

Abstract
Memory impairment following West Nile virus neuroinvasive disease (WNND) is associated with loss of hippocampal synapses with lack of recovery. Adult neurogenesis and synaptogenesis are fundamental features of hippocampal repair, suggesting viruses impact these processes. Here, using an established model of WNND-induced cognitive dysfunction, transcriptional profiling revealed alterations in gene expression that limit adult neurogenesis, including interleukin (IL)-1. WNND-recovered animals exhibit decreased neuroblasts and increased astrogenesis, without recovery of hippocampal neurogenesis at thirty days. Analysis of cytokine production in ex vivo isolated microglia and astrocytes revealed the latter to be the predominant source of IL-1. IL-1R1-deficient, WNND-recovered mice exhibit normal neurogenesis, recovery of presynaptic termini, and resistance to spatial learning defects, the latter of which likewise occurred after treatment with IL-1R1 antagonist. Thus, preferential generation of proinflammatory astrocytes impairs neuronal progenitor cell homeostasis via expression of IL-1, which may underlie long-term cognitive consequences of WNND, but provides a therapeutic target.
Introduction
Members of the Flavivirus genus, which include West Nile (WNV), Japanese encephalitis (JEV), and Zika (ZIKV) viruses, are the most important arthropod-borne viruses causing encephalitis in humans1. Acutely, patients suffering from WNV neuroinvasive disease (WNND) can experience confusion, fatigue, loss of motor control, memory loss, coma, and a mortality rate of 5–10%1. WNV is a (+)-sense single-stranded RNA virus that targets fully differentiated neurons, but may be cleared by immune-mediated processes, even after infection of the central nervous system (CNS)2. However, approximately half of survivors experience debilitating, long-term cognitive sequelae, including defects in verbal and visuospatial learning, for months to years beyond the acute infectious event3,4. Animal studies have identified multiple cytokines that play critical roles in cell-mediated antiviral immunity, including tumor necrosis factor (TNF)5, type I, II and III interferons (IFNs)6–9, and interleukin (IL)-110,11, which improve survival. Prior studies have determined that human and murine neurons are the target of WNV in vivo12–14. Importantly, studies in which critical cytokines have been deleted via genetic approaches have not led to expanded tropism of WNV to nonneuronal cells within the CNS10,15. While neuronal death is associated with high mortality in human and murine cases of WNV encephalitis16, survivors may exhibit limited neuronal loss14,17, suggesting inflammatory processes triggered acutely contribute to long-term memory dysfunction. That many patients recovering from WNND experience memory impairments for months to years beyond viral clearance indeed suggests a chronic condition with either sustained damage or limited repair.
In a murine model of recovery from WNND, in which intracranial inoculation of a mutant WNV (WNV-NS5-E218A) leads to high survival rates with visuospatial learning defects, hippocampi exhibit up-regulation of genes involved in microglial-mediated synaptic remodeling, including drivers of phagocytosis and the classical complement pathway, and decreased expression of synaptic scaffolding proteins and glutamate receptors14. Complement-mediated synapse elimination has been reported to occur in numerous neuroinflammatory diseases, including multiple sclerosis18, Alzheimer’s disease19, and schizophrenia20, suggesting this may be a general mechanism underlying inflammation-associated disruption of neural circuitry. The hippocampus, essential for spatial and contextual memory formation, receives input from the entorhinal cortex, which relays through the dentate gyrus, CA3, and CA121. WNV-NS5-E218A-recovered mice with poor spatial learning show persistence of phagocytic microglia engulfing presynaptic terminals within the hippocampal CA3 acutely, and during recovery14. While this provides molecular explanations for poor spatial learning in WNV-recovered animals, it does not explain why other hippocampal correlates of learning, such as adult neurogenesis, are not able to restore spatial learning.
Adult neurogenesis occurs within the hippocampal dentate gyrus (DG) and the subventricular zone (SVZ)22. Within the DG, adult neural stem cells (NSCs) give rise to astrocytes and intermediate neuronal progenitors, the latter of which proliferate and differentiate into neuroblasts that mature into granule cell neurons and integrate into the hippocampal circuit over the course of a few weeks23. This process is regulated by intrinsic and extrinsic factors, including local signaling molecules, exercise, aging and inflammation24. A variety of endogenous factors play critical roles in the generation and integration of newly generated neurons in the adult hippocampus. These include morphogens, such as neurogenic locus notch homolog (Notch) proteins, sonic hedgehog (Shh), wingless integrated signals (Wnts), and bone morphogenic proteins (BMPs), and neurotrophic factors, such as brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF), insulin-like growth factor-1 (IGF-1), and vascular endothelium growth factor (VEGF)25. Recently, proinflammatory pathways, including those triggered by systemic accumulations of TNF, interleukin (IL)-1β, and IL-6, and microglial activation have been implicated in the regulation of neural correlates of memory including adult neurogenesis, synaptic plasticity and modulation of long-term potentiation14,26–29.
IL-1, in particular, has gained attention for its impact on cognitive function in the context of neuroinflammation. IL-1 signaling is mediated by a family of proteins comprised of IL-1α, IL-1β, and IL-1 receptor antagonist (IL-1ra) primarily through type I IL-1 receptor (IL-1R1). IL-1β is generated via proteolytic cleavage of pro-IL-1β by Caspase 1 during inflammasome activation30. IL-1 is highly expressed in vivo by infiltrating myeloid cells during WNV encephalitis, where it critically regulates antiviral effector T cell responses10,11. While IL-1 is a key player in orchestrating CNS immune responses, including onset of fever27, it also plays a role in spatial learning and memory-related behavior28. Indeed, injection of IL-1β into the brain impairs spatial learning, contextual fear memory, and adult neurogenesis29–31. Although several studies have investigated impacts of IL-1 on hippocampal-based learning and behavior in neurologic diseases31–35, none have done so in a setting of IL-1 induction during CNS viral infection.
Here, using an established model of post-infectious cognitive dysfunction from WNND in which animals display defects in spatial learning, we investigated the regulation of neurogenesis during repair and recovery. We identified a novel feed forward mechanism in which IL-1 contributes to spatial learning defects via derailment of hippocampal neurogenesis to generate proinflammatory astrocytes, which exhibit a previously unidentified role in IL-1-mediated cognitive dysfunction, and that this pathway may be successfully targeted for the prevention of learning defects during recovery from viral encephalitis.
Results
WNND-recovered mice exhibit genetic signatures of derailed neurogenesis
Our previous microarray study of hippocampal gene expression in WNV-NS5-E218A-recovered mice identified several significantly altered pathways in mice with impaired spatial memory including axon guidance, Wnt signaling, and p53 signaling14, which point to potential effects on adult neurogenesis. To further examine these pathways, we grouped the genes altered between Mock and WNV-NS5-E218A-recovered animals into those that promote or inhibit neurogenesis, here shown as gene expression heatmaps (Fig. 1a, b). Expression of genes associated with inflammation and/or prohibition of neurogenesis (Casp1, Il1a, Tnf, Tnfrsf1a) was higher in WNV-NS5-E218A-recovered (Fig. 1a) compared with mock-infected mice, which exhibited higher expression of genes that promote proliferation and differentiation of neuroblasts (Epha5, Wnt2, Nrg3) and axon guidance (Robo2, Sema3) (Fig. 1b). Markers for recently identified subgroups of reactive astrocytes36 deemed A2 and A1, the latter of which are proposed to be induced by activated microglia and lose the ability to promote neuronal survival, outgrowth, were also significantly elevated (Fig. 1c, d). Alterations in genes that impact neurogenesis after infection with WNV-NS5-E218A and markers of reactive astrocytes, including additional panreactive astrocyte markers36 were validated in an independent set of hippocampal samples by qPCR (Fig. 1e–h). These data suggest that WNND might limit adult neurogenesis in favor of astrogenesis.
Fig. 1. Gene transcripts that impact neurogenesis and markers of proinflammatory astrocytes are altered in WNV-recovered mice.
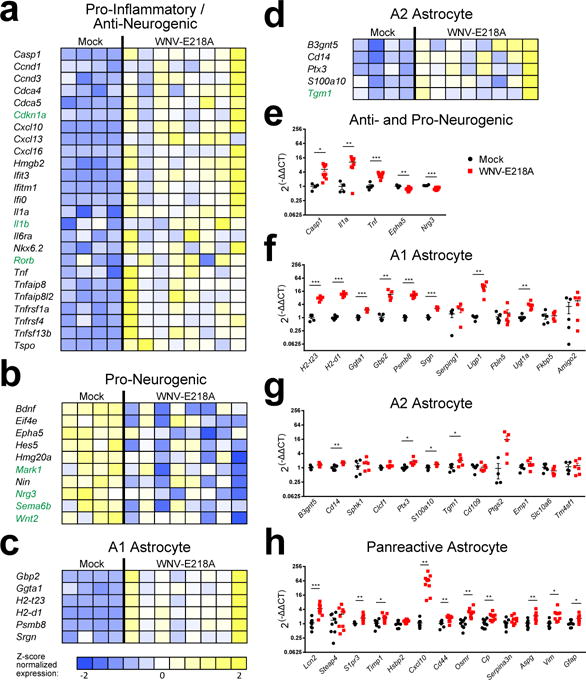
(a–d) Heat maps show relative expression by z-score of altered genes generated by microarray analysis of hippocampal RNA collected at 25 days post infection (d.p.i.) from Mock, and WNV-NS5-E218A infected, recovering mice. All genes listed in black font show significant fold change (P < 0.05 by two-tailed Student’s t-test) while those in green are trending toward significance (0.05 < P < 0.1 by two-tailed Student’s t-Test). (e–h) Validation of microarray hits by QPCR in an independent sample of hippocampal RNA collected at 25 d.p.i. from mock and WNV-NS5-E218A infected, recovering mice. *, P < 0.05; ** P < 0.01, *** P < 0.001, by two-tailed Student’s t-test. Each symbol represents an individual mouse; data are mean ± s.e.m. Data are pooled from 2 independent experiments with 4–10 mice per condition in each. Panels of gene-expression markers include genes that are pro and anti-neurogenic (e), A1 & A2 astrocyte markers (f–g) and panreactive astrocyte markers (h).
WNND induces acute loss of adult neurogenesis in the hippocampus
Given that genetic signatures in the hippocampi of WNV-recovered mice are consistent with pathways that negatively impact adult neurogenesis, we evaluated the generation of new neurons during WNV encephalitis. We administered BrdU during the peak of WNV encephalitis for a period of 4 days (days 3–7), allowed mice to recover for 45 days and then evaluated numbers of newly generated neurons (Fig. 2a–d). WNV-recovered mice exhibited fewer BrdU-labeled neurons within the DG granule cell layer than mock-infected controls (Fig. 2a, c, d). To determine whether reduction in newly generated neurons in WNV-recovered mice is due to alteration in the rate of neuronal progenitor cell proliferation, mock-infected versus WNV-NY99-infected or WNV-NS5-E218A-infected mice were administered BrdU during the peak of encephalitis, followed by either immunohistochemical (Fig. 3a, b) or flow cytometric (Supplementary Fig. 1) evaluation of neuronal progenitors, which express doublecortin (DCX+), in the SVZ or hippocampi, as described previously37. Infections with wild-type WNV (NY-99) via either the peripheral footpad or intracranial route, or attenuated WNV-NS5-E218A all led to fewer BrdU-labeled neuroblasts at 6–8 days post-infection (Fig. 3c). This reduction in generation of neuroblasts was assessed over the course of recovery from WNND, at days 6, 15, and 30 post-infection (d.p.i.) as compared to age-matched mock-infected controls. This analysis revealed that significant deficits persist until day 30 in the hippocampus, with a trend towards recovery observed in the SVZ (Fig. 3d). Taken together, these data indicate that hippocampal neuronal repair is defective after WNND.
Fig 2. Fewer new neurons are born within the dentate gyrus during WNV-NS5-E218A recovery.
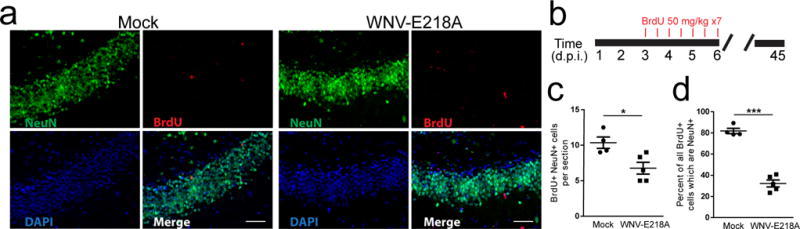
(a) Representative image of the dentate gyrus of Mock and WNV-NS5-E218A infected animals at 45 d.p.i., following in vivo BrdU labeling during acute infection. BrdU (red), NeuN (green), and DAPI (blue). (b) Experimental design. Mice were given BrdU by intraperitoneal injection every 12 hours for 3.5 days (7 injections) beginning at 4 d.p.i., then animals were sacrificed at 45 d.p.i. for immunohistochemical analysis. (c, d) Quantification of BrdU+NeuN+ cells per section of dentate gyrus depicted in panel a (c), and normalized to the number of BrdU+ cells. *, P < 0.05 by two-tailed, Student’s t-test. ***, P < 0.001. Each symbol represents an individual mouse; small horizontal lines indicate the mean (± s.e.m). Data are representative of one experiment.
Fig 3. Deficits in adult neurogenesis during WNV infection.
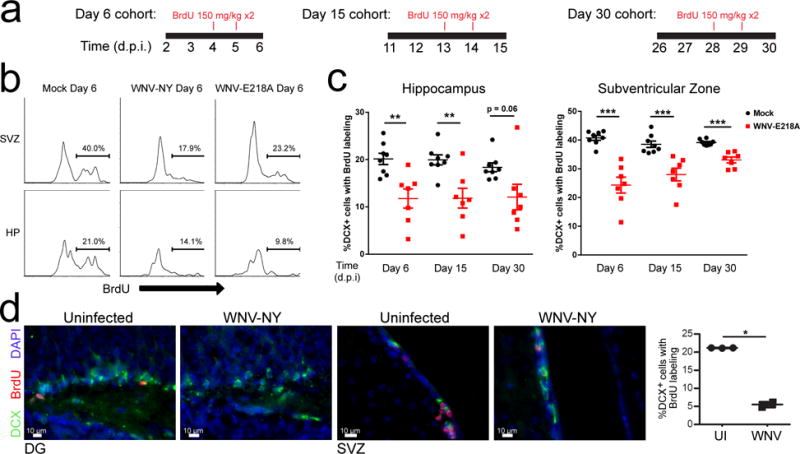
(a) Experimental design. Mock, WNV-NY99 or WNV-NS5-E218A mice were given an i.p. BrdU injection (100 mg/kg) at 24 and 48 hours prior to harvest at 6, 15, or 30 d.p.i. (b) Cells were isolated from subventricular zone (SVZ) or hippocampus (HP) and proliferation rates of neuroblasts (% BrdU incorporation) was measured by flow cytometry. Representative plots showing BrdU+ cells after gating on doublecortin+ (DCX) cells (see Supplementary Fig. 1) in mock, WNV-NY-99 and WNV-NS5-E218A intracranially infected animals. (c) Quantification of proliferating neuroblasts (%DCX+ cells labeled with BrdU) in the hippocampus and subventricular zone at 6, 15, and 30 d.p.i. following in vivo BrdU labeling as shown in a. (d) Representative images and quantification of immunostaining for proliferating neuroblasts in the SVZ and HP following footpad infection with WNV-NY99. *, P < 0.05, **, P < 0.005, ***, P < 0.001 by two-tailed student’s t-test. Quantification in c and d was normalized to age-matched mock-infected controls to compensate for age-related alterations in neurogenesis. Each symbol represents an individual mouse; small horizontal lines indicate the mean (± s.e.m.). Data are pooled from 2 independent experiments with 3–5 mice per condition per time point (c) or representative of one experiment with 5 mice (d).
WNV does not target NSCs or neuronal progenitors
One possible explanation of fewer new neurons after the recovery period of WNV encephalitis is that neuroblasts are dying before reaching maturity. In agreement with previous studies, we confirmed that neural stem cells and intermediate neuronal progenitors were not permissive to WNV infection36, and observed that less than 1% of DCX+ neuroblasts were infected in vivo in both the SVZ and DG (Supplementary Fig. 2a). Furthermore rates of neuroblast apoptosis during acute WNND were equivalent to mock-infected controls (data not shown). The few infected neuroblasts that were observed within the DG were located within the granule cell layer, suggesting that these may be late-stage DCX+ cells during their transition into immature neurons. Late-stage neuroblasts exhibit many of the same properties as neurons including, receptors, filament proteins, and cellular processes (e.g. axon and dendrite formation), thus potentially explaining the ability of WNV to infect them. Alterations in the proliferation rates of neural progenitors could result in changes to the overall pool of stem cells over time. Using mice expressing GFP under the Nestin promoter, we counted the numbers of Nestin and GFAP-double positive neural stem cells remaining within the hippocampal DG at 45 days post-infection, but found no differences in the numbers of these cells present in mock and WNV-NS5-E218A recovered mice (Supplementary Fig. 2b). Thus, WNV does not target or alter the numbers of NSCs, nor does it infection neuroblasts.
Astrocytes are the primary source of IL-1β in the recovering CNS
To further investigate the potential for an alteration in cell fate of early stage progenitors, we determined whether fewer neuronal progenitors were produced in favor of more glial progenitors within the hippocampus of WNV-infected mice. To test this, mock- versus WNV-NY- or WNV-NS5-E218A-infected mice were administered BrdU during the peak of encephalitis followed by flow cytometric evaluation of hippocampal neuronal progenitors and astrocytes 48 hours later (Fig. 4a, b). Infections with either wild type WNV (NY-99) or WNV-NS5-E218A led to more BrdU-labeled GFAP-expressing astrocytes in the hippocampus at 7 days post infection than mock-infected controls (Fig. 4b). Although GFAP expression varies among astrocyte subpopulations, recent work has demonstrated that GFAP is the most highly expressed transcript and protein in astrocytes isolated from the hippocampus38. Next, we determined whether an increase in astrocyte genesis during acute infection significantly contributes to alterations in the CNS immune profile after viral clearance. Analysis of ex vivo-isolated astrocytes at 25 d.p.i. demonstrated an increase in markers of A1, but not A2, reactive astrocyte markers, (Fig. 4c, c′). Thus, the slight increases in neuroprotective markers previously detected in whole hippocampal samples at the same time point (Fig. 1c–e) are likely contributed by other cell types. Analysis of panreactive markers36 also detected increased expression of GFAP (Fig. 4c′). Identification of cellular sources of anti-neurogenic and proinflammatory cytokines in the CNS via transcriptional analysis of ex vivo-isolated astrocytes and microglia at 25 d.p.i. revealed astrocytes as the primary source of IL-1β, Caspase-1, and TNF (Fig. 4d). The purity of those cellular sources was confirmed by transcriptional analysis of cell type-specific markers, as previously reported39. Isolated astrocytes demonstrated significant enrichment of the astrocyte Gfap gene expression while isolated microglia show significant enrichment of microglial Cx3cr1 and Trem2 expression. Both ASCA-2 positive astrocytes and CD11b positive microglia had negligible expression of the neuronal gene Rbfox3 (Supplementary Fig. 3). Kinetic analysis of Il1b expression within hippocampi of WNV-NS5-E218A-infected mice showed persistent elevation of this anti-neurogenic cytokine at 25 d.p.i. (Fig. 4e). Immunohistochemical analysis of mock- and WNV-NS5-E218A-infected tissue at 25 d.p.i. (Fig. 4f) confirmed increases in numbers of activated, GFAP+ astrocytes (Fig. 4g) that are the primary cellular source of IL-1β protein expression in the recovering hippocampus (Fig. 4h, i). Taken altogether, these data indicate that WNND promotes the generation of A1 reactive astrocytes that express anti-neurogenic cytokines.
Fig 4. Greater numbers of astrocytes are born within the hippocampus during acute WNV encephalitis which adopt a proinflammatory phenotype and express IL-1β.
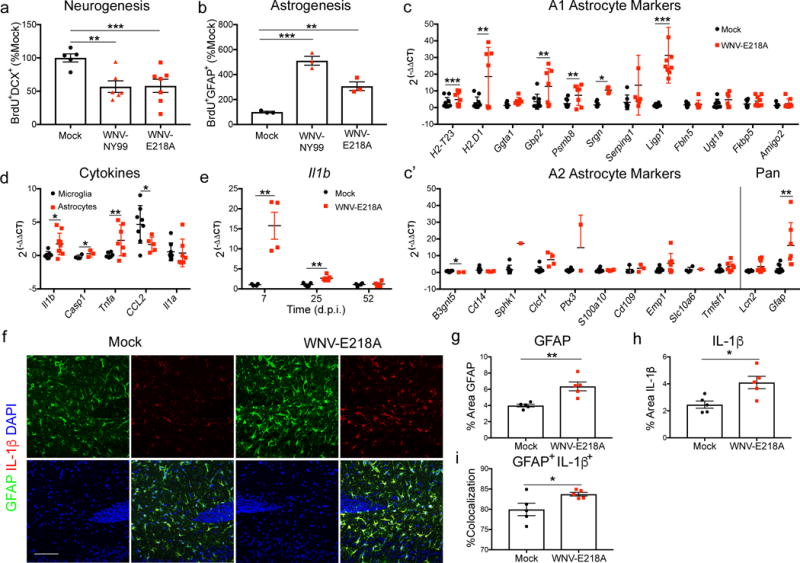
(a,b) Mock, WNV-NY99 and WNV-NS5-E218A infected mice were given an i.p. BrdU injection at 24 and 48 hours prior to harvest at 6 d.p.i. Cells were isolated from dissected hippocampi and stained for flow cytometry. Quantification of neurogenesis (BrdU+DCX+) and astrogenesis (BrdU+GFAP+) was normalized to mock. Gene expression analysis of (c) A1, and (c′) A2 and panreactive astrocyte markers in ex vivo isolated astrocytes from mock and WNV-NS5-E218A whole brains by qPCR. (d) Gene expression analysis of cytokines in ex vivo isolated microglia and astrocytes from WNV-NS5-E218A whole brains by qPCR, normalized to mock-infected samples of the respective cell types. (e) Gene expression of Il1b by qPCR of RNA isolated from hippocampal tissue collected at 7, 25, and 52 d.p.i. from mock and WNV-NS5-E218A infected animals. (f–i) Immunostaining for IL-1β in hippocampus of mock and WNV-NS5-E218A infected animals collected at 25 d.p.i. Representative images are shown in f, percent area of GFAP in g, percent area of IL-1β in h, and percent area of Il-1b +GFAP+ normalized to total IL-1β+ area in i. *, P < 0.05, **, P < 0.005, ***, P < 0.001 by two-tailed student’s t-test. Each symbol represents an individual mouse; small horizontal lines or bar height represent the mean (± s.e.m.). Data are representative of one experiment (a,b,e–i), or pooled from three independent experiments with at least 3 mice per group (c, c′, d).
Il1r1−/− mice resist alterations in cell fate of early progenitors
To determine whether WNV-mediated reduction in neuroblast proliferation requires IL-1R1 signaling, mock- and WNV-NS5-E218A-infected Il1r1−/− mice were administered BrdU during the peak of encephalitis, followed by flow cytometric evaluation of hippocampal neuronal progenitors. In contrast to the reduction in neuroblast proliferation observed in neurogenic zones of wild type animals, Il1r1−/− mice exhibited normal neurogenesis in both the hippocampus (Fig. 5a) and SVZ (Fig. 5b). In addition, Il1r1−/− mice did not undergo the substantial increase in proliferating astrocytes observed in wild-type animals (Fig. 5c). Immunohistochemical identification of Ki67+ proliferating neural progenitor cells in conjunction with Mash1, which identifies early neural progenitor cells with neurogenic potential40, within the DG revealed acute WNV-mediated decreases in wild-type animals, but not in animals deficient in IL-1R1 or Caspase 1 (Casp1−/−), which cleaves pro-IL-1β to produce mature IL-1β (Fig. 5d). Acute viral loads did not differ between wild-type and Il1r1−/− animals at 6 d.p.i. in various brain regions (Supplementary Fig. 4a–c), and there was no difference in persistent viral RNA at 25 d.p.i. in the hippocampus (Supplementary Fig. 4d), confirming that virologic control of WNV-NS5-E218A was intact in Il1r1−/− animals. In addition, flow cytometric analysis of cells isolated from the hippocampus demonstrated similar numbers of various subpopulations of resident and infiltrating CD45+ cells in WNV-NS5-E218A infected wildtype and Il1r1−/− animals (Supplementary Fig. 4e–l). These data support the notion that IL-1R1 signaling underlies derailment of neurogenesis during WNND.
Fig 5. IL-1R1−/− mice resist WNV-mediated alterations in neuroblast proliferation and recover synapses earlier than wildtype controls.
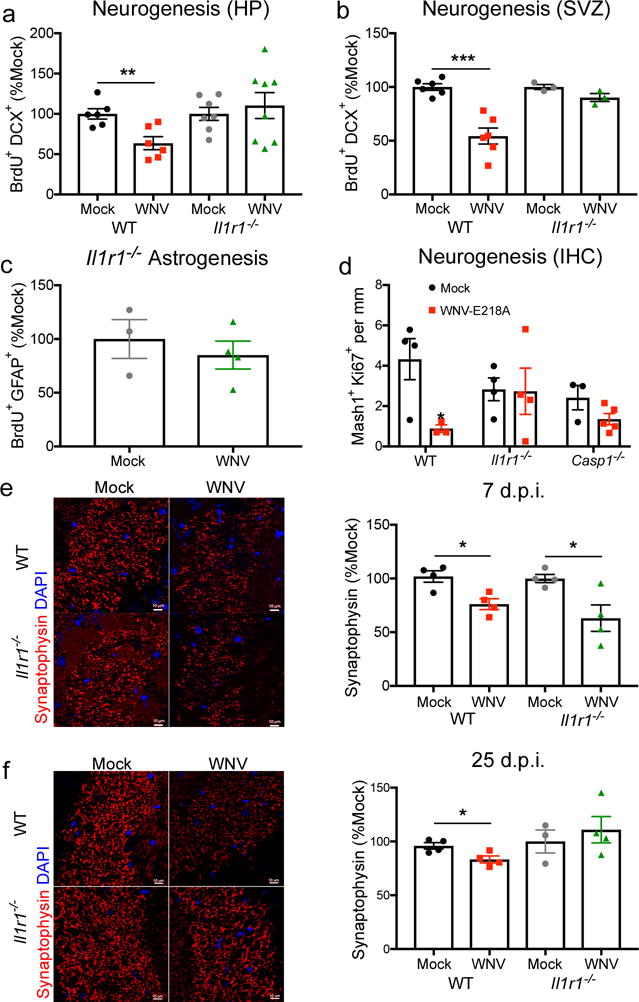
(a,b) Mock and WNV-NS5-E218A infected wild-type (WT) and Il1r1−/− animals were assessed for alterations in neurogenesis at 6 d.p.i. following in vivo BrdU labeling at 24 and 48 hours prior to harvest. Quantification of BrdU+DCX+ cells by flow cytometry are shown for the hippocampus (HP) in a and the subventricular zone (SVZ) in b, normalized to age-matched mock infected controls. (c) Mock and WNV-NS5-E218A infected Il1r1−/− animals were assessed for alterations in astrogenesis at 6 d.p.i. following in vivo BrdU labeling at 24 and 48 hours prior to harvest. Quantification of BrdU+GFAP+ cells by flow cytometry are shown for the hippocampus, normalized to age-matched mock infected controls. (d) Hippocampal neurogenesis in Mock and WNV-NS5-E218A infected WT, Il1r1−/−, and Caspase-1−/− (Casp1−/−) was analyzed by immunohistochemistry (IHC) at 6 d.p.i. Quantification of the number of Mash1+Ki67+ cells per mm of dentate gyrus analyzed. (e–f) Immunostaining of synapses in mock and WNV-NS5-E218A infected WT at Il1r1−/− animals at 7 d.p.i. in e and 25 d.p.i. in f. Quantification shows percent area of synaptophysin in the CA3 of the hippocampus. Synaptophysin (red) and DAPI (blue). *, P < 0.05, **, P < 0.005, ***, P < 0.001 by two-tailed student’s t-test. Each symbol represents an individual mouse; height of bar indicates the mean (± s.e.m.). Data are pooled from 2 independent experiments (a,b) or representative of one experiment (e–f) with 4–5 mice/group.
Il1r1−/− mice exhibit synapse recovery and normal spatial learning
Given that Il1r1−/− mice are protected from derailed neurogenesis following WNV infection, we hypothesized that IL-1R1-deficient animals would exhibit improved synapse recovery following infection with WNV-NS5-E218A. Both WNV-NS5-E218A-infected wild-type and Il1r1−/− mice exhibit acute loss of presynaptic terminals at 7 d.p.i. (Fig. 5e), and wild-type animals continued to show decreased numbers of presynaptic terminals at 25 d.p.i. In contrast, Il1r1−/− animals displayed recovery of synapses at this time point (Fig. 5f). Given that adult neurogenesis and synaptic plasticity are critical for spatial learning, we allowed WNV-NS5-E218A-infected, Il1r1−/− mice to recover for a month beyond viral clearance (46 d.p.i.) and tested their ability to spatially locate and remember the location of a target hole in a Barnes maze over the course of 10 trials held twice daily for 5 days (Fig. 6a). While WNV-recovered, wild-type mice display significant deficits in spatial learning14 (Fig. 6b), the performance of WNV-recovered Il1r1−/− mice was indistinguishable from mock-infected animals (Fig 6c). Area under the curve (AUC) analysis for each animal, which allows a comprehensive view of how individuals within the group performed across all 5 days of testing on the Barnes Maze, demonstrated that wild-type animals recovering from WNND had more severe memory impairments than mock-infected wild-type controls and WNND recovered Il1r1−/− animals (Fig 6d). To assess differences in exploratory behavior that may affect performance on the Barnes Maze spatial learning task, we performed Open Field testing (OFT) at 45 d.p.i., 1 day prior to the Barnes Maze (Fig. 6a). While the number of lines crossed during OFT were similar for mock- and WNV-infected mice of both genotypes, mock-infected Il1r1−/− animals crossed fewer lines than mock-infected wild-type controls (Fig. 6e). No differences in the number of center crosses were observed between all groups (Fig. 6f). These studies demonstrate a critical role for IL-1R1 in limiting synapse and cognitive recovery.
Fig 6. Il1r1−/− WNV-NS5-E218A-infected mice are protected from virus-induced spatial learning deficits on the Barnes Maze behavior task.
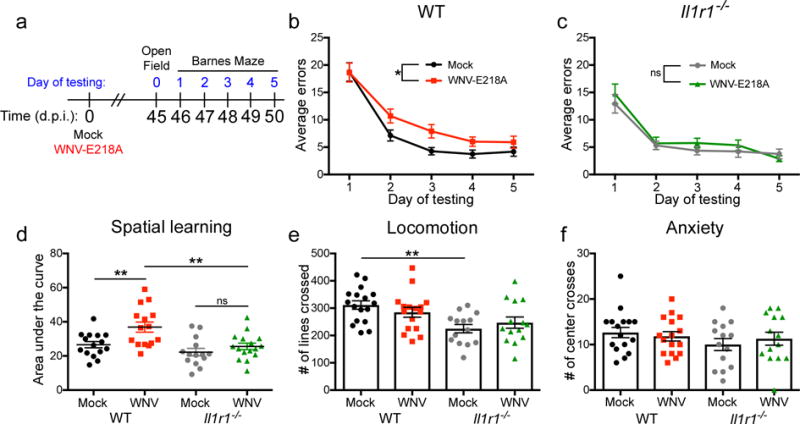
(a) Experimental design. Mock or WNV-NS5-E218A infected wild-type (WT) and Il1r1−/− underwent behavioral testing beginning at 45 d.p.i. Open field testing was performed at 45 d.p.i., followed by 5 consecutive days of Barnes Maze testing. (b) Barnes maze performance of WT mice, showing average errors per trial of each group for each day of testing. (c) Barnes maze performance of Il1r1−/− mice. (d) Barnes Maze performance of mock and WNV-NS5-E218A infected mice, quantified as area under the curve for each individual animal within each group. (e) Open Field testing showing the number of lines crossed in 5 minutes of testing for each animal. (f) Open Field testing showing the number of times an animal crossed the center of the arena in 5 minutes of testing for each animal. For Barnes Maze, ns = not significant (P > 0.05), *, P < 0.05 by two-way ANOVA for effect of WNV. ns = not significant (P > 0.05), *, P < 0.05, **, P < 0.005 by one-way ANOVA with Bonferroni’s multiple comparisons test to compare groups. Each symbol represents an individual mouse; small horizontal lines or height of bars represents the mean (± s.e.m.). Data are pooled composite experiments with at least 4 animals per group.
IL-1R antagonism prevents WNV-induced spatial learning deficits
In order to explore the potential for therapeutic intervention, we opted to test cognitive recovery in wild-type animals treated during acute infection with either vehicle or the IL-1R antagonist, Anakinra. As synapse elimination14 and loss of neurogenesis occurs during acute infection, which is also when IL-1 expression was highest, we began treatment at 10 d.p.i., a time point during viral clearance14 when the blood brain barrier was still permeable (Supplementary Fig. 7). Mock- or WNV-NS5-E218A-infected animals received 5 daily intra-peritoneal doses of vehicle or Anakinra, and then recovered for 30 days prior to Barnes Maze testing at 46 d.p.i. (Fig. 7a). As expected, vehicle treated WNND recovered animals were significantly impaired in their ability to identify the location of the target hole during Barnes Maze testing (Fig. 7b). In contrast, animals treated with Anakinra were protected from WNV-induced spatial learning impairment (Fig. 7c). AUC analysis confirmed that WNND-recovered animals treated with Anakinra had improved performance on the Barnes Maze compared with vehicle treated animals (Fig. 7d). Finally, OFT at 45 d.p.i. demonstrated that there were no differences between any of the groups in exploratory behavior, locomotion, or number of center crosses (Fig. 7f). These studies identify a potential therapeutic target for the prevention of spatial learning defects that occur during recovery from WNND.
Fig 7. WNV-NS5-E218A-infected mice treated with Anakinra are protected from virus-induced spatial learning deficits on the Barnes Maze behavior task.
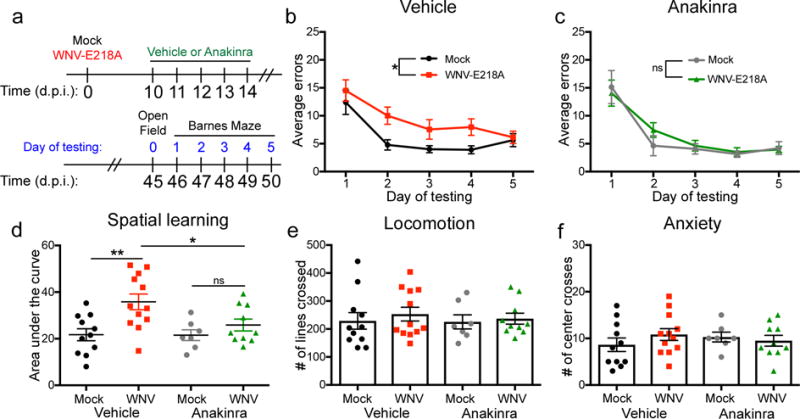
(a) Experimental design. Mock or WNV-NS5-E218A infected wild-type (WT) mice were treated with 5 consecutive doses of 100mg/kg/day of Anakinra or vehicle by intraperitoneal (ip) injection beginning at 10d.p.i. Animals were allowed to recover for an additional 30 days post treatment, followed by behavior testing at 45 d.p.i. Open Field testing was performed at 45 d.p.i., followed by 5 consecutive days of Barnes Maze testing. (b) Barnes maze performance of vehicle treated WT mice, showing average errors per trial of each group for each day of testing. (c) Barnes maze performance of Anakinra treated wildtype mice. (d) Barnes Maze performance of mock and WNV-NS5-E218A infected mice, quantified as area under the curve for each individual animal within each group. (e) Open Field testing showing the number of lines crossed in 5 minutes of testing for each animal. (f) Open Field testing showing the number of times an animal crossed the center of the arena in 5 minutes of testing for each animal. For Barnes Maze, ns = not significant (P > 0.05), *, P < 0.05 by two-way ANOVA for effect of WNV. ns = not significant (P > 0.05), *, P < 0.05, **, P < 0.005 by one-way ANOVA with Bonferroni’s multiple comparisons test to compare groups. Each symbol represents an individual mouse; small horizontal lines or height of bars represents the mean (± s.e.m.). Data are pooled composites of 3 independent experiments, with at least 3 animals per group.
Discussion
The original designation of cytokines as immune modulators has expanded to include a variety of functions in nonlymphoid tissues, especially the CNS, where they play critical roles in neurodevelopment. Regulation of signaling by the gp130 family of cytokines through Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathways maintains the pool of neural stem and progenitor cells (NPCs), promoting neurogenesis of the latter41. During development, IL-1R1 is also expressed on proliferating NPCs and IL-1β exerts an anti-proliferative, anti-neurogenic and pro-gliogenic effect on embryonic hippocampal NPCs in vitro42,43. Studies of viral encephalitis have demonstrated critical roles for innate cytokines expressed by infiltrating immune cells in T cell-mediated clearance of pathogen. However, their impact on CNS recovery and repair after viral clearance via actions on NPCs has not been elucidated. Our findings suggest that during the acute phase of viral infection, myeloid cell-derived IL-1 (ref.10) alters the proliferation and differentiation fates of neural progenitor cells, leading to a shift from neurogenesis to astrogenesis. Proinflammatory astrocytes then become the predominant source of the cytokine, which continues to inhibit neurogenesis, after myeloid cells retreat from the CNS14. Accordingly, mice deficient in IL-1R signaling are resistant to both derailment of neurogenesis as well as spatial memory impairments, which are observed in wild-type WNV-recovered mice. Because loss of neurogenesis is detected early in the course of infection, administration of IL-1R antagonist at 10 d.p.i., a time-point when the BBB is still permeable, was able to reverse the effects of IL-1 and improve spatial learning. Our data indicate that the combinatorial effect of synapse loss14 and reduced neurogenesis can negatively impact hippocampal spatial learning and memory long beyond the initial episode of infection via a shift in sources of cytokines to neural cells.
Other neurotropic flaviviruses, including Japanese Encephalitis and Zika Virus, have been shown to directly infect neural progenitors, causing apoptosis of progenitors and progeny44,45. In agreement with previous studies46, we found very few neural progenitor cells infected with WNV. However, transcriptional profiling detected altered expression of genes implicated in hippocampal neurogenesis during WNND recovery. These included increased expression of genes that encode CDKN1a, CDCA4, and CCND1, cell cycle progression inhibitors, and decreased expression of genes that encode Epha5 and Sema6B, which regulate hippocampal axon pathfinding during neural development47,48, were detected during WNND recovery. We also detected alterations to the Wnt signaling pathway, which can impact neural progenitor cell proliferation and motility, but also regulates synapse formation49 and plasticity50, potentially contributing to the failure of mice to recover from both neurogenesis derailment as well as WNV-mediated synapse loss14. The genetic signatures present during WNV recovery describes an environment which both impedes neural progenitor cell proliferation and the ability of immature and mature neurons to form new synapses.
While previous studies have detected IL-1R1 within the hippocampus51, recent research highlighting astrocyte heterogeneity has not identified these cells as a first target of IL-152. In contrast, numerous studies have implicated neural progenitor cells as a significant target of IL-1. IL-1α and IL-1β have both been shown to induce neural stem and progenitor cells to favor the astrocyte rather than neuronal lineage in vitro42. TNF, which is highly expressed in WNV-infected brains5 in an IL-1R-dependent manner10, has been shown in other contexts to direct neuronal progenitors towards an astrocyte fate via downstream STAT3 induction53, suggesting that these may be part of the same anti-neurogenic pathway. In vivo studies using overexpression and stress models have shown that IL-1β decreases neurogenesis43,54,55, and influences synaptic plasticity56,57, processes that are vital for the development and retention of spatial memory. We found that abundance of IL-1β during acute infection altered lineage fate of neural stem cells within neurogenic zones of the CNS favoring astrocyte genesis. This led to a feed-forward cycle of inflammation, as astrocytes became the primary source of IL-1β in the recovering hippocampus. IL-1R1-deficient mice were protected from decreased neurogenesis following WNV infection, and retained the ability to learn a spatial memory task. Importantly, we demonstrated that the effect was specific to inflammasome-activated IL-1β by confirming that Casp1−/− animals were also protected from decreased neurogenesis. In studies using cultured NPCs, IL-1β and TNF are both capable of altering lineage-fate to favor greater numbers of astrocytes and both of these pathways are dependent on downstream STAT3 activation58. While we focused on the role and necessity of IL-1R signaling, TNF, which was also upregulated by astrocytes at 25 d.p.i., may also contribute to the observed lineage fate-altering phenotype.
While multiple studies have described effects of cytokines released from astrocytes59 and microglia60 on memory and hippocampal neurogenesis, the precise contributions of each cell type in vivo remain ill-defined. In the current study, we found that astrocytes within WNV-recovered CNS express increased expression of Il1b, Casp1 and Tnf, whereas microglia increased expression of Ccl2. Astrocytes display a regional heterogeneity38,52, which can also reflect differences in homeostatic function, such as synaptogenesis52, and response to infection, where cerebellar astrocytes are poised to quickly mount antiviral programs61. In addition to developmentally determined sub-populations62,63, astrocytes display a remarkable plasticity and molecular identity determined in part by neuronal cues64. In response to different types of injury, reactive astrocytes can develop a recently described polarized phenotype termed A1 (infection induced, pro-inflammatory astrocytes) or A2 (ischemia induced, promoting tissue repair) that are distinguished by distinct genetic signatures36. In this study, A1 astrocytes were induced in vivo by lipopolysaccharide injection and the combination of IL-1α, TNF, and C1q was observed to promote A1 polarization in vitro. Our model of viral infection also exhibits elevated production of IL-1, TNF, and C1q14. Consistent with this, we also observed the in vivo development and persistence of astrocytes with panreactive and A1 markers. As proinflammatory astrocytes lose the ability to phagocytose due to a decrease in mRNA expression of the phagocytic receptors Mertk and Megf10, and fail to support synaptogenesis in vitro due to decreases in expression of Gpc6 and Sparcl1, factors that normally promote excitatory synapse formation36. Thus, the lack of recovery of synapses in our model could be explained by the generation of this subset of reactive astrocytes. Alternatively, lack of recovery could also be due to the loss of a specific astrocyte population that normally promotes synaptogenesis52. Although expansion of a synapse-promoting astrocyte population leads to seizures in the context of a reactive glioma52, this population may be crucial for CNS repair in the setting of the diverse array of diseases recently associated with loss of synapses in addition to WNV encephalitis, including Alzheimer’s disease19, schizophrenia20, and lupus65. Further studies are needed to more fully understand how homeostatic astrocyte populations respond to CNS injury, and whether this correlates with regional or functional identity. The discovery that newly derived, reactive astrocytes of a recently identified proinflammatory subset36 are responsible for persistently decreased adult neurogenesis during WNV recovery also suggests cell-type specific studies will be required to elucidate these pathways in disease models.
There is a growing body of experimental evidence demonstrating the importance of homeostatic neuroimmune interactions for normal cognitive function (recently reviewed by66,67). However, the effects of immune mediators on brain function depend on their levels and locations; this phenomenon was classically demonstrated as a U-shaped curve relating the effect of IL-1 signaling on cognitive function in which either overexpression or complete blockade of IL-1 signaling both negatively impacted spatial learning68. Thus, in the context of neuroinflammation, increased expression of immune molecules may similarly lead to altered cognitive performance. Of note, we observed a trend towards recovery of neurogenesis in the SVZ that was absent in the hippocampus, an effect that has been reported previously in response to ionizing radiation69, suggesting a differential responses to injury in these neurogenic niches. Interestingly, in a study of hypoxia driven neuroinflammation, hippocampal neurogenesis decreased, while SVZ neurogenesis increased, an effect that was linked to IL-6 signaling70. Further work addressing the mechanism underlying differential responsiveness to cytokine signaling in neurogenic niches could shed light on basic aspects of neural precursor cell biology.
Materials and methods
Animals
5–8 week-old male mice were used at the outset of all experiments. C57BL/6J mice were obtained from Jackson Laboratories. Il1r1−/− mice (> 10 generations backcrossed to C57BL/6) were obtained from Jackson Laboratories. Nestin-GFP mice (> 10 generations backcrossed to C57BL/6) were obtained from G. Enikolopov (Cold Spring Harbor Laboratories). All experimental protocols were performed in compliance with the Washington University School of Medicine Animal Safety Committee (protocol# 20140122).
Mouse models of WNV infection
Footpad
(WNV-NY99) The WNV strain 3000.0259 was isolated in New York in 2000 (Ebel et al., 2001) and passaged once in C6/36 Aedes albopictus cells to generate an insect cell–derived stock. 100 plaque-forming units (pfu) of WNV-NY99 were delivered in 50 μl to the footpad of anaesthetized mice.
Intracranial
“WNV-NS5-E218A,” which harbors a single point mutation in the 2′O-methyl-transferase gene, was obtained from M. Diamond (Washington University) and passaged in Vero cells as described previously41. Deeply anaesthetized mice were administered 104 pfu of WNV-NS5-E218A or 10 pfu of WNV-NY99 in 10 μl of 0.5% FBS in HBSS into the brain’s third ventricle via a guided 29-gauge needle. “Mock” infected animals were deeply anesthetized and administered 10 μl of 0.5% FBS in HBSS into the brain’s third ventricle via a guided 29-gauge needle.
Stock titers of all viruses were determined by using BHK21 cells for viral plaque assay as previously described42.
Microarray analysis
Further analysis was performed on previously published hippocampal microarray gene expression data14 (Gene Expression Omnibus #GSE72139) from Mock-infected and WNV-NS5-E218A animals at 25 days post infection. Expression data from selected genes (P < 0.05 or P < 0.1 for italicized genes, Two-tailed Students t-test) were converted to Z-scores for each individual gene and animal and are displayed as colorimetric heatmaps.
In vivo bromodeoxyuridine (BrdU) labeling
BrdU (Sigma Aldrich) in sterile PBS was injected intraperitoneally (IP) for all experiments. For acute Immunohistochemical studies (harvests day 7–8 post-infection) mice were given 150 mg/kg of BrdU at 48 h prior to tissue harvest. For flow cytometric analyses, mice were given 100 mg/kg of BrdU at 48 h and again at 24 h prior to tissue harvest. For neuronal BrdU labeling, mice were given 75 mg/kg every 12 h beginning at 3 days post-infection and ending at 7 days post-infection for a total of 7 doses.
Antibodies
WNV (1:100 described previously8), NeuN (1:100, Cell Signaling, Cat 12943S, Clone D3S3I), BrdU (1:200, Abcam, Cat ab1893, polyclonal), CD45 (Biolegend, Cat 103114, Clone 30-F11), Doublecortin (1:150, Cell Signaling, Cat 4604S, polyclonal), GFAP (1:50 for flow cytometry; 1: 200 for IHC, BD, Cat 561483, Clone 1B4), IL-1β (R&D, Cat AF-401, polyclonal), Mash1 (BD, Cat 556604, Clone 24B72D11.1), Ki67 (Abcam, Cat AB15580, polyclonal), Synaptophysin (1:250, Synaptic Systems, Cat 101004, polyclonal). Secondary antibodies conjugated to Alexa-488, Alexa-555, or Alexa-647 (Invitrogen) were used at a1:400 dilution.
Immunohistochemistry
Following perfusion with ice-cold PBS and 4% paraformaldehyde (PFA), brains were immersion-fixed overnight in 4% PFA, followed by cryoprotection in 2 exchanges of 30% sucrose for 72 h, then frozen in OCT (Fisher). 9 μm-thick fixed-frozen coronal brain sections were washed with PBS and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich), and nonspecific Ab was blocked with 5–10% normal goat serum (Santa Cruz Biotechnology) for 1 h at 23°C. Mouse on Mouse kit (MOM basic kit, Vector) was used per manufacturers protocol when detecting synaptophysin (mouse, DAKO) to reduce endogenous mouse Ab staining. After block, slides were exposed to primary Ab or isotype matched IgG overnight at 4°C, washed with 0.2% FSG in PBS and incubated with secondary Abs for 1 h at 23°C. Nuclei were counterstained with DAPI (Invitrogen) and coverslips were applied with vectashield (Vector). Immunofluorescence was analyzed using a Zeiss LSM 510 laser-scanning confocal microscope and accompanying software (Zeiss). For each animal, 6–8 images were taken from 2–3 different coronal sections spaced at least 50 μm apart. Positive immunofluorescent signals were quantified using the public domain NIH Image analysis software, ImageJ.
Flow cytometry
Cells were isolated from brains of WT mice at day 6, 15, or 30 days post-infection and stained with fluorescently conjugated antibodies to CD45, BrdU, and Doublecortin as previously described37. Briefly, animals were deeply anesthetized with a ketamine/xylazine mixture and perfused intracardially with ice cold dPBS (Gibco). Brains were aseptically removed, minced and enzymatic digested in HBSS (Gibco) containing collagenase D (Sigma, 50mg/ml), TLCK trypsin inhibitor (Sigma, 100 μg/ml), DNase I (Sigma, 100U/μl), HEPES ph 7.2 (Gibco, 1M), for 1hr at 23°C while shaking. The tissue was pushed through a 70 μm strainer and spun down at 500g for 10min. The cell pellet was resuspended in a 37% percoll solution and spun at 1200g for 30min to remove myelin debris. Cells were washed in PBS, then resuspended in FACS buffer. Cells were blocked with TruStain fcX anti-mouse CD16/32 (Biolegend) for 5 min on ice, followed by incubation with fluorescently conjugated antibodies for 30 min on ice. Cells were then washed 2 times in PBS, fixed with 4% PFA for 10 min at 23°C and resuspended in FACS buffer. Data collection was performed with LSR-II (BD Biosciences) and analyzed with Flowjo software.
Ex vivo isolation of microglia and astrocytes with microbeads
Animals were deeply anesthetized with a ketamine/xylazine mixture and perfused intracardially with ice cold dPBS (Gibco). Brains were aseptically removed, minced and enzymatic digested in HBSS (Gibco) containing collagenase D (Sigma, 50mg/ml), TLCK trypsin inhibitor (Sigma, 100 μg/ml), DNase I (Sigma, 100 U/μl), HEPES 7.2 (Gibco, 1M), for 1hr at 23°C while shaking. The tissue was pushed through a 70 μm strainer and spun down at 500g for 10min. The cell pellet was resuspended in a 37% percoll solution and spun at 1200g for 30 min to remove myelin debris. Cells were washed in PBS then resuspended in MACS buffer. CD11b+ Microglia and ACSA-2+ astrocytes were isolated using manual MACS sorting according to the manufacturer’s instructions (Miltenyi Biotec). Non-specific labeling of CD11b+ microglia was prevented by incubating the cells with FcR blocking reagent prior to labeling the cells with ACSA-2 antibodies for magnetic isolation. The purified microglia and astrocytes were processed for real-time quantitative RT-PCR as described below.
Real-time quantitative RT-PCR
cDNA was synthesized using random hexamers, oligodT15, and MultiScribe reverse transcriptase (Applied Biosystems). A single reverse transcription master mix was used to reverse transcribe all samples in order to minimize differences in reverse transcription efficiency. The following conditions were used for reverse transcription: 25°C for 10 min, 48°C for 30 min, and 95°C for 5 min. Real-time quantitative RT-PCR was performed as previously described14.
Behavioral Testing
Open field and Barnes Maze testing was performed as previously describe14. Briefly, mice were tested on the Barnes Maze over the course of 5 consecutive days, receiving two trials per day, spaced 30 minutes apart. For each trial, the mouse was given 3 minutes to explore the maze and find the target hole. Mice that did not enter the target hole within 3 minutes were gently guided into the hole. After each trial, the mouse remained in the target hole for exactly 1 minute, and then was returned to its home cage. The maze was decontaminated with 70% ethanol between each trial. The numbers of errors (nose pokes over non-target holes) were measured. One day prior to Barnes Maze testing, mice were tested via open field testing to monitor differences in exploratory behavior. Each animal was given 5 minutes to explore an open field arena before returning to its home cage. Behavior was recorded using a camera (Canon Powershot SD1100IS), and a blinded experimenter scored the trials.
Anakinra Treatment
Anakinra (Kineret®, Sobi) was diluted in PBS to 10 mg/mL and animals were treated with 100 mg/kg/day of Anakinra or Vehicle (PBS) for 5 consecutive days by intraperitoneal (ip) injection. Animals were weighed daily to monitor for weight loss or adverse events, none of which were observed during the course of treatment.
Statistical Analysis
Statistical analyses were performed using Prism 7.0 (GraphPad Software). All data were analyzed using an unpaired student’s t-test, one-way or two-way ANOVA with Bonferroni post-test to correct for multiple comparisons as indicated in the corresponding figure legends. A P value of ≤ 0.05 was considered significant.
Data Availability
Data supporting the findings in this study are available upon request from the corresponding author. The accession code is GSE72139.
Supplementary Material
Acknowledgments
This work was supported by NIH grants U19 AI083019, R01 NS052632, HDTRA11510032, (all to RSK).
Experimental support was provided by the Speed Congenics Facility of the Rheumatic Diseases Core Center. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the National Institutes of Health, under Award Number P30AR048335. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Contributions
C.G., M.J.V., and R.S.K. designed the experiments; C.G. and M.J.V. did most of the experiments, compiled and analyzed the data; C.G., M.J.V., and R.S.K. prepared the figures; L.L.V., T.S. and X.J. were involved in specific experiments; C.G., M.J.V., and R.S.K. analyzed the data and wrote the manuscript.
Competing Interests
The authors declare no competing interests.
References
- 1.Salimi H, Cain MD, Klein RS. Encephalitic Arboviruses: Emergence, Clinical Presentation, and Neuropathogenesis. Neurotherapeutics. 2016;13:514–534. doi: 10.1007/s13311-016-0443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lazear HM, Diamond MS. New insights into innate immune restriction of West Nile virus infection. Curr Opin Virol. 2015;11:1–6. doi: 10.1016/j.coviro.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambert SL, Aviles D, Vehaskari VM, Ashoor IF. Severe West Nile virus meningoencephalitis in a pediatric renal transplant recipient: successful recovery and long-term neuropsychological outcome. Pediatr Transplant. 2016;20:836–839. doi: 10.1111/petr.12768. [DOI] [PubMed] [Google Scholar]
- 4.Patel H, Sander B, Nelder MP. Long-term sequelae of West Nile virus-related illness: a systematic review. Lancet Infect Dis. 2015;15:951–959. doi: 10.1016/S1473-3099(15)00134-6. [DOI] [PubMed] [Google Scholar]
- 5.Shrestha B, Zhang B, Purtha WE, Klein RS, Diamond MS. Tumor necrosis factor alpha protects against lethal West Nile virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J Virol. 2008;82:8956–8964. doi: 10.1128/JVI.01118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lazear HM, et al. Interferon-lambda restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med. 2015;7:284ra259. doi: 10.1126/scitranslmed.aaa4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lazear HM, Pinto AK, Vogt MR, Gale M, Jr, Diamond MS. Beta interferon controls West Nile virus infection and pathogenesis in mice. J Virol. 2011;85:7186–7194. doi: 10.1128/JVI.00396-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shrestha B, et al. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J Virol. 2006;80:5338–5348. doi: 10.1128/JVI.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durrant DM, Robinette ML, Klein RS. IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J Exp Med. 2013;210:503–516. doi: 10.1084/jem.20121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramos HJ, et al. IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 2012;8:e1003039. doi: 10.1371/journal.ppat.1003039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guarner J, et al. Clinicopathologic study and laboratory diagnosis of 23 cases with West Nile virus encephalomyelitis. Hum Pathol. 2004;35:983–990. doi: 10.1016/j.humpath.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Leyssen P, et al. Acute encephalitis, a poliomyelitis-like syndrome and neurological sequelae in a hamster model for flavivirus infections. Brain Pathol. 2003;13:279–290. doi: 10.1111/j.1750-3639.2003.tb00028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vasek MJ, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534:538–543. doi: 10.1038/nature18283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang B, Patel J, Croyle M, Diamond MS, Klein RS. TNF-alpha-dependent regulation of CXCR3 expression modulates neuronal survival during West Nile virus encephalitis. J Neuroimmunol. 2010;224:28–38. doi: 10.1016/j.jneuroim.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ceccaldi PE, Lucas M, Despres P. New insights on the neuropathology of West Nile virus. FEMS Microbiol Lett. 2004;233:1–6. doi: 10.1016/j.femsle.2004.01.035. [DOI] [PubMed] [Google Scholar]
- 17.Szretter KJ, et al. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012;8:e1002698. doi: 10.1371/journal.ppat.1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michailidou I, et al. Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann Neurol. 2015;77:1007–1026. doi: 10.1002/ana.24398. [DOI] [PubMed] [Google Scholar]
- 19.Hong S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sekar A, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–183. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jarrard LE. On the role of the hippocampus in learning and memory in the rat. Behav Neural Biol. 1993;60:9–26. doi: 10.1016/0163-1047(93)90664-4. [DOI] [PubMed] [Google Scholar]
- 22.Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mishra BB, Gundra UM, Teale JM. Expression and distribution of Toll-like receptors 11-13 in the brain during murine neurocysticercosis. J Neuroinflammation. 2008;5:53. doi: 10.1186/1742-2094-5-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lieberwirth C, Pan Y, Liu Y, Zhang Z, Wang Z. Hippocampal adult neurogenesis: Its regulation and potential role in spatial learning and memory. Brain Res. 2016;1644:127–140. doi: 10.1016/j.brainres.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheffer A, Tarnok A, Ulrich H. Cell cycle regulation during neurogenesis in the embryonic and adult brain. Stem Cell Rev. 2013;9:794–805. doi: 10.1007/s12015-013-9460-5. [DOI] [PubMed] [Google Scholar]
- 26.Riazi K, et al. Microglia-dependent alteration of glutamatergic synaptic transmission and plasticity in the hippocampus during peripheral inflammation. J Neurosci. 2015;35:4942–4952. doi: 10.1523/JNEUROSCI.4485-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu MD, Montgomery SL, Rivera-Escalera F, Olschowka JA, O’Banion MK. Sustained IL-1beta expression impairs adult hippocampal neurogenesis independent of IL-1 signaling in nestin+ neural precursor cells. Brain Behav Immun. 2013;32:9–18. doi: 10.1016/j.bbi.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.del Rey A, Balschun D, Wetzel W, Randolf A, Besedovsky HO. A cytokine network involving brain-borne IL-1beta, IL-1ra, IL-18, IL-6, and TNFalpha operates during long-term potentiation and learning. Brain Behav Immun. 2013;33:15–23. doi: 10.1016/j.bbi.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 29.Belarbi K, Rosi S. Modulation of adult-born neurons in the inflamed hippocampus. Front Cell Neurosci. 2013;7:145. doi: 10.3389/fncel.2013.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 31.Ben-Menachem-Zidon O, Ben-Menahem Y, Ben-Hur T, Yirmiya R. Intra-hippocampal transplantation of neural precursor cells with transgenic over-expression of IL-1 receptor antagonist rescues memory and neurogenesis impairments in an Alzheimer’s disease model. Neuropsychopharmacology. 2014;39:401–414. doi: 10.1038/npp.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hein AM, et al. Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun. 2010;24:243–253. doi: 10.1016/j.bbi.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larson SJ, Hartle KD, Ivanco TL. Acute administration of interleukin-1beta disrupts motor learning. Behav Neurosci. 2007;121:1415–1420. doi: 10.1037/0735-7044.121.6.1415. [DOI] [PubMed] [Google Scholar]
- 34.Cunningham C, Sanderson DJ. Malaise in the water maze: untangling the effects of LPS and IL-1beta on learning and memory. Brain Behav Immun. 2008;22:1117–1127. doi: 10.1016/j.bbi.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heneka MT, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun T, Vasek MJ, Klein RS. Congenitally acquired persistent lymphocytic choriomeningitis viral infection reduces neuronal progenitor pools in the adult hippocampus and subventricular zone. PLoS One. 2014;9:e96442. doi: 10.1371/journal.pone.0096442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chai H, et al. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron. 2017;95:531–549 e539. doi: 10.1016/j.neuron.2017.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feldmann M, Pathipati P, Sheldon RA, Jiang X, Ferriero DM. Isolating astrocytes and neurons sequentially from postnatal murine brains with a magnetic cell separation technique. Journal of Biological Methods. 2014;1:1–7. [Google Scholar]
- 40.Kim EJ, Ables JL, Dickel LK, Eisch AJ, Johnson JE. Ascl1 (Mash1) defines cells with long-term neurogenic potential in subgranular and subventricular zones in adult mouse brain. PLoS One. 2011;6:e18472. doi: 10.1371/journal.pone.0018472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 42.Green HF, et al. A role for interleukin-1beta in determining the lineage fate of embryonic rat hippocampal neural precursor cells. Mol Cell Neurosci. 2012;49:311–321. doi: 10.1016/j.mcn.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, et al. Interleukin-1beta mediates proliferation and differentiation of multipotent neural precursor cells through the activation of SAPK/JNK pathway. Mol Cell Neurosci. 2007;36:343–354. doi: 10.1016/j.mcn.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 44.Das S, Basu A. Japanese encephalitis virus infects neural progenitor cells and decreases their proliferation. J Neurochem. 2008;106:1624–1636. doi: 10.1111/j.1471-4159.2008.05511.x. [DOI] [PubMed] [Google Scholar]
- 45.Tang H, et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell. 2016;18:587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shrestha B, Gottlieb D, Diamond MS. Infection and injury of neurons by West Nile encephalitis virus. J Virol. 2003;77:13203–13213. doi: 10.1128/JVI.77.24.13203-13213.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tawarayama H, Yoshida Y, Suto F, Mitchell KJ, Fujisawa H. Roles of semaphorin-6B and plexin-A2 in lamina-restricted projection of hippocampal mossy fibers. J Neurosci. 2010;30:7049–7060. doi: 10.1523/JNEUROSCI.0073-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yue Y, et al. Mistargeting hippocampal axons by expression of a truncated Eph receptor. Proc Natl Acad Sci U S A. 2002;99:10777–10782. doi: 10.1073/pnas.162354599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bamji SX, et al. Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron. 2003;40:719–731. doi: 10.1016/s0896-6273(03)00718-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci. 2003;6:1169–1177. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]
- 51.Cunningham ET, Jr, et al. In situ histochemical localization of type I interleukin-1 receptor messenger RNA in the central nervous system, pituitary, and adrenal gland of the mouse. J Neurosci. 1992;12:1101–1114. doi: 10.1523/JNEUROSCI.12-03-01101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.John Lin CC, et al. Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci. 2017;20:396–405. doi: 10.1038/nn.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng H, et al. HIV-1-infected and immune-activated macrophages induce astrocytic differentiation of human cortical neural progenitor cells via the STAT3 pathway. PLoS One. 2011;6:e19439. doi: 10.1371/journal.pone.0019439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A. 2008;105:751–756. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu MD, et al. Adult murine hippocampal neurogenesis is inhibited by sustained IL-1beta and not rescued by voluntary running. Brain Behav Immun. 2012;26:292–300. doi: 10.1016/j.bbi.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prieto GA, et al. Synapse-specific IL-1 receptor subunit reconfiguration augments vulnerability to IL-1beta in the aged hippocampus. Proc Natl Acad Sci U S A. 2015;112:E5078–5087. doi: 10.1073/pnas.1514486112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tong L, et al. Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1beta via p38 mitogen-activated protein kinase. J Neurosci. 2012;32:17714–17724. doi: 10.1523/JNEUROSCI.1253-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen E, et al. A novel role of the STAT3 pathway in brain inflammation-induced human neural progenitor cell differentiation. Curr Mol Med. 2013;13:1474–1484. doi: 10.2174/15665240113139990076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vallieres L, Campbell IL, Gage FH, Sawchenko PE. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci. 2002;22:486–492. doi: 10.1523/JNEUROSCI.22-02-00486.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith PL, Hagberg H, Naylor AS, Mallard C. Neonatal peripheral immune challenge activates microglia and inhibits neurogenesis in the developing murine hippocampus. Dev Neurosci. 2014;36:119–131. doi: 10.1159/000359950. [DOI] [PubMed] [Google Scholar]
- 61.Daniels BP, et al. Regionally distinct astrocyte interferon signaling promotes blood-brain barrier integrity and limits immunopathology during neurotropic viral infection. J Clin Invest. 2017 in press. [Google Scholar]
- 62.Muroyama Y, Fujiwara Y, Orkin SH, Rowitch DH. Specification of astrocytes by bHLH protein SCL in a restricted region of the neural tube. Nature. 2005;438:360–363. doi: 10.1038/nature04139. [DOI] [PubMed] [Google Scholar]
- 63.Tsai HH, et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science. 2012;337:358–362. doi: 10.1126/science.1222381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farmer WT, et al. Neurons diversify astrocytes in the adult brain through sonic hedgehog signaling. Science. 2016;351:849–854. doi: 10.1126/science.aab3103. [DOI] [PubMed] [Google Scholar]
- 65.Bialas AR, et al. Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature. 2017;546:539–543. doi: 10.1038/nature22821. [DOI] [PubMed] [Google Scholar]
- 66.Filiano AJ, Gadani SP, Kipnis J. How and why do T cells and their derived cytokines affect the injured and healthy brain? Nat Rev Neurosci. 2017;18:375–384. doi: 10.1038/nrn.2017.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tay TL, Savage JC, Hui CW, Bisht K, Tremblay ME. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J Physiol. 2017;595:1929–1945. doi: 10.1113/JP272134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goshen I, et al. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology. 2007;32:1106–1115. doi: 10.1016/j.psyneuen.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 69.Hellstrom NA, Bjork-Eriksson T, Blomgren K, Kuhn HG. Differential recovery of neural stem cells in the subventricular zone and dentate gyrus after ionizing radiation. Stem Cells. 2009;27:634–641. doi: 10.1634/stemcells.2008-0732. [DOI] [PubMed] [Google Scholar]
- 70.Covey MV, Loporchio D, Buono KD, Levison SW. Opposite effect of inflammation on subventricular zone versus hippocampal precursors in brain injury. Ann Neurol. 2011;70:616–626. doi: 10.1002/ana.22473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings in this study are available upon request from the corresponding author. The accession code is GSE72139.