant differences (Student’s t test was used) (***, p<0.001).
(H) The epigenetic landscape showing different histone modification markers, RNA-Seq, and DNase-Seq for unexpressed or lowly expressed and highly expressed ISGs in hESCs. Examples of lowly expressed ISGs (Lowly exp ISGs) include APOBEC3A, IFIT1 and RSAD2, and examples of highly expressed ISGs (highly exp ISGs) are IFITM1-3, BST2 and MOV10. ChIP-Seq (H3K4me1, H3K4me2, H3K4me3, H3K9me3, H3K9ac, H3K27me3, H3K27ac, y axis represents reads over million unique mapped reads) are shown. The scale for each modification is among different genes. Data are from the Encyclopedia of DNA Elements (ENCODE) Consortium.
See also Table S4 for epigenetic data.
Figure S6, related to Figure 5. Antiviral activity of ISGs in human stem cells.
(A) Representative images showing expression of pluripotent markers in three hESC clones. Cells were fixed and stained with the indicated antibodies. Scale bars: 100 μm.
(B) WT, KO23, and KO123 clones were differentiated ex vivo into cells representing the three germ layers and were fixed and stained with antibodies for the indicated markers. Nuclei were stained with DAPI. Representative merged images are shown. Scale bars: 100 μm.
(C) Equal numbers of WT, KO23, and KO123 clones were plated and at the indicated time points, viable cell numbers were determined using the CellTiter-Glo kit. Shown are the means of relative luciferase units (RLU) ± SD from 3 independent experiments.
(D) WT, KO23, and KO123 clones were infected with GFP-expressing human parainfluenza virus type-3 (hPIV3, MOI=1.0) and after 24 hr the cells were fixed and nuclei stained with DAPI. Representative images showing infection (green, top) and nuclei (blue, bottom) are shown. Scale bars: 100 μm.
(E) WA09 hESCs were transduced with control (CTRL) shRNA or shRNA targeting IFITM3 (sh-1 and sh-2) and puromycin-selected stable clones were analyzed by western blot for the indicated IFITM family members. GAPDH served as a loading control.
(F) Stable clones derived from (E) were fixed and stained with antibodies against the indicated pluripotent markers. Representative merged images are shown. Scale bars: 100 μm.
(G) Stable clones derived from (E) were infected with the indicated viruses (MOI=1.0) and after 48 hr fixed and analyzed by flow cytometry. DENV infected cells were stained with anti-NS3 antibody prior to analysis, while NDV and YFV were GFP tagged. Shown are the means ± SD from 3 independent experiments. Asterisks indicate statistically significant differences (***, p<0.001).
(H) Equal numbers of WT, KO23, and KO123 NSCs were plated and at the indicated time points, cell viable numbers were determined using the CellTiter-Glo kit. Shown are the means of RLU ± SD from 3 independent experiments.
(I) WT and KO123 NSC were differentiated into neurons or astrocytes; after fixation they were stained with antibodies against TUJ1 or GFAP, respectively. Nuclei were stained with DAPI (blue). Representative merged images are shown. Scale bars: 100 μm.
(J) WT and KO123 NSC were infected with the indicated GFP-expressing neurotropic viruses (MOI=1.0). After infection, cells were fixed and stained as follows: infections with VSV and VEEV were fixed at 12 hr p.i. and infection with MeV was fixed at 48 hr p.i. Representative images of infection (green, top) and nuclei (blue, bottom) are shown. Scale bars: 100 μm.
(K–L) hESC-derived NSCs were cultured under conditions where NSCs gave rise to more NSCs as well as differentiated neurons; cultures were then infected with a GFP-expressing YFV (stain 17D). Cells were fixed at 48 hr p.i. and stained with NSC marker SOX2 (red) and neuronal marker TUJ1 (blue). Representative images are shown in (K). Scale bars: 100 μm. Shown in (L) are the mean percentages of infected cells ± SD in the NSC (SOX2+) and neuronal (TUJ1+) populations from 3 independent experiments are plotted.
(M) Primary MSCs were transduced with the indicated shRNAs [control (CTRL), IFITM or p21/CDKN1A)] and 5 days later fixed and stained with the indicated antibodies against MSC markers (CD90/THY1, COL1A1). Nuclei were stained with DAPI (blue). Representative fluorescence microscopy images are shown. Scale bars: 100 μm.
(N) Primary WT MSCs or MSCs transduced with the indicated shRNAs were differentiated into adipocyte cells and analyzed by qRT-PCR for induction of adipocyte markers. Data were normalized to RPS11 expression. Fold changes from expression levels in parental MSCs are shown as the means ± SD from 3 independent experiments.
Asterisks indicate statistically significant differences (Student’s t test was used) (**, p<0.01; ***, p<0.001).
Figure S7, related to Figure 7. ISGs protect stem cells from viral infection during development.
(A–B) Top: Schematic representation of conditions for ex vivo differentiation of HSCs into erythrocyte and macrophage. Bottom: HSCs transduced to express control (CTRL) or IFITM shRNA (KD) were mock infected or exposed to IAV or DENV for 3 hr and then subjected to ex vivo differentiation into erythrocytes (A) or macrophages (B). At the indicated time points the number of cells were counted and normalized to the number present at day 0. The means ± SD of 3 biological replicates are shown per condition. Different colored symbols correspond to 3 different donors.
(C–E) Erythrocyte differentiation conducted in (A) was monitored at the indicated time points by flow cytometry of the indicated surface markers (CD36, CD34, ITGA4, and Band3). (C) Representative FACS plots from 3 biological replicates. (D–E) The percentages of the indicated cell types. CFU-E: colony forming unit-erythroid, defined as CD36+CD34−; BFU-E: burst-forming units-erythroid, defined as CD36−CD34+.
(F) Representative flow cytometry plots showing the composition of human leukocytes in the bone marrow from the transplanted mice described in Figure 7E–H. CD19, surface marker for human B-cells; CD33, surface marker for human cells of myeloid lineage. Shown are representative plots from mice transplanted with mock infected control HSCs. No detectable human hematopoietic cells were seen in mice transplanted with IAV pre-exposed KD HSCs.
(G) Representative flow cytometry plots showing the gating strategies for human and mouse HSCs isolated from mouse bone marrow. Human HSCs, CD34+CD38−; mouse HSCs: c-KIT+Sca-1+.
(H) Bone marrow harvested at 16 weeks form the mice described in Figure 8E–H was analyzed by flow cytometry for the percentages of murine HSCs. Each symbol represents the value obtained in an individual mouse. Bars represent the means ± SD from n=5 mice per group from two donors.
Asterisks indicate statistically significant differences (Student’s t test was used) (*, p<0.05; **, p<0.01; ***, p<0.001).
Table-S1, related to Figure 1. RT-qPCR primers used.
Primer sequences for all primers used for real-time RT-PCR and primers used for generating shRNAs against ISGs.
Table-S2, related to Figure 1. Human and mouse ISG lists.
Detailed information for human and mouse interferon-stimulated genes (ISGs). Entrez Gene IDs and detailed descriptions included.
Table-S3, related to Figure 2. Gene expression data for human HSCs and MSCs.
Transcriptome analysis of human hematopoietic stem cells isolated from four different sources and human mesenchymal stem cells isolated from two different sources.
Table-S4, related to Figure S5. Gene expression data for non-human primate stem cells.
Transcriptional analysis of three mouse embryonic stem cell lines and four induced pluripotent stem cell line from chimpanzee; comparison between mouse, chimpanzee, and human pluripotent stem cells; and processed epigenetic data used for generating Figure S5 (S5A–B).