

Dear Editor,
Ghosal type of hemato-diaphyseal dysplasia (GHDD) is a rare variety of hypoplastic anemia and only limited case reports are available.
A 3 year old male born to 2nd degree consanguineous parents presented with progressive pallor and bowing of lower extremities noticed since 1 year of age. He received packed cell transfusion twice in the last 1 year. There was no history of bone pain or skin bleeds. There were no affected family members. Birth history and development were normal. On examination, the height was 90 cm (15th centile for age), weight was 12.2 kg (15th centile for age) and head circumference was 47.5 cm (between mean and −2SD). Vitals were stable. Lateral bowing of legs and forearms was present along with characteristic palpable diffuse thickening of bones. There were no hand anomalies or dysmorphic features. Fundus examination and hearing was normal. Severe pallor was present. There was no icterus, lymphadenopathy, bony tenderness or skin bleeds. There was no organomegaly. Systemic examination was unremarkable.
Investigations showed hemoglobin of 49 g/L, hematocrit of 16.2%, and platelet count of 22 × 109/L. The white blood cell count was 7.4 × 109/L with a differential count of 15% neutrophils, 82% lymphocytes, and 3% monocytes. The erythrocyte sedimentation rate was 80 mm 1st hour. The mean corpuscular volume (MCV) was 82.2 fl; MCH 25 pg; MCHC 30.3 g/dL and RDW was 19%. The peripheral blood smear revealed normocytic, normochromic red blood cells with reactive lymphocytes; platelet count was reduced with occasional giant forms. Reticulocyte count was 2.4%. Haemoglobin HPLC done prior to packed cell transfusion was normal. HIV ELISA and Mantoux test were negative. Serum immunoglobulin assay showed elevated levels of IgG and IgA. Radiography of long bones revealed diffuse sclerosis of the entire cortices extending up to metaphyses suggestive of meta-diaphyseal dysplasia (Fig. 1). There was no sub periosteal bone formation. Bone marrow aspiration showed dry tap with markedly diluted sinusoidal blood. Histopathological examination of the bone marrow biopsy specimen revealed bony trabeculae enclosing markedly hypo cellular marrow spaces. Genetic workup was not done.
Fig. 1.
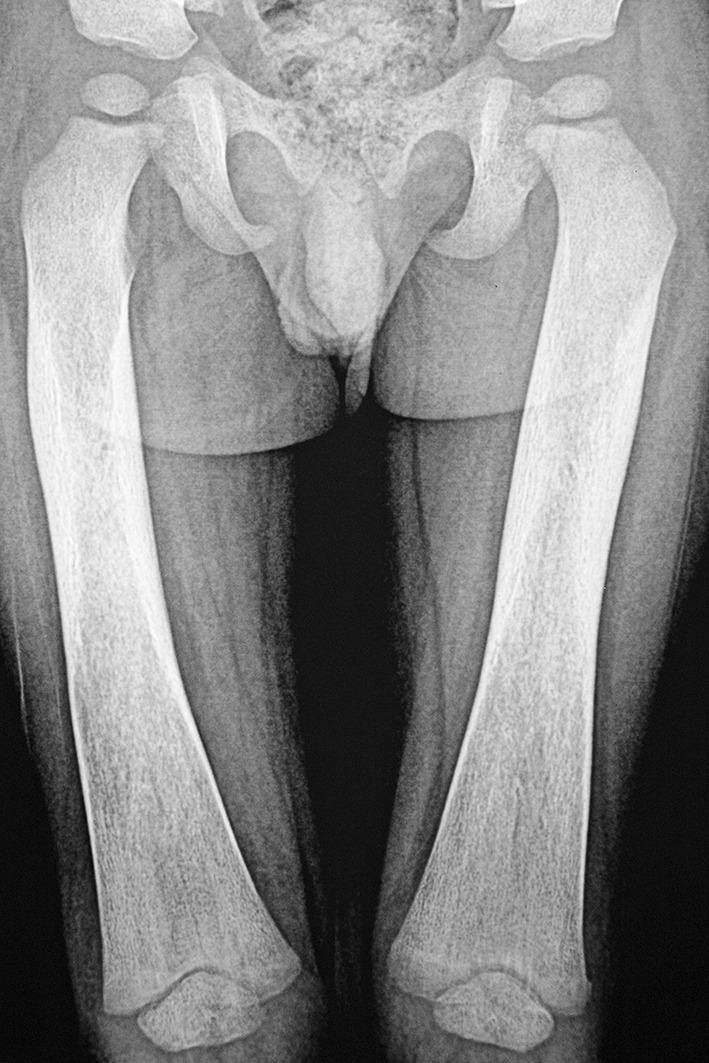
Radiograph of both femurs showing diffuse sclerosis of diaphysis extending up to the metaphysis
He was treated with packed cell transfusion. Oral Prednisolone (1 mg/kg/day) was started along with folic acid supplementation. There was prompt response to the steroid therapy. Repeat haemoglobin and platelet count done after 4 weeks of daily prednisolone showed improving trend (haemoglobin 116 g/L; platelet count 88 × 109/L). Prednisolone was gradually tapered (5 mg/week) and later switched over to alternate day. The child did not require further packed cell transfusion. Currently during the follow up period of 1 year, the child is on 0.5 mg/kg prednisolone on alternate day and is maintaining his haemoglobin between 10 and 11 g/dL. No adverse effects of steroid therapy were noticed during this period.
Ghosal hemato-diaphyseal dysplasia (GHHD OMIM #231095) was initially described by Ghosal [1] in five Indian patients as a rare disorder of refractory anaemia responsive to steroid therapy. The gene responsible for GHHD (TBXAS1 gene) located on chromosome 7q33–34, which encodes thromboxane-A-synthase, an enzyme which synthesizes thromboxane A2 (TA2) [2]. TA2 modulates the expression of TNFSF11 and TNFRSF11B that encode RANKL and osteoprotegerin in osteoblasts promoting osteosclerosis. Though progressive anaemia is the most characteristic feature, some patients with GHHD may present with varying degrees of thrombocytopenia, or pancytopenia.
Radiographically, GHDD is characterized by diaphyseal sclerosis of long bones with widening of medullary cavities and cortical hyperostosis. The other differential diagnoses that need to be considered in a case of anaemia with bony dysplasia are Caffey disease and Camurati–Engelmann disease. Caffey disease (infantile cortical hyperostosis) presents in early infancy with fever and soft tissue swelling in addition to the bony dysplasia. Classical radiological finding includes periosteal reaction progressing to sub-periosteal new bone formation. Camurati–Engelmann dysplasia is characterized by limb pain, gait disturbances, neuromuscular weakness and entrapment neuropathies of cranial nerves in addition to cortical hyperostosis. Radiologically isolated diaphyseal involvement in seen Camurati–Engelmann disease, in contrast to GHHD, where both metaphysis and diaphysis are affected [3]. Steroid therapy is the mainstay of treatment of GHHD. There is no standard steroid regimen designed till date for this steroid sensitive disorder. Most of the authors have reported good response to maintenance therapy with low dose oral prednisolone throughout life [4, 5].
Thus GHHD should to be considered as a possible diagnosis children presenting with progressive pallor and diffuse palpable thick deformed bones. Though mutational analysis is confirmatory, the classical clinical and radiological picture helps to differentiate from other sclerosing bony dysplasias. This disorder responds to steroids thus obviating the need for frequent packed cell transfusion.
Compliance with Ethical Standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Ghosal SP, Mukherjee AK, Mukherjee D et al (1988) Diaphyseal dysplasia associated with anemia. J Pediatr 113:49–57. Erratum in: J Pediatr 1988; 113(2):410 [DOI] [PubMed]
- 2.Geneviève D, Proulle V, Isidor B, et al. Thromboxane synthase mutations in an increased bone density disorder (Ghosal syndrome) Nat Genet. 2008;40(3):284–286. doi: 10.1038/ng.2007.66. [DOI] [PubMed] [Google Scholar]
- 3.Arora R, Aggarwal S, Deme S. Ghosal hematodiaphyseal dysplasia—a concise review including an illustrative patient. Skeletal Radiol. 2015;44:447–450. doi: 10.1007/s00256-014-1989-0. [DOI] [PubMed] [Google Scholar]
- 4.Mondal R, Sil A, Nag SS, et al. Ghosal syndrome—ten years follow-up. Indian J Pediatr. 2015;82(6):568–569. doi: 10.1007/s12098-014-1654-6. [DOI] [PubMed] [Google Scholar]
- 5.John RR, Boddu D, Chaudhary N, et al. Steroid-responsive anemia in patients of Ghosal hematodiaphyseal dysplasia: simple to diagnose and easy to treat. J Pediatr Hematol Oncol. 2015;37(4):285–289. doi: 10.1097/MPH.0000000000000279. [DOI] [PubMed] [Google Scholar]