

ABSTRACT
Protozoan parasites, including the apicomplexan pathogens Plasmodium falciparum (which causes malaria) and Toxoplasma gondii (which causes toxoplasmosis), infect millions of people worldwide and represent major human disease burdens. Despite their prevalence, therapeutic strategies to treat infections caused by these parasites remain limited and are threatened by the emergence of drug resistance, highlighting the need for the identification of novel drug targets. Recently, homologues of the core autophagy proteins, including Atg8 and Atg3, were identified in many protozoan parasites. Importantly, components of the Atg8 conjugation system that facilitate the lipidation of Atg8 are required for both canonical and parasite-specific functions and are essential for parasite viability. Structural characterization of the P. falciparum Atg3-Atg8 (PfAtg3-Atg8) interaction has led to the identification of compounds that block this interaction. Additionally, many of these compounds inhibit P. falciparum growth in vitro, demonstrating the viability of this pathway as a drug target. Given the essential role of the Atg8 lipidation pathway in Toxoplasma, we sought to determine whether three PfAtg3-Atg8 interaction inhibitors identified in the Medicines for Malaria Venture Malaria Box exerted a similar inhibitory effect in Toxoplasma. While all three inhibitors blocked Toxoplasma replication in vitro at submicromolar concentrations, they did not inhibit T. gondii Atg8 (TgAtg8) lipidation. Rather, high concentrations of two of these compounds induced TgAtg8 lipidation and fragmentation of the parasite mitochondrion, similar to the effects seen following starvation and monensin-induced autophagy. Additionally, we report that one of the PfAtg3-Atg8 interaction inhibitors induces Toxoplasma egress and provide evidence that this is mediated by an increase in intracellular calcium in response to drug treatment.
KEYWORDS: parasites, Plasmodium, Toxoplasma, antiparasitic agents, apicomplexan parasites, autophagy
INTRODUCTION
In eukaryotic cells, macroautophagy (referred to here as autophagy) is a catabolic process wherein cytoplasm components are engulfed by an elongating membrane structure referred to as the phagophore. This vesicle matures into an enclosed, dual-membrane vesicle, termed the autophagosome, and is trafficked to the lysosome/vacuole for degradation of the engulfed material (1). This process is induced in response to nutrient starvation (2, 3) but also plays a significant role in cellular development and differentiation (4), proteostasis (5), and clearance of damaged organelles and as a defense mechanism against invading pathogens (6, 7). While over 30 autophagy-related (ATG) genes have been identified in Saccharomyces cerevisiae and mammalian systems, a subset of these genes comprises the core autophagy machinery. These core ATG genes are required for de novo autophagosome formation (8) and are especially well conserved across eukaryotes.
Approximately half of the core autophagy proteins are directly or indirectly involved in facilitating the conjugation of the cytosolic ubiquitin-like protein Atg8 (LC3/GABARAP in mammals) to phosphatidylethanolamine (PE), which recruits Atg8 to the phagophore membrane. During autophagosome biogenesis, Atg8 is critical for elongation and fusion of the phagophore membrane. Atg8 can also bind with substrate and adapter proteins containing the Atg8-interacting motif (AIM) to selectively recruit proteins to the autophagosome for degradation (9, 10). Conjugation of Atg8 to PE is carried out via the E1-like enzyme Atg7 and the E2-like enzyme Atg3, with the oligomeric Atg5-Atg12-Atg16 complex providing E3-like enzymatic function to stimulate the transfer of Atg8 from Atg3 to PE (11). Additionally, the cysteine proteinase Atg4 is responsible both for preprocessing of Atg8 to expose the C-terminal glycine for lipidation and for cleavage of the amide bond between Atg8 and PE to liberate Atg8 from the autophagosome outer membrane during autophagosome maturation (11, 12).
Recently, a conserved subset of the autophagy machinery has been identified in multiple species of early-branching eukaryotic pathogens (13). While homologues of many ATG genes are either highly divergent or completely absent, the Atg8 conjugation machinery is well conserved in many protozoans. Characterization of the components of the Atg8 conjugation machinery in the apicomplexan parasites Plasmodium spp. and Toxoplasma gondii revealed that this pathway is functionally conserved and essential for survival (14–16). In Toxoplasma, Atg8 lipidation is induced in response to starvation or drug treatment, as visualized by the formation of Atg8-containing double-membrane vesicles and/or green fluorescent protein (GFP)-T. gondii Atg8 (TgAtg8) punctae that resemble putative autophagosomes (14, 17). Although autophagy may be induced to allow the parasite to mitigate potentially harmful environmental stresses, prolonged exposure to autophagy-inducing stressors results in a loss of parasite viability with features resembling an autophagy-like cell death process (17, 18). Nonetheless, Atg8 also has unique functions beyond autophagy in apicomplexan parasites. Atg8 homologues in Plasmodium and Toxoplasma localize to the outer membrane of the apicoplast (14, 19, 20), a nonphotosynthetic plastid organelle that plays an essential role in fatty acid metabolism (21). In Toxoplasma, Atg8 tethers the apicoplast to centrosomes, either directly or indirectly, to ensure proper division of this organelle during parasite replication (22). In both Toxoplasma and Plasmodium, localization of Atg8 to the apicoplast is dependent upon its conjugation to PE, highlighting the importance of a functional Atg8 conjugation pathway in apicomplexan-specific biological processes.
The conservation of a functional subset of the autophagy machinery in protozoans and the parasite-specific roles of the members of this subset suggest that this pathway may be an ideal target for the development of novel antiparasitics. However, many of the upstream regulatory proteins targeted by autophagy-manipulating drugs used in higher eukaryotes are absent or highly divergent in protozoans, rendering these compounds ineffective (23, 24). Recent structural characterization of Plasmodium falciparum Atg8 (PfAtg8) identified differences in the electrostatic surface potential and amino acid composition between PfAtg8 and the human Atg8 homologues LC3 and GATE-16 (25). These differences were significant enough to result in an ∼30-fold preference for the binding of small-molecule inhibitors for PfAtg8 compared to human LC3 (26). Moreover, high-throughput screens have identified numerous compounds that inhibit the interaction between recombinant P. falciparum Atg3 (PfAtg3) and PfAtg8 in vitro, including a group of compounds from the Medicines for Malaria Venture (MMV) Malaria Box with antimalarial activity in tissue culture (25–27). These findings illustrate the ability to specifically target the parasite Atg3-Atg8 interaction and suggest that the Atg3-Atg8 interaction may be an attractive new drug target in apicomplexan parasites. In addition to serving as novel therapeutics, Atg3-Atg8 interaction inhibitors would be powerful tools with which to assess the Atg8 function in apicomplexan parasite biology. While the results of preliminary studies in Plasmodium are promising, the utility of these compounds in other apicomplexans has not yet been evaluated.
In this study, three PfAtg3-Atg8 inhibitors were assessed to identify their activities and mechanisms of action in Toxoplasma. All three inhibitors blocked Toxoplasma growth in vitro at submicromolar concentrations. Interestingly, treatment with two of the three compounds induced TgAtg8 lipidation to a similar extent as induction of autophagy by treatment with monensin or extracellular starvation. Further, we found that treatment with the third compound resulted in increased levels of intracellular calcium and the induction of parasite egress from host cells. Together, our results show that the reported PfAtg3-Atg8 interaction inhibitors are active against Toxoplasma in vitro but are active through unique and previously unreported mechanisms of action.
RESULTS
Plasmodium Atg8-Atg3 inhibitors impair Toxoplasma replication in vitro.
To determine whether the three PfAtg3-Atg8 interaction inhibitors MMV007907 (MMV1), MMV001246 (MMV2), and MMV665909 (MMV3) (Table 1) had activity against Toxoplasma, parasites were inoculated onto human foreskin fibroblast (HFF) monolayers and allowed to replicate for 5 days in the presence of either 10 μM drug or the dimethyl sulfoxide (DMSO) vehicle. Treatment with all three MMVs showed a significant reduction in the area of host cell lysis, suggesting that Toxoplasma progression through the lytic cycle was impaired by drug treatment (Fig. 1A). This growth inhibition was further confirmed using an established colorimetric assay (28) that utilizes a Toxoplasma strain stably expressing the Escherichia coli β-galactosidase enzyme to generate quantitative inhibition curves for MMVs 1 to 3 (Fig. 1B). All three compounds exhibited submicromolar 50% effective concentrations (EC50s), in agreement with the range of EC50s reported for these compounds in P. falciparum and other protozoan parasites (Table 1) (29–33).
TABLE 1.
Reported activity of PfAtg3-Atg8 inhibitors against protozoan pathogensb
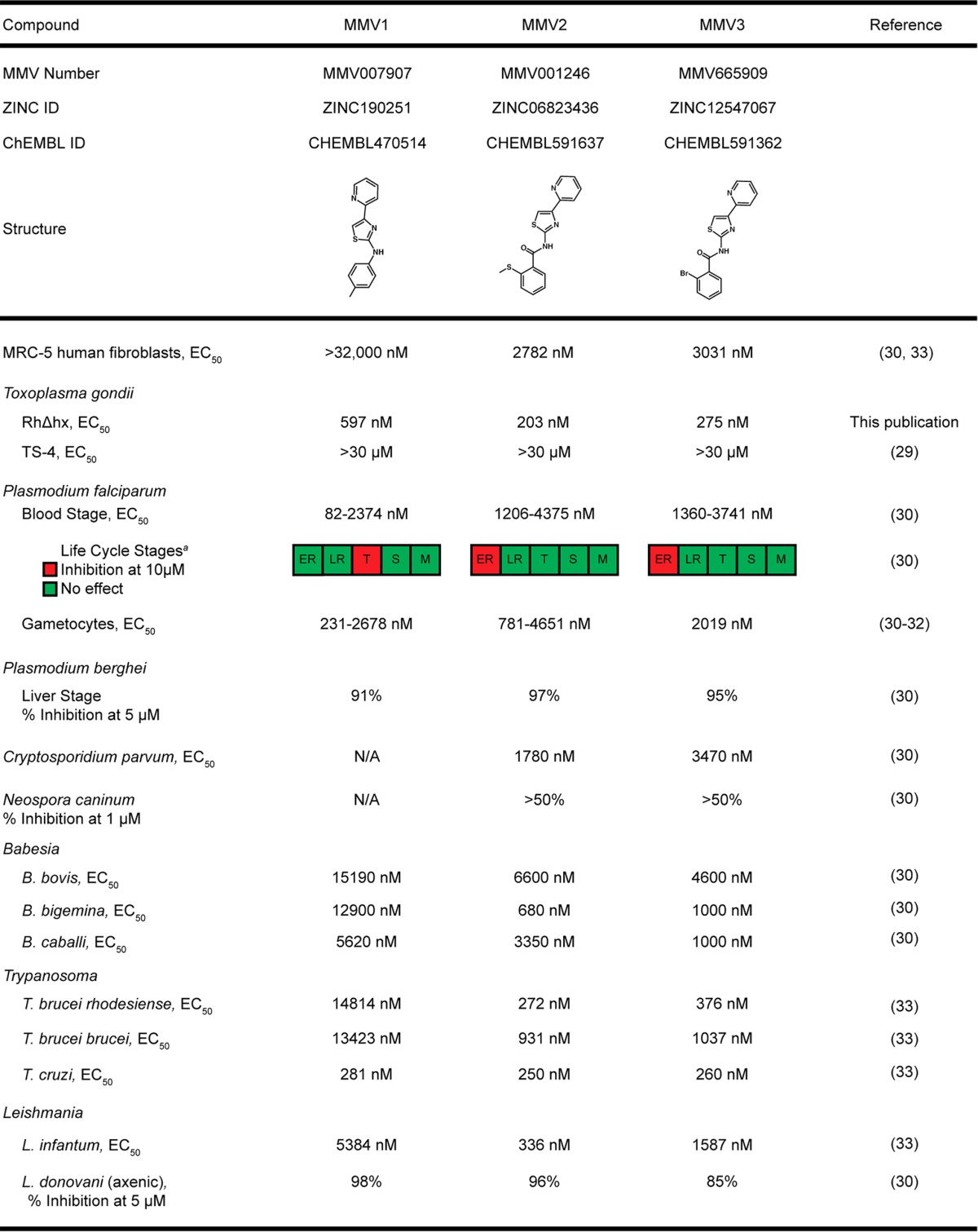
a ER, early ring; LR, late ring; T, trophozoite; S, schizont; M, merozoite.
ID, accession number; N/A, not available.
FIG 1.

PfAtg3-Atg8 inhibitors block Toxoplasma growth in vitro. (A) Representative results of a plaque assay showing impaired parasite growth over a 5-day period in the presence of MMV compounds at 10 μM compared to the growth of DMSO vehicle-treated controls. (B) Growth inhibition curves were generated to determine the EC50 of each of the three MMV compounds. Parasites expressing β-galactosidase were cultured in the presence of drug or DMSO for 4 days. Parasite growth was assessed by colorimetric assay following a 24-h incubation with the β-galactosidase substrate CPRG. Percent inhibition was calculated by normalization to the levels of inhibition for wells with uninfected HFFs (100% inhibition) and DMSO vehicle-treated infected HFFs (0% inhibition). Curves represent the averages from three independent experiments and are plotted with error bars, representing SEMs. EC50s were calculated by nonlinear regression using Prism software, and the EC50 of each compound is included below the respective curve. (C) After 24 h of treatment with MMVs 1 to 3, the inhibitors showed a concentration-dependent decrease in the number of parasites per vacuole (indicated in the key) compared to that for the DMSO vehicle-treated controls. Data represent means ± SEMs (n = 3).
Interestingly, MMVs 1 to 3 were not identified as having activity against Toxoplasma in a recent high-throughput screen of the Malaria Box compounds (29). To address this discrepancy and to confirm the validity of our experimental approach, growth inhibition assays were also conducted using pyrimethamine. From these assays, we obtained an EC50 of 1,084 nM, in agreement with previously reported EC50s for pyrimethamine in RH strain parasites (34). Additionally, the activities of MMVs 1 to 3 against Toxoplasma were further confirmed using parasite doubling assays, which showed that all three compounds impaired parasite replication in a concentration-dependent manner (Fig. 1C). Together, our results show that all three PfAtg3-Atg8 interaction inhibitors are active against Toxoplasma, even though they were not identified in a previous screen of the Malaria Box compounds to have activity against Toxoplasma.
Virtual docking suggests that MMV compounds are unable to bind within TgAtg8 W- and L-site pockets.
Having shown that MMVs 1 to 3 inhibit Toxoplasma growth, we next sought to determine whether the mechanism of action for these compounds was conserved between Plasmodium falciparum and Toxoplasma. We first assessed the conservation of amino acid residues within the W- and L-site binding pockets between PfAtg8 and TgAtg8, as these regions represent the putative sites for MMV 1 to 3 binding to PfAtg8 (26). Alignment of the PfAtg8 and TgAtg8 amino acid sequences showed a high level of conservation (66% identity, 83% similarity), including 17 out of the 19 residues comprising the PfAtg8 W- and L-site binding pockets (Fig. 2A). This significant level of conservation suggested that MMVs 1 to 3 may bind to TgAtg8 in a manner similar to that reported for PfAtg8.
FIG 2.

P. falciparum Atg3-Atg8 inhibitors are not predicted to bind to TgAtg8, despite the conservation of residues comprising the W- and L-site pockets. (A) Comparative analysis of the PfAtg8 and TgAtg8 amino acid sequences, highlighting the conservation of residues in the W site (blue), L site (red), and apicomplexan-specific loop (yellow). Two residues (F48 and L49) contributing to both the W and L sites are highlighted in purple. Numbers below the sequence alignments indicate the number of conserved residues in each region. (B) The TgAtg8 protein structure was predicted using the I-TASSER server and was used for in silico docking studies for MMVs 1 to 3. (Insets) Predicted binding sites for each of the MMV compounds. The residues located in the W- and L-site binding pockets, as well as the apicomplexan loop, on the protein surface are highlighted using the coloring scheme described above.
To test this hypothesis, we modeled the TgAtg8 protein structure using the I-TASSER server (35) and used the predicted structure for docking analyses with MMVs 1 to 3 (Fig. 2B). In parallel, we performed docking of all three compounds with the solved PfAtg8 crystal structure to confirm that the predictions generated by the docking program used in our studies (SwissDock) were in agreement with those generated in previous docking studies that used the OpenEye docking program (26). Interestingly, while all three compounds were predicted to bind within the W- and L-site pockets in PfAtg8, only MMV1 was predicted to bind to this region in TgAtg8 (Fig. 2B). Together, these results suggest that the ability of the PfAtg3-Atg8 inhibitors to bind within the TgAtg8 W- and L-site pockets may not be conserved, despite the high levels of similarity between PfAtg8 and TgAtg8 in both amino acid sequence and predicted protein structure.
MMVs 1 to 3 do not block the lipidation of TgAtg8.
Despite these potential differences in binding, we next sought to experimentally assess whether the activities of MMVs 1 to 3 in Toxoplasma were attributable to their reported mechanism of action in inhibiting the Atg3-Atg8 interaction and, thus, preventing Atg8 lipidation. Genetic manipulation of TgAtg8 lipidation by conditional knockdown of T. gondii Atg3 (TgAtg3) (14) and T. gondii Atg4 (TgAtg4) (15) resulted in defects in the maintenance of the mitochondrion and apicoplast organelles. We therefore hypothesized that if the mechanism of action for MMVs 1 to 3 was conserved in Toxoplasma, treatment with the MMV compounds would similarly affect these organelles. Interestingly, while conducting these assays, we noted that treatment with MMV1 at concentrations greater than 1 μM caused parasite egress from the host cell. This effect was unique to MMV1, as MMV2 and MMV3 did not induce egress at any of the concentrations tested. Therefore, our characterization of the effects of MMV1 treatment on the mitochondrion and apicoplast was limited to concentrations below 1 μM. In contrast, MMV2 and MMV3 were tested at concentrations up to 10 μM, as the effects of PfAtg3-Atg8 inhibitors on PfAtg8 lipidation were observed only at concentrations significantly higher than their observed EC50s (26).
No alterations in mitochondrial morphology were observed in parasites treated with MMV2 or MMV3 at 1 μM (Fig. 3A), a concentration at which both compounds significantly inhibited parasite growth (Fig. 1). However, upon treatment of intracellular parasites with 10 μM MMV2 and MMV3, we observed significant mitochondrial fragmentation (Fig. 3A). The effects on the mitochondrion were observed within the first 6 h of treatment (mean ± standard error of the mean [SEM] mitochondrial fragmentation, 65.0% ± 13.0% and 84.7% ± 8.2% for MMV2 and MMV3, respectively; n = 3), with nearly all vacuoles containing parasites with fragmented mitochondria after 24 h of treatment (mean ± SEM mitochondrial fragmentation, 94.4% ± 3.2% and 97.7% ± 0.7% for MMV2 and MMV3, respectively; n = 3) (Fig. 3B). Nonetheless, since this concentration is significantly higher than the lethal concentrations at which we did not observe mitochondrial alterations, inhibition of parasite growth by MMV2 and MMV3 is likely independent of their ability to induce mitochondrial fragmentation. Additionally, whereas the effects of monensin treatment and starvation on mitochondrial fragmentation were prevented by cotreatment with the autophagy inhibitor 3-methyladenine (3-MA) (17, 18), no such protection was observed upon cotreatment of MMV2 or MMV3 with 3-MA (Fig. 3A). Unlike the mitochondrion, the apicoplast remained intact under all treatment conditions, although defects in the positioning and segregation of this organelle into dividing parasites were frequently observed (Fig. 3C, arrowheads). The mitochondrion and apicoplast were unaffected by treatment with concentrations of MMV1 that did not induce parasite egress (Fig. 3B and C).
FIG 3.

Effects of MMV treatment on autophagy-related organelles. (A) Representative IFA images of parasites treated with DMSO, 10 mM 3-MA, MMV2, or MMV3 for 24 h and stained for the mitochondrial matrix protein F1β-ATPase (red) and with DAPI (blue). Mitochondrial fragmentation was observed with 10 μM treatment and was not protected by cotreatment with 3-MA. (B) Representative IFA images following 6 and 24 h of treatment with the DMSO vehicle or MMVs stained for F1β-ATPase (red) to visualize the mitochondrion. (C) Results of IFAs of DMSO- or MMV-treated parasites stained with ATrx1 (red) to visualize the apicoplast and DAPI (blue). The apicoplast remains intact during MMV treatment; however, organelle positioning defects are observed at 24 h (arrowheads), likely due to the observed defects in cytokinesis. Bars = 3 μm.
We next sought to directly assess whether treatment with MMVs 1 to 3 altered TgAtg8 lipidation status. In yeast and mammalian systems, Atg8 lipidation is routinely monitored by visualization of a GFP-Atg8 fusion protein, which relocalizes to cytosolic punctae representing autophagosomes following autophagy induction (36). A similar approach has been used to monitor the induction of Atg8 lipidation in Toxoplasma in response to drug treatment and starvation (14, 17). However, TgAtg8 also localizes to the outer membrane of the apicoplast in a lipidation-dependent manner (15). Given the reported function of MMVs 1 to 3 as Atg3-Atg8 interaction inhibitors, we hypothesized that treatment of intracellular parasites with MMVs 1 to 3 would result in a loss of TgAtg8 localization to the apicoplast, as was observed in TgAtg8 mutants lacking the C-terminal glycine required for lipidation (15). However, we experienced difficulties in testing this hypothesis due to the high levels of GFP-TgAtg8 present in the cytosol as the result of its overexpression (14). Thus, to facilitate the monitoring of TgAtg8 lipidation and localization in response to intracellular MMV treatment, we generated a parasite strain in which the endogenous TgAtg8 gene was replaced with the TgAtg8 cDNA sequence containing an N-terminal c-Myc epitope tag driven by the endogenous TgAtg8 promoter (Fig. 4A). Correct integration was confirmed by PCR of the TgAtg8 locus from recovered clones (Fig. 4B). Immunoblotting revealed that, as reported for endogenous TgAtg8 (14), c-Myc–TgAtg8 migrates as two bands representing cytosolic and lipidated Atg8 (Atg8-PE) (Fig. 4C). Additionally, immunofluorescence staining showed that c-Myc–Atg8 correctly localized to the apicoplast, as shown by the colocalization of c-Myc–Atg8 with the DAPI (4′,6-diamidino-2-phenylindole)-stained apicoplast immediately apical of the parasite nucleus (Fig. 4D). These results demonstrate that endogenously tagged c-Myc–TgAtg8 undergoes lipidation and localization to the apicoplast in a manner similar to that previously reported for endogenous TgAtg8 and GFP-TgAtg8 (14).
FIG 4.

Generation of the c-Myc–Atg8 parasite strain. (A) Diagram of the construct used to target the TgAtg8 genomic locus for replacement with a c-Myc epitope-tagged TgAtg8 cDNA sequence. Arrows indicate the primer pairs used to confirm proper integration at the endogenous TgAtg8 locus. The blue primer pair amplifies a fragment unique to the parental strain, the red pair amplifies a unique fragment in the recombined strain, and the black pair amplifies a 1,791-bp fragment in the parental strain and a 372-bp fragment in the recombined strain. (B) PCR products from genomic DNA (gDNA) isolated from c-Myc–TgAtg8 and parental (RH Δhx Δku80) parasites. The blue, red, and black arrows refer to the primer pairs described in panel A. Numbers to the right of the gel are molecular sizes (in base pairs). (C) Western blot (WB) of lysates from freshly egressed parental RH Δhxgprt Δku80 and c-Myc–TgAtg8 parasites, probed with antibodies recognizing the c-Myc epitope, and Toxoplasma β-tubulin as a loading control. Two bands are observed for c-Myc–TgAtg8 and represent the unmodified (Atg8) and lipidated (Atg8-PE) forms of TgAtg8. (D) Immunofluorescence assay of c-Myc–TgAtg8 parasites stained with DAPI (blue) and for c-Myc (red), showing that c-Myc–TgAtg8 localizes predominantly to the apicoplast. Bar = 3 μm.
We next used these parasites to test the hypothesis that treatment with the MMV compounds would result in a shift in TgAtg8 localization from the apicoplast to the cytosol. Intracellular parasites were treated for 6 and 24 h with 500 nM MMV1, 1 μM (data not shown) or 10 μM MMV2 and MMV3, or DMSO. Under all treatment conditions, TgAtg8 retained its punctate staining pattern and remained predominantly localized to the apicoplast (Fig. 5A). Further, immunoblotting revealed that rather than inhibiting TgAtg8 lipidation, treatment with 10 μM MMV2 and MMV3 for 6 h resulted in an increase in the lipidated form of TgAtg8, similar to the effects of autophagy induction by starvation and monensin treatment on endogenous TgAtg8 (Fig. 5B).
FIG 5.

Treatment with MMVs does not alter Atg8 localization at the apicoplast or prevent Atg8 lipidation. (A) Representative results of IFAs of c-Myc–TgAtg8 parasites after 6 and 24 h of treatment with DMSO or MMVs stained with DAPI (blue) and for c-Myc (red). c-Myc–TgAtg8 retains its apicoplast localization following MMV treatment. Bars = 3 μm. (B) Representative immunoblots of lysates from c-Myc–Atg8 parasites treated for 6 h with DMSO or MMVs. Treatment with 10 μM MMV2 and MMV3 increased the ratio of lipidated TgAtg8 (Atg8-PE) to unlipidated TgAtg8 (Atg8). No change in lipidation state was observed by treatment with 500 nM MMV1. The increased levels of Atg8-PE were similar to the increase in the level of endogenous TgAtg8 lipidation in RH Δhxgprt parasites starved extracellularly (HBSS) or treated with 1 ng/ml monensin for 6 h. Blots were probed with anti-c-Myc or anti-TgAtg8 and with anti-SAG1 as a loading control. (C) (Left) Representative IFA images of GFP-Atg8 parasites before (Control) and after 8 h of extracellular starvation in HBSS. (Right) Cotreatment with MMV1 failed to reduce the number of parasites with GFP-Atg8 punctae. Data represent means ± SEMs (n = 3). ***, P < 0.001. Bar = 3 μm.
As before, the induction of egress by MMV1 treatment limited the concentrations of this compound that we were able to assess to concentrations well below those reported to affect Atg8 lipidation in Plasmodium falciparum (26). Therefore, we could not determine whether MMV1 could block TgAtg8 lipidation in intracellular parasites. To overcome this limitation, we used established methods to monitor GFP-TgAtg8 lipidation following induction of autophagy by extracellular starvation and tested whether MMV1 could prevent the lipidation-dependent relocalization from the cytosol to punctate structures (14). As shown in Fig. 5C, no reduction of GFP-TgAtg8 punctum formation was observed by cotreatment with either 1 or 10 μM MMV1. Together, these findings show that the MMV compounds do not inhibit Atg8 lipidation in Toxoplasma and are unlikely to act as the TgAtg3-TgAtg8 interaction inhibitors reported for Plasmodium.
MMV2 and MMV3 impair Toxoplasma cytokinesis.
Given our results suggesting that MMVs 1 to 3 do not act as Atg3-Atg8 interaction inhibitors in Toxoplasma, we next sought to examine their mechanisms of action in Toxoplasma. Our initial characterization of MMVs 1 to 3 revealed that they blocked progression through the lytic cycle, as shown by the decreased area of lysis of the host cell monolayer in the presence of drug (Fig. 1B). Further studies showed that MMVs 1 to 3 impaired parasite replication in a concentration-dependent manner (Fig. 1C). Strikingly, replication was completely blocked by treatment with 2.5 μM MMV2 or MMV3, as all vacuoles contained two parasites or less after 24 h of treatment, while ∼50% of DMSO vehicle-treated vacuoles contained eight or more parasites (Fig. 1C). Importantly, the effects of MMV2 and MMV3 on replication were observed at concentrations below those found to induce mitochondrial fragmentation (Fig. 3C) or TgAtg8 lipidation (Fig. 5B). These results suggest that MMV2 and MMV3 inhibit parasite replication at low concentrations, while treatment with high concentrations results in mitochondrial fragmentation and the induction of autophagy.
While characterizing the effects of MMV2 and MMV3 on parasite replication, we observed that parasites treated with MMV2 and MMV3 at concentrations of ≥2.5 μM exhibited an abnormal, rounded morphology with a loss of uniform parasite size and shape (Fig. 6B). Staining with DAPI to visualize the parasite nuclei revealed that the parasites within these abnormal vacuoles appeared to have nuclei that had duplicated but failed to properly divide. Together, these changes suggest that the effects of MMV2 and MMV3 on parasite replication may involve impairment of cytokinesis, as the phenotypes are similar to those observed in parasites with defects in the late stages of cell division (37, 38).
FIG 6.

MMV2 and MMV3 block Toxoplasma replication and impair cytokinesis. (A) (Left) IFA images of parasites treated with 10 μM MMV2 and MMV3 for 6 h showing representative images of normal vacuoles (vacuoles treated with DMSO) and abnormal vacuoles observed with MMV2 and MMV3 treatment. Visualization of the nuclei by staining with DAPI revealed that MMV2- and MMV3-treated parasites contained distorted parasites with apparent defects in nuclear division. (Right) The percentage of abnormal vacuoles did not increase at between 6 and 24 h of treatment. Data represent means ± SEMs (n = 3). (B) Representative IFA images of normal vacuoles (vacuoles treated with DMSO) and abnormal vacuoles (vacuoles treated with MMV2 or MMV3) stained for IMC3 (red) to visualize the inner membrane complex of daughter cells and DAPI (blue). MMV-treated parasites display nuclei that are not surrounded by an IMC3-positive cytoskeleton and IMC3 structures that do not contain nuclei or apicoplasts, as observed by DAPI staining. Bars = 3 μm.
Quantification of abnormal vacuoles showed that approximately 40% of the vacuoles displayed these morphological abnormalities, and the percentage of abnormal vacuoles did not increase at between 6 and 24 h of treatment. These replication defects were further characterized by staining with an antibody against the protein inner membrane complex 3 (IMC3), a component of the parasite cytoskeleton that, together with the plasma membrane, comprises the parasite pellicle (39). While the nuclei in DMSO-treated parasites appeared uniform in size and shape and were correctly segregated into a single IMC3-stained pellicle, parasites treated with MMV2 and MMV3 contained duplicated nuclei surrounded by a single cytoskeleton, as well as IMC3 structures containing no nuclei (Fig. 6B). Together, these results suggest that the growth-inhibitory effects of MMV2 and MMV3 are a result of a rapid block of Toxoplasma replication involving disruption of cell division due to impaired daughter bud formation and nuclear division.
MMV1 induces egress by inducing calcium release from intracellular stores.
We next sought to characterize the mechanism by which MMV1 induces parasite egress from the host cells. The induction of egress by MMV1 followed kinetics similar to those observed upon treatment with 1 μM the calcium ionophore A23187, which induces nearly 100% egress within 2 min (Fig. 7A). To quantitatively assess MMV1-induced egress, we utilized an established assay to measure the release of lactate dehydrogenase (LDH) caused by lysis of the host cells upon parasite egress (40). The induction of egress by MMV1 was concentration dependent, with the egress EC50 being ∼1,150 nM (Fig. 7B).
FIG 7.

MMV1 induces parasite egress by increasing intracellular calcium levels. (A) Infected HFF monolayers were treated for 2 min with 10 μM MMVs, the DMSO vehicle, or 1 μM the calcium ionophore A23187. The percentage of egressed vacuoles was calculated by scoring at least 100 vacuoles as intracellular or egressed, Data represent means ± SEMs (n = 3). (B) The induction of egress by MMV1 was quantitatively assessed to determine an EC50 for egress by measuring the amount of lactate dehydrogenase (LDH) released from the host cells upon parasite egress. Percent egress was normalized to that achieved in DMSO-treated (0%) and 10 μM MMV1-treated (100%) wells. Data represent means ± SEMs (n = 3). CI, confidence interval. (C) Parasites lacking the calcium-dependent protein kinase TgCDPK3 fail to egress following 2 min of treatment with MMV1. Data represent means ± SEMs (n = 3) ***, P < 0.0001. WT, wild type. (D) Kinetic traces of Fluo-4 AM fluorescence (in relative fluorescence units [RFU]) in response to A23187 or MMV1. Extracellular parasites were loaded with the calcium indicator dye Fluo-4 AM to measure changes in intracellular calcium concentrations following drug treatment. Baseline fluorescence was measured for 60 s, at which time the compounds were injected into the wells (indicated by the arrow). The traces represent the average for three technical replicates, with the shaded areas representing the standard errors of the means. (E) Change in fluorescence intensity between the final baseline measurement of the number of relative fluorescence units (60 s) and the first measurement following injection of compounds (67 s). Parasites treated with the calcium chelator BAPTA-AM showed no change in intracellular calcium levels following treatment with MMV1. Data represent means ± SEMs (n = 3). (F) Representative results of a plaque assay used to assess parasite viability following treatment of extracellular parasites for 2 h with DMSO or MMV1. Control parasites were added to an HFF monolayer immediately following manual release from host cells and prior to 2 h of treatment.
We next examined whether MMV1 induces egress by a mechanism similar to that for calcium ionophores, which depends on the calcium-dependent protein kinase T. gondii CDPK3 (TgCDPK3) (41, 42). Interestingly, MMV1 failed to induce egress in parasites lacking TgCDPK3 (Fig. 7C), indicating that MMV1 induces egress by a mechanism similar to that used by calcium ionophores. We therefore hypothesized that treatment of extracellular parasites with MMV1 would result in the release of intracellular calcium, as has been shown for ionophores (43). To test this, freshly egressed parasites were loaded with the calcium indicator dye Fluo-4 acetoxymethyl ester (AM) and the fluorescence intensities were monitored prior to and immediately following treatment with MMV1, A23187, or the DMSO vehicle. Treatment with MMV1 resulted in increased Fluo-4 AM fluorescence, with the kinetics and peak intensities being similar to those obtained by treatment with A23187 (Fig. 7D). Treatment with the cell-permeant calcium chelator BAPTA-AM [1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester] completely blocked the increase in Fluo-4 AM fluorescence in response to MMV1 treatment (Fig. 7E). Lastly, we assessed whether the effect of prolonged treatment with MMV1 is a recapitulation of the loss of parasite viability observed with extracellular ionophore treatment, termed ionophore-induced death (44). Indeed, treatment of extracellular parasites with MMV1 for 2 h dramatically reduced parasite viability, as illustrated by the reduction in growth compared to that of the DMSO-treated controls, as measured by plaque assay (Fig. 7F). Together, our results suggest that MMV1-induced egress involves the release of calcium from intracellular stores and activation of the calcium-mediated signaling pathways that have previously been implicated in parasite egress.
DISCUSSION
In this study, we assessed three P. falciparum Atg3-Atg8 interaction inhibitors to determine whether they acted in a similar manner in Toxoplasma gondii. Our results show that despite high levels of conservation of the proposed binding sites for these compounds on TgAtg8, these compounds do not block TgAtg8 lipidation. Despite the lack of a conserved mechanism of action, all three compounds were found to inhibit Toxoplasma growth in a dose-dependent manner, with EC50s being in the submicromolar range. We identified that two of these compounds (MMV001246 [MMV2] and MMV665909 [MMV3]) inhibit Toxoplasma replication by disrupting daughter bud formation and nuclear segregation during parasite replication. Additionally, we found that treatment with the third compound (MMV007907 [MMV1]) resulted in the release of calcium from the intracellular stores, resulting in parasite egress from the host cell.
Numerous PfAtg3-Atg8 inhibitor compounds have been identified using biophysical approaches (25–27); however, we chose to focus on MMVs 1 to 3 for the following reasons. First, MMV1 is one of the few PfAtg3-Atg8 inhibitors that have been found to alter PfAtg8 lipidation status when parasites are treated in culture (26). Second, all three of the compounds tested herein were identified from the Medicines for Malaria Venture Malaria Box, a collection of 200 drug-like and 200 probe-like molecules that is available for researchers free of charge. Importantly, MMVs 1 to 3 are active against other important human parasites, including Cryptosporidium spp., Trypanosoma spp., and Leishmania spp. (Table 1). Collectively, these pathogens represent a significant disease burden in humans, and treatment of infections caused by these pathogens is threatened by the development of resistance to the limited number of therapeutics currently available. Therefore, the activity of MMVs 1 to 3 against multiple protozoans suggested that disruption of the Atg3-Atg8 interaction in these parasites may represent a potential avenue for the development of novel broad-spectrum antiparasitics.
To address the possibility of a conserved mechanism of action for MMVs 1 to 3 against organisms other than Plasmodium, we evaluated these compounds for their ability to similarly disrupt the Atg3-Atg8 interaction in the closely related apicomplexan Toxoplasma. We hypothesized that the similarity between P. falciparum and Toxoplasma Atg8 proteins would allow the mechanism of action to be conserved between the two species. Interestingly, our in silico docking experiments failed to identify the predicted interactions of MMV2 and MMV3 with the TgAtg8 W- and L-site pockets, suggesting that these compounds may not bind to TgAtg8 in a fashion similar to that described in P. falciparum (Fig. 2B). It is possible that the absence of predicted docking at the conserved binding sites could be due to limitations of the experimental approach that we used in our study, including using a predicted TgAtg8 structure and performing docking with a single, rigid TgAtg8 conformation. However, even in the absence of a more robust docking analysis, the results of our in vitro studies are consistent with the conclusion that these compounds do not inhibit TgAtg8 lipidation by interfering with TgAtg8 interactions through the W- and L-site binding pockets. Further, although the residues that comprise the W- and L-site binding pockets are highly conserved between PfAtg8 and TgAtg8, it is possible that amino acid substitutions in the residues proximal to these binding pockets could prevent the binding of these compounds to TgAtg8, as shown for P. yoelii Atg8 (26). The PfAtg8 W- and L-site pockets targeted by MMVs 1 to 3 were proposed to be ideal targets for small-molecule inhibitors due to the differences in the structure and electrostatic potential of these pockets from those of human Atg8 homologues (25). However, an additional set of compounds that inhibits the PfAtg3-Atg8 interaction by binding to the apicomplexan loop structure on PfAtg8 has recently been identified (27). As this structure is unique to apicomplexan Atg8 proteins, it provides an additional avenue for targeted disruption of the parasite Atg3-Atg8 interaction while avoiding effects on the host. Whether any of the compounds that target the PfAtg8 apicomplexan loop have activity against Toxoplasma or other apicomplexans remains untested. However, given the differences in the amino acid sequence present in this loop in Toxoplasma from the sequence present in this loop in Plasmodium (Fig. 2A), identification of bona fide TgAtg3-Atg8 inhibitors may require structural resolution of the TgAtg3-Atg8 interaction and/or the development of biophysical screening approaches like those used in P. falciparum.
Our characterization of MMVs 1 to 3 shows that all three compounds inhibit Toxoplasma at submicromolar concentrations. This is in contrast to the findings of a previous high-throughput screen that failed to identify any of the three MMVs as having activity against Toxoplasma (29). However, as noted by other authors (29), the EC50s of certain 2,4-diamino-quinazoline-based compounds that they observed were significantly higher than the values reported in a separate study with Toxoplasma (45). The authors attribute these differences to their use of the TS-4 mutant derived from the standard RH strain, which could also explain the differences between the activities observed in their study and activities of MMVs 1 to 3 that we observed. Importantly, the TS-4 mutants were also less sensitive to pyrimethamine (EC50 = 3.62 μM) than the RH parasites (EC50 = 0.9 μM) (34), suggesting that the TS-4 strain is less susceptible to a variety of compounds. Additionally, as shown in Table 1, the EC50s obtained from high-throughput screens are highly variable, likely due to differences in the methods used to assess compound efficacy between studies. In contrast, our study characterized the effects of MMVs 1 to 3 on multiple aspects of Toxoplasma biology using standard Toxoplasma strains and assays. Our findings illustrate the importance of using multiple, independent methodologies for screening and validating the activity of the Malaria Box compounds across species.
Our studies revealed that inhibition of Toxoplasma by MMV2 and MMV3 involves impairment of parasite replication, as shown by the changes in parasite morphology and defects in nuclear segregation into daughter parasites in Fig. 6. Additionally, at higher concentrations, MMV2 and MMV3 induced fragmentation of the mitochondrion and increased lipidation of TgAtg8 (Fig. 5), similar to the effects of monensin treatment and starvation (17, 18). These findings provide further evidence for an autophagic cell death pathway in Toxoplasma that can be induced in response to various stressors, including drug treatment. However, unlike monensin and starvation, the autophagy inhibitor 3-MA did not block the effects of MMV2 and MMV3 on the mitochondrion. One possible explanation for this observation is that the induction of autophagy may occur downstream of mitochondrial fragmentation. In this scenario, mitochondrial disruption could be induced by several stimuli, including some that are blocked by 3-MA's inhibition of phosphoinositide 3-kinase (PI3K) (including starvation and monensin), as well as others that target the mitochondria through alternative, PI3K-independent mechanisms (like MMV2 and MMV3). We propose that in the case of MMV2 and MMV3, autophagy is likely induced following mitochondrial fragmentation, and the subsequent induction of TgAtg8 lipidation may represent cell death with autophagy rather than cell death by autophagy (46).
In addition to the novel mechanisms of action identified for MMV2 and MMV3, we also found that MMV1 can induce parasite egress from the host cells. This effect is comparable to the activity of the calcium ionophore A23187, and like the A23187-induced egress, the MMV1-induced egress is dependent on signaling through TgCDPK3. As shown in Fig. 7D and E, MMV1 triggers the release of calcium from intracellular stores; however, the exact mechanism by which this occurs remains unclear. The chemical structure of MMV1 precludes it from coordinating with calcium ions and transporting them across membranes in a manner similar to that for A23187. Instead, MMV1 may bind directly to receptors or ion channels to trigger calcium release, although such receptors and channels remain to be identified in Toxoplasma. Alternatively, MMV1 may act similarly to other small molecules that stimulate calcium release and parasite egress through activation of the cGMP-dependent protein kinase G (PKG) signaling pathway (40, 47). Further studies are needed to determine whether the effects of MMV1 are dependent on PKG signaling and to determine which intracellular stores release calcium in response to MMV1 treatment.
Together, our studies revealed that MMVs 1 to 3 inhibit Toxoplasma at concentrations below those reported to have cytotoxic effects in human fibroblasts (Table 1) (30, 33). However, it is worth noting that the human cytotoxicity data were obtained from experiments in which subconfluent MRC-5 fibroblasts were grown for 72 h in the presence of each compound. As a result, the values obtained from these experiments could also be attributed to inhibition of cell proliferation by these compounds. Other studies exploring the cytotoxic effects of MMVs 1 to 3 showed that while MMV1 was well tolerated, MMV2 and MMV3 displayed cytotoxic effects on zebrafish embryos and human liver hepatocytes at low micromolar concentrations (30). It is possible that the inhibitory effects of MMV2 and MMV3 on cell division observed in Toxoplasma are conserved in higher eukaryotes and contribute to the cytotoxicity of these compounds. Interestingly, we did not observe any negative effects on HFFs exposed to MMV2 and MMV3 at 10 μM, suggesting that these compounds may be selectively toxic to cells undergoing active proliferation.
Preliminary pharmacokinetic studies in mice suggest that MMVs 1 to 3 are highly bound to plasma proteins (>95%) (30). Additionally, mice given an oral dose of 140 μg/kg of body weight failed to show plasma concentrations greater than 1 μM within 9 h of administration, suggesting that the efficacy of these compounds in vivo may be limited due to poor bioavailability (30). However, whether these compounds can be administered at dosages that are efficacious in an in vivo model of toxoplasmosis remains untested. While MMVs 1 to 3 may currently be limited to use in vitro for target identification, future structure-activity relationship studies may lead to the development of derivatives of these compounds with better pharmacological properties. As MMVs 1 to 3 already display activity against a variety of protozoan pathogens, further characterization of their mechanisms of action and identification of their specific targets hold promise toward the development of compounds for use as broad-spectrum antiparasitics.
MATERIALS AND METHODS
Chemicals and reagents.
The compounds used for the initial screens against Toxoplasma were from the Malaria Box compound library from the Medicines for Malaria Venture (48–50). For further studies, MMV007907 (referred to here as MMV1) was obtained from Hit2Lead (compound identification number 5664106), while MMV001246 (MMV2) and MMV665909 (MMV3) were obtained from Enamine Ltd. (catalogue numbers Z29466516 and Z29467539, respectively). A23187 (Sigma-Aldrich) was dissolved in DMSO at 1 mM and stored in single-use aliquots at −20°C. The cell-permeant calcium chelator BAPTA-AM (Sigma-Aldrich) was dissolved in DMSO at 50 mM. The antibodies used in this study included mouse anti-F1β (1:2,000), mouse anti-c-Myc (1:1,000; clone 9B11; Cell Signaling Technology), mouse anti-SAG1 (1:2,000; GenWay Biotech), rat anti-IMC3 (1:2,000) (39), mouse anti-ATrx1 (1:2,000) (51), and rabbit anti-toxoplasma β-tubulin (1:2,000) (37). Fluorophore-conjugated secondary antibodies for immunofluorescence assays (IFAs) (Alexa Fluor; Thermo Fisher) were used at 1:2,000 dilutions. Horseradish peroxidase-conjugated secondary antibodies (GE Healthcare) were used for immunoblotting at 1:2,000 dilutions.
Bioinformatics and modeling of TgAtg8.
Amino acid sequence alignments were performed using the Clustal Omega program (52). To generate a structural model of TgAtg8, the TgAtg8 protein sequence was provided as input to the I-TASSER online server (35) using default settings and no additional restraints or templates. Docking simulations were done using the SwissDock server (53, 54), providing the solved PfAtg8 structure (PDB accession number 4EOY) (25) or the output of the I-TASSER TgAtg8 model as the input for the target and the ZINC accession numbers for the MMV compounds (Table 1) as the input for the ligands. All docking was done using the default/accurate specifications and without defining a region of interest. Docking results were visualized and screened using the ViewDock tool in the UCSF Chimera program (55, 56).
Parasite culture.
Parasites were maintained by serial passage in human foreskin fibroblasts (HFFs) in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% 100 U/ml penicillin plus 100 μg/ml streptomycin (PenStrep), and 2 mM l-glutamine and incubated at 37°C in 5% CO2. All drug treatment experiments were conducted in DMEM supplemented with 1% FBS, 1% PenStrep, and 2 mM l-glutamine. The type I RH Δhxgprt parasite strain (57) was used to assess parasite doubling time, plaque formation, organelle integrity, and intracellular calcium levels in response to MMV treatment. Egress assays were performed using RH Δhxgprt and TgCDPK3 knockout parasites generated in the type II Pru Δku80 background strain (58) and the vector and cloning approach for generation of the type I TgCDPK3 knockout strain described in reference 41. Extracellular starvation assays were done using RH Δhxgprt parasites ectopically expressing GFP-TgAtg8 (14). Endogenous c-Myc epitope tagging of TgAtg8 was performed using RH Δhxgprt Δku80 parasites (59).
Assessment of parasite viability in vitro.
For plaque assays, 500 syringe-lysed parasites were added to a confluent HFF monolayer in a 12-well plate and allowed to propagate for 5 days at 37°C in 5% CO2. For plaque assays assessing the effect of MMV1 treatment on extracellular parasites, parasites were added to the HFF monolayer immediately upon syringe lysis or following a 2-h incubation in 1% FBS medium containing MMV1 or the DMSO vehicle. After 5 days of incubation, the cells were fixed with methanol and stained using crystal violet (Sigma-Aldrich) to observe regions of host cell lysis. To assess the parasite doubling time, syringe-lysed parasites were inoculated into confluent HFF monolayers in 12-well plates and allowed to invade for 2 h. The monolayers were then washed three times with medium to remove uninvaded parasites, and medium containing drug or vehicle was added to the wells. At 24 h postinfection, the cells were fixed with methanol and stained with stain from a Differential Quik stain kit (Polysciences, Inc.). For each treatment, at least 100 vacuoles from three biological replicates were assessed for the number of parasites per vacuole. Graphs were generated using Prism software (v7.0a; GraphPad).
To generate growth inhibition curves to determine the EC50s of the MMV compounds, HFF monolayers in 96-well plates were inoculated with 100 syringe-lysed β-galactosidase-expressing parasites (28). At 4 days after inoculation, chlorophenol red-β-d-galactopyranoside (CPRG) was added to each well at a final concentration of 100 μM. The plates were read 24 h after the addition of CPRG on a Synergy H1 plate reader (BioTek) with excitation at 570 nm and emission at 630 nm. Averages from three biological replicates were used to generate growth inhibition curves using Prism software, and the results were normalized to those for wells with uninfected HFFs (100% inhibition) and the dimethyl sulfoxide (DMSO) vehicle (0% inhibition). Results from three independent replicates were plotted in Prism software, and curves were fit to the log-transformed (x axis) data by nonlinear regression, using the four-parameter, variable slope settings.
Analysis of parasite egress.
Freshly lysed parasites were inoculated into confluent HFF monolayers and allowed to replicate overnight. On the following day, the monolayers were washed with Hanks' buffered saline solution (HBSS), after which HBSS containing drug or DMSO as a vehicle control was added to the cells. Following a 2-min incubation at 37°C, the plates were fixed with methanol and stained with Differential Quik stain. Percent egress was determined by counting at least 100 vacuoles from biological triplicates under each condition.
Induction of egress by MMV1 was quantitatively measured using a CytoTox-One homogeneous membrane integrity assay (Promega). This assay measures the amount of lactate dehydrogenase (LDH) released from host cells as a result of parasite egress (40). Confluent HFF monolayers were infected with 5 × 104 parasites per well in 96-well format. At 24 h postinfection, the monolayers were washed with DMEM supplemented with 1% FBS and were incubated in the same medium with drug (MMV1) in a 2-fold serial dilution or vehicle (DMSO), with each treatment being done in technical triplicate. After a 15-min incubation at 37°C, 50 μl of CytoTox-One assay buffer with substrate was added to each well and the plate was incubated for 5 min at 25°C. The reaction was stopped by the addition of 25 μl stop solution to each well, and the plates were read on a plate reader with excitation at 560 nm and emission at 590 nm. The values were analyzed using Prism software and were normalized to the results obtained with 10 μM MMV1 (100%) inhibition and DMSO (0% inhibition). Results represent averaged values from three independent biological replicates, and the log-transformed (x axis) data were fit using nonlinear regression (four parameters, variable slope) to determine EC50s.
Endogenous tagging of TgAtg8.
Total RNA from RH Δhxgprt Δku80 parasites was isolated using an RNeasy minikit (Qiagen), and 2 μg of total RNA was reverse transcribed using an Omniscript reverse transcription kit (Qiagen) to generate cDNA. The TgAtg8 coding sequence (EuPathDB GeneID TGGT1_254120) was amplified using primers 1 and 2, and primers 3 to 5 were used to introduce an N-terminal c-Myc epitope tag and overhangs for In-Fusion cloning (see Table S1 in the supplemental material). Primers 6 to 9 were used to amplify the 5′ and 3′ homologous sequences from RH Δhxgprt Δku80 genomic DNA isolated using a DNeasy kit (Qiagen). The SAG1 3′untranslated region (UTR) was amplified using primers 10 and 11. These fragments were cloned into a modified pBluescript KS(−) vector containing the hypoxanthine-xanthine-guanine phosphoribosyltransferase (HXGPRT) selection cassette using an In-Fusion HD cloning kit (Clontech) to generate the final plasmid depicted in Fig. 4A.
For the generation of c-Myc–Atg8 parasite strains, 100 μg of BglI-linearized plasmid DNA was transfected into freshly lysed RH Δhxgprt Δku80 parasites. Parasites were subjected to selection with 25 μg/ml mycophenolic acid with 25 μg/ml xanthine for three passages, and surviving parasites were cloned by limiting dilution into 96-well plates. Single, independent clones were screened for proper integration by genomic PCR using primers 1, 2, 10, and 12 to 14, and positive clones were confirmed by immunoblotting and immunofluorescence assays by staining for c-Myc.
Induction of autophagy.
To assess TgAtg8 lipidation status in response to drug treatment, confluent HFF monolayers in T-175 tissue culture flasks were inoculated with 2 × 107 freshly lysed parasites. At 36 h postinfection, the cells were washed with 1% FBS medium and replaced with 1% FBS medium containing vehicle or drug. After 6 h, the cells were washed twice with medium to remove extracellular parasites. Intracellular parasites were then scraped and passed through a 27-gauge needle to release the parasites from the host cells, and the recovered parasites were washed twice with ice-cold phosphate-buffered saline (PBS). Assessment of the endogenous TgAtg8 lipidation status in response to extracellular starvation was done as was described above for the GFP-TgAtg8 starvation experiments, but intracellular parasites that were manually released from cells prior to HBSS starvation were used.
For the assessment of GFP-TgAtg8 punctum formation, freshly lysed GFP-TgAtg8 parasites were washed in HBSS and resuspended in HBSS with the DMSO vehicle or drug. The parasites were incubated for 8 h at 37°C in 5% CO2 and were added to poly-l-lysine-coated coverslips for imaging. At least 100 parasites from three biological replicates were counted.
Immunoblotting.
Parasite pellets were lysed for 30 min in cold radioimmunoprecipitation assay (RIPA) buffer containing 1% sodium deoxycholate and supplemented with a cOmplete Mini protease inhibitor tablet (Roche), sonicated with a microtip sonicator once for 10 s, and centrifuged for 5 min at 16,000 × g to remove insoluble debris. The cleared lysate equivalent of 1 × 107 parasites per load was separated on 4 to 20% Mini-Protean TGX gels (Bio-Rad) and transferred to a nitrocellulose membrane for immunoblotting. Membranes were blocked for 30 min in a solution of 5% milk–Tris-buffered saline with 0.1% Tween 20 (TBST) and probed with primary antibodies overnight at 4°C. Following three 10-min washes in TBST, secondary antibodies were incubated for 45 min at room temperature in milk-TBST. The membranes were again washed three times for 10 min each time in TBST, and proteins were detected with SuperSignal West Femto substrate (Thermo Fisher) and imaged on a FluorChem R imager (Bio-Techne).
Immunofluorescence assays.
For all IFAs, infected HFFs (grown on coverslips) were washed with PBS, fixed with 4% paraformaldehyde in PBS for 15 min, and washed two additional times with PBS. The fixed, infected monolayers were then incubated for 30 min with 3% bovine serum albumin (BSA)–PBS to block and permeabilized for 15 min in BSA–PBS with 0.2% Triton X-100 (PBS-T). Samples were then incubated overnight with primary antibodies in BSA–PBS-T at 4°C, washed 3 times for 10 min each time with PBS, and incubated in secondary antibodies for 45 min in BSA-PBS. Coverslips were then washed twice with PBS for 10 min, stained with DAPI (4′,6-diamidino-2-phenylindole) in PBS for 10 min, and washed two more times with PBS for 10 min. The coverslips were then mounted using Vectashield antifade mounting medium (Vector Laboratories) and imaged on a Nikon Eclipse 80i microscope.
Cytosolic Ca2+ measurements.
Freshly egressed parasites were passed through a 3-μm-pore-size filter to remove host cell debris and were washed once with intracellular buffer (IB; 5 mm NaCl, 144 mm KCl, 5.6 mm d-glucose, 1 mm MgCl2, 2 mm EGTA, 25 mm HEPES, pH 7.4). The parasites were then loaded with 2.5 μM Fluo-4 AM calcium indicator dye (Thermo Fisher) with or without 50 μM BAPTA-AM in IB for 1 h at room temperature. Following a 30-min wash with IB to remove extracellular dye, the parasites were resuspended in IB to a final concentration of 107 parasites per ml, and 100 μl was transferred to each well of a 96-well plate. A Synergy H1 (BioTek) plate reader was used to monitor the Fluo-4 AM fluorescence at 488-nm excitation and 524-nm emission wavelengths. Experiments were performed under each treatment condition in triplicate, with measurements being taken every 7 s. After a 1-min period to establish the baseline fluorescence levels, 10 μl of HBSS (calcium and magnesium free) containing DMSO vehicle, A23187, or MMV1 was added to each well using the Synergy H1 injectors, and measurements were taken every 5 s for 10 min. Data were analyzed using Prism software, with graphs representing the Fluo4-AM fluorescence intensities after DMSO subtraction.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Sibley, Marc-Jan Gubbels, and Peter Bradley for providing antibodies and Vern Carruthers for providing the RH Δhxgprt Δku80 parasite strain. We thank Chunlin Yang for the generation of the Pru Δku80 ΔCDPK3 parasites used in this study. We also thank the W. J. Sullivan, Jr., and G. Arrizabalaga labs and members of the IUSM Biology of Intracellular Pathogens group for their helpful discussions and suggestions.
This research was supported by grants from the National Institutes of Health (AI125822 to G.A. and W.J.S. and R21AI119516 to G.A.) and the PhRMA Foundation (a predoctoral fellowship in pharmacology/toxicology to J.M.V.). K.A.L. has been funded by a National Institutes of Health training grant (AI060519) and a fellowship from the American Heart Association (16PRE27260042).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01489-17.
REFERENCES
- 1.Feng Y, He D, Yao Z, Klionsky DJ. 2014. The machinery of macroautophagy. Cell Res 24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N, Klionsky DJ. 2007. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 27:19–40. doi: 10.1146/annurev.nutr.27.061406.093749. [DOI] [PubMed] [Google Scholar]
- 3.Russell RC, Yuan H-X, Guan K-L. 2014. Autophagy regulation by nutrient signaling. Cell Res 24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizushima N, Levine B. 2010. Autophagy in mammalian development and differentiation. Nat Cell Biol 12:823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaushik S, Cuervo AM. 2015. Proteostasis and aging. Nat Med 21:1406–1415. doi: 10.1038/nm.4001. [DOI] [PubMed] [Google Scholar]
- 6.Choi J, Park S, Biering SB, Selleck E, Liu CY, Zhang X, Fujita N, Saitoh T, Akira S, Yoshimori T, Sibley LD, Hwang S, Virgin HW. 2014. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity 40:924–935. doi: 10.1016/j.immuni.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colombo MI. 2005. Pathogens and autophagy: subverting to survive. Cell Death Differ 12:1481–1483. doi: 10.1038/sj.cdd.4401767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin M, Klionsky DJ. 2013. The core molecular machinery of autophagosome formation, p 25–45. In Wang H-G. (ed), Autophagy and cancer. Springer, New York, NY. [Google Scholar]
- 9.Mizushima N, Yoshimori T, Ohsumi Y. 2011. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 10.Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z. 2011. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 20:444–454. doi: 10.1016/j.devcel.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, Ohsumi Y. 2000. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol 151:263–276. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki K, Ohsumi Y. 2007. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett 581:2156–2161. doi: 10.1016/j.febslet.2007.01.096. [DOI] [PubMed] [Google Scholar]
- 13.Duszenko M, Ginger ML, Brennand A, Gualdrón-López M, Colombo M- I, Coombs GH, Coppens I, Jayabalasingham B, Langsley G, de Castro SL, Menna-Barreto R, Mottram JC, Navarro M, Rigden DJ, Romano PS, Stoka V, Turk B, Michels PA. 2011. Autophagy in protists. Autophagy 7:127–158. doi: 10.4161/auto.7.2.13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Besteiro S, Brooks CF, Striepen B, Dubremetz J-F. 2011. Autophagy protein Atg3 is essential for maintaining mitochondrial integrity and for normal intracellular development of Toxoplasma gondii tachyzoites. PLoS Pathog 7:e1002416. doi: 10.1371/journal.ppat.1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong-Hap MA, Mouammine A, Daher W, Berry L, Lebrun M, Dubremetz J-F, Besteiro S. 2013. Regulation of ATG8 membrane association by ATG4 in the parasitic protist Toxoplasma gondii. Autophagy 9:1334–1348. doi: 10.4161/auto.25189. [DOI] [PubMed] [Google Scholar]
- 16.Walker DM, Mahfooz N, Kemme KA, Patel VC, Spangler M, Drew ME. 2013. Plasmodium falciparum erythrocytic stage parasites require the putative autophagy protein PfAtg7 for normal growth. PLoS One 8:e67047. doi: 10.1371/journal.pone.0067047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavine MD, Arrizabalaga G. 2012. Analysis of monensin sensitivity in Toxoplasma gondii reveals autophagy as a mechanism for drug induced death. PLoS One 7:e42107. doi: 10.1371/journal.pone.0042107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh D, Walton JL, Roepe PD, Sinai AP. 2012. Autophagy is a cell death mechanism in Toxoplasma gondii. Cell Microbiol 14:589–607. doi: 10.1111/j.1462-5822.2011.01745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitamura K, Kishi-Itakura C, Tsuboi T, Sato S, Kita K, Ohta N, Mizushima N. 2012. Autophagy-related Atg8 localizes to the apicoplast of the human malaria parasite Plasmodium falciparum. PLoS One 7:e42977. doi: 10.1371/journal.pone.0042977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomlins AM, Ben-Rached F, Williams RA, Proto WR, Coppens I, Ruch U, Gilberger TW, Coombs GH, Mottram JC, Müller S, Langsley G. 2013. Plasmodium falciparum ATG8 implicated in both autophagy and apicoplast formation. Autophagy 9:1540–1552. doi: 10.4161/auto.25832. [DOI] [PubMed] [Google Scholar]
- 21.Sheiner L, Vaidya AB, McFadden GI. 2013. The metabolic roles of the endosymbiotic organelles of Toxoplasma and Plasmodium spp. Curr Opin Microbiol 16:452–458. doi: 10.1016/j.mib.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lévêque MF, Berry L, Cipriano MJ, Nguyen H-M, Striepen B, Besteiro S. 2015. Autophagy-related protein ATG8 has a noncanonical function for apicoplast inheritance in Toxoplasma gondii. mBio 6:e01446-. doi: 10.1128/mBio.01446-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cervantes S, Bunnik EM, Saraf A, Conner CM, Escalante A, Sardiu ME, Ponts N, Prudhomme J, Florens L, Le Roch KG. 2014. The multifunctional autophagy pathway in the human malaria parasite, Plasmodium falciparum. Autophagy 10:80–92. doi: 10.4161/auto.26743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brennand A, Gualdrón-López M, Coppens I, Rigden DJ, Ginger ML, Michels PAM. 2011. Autophagy in parasitic protists: unique features and drug targets. Mol Biochem Parasitol 177:83–99. doi: 10.1016/j.molbiopara.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Hain AUP, Weltzer RR, Hammond H, Jayabalasingham B, Dinglasan RR, Graham DRM, Colquhoun DR, Coppens I, Bosch J. 2012. Structural characterization and inhibition of the Plasmodium Atg8-Atg3 interaction. J Struct Biol 180:551–562. doi: 10.1016/j.jsb.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hain AUP, Bartee D, Sanders NG, Miller AS, Sullivan DJ, Levitskaya J, Meyers CF, Bosch J. 2014. Identification of an Atg8-Atg3 protein-protein interaction inhibitor from the Medicines for Malaria Venture Malaria Box active in blood and liver stage Plasmodium falciparum parasites. J Med Chem 57:4521–4531. doi: 10.1021/jm401675a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hain AUP, Miller AS, Levitskaya J, Bosch J. 2016. Virtual screening and experimental validation identify novel inhibitors of the Plasmodium falciparum Atg8-Atg3 protein-protein interaction. ChemMedChem 11:900–910. doi: 10.1002/cmdc.201500515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McFadden DC, Seeber F, Boothroyd JC. 1997. Use of Toxoplasma gondii expressing beta-galactosidase for colorimetric assessment of drug activity in vitro. Antimicrob Agents Chemother 41:1849–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyom FF, Fokou PVT, Tchokouaha LRY, Spangenberg T, Mfopa AN, Kouipou RMT, Mbouna CJ, Donfack VFD, Zollo PHA. 2014. Repurposing the open access Malaria Box to discover potent inhibitors of Toxoplasma gondii and Entamoeba histolytica. Antimicrob Agents Chemother 58:5848–5854. doi: 10.1128/AAC.02541-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Voorhis WC, Adams JH, Adelfio R, Ahyong V, Akabas MH, Alano P, Alday A, Alemán Resto Y, Alsibaee A, Alzualde A, Andrews KT, Avery SV, Avery VM, Ayong L, Baker M, Baker S, Ben Mamoun C, Bhatia S, Bickle Q, Bounaadja L, Bowling T, Bosch J, Boucher LE, Boyom FF, Brea J, Brennan M, Burton A, Caffrey CR, Camarda G, Carrasquilla M, Carter D, Belen Cassera M, Chih-Chien Cheng K, Chindaudomsate W, Chubb A, Colon BL, Colón-López DD, Corbett Y, Crowther GJ, Cowan N, D'Alessandro S, Le Dang N, Delves M, DeRisi JL, Du AY, Duffy S, Abd El-Salam El-Sayed S, Ferdig MT, Fernández Robledo JA, Fidock DA, et al. 2016. Open source drug discovery with the Malaria Box compound collection for neglected diseases and beyond. PLoS Pathog 12:e1005763. doi: 10.1371/journal.ppat.1005763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duffy S, Avery VM. 2013. Identification of inhibitors of Plasmodium falciparum gametocyte development. Malar J 12:408. doi: 10.1186/1475-2875-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lucantoni L, Fidock DA, Avery VM. 2016. Luciferase-based, high-throughput assay for screening and profiling transmission-blocking compounds against Plasmodium falciparum gametocytes. Antimicrob Agents Chemother 60:2097–2107. doi: 10.1128/AAC.01949-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaiser M, Maes L, Tadoori LP, Spangenberg T, Ioset J-R. 2015. Repurposing of the open access Malaria Box for kinetoplastid diseases identifies novel active scaffolds against trypanosomatids. J Biomol Screen 20:634–645. doi: 10.1177/1087057115569155. [DOI] [PubMed] [Google Scholar]
- 34.Reynolds MG, Oh J, Roos DS. 2001. In vitro generation of novel pyrimethamine resistance mutations in the Toxoplasma gondii dihydrofolate reductase. Antimicrob Agents Chemother 45:1271–1277. doi: 10.1128/AAC.45.4.1271-1277.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mizushima N, Yoshimorim T, Levine B. 2010. Methods in mammalian autophagy. Res Cell 140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrissette NS, Sibley LD. 2002. Disruption of microtubules uncouples budding and nuclear division in Toxoplasma gondii. J Cell Sci 115(Pt 5):1017–1025. [DOI] [PubMed] [Google Scholar]
- 38.Varberg JM, Padgett LR, Arrizabalaga G, Sullivan WJ. 2016. TgATAT-mediated α-tubulin acetylation is required for division of the protozoan parasite Toxoplasma gondii. mSphere 1(1):e00088-. doi: 10.1128/mSphere.00088-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gubbels M-J, Wieffer M, Striepen B. 2004. Fluorescent protein tagging in Toxoplasma gondii: identification of a novel inner membrane complex component conserved among Apicomplexa. Mol Biochem Parasitol 137:99–110. doi: 10.1016/j.molbiopara.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Lourido S, Tang K, Sibley LD. 2012. Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J 31:4524–4534. doi: 10.1038/emboj.2012.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garrison E, Treeck M, Ehret E, Butz H, Garbuz T, Oswald BP, Settles M, Boothroyd J, Arrizabalaga G. 2012. A forward genetic screen reveals that calcium-dependent protein kinase 3 regulates egress in Toxoplasma. PLoS Pathog 8:e1003049. doi: 10.1371/journal.ppat.1003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Treeck M, Sanders JL, Gaji RY, LaFavers KA, Child MA, Arrizabalaga G, Elias JE, Boothroyd JC. 2014. The calcium-dependent protein kinase 3 of Toxoplasma influences basal calcium levels and functions beyond egress as revealed by quantitative phosphoproteome analysis. PLoS Pathog 10:e1004197. doi: 10.1371/journal.ppat.1004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carruthers VB, Sibley LD. 1999. Mobilization of intracellular calcium stimulates microneme discharge in Toxoplasma gondii. Mol Microbiol 31:421–428. doi: 10.1046/j.1365-2958.1999.01174.x. [DOI] [PubMed] [Google Scholar]
- 44.Black MW, Arrizabalaga G, Boothroyd JC. 2000. Ionophore-resistant mutants of Toxoplasma gondii reveal host cell permeabilization as an early event in egress. Mol Cell Biol 20:9399–9408. doi: 10.1128/MCB.20.24.9399-9408.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bessoff K, Spangenberg T, Foderaro JE, Jumani RS, Ward GE, Huston CD. 2014. Identification of Cryptosporidium parvum active chemical series by repurposing the open access Malaria Box. Antimicrob Agents Chemother 58:2731–2739. doi: 10.1128/AAC.02641-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kroemer G, Levine B. 2008. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9:1004–1010. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sidik SM, Triana MAH, Paul AS, Bakkouri ME, Hackett CG, Tran F, Westwood NJ, Hui R, Zuercher WJ, Duraisingh MT, Moreno SNJ, Lourido S. 2016. Using a genetically encoded sensor to identify inhibitors of Toxoplasma gondii Ca2+ signalling. J Biol Chem 291:9566–9580. doi: 10.1074/jbc.M115.703546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jiménez-Díaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, Mazouni FE, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, DeRisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. 2010. Chemical genetics of Plasmodium falciparum. Nature 465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gagaring K, Borboa R, Francek C, Chen Z, Buenviaje J, Plouffe D, Winzeler E, Brinker A, Diagana T, Taylor J, Glynne R, Chatterjee A, Kuhen K. 2012. Novartis-GNF Malaria Box. Genomics Institute of the Novartis Research Foundation (GNF), San Diego, CA, and Novartis Institute for Tropical Disease, Singapore https://www.ebi.ac.uk/chemblntd. [Google Scholar]
- 50.Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TNC, Willis P. 2013. The open access Malaria Box: a drug discovery catalyst for neglected diseases. PLoS One 8:e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeRocher AE, Coppens I, Karnataki A, Gilbert LA, Rome ME, Feagin JE, Bradley PJ, Parsons M. 2008. A thioredoxin family protein of the apicoplast periphery identifies abundant candidate transport vesicles in Toxoplasma gondii. Eukaryot Cell 7:1518–1529. doi: 10.1128/EC.00081-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grosdidier A, Zoete V, Michielin O. 2011. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res 39:W270–W277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grosdidier A, Zoete V, Michielin O. 2011. Fast docking using the CHARMM force field with EADock DSS. J Comput Chem 32:2149–2159. doi: 10.1002/jcc.21797. [DOI] [PubMed] [Google Scholar]
- 55.Lau CD, Levesque MJ, Chien S, Date S, Haga JH. 2010. ViewDock TDW: high-throughput visualization of virtual screening results. Bioinformatics 26:1915–1917. doi: 10.1093/bioinformatics/btq296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 57.Donald RG, Roos DS. 1998. Gene knock-outs and allelic replacements in Toxoplasma gondii: HXGPRT as a selectable marker for hit-and-run mutagenesis. Mol Biochem Parasitol 91:295–305. doi: 10.1016/S0166-6851(97)00210-7. [DOI] [PubMed] [Google Scholar]
- 58.Fox BA, Ristuccia JG, Gigley JP, Bzik DJ. 2009. Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot Cell 8:520–529. doi: 10.1128/EC.00357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huynh M-H, Carruthers VB. 2009. Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot Cell 8:530–539. doi: 10.1128/EC.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.