

ABSTRACT
β-Lactams are often used to treat Helicobacter cinaedi infections; however, the mechanism underlying β-lactam resistance is unknown. In this study, we investigated β-lactam resistance in an H. cinaedi strain, MRY12-0051 (MICs of amoxicillin [AMX] and ceftriaxone [CRO], 32 and 128 μg/ml; obtained from human feces). Based on a comparative whole-genome analysis of MRY12-0051 and the CRO-susceptible H. cinaedi strain MRY08-1234 (MICs of AMX and CRO, 1 and 4 μg/ml; obtained from human blood), we identified five mutations in genes encoding penicillin-binding proteins (PBPs), including two in pbpA, one in pbp2, and two in ftsI. Transformation and penicillin binding assays indicated that CRO resistance was mainly associated with mutations in pbpA; mutations in ftsI also led to increased resistance to AMX. Knocking out cmeB and cmeD, which encode resistance-nodulation-division-type efflux pump components, in H. cinaedi type strain CCUG18818 (AMX MIC, 4 to 8 μg/ml) resulted in 8- and 64-fold decreases, respectively, in the AMX MIC. Hence, MICs of AMX in H. cinaedi become similar to those of Helicobacter pylori isolates in the absence of cmeD. In conclusion, the difference in susceptibility to β-lactams between H. pylori and H. cinaedi is explained by differences in efflux pump components. Mutations in pbpA are the primary determinant of high resistance to β-lactams in H. cinaedi.
KEYWORDS: Helicobacter cinaedi, amoxicillin, β-lactams, ceftriaxone, cmeB, cmeD, efflux pumps, penicillin-binding proteins
INTRODUCTION
Helicobacter cinaedi is a Gram-negative spiral bacterium that inhabits humans and animals (1–3). Helicobacter species can be divided into two groups: gastric and enterohepatic. H. cinaedi is the major enterohepatic Helicobacter species isolated from humans. This bacterium is isolated mainly from the blood of immunocompromised patients with bacteremia (4, 5). Further, H. cinaedi causes cellulitis, arthritis, meningitis, endocarditis, and abdominal aortic aneurysm (6–10). It has also been isolated from healthy subjects (6, 7, 11). Recently, the incidence of H. cinaedi isolation has increased, and this can be attributed to both increasing awareness of this species and advances in clinical microbiology tools (12).
Unlike for H. pylori infections, a standard treatment strategy for H. cinaedi infections has not been established. Response to treatment with β-lactam antibiotics is good in many cases (9, 13–15); however, the treatment period tends to be long owing to frequently recurring symptoms (16–18). We previously determined antimicrobial susceptibilities of H. cinaedi isolates from Japan and reported a MIC90 of amoxicillin (AMX) of 8 μg/ml (19). AMX is the major antimicrobial agent used in standard eradication therapy of Helicobacter pylori; AMX MICs in most clinical H. pylori isolates from Japan are less than 0.125 μg/ml (20). Hence, the antimicrobial susceptibility profile of H. cinaedi is different from that of H. pylori.
Bacterial antimicrobial resistance may be associated with the acquisition of genes conferring resistance (e.g., genes encoding β-lactamases), alterations of drug targets (e.g., mutations in pbp genes), decreased uptake (e.g., porin alterations), and/or increased efflux (e.g., increased expression of multidrug transporter genes). In H. pylori, resistance to antimicrobial agents is often caused by alterations (mutations) in the target; e.g., clarithromycin resistance is caused by a mutation in 23S rRNA (21).
The mechanism underlying β-lactam resistance in H. cinaedi has not been investigated. In the current study, we identified specific mutations responsible for the high β-lactam resistance of a clinical H. cinaedi isolate, strain MRY12-0051. This strain belongs to sequence type 10 (ST10) based on multilocus sequence typing (19); another ST10 strain, MRY08-1234, characterized by low β-lactam MICs and isolated from the same hospital as MRY12-0051, was chosen as a reference strain. We generated and compared whole-genome sequences of the two isolates and used transformation assays to determine the mechanisms of β-lactam resistance in H. cinaedi. Additional resistance mechanisms, including efflux pumps, were also analyzed to comprehensively delineate the mechanisms of resistance.
RESULTS
Antimicrobial susceptibilities of H. cinaedi clinical isolates.
The susceptibilities of H. cinaedi strains to AMX, ceftriaxone (CRO), and imipenem (IPM) are shown in Table 1. MRY12-0051 was more highly resistant to AMX and CRO than were other strains.
TABLE 1.
Susceptibilities to amoxicillin, ceftriaxone, and imipenem and point mutations in PBPA, PBP2, and FtsI in clinical H. cinaedi isolates and ceftriaxone-resistant mutants
| Strain | Sequence type (clonal complex) | MIC (μg/ml)a |
Amino acid at indicated positionb |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PBPA |
PBP2 |
FtsI |
||||||||||||||
| CRO | AMX | IPM | 22 | 423 | 447 | 2 | 121 | 416 | 86 | 308 | 351 | 419 | 503 | 516 | ||
| Clinical isolates | ||||||||||||||||
| MRY08-1234 | 10 (9) | 4 | 1 | 0.031 | V | S | T | M | E | L | L | Y | R | E | Q | S |
| MRY12-0027 | 15 (7) | 4 | 2 | 0.063 | V | S | T | M | E | L | F | H | R | E | Q | A |
| MRY12-0045 | 15 (7) | 4 | 4 | 0.125 | V | S | T | M | E | L | F | H | R | E | Q | A |
| MRY08-1236 | 10 (9) | 8 | 2 | 0.031 | V | S | T | M | E | L | L | Y | R | E | Q | S |
| MRY08-1238 | 10 (9) | 8 | 1 | 0.031 | V | S | T | M | E | L | L | Y | R | E | Q | S |
| MRY08-1235 | 11 (9) | 16 | 4 | 0.031 | V | S | T | M | E | L | L | Y | R | E | Q | A |
| MRY08-1240 | 2 (1) | 8–16 | 8 | 0.063 | A | S | T | M | E | L | F | Y | R | E | R | A |
| MRY08-1241 | 11 (9) | 16 | 8 | 0.125 | V | S | T | del | K | L | L | Y | R | E | Q | A |
| MRY08-1243 | 11 (9) | 16 | 8 | 0.063 | V | S | T | M | K | L | L | Y | R | E | Q | A |
| MRY12-0022 | 11 (9) | 16 | 4 | 0.125 | V | S | T | M | E | F | L | Y | R | E | Q | A |
| MRY12-0023 | 4 (4) | 16 | 4 | 0.063 | V | S | T | M | E | L | L | Y | R | E | Q | A |
| MRY12-0043 | 10 (9) | 16 | 2 | 0.063 | V | S | T | M | E | L | L | Y | R | E | Q | S |
| MRY12-0048 | 16 (16) | 16 | 8 | 0.063 | V | S | T | M | E | L | F | Y | R | E | Q | A |
| MRY12-0059 | 10 (9) | 16 | 4 | 0.063 | V | S | T | M | E | L | L | Y | R | E | Q | A |
| MRY12-0061 | 10 (9) | 16 | 4 | 0.063 | V | S | T | M | E | L | L | Y | R | E | Q | A |
| MRY12-0099 | 10 (9) | 16 | 4 | 0.063 | V | S | T | M | E | L | L | Y | R | E | Q | S |
| MRY12-0053 | 10 (9) | 32 | 4 | 0.063 | V | S | T | M | E | L | L | Y | R | E | Q | S |
| MRY12-0033 | 10 (9) | 64 | 4 | 0.063 | V | S | M | M | E | L | L | Y | R | E | Q | S |
| MRY12-0051 | 10 (9) | 128 | 32 | 0.25 | V | R | M | M | K | L | L | Y | H | K | Q | S |
| Transformantsc | ||||||||||||||||
| MRY08-1234_pbpA | 32 | 8 | 0.125 | V | R | M | M | E | L | L | Y | R | E | Q | S | |
| MRY08-1234_ftsI | 16 | 4 | 0.125 | V | S | T | M | E | L | L | Y | H | K | Q | S | |
| MRY08-1234_pbpA_ftsI | 32 | 16 | 0.125 | V | R | M | M | E | L | L | Y | H | K | Q | S | |
Abbreviations: CRO, ceftriaxone; AMX, amoxicillin; IPM, imipenem.
The amino acid numbers for PBPA, PBP2, and FtsI are based on those of MRY08-1234.
Transformants were created using the pbpA and ftsI genes from MRY12-0051. Transformants using the pbp2 gene of MRY12-0051 were not obtained.
Identification of single-nucleotide polymorphisms (SNPs) in the highly β-lactam-resistant H. cinaedi strain MRY12-0051.
MiSeq and PacBio RSII whole-genome sequencing of the reference strain MRY08-1234 generated 1,337,838 reads (259 Mb) and 81,221 reads (625 Mb), respectively. The de novo assembly of the PacBio sequence reads resulted in a single circular contig (2.2 Mb). MiSeq sequence reads were mapped onto the contig to correct any errors, and the corrected contig was confirmed as the complete genome of MRY08-1234.
MiSeq whole-genome sequencing of MRY12-0051 generated 5,256,956 reads (1.1 Gb). When the reads from MRY12-0051 were mapped onto the complete MRY08-1234 genome, 31 nonsynonymous SNPs were identified in 23 genes (see Table S1 in the supplemental material). Of these, five SNPs were in genes encoding penicillin (PEN)-binding proteins (PBPs): two in pbpA (HC081234_0020020), one in pbp2 (HC081234_0022110), and two in ftsI (HC081234_008570). Other SNPs were detected in the gyrA and gyrB genes encoding DNA gyrase, various genes encoding methyl-accepting chemotaxis signal transduction proteins, and cmeC, which encodes an efflux component.
Comparison of the predicted PBPA, PBP2, and FtsI sequences in clinical isolates from Japan.
In addition to MRY08-1234 and MRY12-0051, predicted amino acid sequences of PBPA, PBP2, and FtsI proteins from H. cinaedi isolates from Japan (n = 16) were analyzed, including MRY12-0033, a strain with a high level of CRO resistance (MIC, 64 μg/ml). Substitutions in the complete sequences of each PBP are presented in Table 1. Of the five point mutations identified in PBPs of MRY12-0051, T447M in PBPA was also detected in MRY12-0033. The remaining alterations (S423R in PBPA and R351H and E419K in FtsI) were not present in MRY12-0033 or other H. cinaedi isolates. On the other hand, E121K identified in PBP2 in MRY12-0051 was also present in two H. cinaedi isolates; the CRO MICs of both these isolates were 16 μg/ml.
Transformation analysis of the CRO-susceptible strain MRY08-1234.
To elucidate the roles of the SNPs in PBP-encoding genes in the β-lactam resistance of H. cinaedi, MRY08-1234 mutants were constructed by transformation. Every PBP-encoding gene of MRY08-1234 was replaced with its MRY12-0051 counterpart, and the MICs of β-lactams of the transformants were determined (Table 1). When pbpA was replaced, the MICs of the transformants increased 8-fold. Exchange of the pbp2 gene was not successful. When ftsI was replaced, MICs of the transformants increased 4-fold. Finally, when both pbpA and ftsI were replaced, the transformants became highly resistant to AMX and CRO.
Binding of CRO to PBPs in strains MRY08-1234 and MRY12-0051.
To analyze the affinity of CRO for PBPs of H. cinaedi, MRY08-1234 and MRY12-0051 were used in penicillin binding assays. The molecular masses of PBPs deduced from the sequences of the pbp1C, pbpA, pbp2, and ftsI genes were approximately 86.4, 73.2, 67.4, and 66.2 kDa, respectively. As shown in Fig. 1, four bands were detected between 75 kDa and 50 kDa, with predicted molecular masses of 68.8 (band A), 61.0 (band B), 56.6 (band C), and 52.1 (band D) kDa. No band was greater than 75 kDa, indicating that the protein encoded by the pbpIC gene was not detected. Since band A was the largest and became undetectable at high concentrations of CRO and IPM, it was predicted to be PBPA. It is possible that bands B and C correspond to PBPs; however, the bands were present even when a high concentration of ceftriaxone was used. Band D weakened as ceftriaxone increased; however, the band was too small to be assigned to Pbp2 or FtsI, suggesting that it represents a proteolytic fragment of a PBP. Taken together, the results indicate that we were not able to detect Pbp2 or FtsI in this assay.
FIG 1.

Binding of ceftriaxone (A) and imipenem (B) to PBPA in H. cinaedi clinical isolates MRY08-1234 and MRY12-0051 and pbpA mutant strain MRY08-1234_pbpA. Membrane proteins were treated with various concentrations of ceftriaxone and imipenem and subsequently labeled with Bocillin FL. Arrows indicate the bands corresponding to PBPA. Images are representative of results from two independent experiments. Gel band quantitation is shown with the standard deviations from two independent experiments.
Gel band quantitation indicated a higher affinity of PBPA of MRY08-1234 for CRO and IMP than those of MRY12-0051 and MRY08-1234 pbpA transformants. Hence, PBPA mutations (S423R and T447M) decreased the affinity for CRO and IMP.
Analysis of pbp1C knockout mutants.
Based on genome information from several H. cinaedi strains, H. cinaedi possessed PBPs encoded by the following four genes: pbpIC, pbpA, pbp2, and ftsI. Three of these genes corresponded to HP0597, HP1565, and HP1556 in H. pylori strain 26695, as determined using Basic Local Alignment Search Tool (BLAST; March 2017); no gene corresponding to pbpIC was identified in H. pylori. Nonsynonymous SNPs were not identified in pbpIC of MRY12-0051 in a comparison with MRY08-1234.
To evaluate the involvement of pbpIC in β-lactam resistance in H. cinaedi, a pbpIC knockout mutant was created. Multiple attempts to create pbpIC knockout mutants in MRY12-0051 and MRY08-1234 were unsuccessful, for unknown reasons. Both isolates belong to clonal complex 9 (CC9) (and ST10) (19). Similarly, mutants of other ST10 isolates, including MRY12-0033, could not be generated using this method; we speculate that knockout mutants of ST10 strains cannot be created using the employed cat cassette approach. Knockout mutants of isolates belonging to other STs were successfully generated; subsequently, pbpIC knockout mutants were created in the CCUG18818 (ST1) and clinical isolate MRY08-1240 (ST2) backgrounds. Knocking out the pbpIC gene did not affect β-lactam susceptibility in either strain (MRY08-1240_pbp1CΔcat and CCUG18818_pbp1CΔcat) (Table 2).
TABLE 2.
β-Lactam susceptibilities of H. cinaedi knockout mutants targeting pbp1C and efflux pump component genes
| H. cinaedi strain | MIC (μg/ml)a |
|
|---|---|---|
| AMX | CRO | |
| CCUG18818 | 4–8 | 8 |
| CCUG18818_pbpICΔcat | 8 | 8 |
| CCUG18818_cmeCΔcat | 4–8 | 8 |
| CCUG18818_macAΔcat | 4–8 | 8–16 |
| CCUG18818_cmeBΔcat | 0.5–1 | 0.5–1 |
| CCUG18818_cmeDΔcat | 0.063–0.125 | 0.063 |
| CCUG18818_cmeFΔcat | 4–8 | 8 |
| CCUG18818_cmeGΔcat | 4–8 | 8 |
| MRY08-1240 | 8 | 8–16 |
| MRY08-1240_pbpICΔcat | 8 | 16 |
| MRY08-1240_cmeCΔcat | 4–8 | 8 |
| MRY08-1240_macAΔcat | 4–8 | 8–16 |
| MRY08-1240_cmeBΔcat | 0.5 | 0.5–1 |
| MRY08-1240_cmeDΔcat | 0.063 | 0.5–1 |
| MRY08-1240_cmeFΔcat | NO | NO |
| MRY08-1240_cmeGΔcat | NO | NO |
| MRY12-0027 | 2–4 | 2 |
| MRY12-0027_pbpICΔcat | NT | NT |
| MRY12-0027_cmeCΔcat | 4 | 2 |
| MRY12-0027_macAΔcat | 4 | 2 |
| MRY12-0027_cmeBΔcat | 0.25 | 0.125 |
| MRY12-0027_cmeDΔcat | 0.031 | 0.016 |
| MRY12-0027_cmeFΔcat | 2–4 | 2 |
| MRY12-0027_cmeGΔcat | 2–4 | 2 |
Abbreviations: NO, not obtained; NT, not tested.
Analysis of efflux pump knockout mutants.
As identified previously in another strain (22), the whole-genome analysis of MRY08-1234 included several genes encoding efflux pumps, i.e., the cmeB-cmeA operon (HC081234_017630 and HC081234_017640, encoding components of the resistance-nodulation-division [RND]-type efflux pump), the cmeC-macA-macB-macB′ operon (HC081234_010300 to HC081234_010270, encoding components of the outer membrane protein and ATP-binding cassette [ABC]-type efflux pump), the cmeD-cmeE-cmeF operon (HC081234_006370, HC081234_006380, and HC081234_006390, encoding the RND-type efflux pump), and cmeG (HC081234_007910, a major facilitator family [MF]-type transporter). To elucidate the roles of these efflux pumps in the resistance of H. cinaedi to β-lactams, appropriate knockout mutants were created. We were not able to create a knockout mutant using ST10 isolates; instead, we used CCUG18818 (ST1), MRY08-1240 (ST2), and MRY12-0027 (ST15), which were transformable. As shown in Table 2, compared to those of the wild types, the AMX susceptibilities of cmeB and cmeD knockout mutants decreased 8- to 16-fold and 64- to 128-fold, respectively. Strikingly, the AMX MICs for cmeD knockout mutants were 0.031 to 0.125 μg/ml, which was similar to the MIC range for H. pylori clinical isolates (20, 23). Knocking out genes encoding other efflux pump components, including cmeC, macA, cmeF, and cmeG, did not affect strain susceptibilities to β-lactam antibiotics.
Analysis of the expression of efflux pump- and porin-encoding genes.
To elucidate the roles of efflux pumps in β-lactam resistance, the expression of efflux pump-encoding genes in H. cinaedi isolates was evaluated. As shown in Fig. 2, most of these genes were more highly expressed in MRY12-0051 (CRO MIC, 128 μg/ml) and MRY12-0033 (CRO MIC, 64 μg/ml) isolates than in CCUG18818 (CRO MIC, 8 μg/ml), MRY08-1240 (CRO MIC, 8 to 16 μg/ml), and MRY12-0027 (CRO MIC, 4 μg/ml). In addition, the expression level of these genes in MRY08-1234 (CRO MIC, 4 μg/ml) was higher than those in CCUG18818, MRY08-1240, and MRY12-0027. All strains exhibiting high expression levels of these genes belonged to the same sequence type (ST10). The expression of the porA gene, which encodes a porin, was almost four times lower in MRY08-1234, MRY12-0033, and MRY12-0051 than in CCUG18818, MRY08-1240, and MRY12-0027.
FIG 2.

Expression of genes encoding efflux pump components and a porin in H. cinaedi isolates. Transcript levels of the specified target genes were normalized to the transcript level of the recA reference gene; the relative gene expression levels are reported as the fold change relative to the expression in the CCUG18818 strain, determined using the threshold cycle (CT) method (33).
DISCUSSION
β-Lactams, such as AMX and CRO, are an important class of antimicrobial agents used to treat H. cinaedi infections, and yet the mechanisms underlying H. cinaedi resistance to these antibiotics are unknown. We focused on MRY12-0051, a clinical isolate that is highly resistant to CRO. Based on a transformation assay, the MICs of both AMX and CRO for pbpA transformants increased 8 times and those for ftsI transformants increased 4 times. MICs of CRO in pbpA transformants did not increase by additional mutations in ftsI. Hence, the contribution of pbpA mutations to CRO resistance is much greater than that of ftsI mutations. In addition, MRY12-0033, which possessed a mutation in pbpA but not in pbp2 or ftsI, exhibited high resistance to CRO (MIC, 64 μg/ml) but moderate susceptibility to AMX (MIC, 4 μg/ml). Therefore, alterations in PBPA are presumably the major determinant of CRO resistance in H. cinaedi. FtsI alterations are probably needed for high AMX resistance in H. cinaedi.
To investigate the positions of the altered amino acids in detail, three-dimensional (3D) structures of PBPA and FtsI of MRY08-1234 were predicted using Robetta (http://robetta.bakerlab.org/). Amino acid residues 423 and 447 in PBPA and amino acid residues 351 and 419 in FtsI were predicted to be in the vicinity of the PEN-binding pocket (Fig. 3). In addition, the T447M PBPA substitution detected in MRY12-0051 was also present in the MRY12-0033 strain, which was highly resistant to CRO. Taken together, the results show that the T447M substitution in PBPA may be the major alteration conferring CRO resistance in H. cinaedi.
FIG 3.
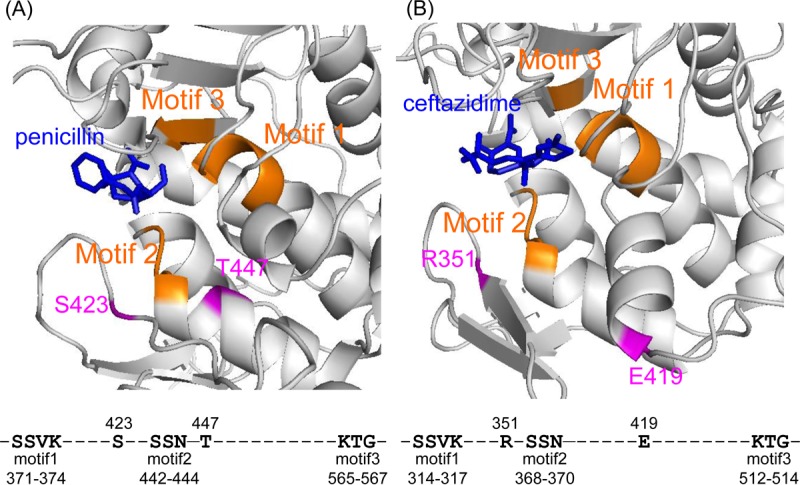
Predicted structures of H. cinaedi PBPA and FtsI. The structural predictions for PBPA (A) and FtsI (B) of the MRY08-1234 isolate were obtained using the Robetta server (http://robetta.bakerlab.org). The obtained structures were overlaid onto a crystal structure of Acinetobacter baumannii PBP1a in complex with penicillin G (PDB code 3UDI) and a crystal structure of Pseudomonas aeruginosa PBP3 in complex with ceftazidime (PDB code 3PBO), respectively, to visualize the β-lactam binding site. The ligands (penicillin G and ceftazidime) are shown in blue. Motifs 1, 2, and 3, shown in orange, are penicillin-binding motifs predicted based on the comparison with PBPA and FtsI of H. pylori, as appropriate (23). Mutations identified in MRY08-1234 are shown in pink. The amino acid numbers for PBPA and FtsI are based on the amino acid sequences of PBPA and FtsI in MRY08-1234.
Since the level of resistance was not completely recovered by the introduction of mutations in both the pbpA and ftsI genes, another mechanism may also contribute to the resistance phenotype. In addition, the higher β-lactam susceptibilities of H. cinaedi than H. pylori suggest that an unknown mechanism contributes to the decreased susceptibility in H. cinaedi. Since H. cinaedi possessed pbpIC, whose homolog is not present in H. pylori, a knockout mutant of the pbpIC gene in H. cinaedi was created in this study. However, this knockout did not affect the β-lactam MIC. H. cinaedi PBP1C has a sequence identity of 35% with Escherichia coli PBP1C, which is a penicillin-insensitive class A PBP that binds to specific β-lactams (24). These data indicate that pbpIC in H. cinaedi might encode a PEN-insensitive transpeptidase that is not targeted by β-lactams.
To elucidate the role of efflux pumps in β-lactam resistance, knockout mutants of genes encoding efflux pump components were created. Knocking out cmeB and cmeD resulted in increased strain susceptibility to AMX and CRO. The AMX MICs of cmeD knockout mutants decreased 64- to 128-fold and were similar to MICs of H. pylori isolates (20). Hence, the difference in β-lactam susceptibilities between H. cinaedi and H. pylori are associated with the activity of efflux pumps. In Campylobacter jejuni, a homolog of CmeC is encoded by the operon containing cmeA and cmeB, and the efflux pump of CmeABC contributes to β-lactam, fluoroquinolone, and macrolide resistance (23, 24). Homologs of CmeDEF were also present in C. jejuni and have a moderate contribution to antimicrobial resistance compared to that of CmeABC (25). Interestingly, although cmeD lies in the cmeDEF operon, cmeF knockout did not affect strain susceptibilities to β-lactams in H. cinaedi. An RND-type efflux pump is composed of an RND drug transporter, membrane fusion protein, and outer membrane protein, and both CmeB and CmeF are RND drug transporters (22). Substrate specificities of RND-type efflux pumps are usually determined by the RND drug transporter; hence, β-lactams could be substrates of CmeB, but not CmeF, in H. cinaedi. We were unable to construct knockout mutants of MRY12-0051 and MRY12-0033 strains; therefore, to elucidate their roles in resistance, the expression of genes encoding these efflux pumps was examined. As anticipated, the expression levels of most genes encoding efflux pump components analyzed in this study were high in MRY12-0051 and MRY12-0033; the expression of porA, encoding a porin, was low. Interestingly, the expression of these genes in MRY08-1234, which is susceptible to AMX and CRO, showed a similar trend. Hence, high expression levels of these efflux pumps do not fully explain H. cinaedi resistance to AMX and CRO. MRY08-1234 belonged to ST10, the same ST as MRY12-0051 and MRY12-0033. Therefore, an increase in efflux pump gene expression and low porin expression may facilitate β-lactam resistance in certain H. cinaedi strains. Further studies are needed to elucidate the mechanism underpinning the high expression of efflux pumps and to predict strains that are likely to become resistant to β-lactams. Further studies are also needed to elucidate additional mechanisms involved in resistance to β-lactams.
In conclusion, PBPA alterations are major determinants of resistance to CRO in H. cinaedi; FtsI alterations also led to increased resistance to AMX. PBP alterations result in high CRO MICs, sufficient to resist therapy. The use of CRO should hence be carefully monitored. PEN and carbapenems might be a better choice for the treatment of H. cinaedi infections.
MATERIALS AND METHODS
Strains and culture.
All clinical H. cinaedi strains used in this study were isolated from patients with H. cinaedi bacteremia between 2008 and 2012 in Sapporo City General Hospital (Sapporo, Japan). These strains were described previously (19, 26). CCUG18818 was used as the type strain. All strains were stored at −80°C in brucella broth (Becton Dickinson, Franklin Lakes, NJ) containing 30% (vol/vol) glycerol and subcultured in brucella agar (Becton Dickinson) containing 5% (vol/vol) horse blood. The bacteria were cultured at 37°C under microaerobic conditions with hydrogen generated by the gas replacement method using an anaerobic gas mixture (H2, 10%; CO2, 10%; and N2, 80%) (19).
Antimicrobial susceptibility testing.
MICs of ampicillin, AMX, CRO, and IMP were determined using the agar dilution method, as previously described (19).
Whole-genome sequencing.
Whole-genome sequencing of H. cinaedi strain MRY08-1234 was performed using the MiSeq system (2 × 300 paired-end run; Illumina, Inc., San Diego, CA) and PacBio RSII (Pacific Biosciences, Menlo Park, CA). PacBio sequence reads were assembled de novo using SMRT Analysis v. 2.2.0 by applying the DNA Data Bank of Japan Read Annotation Pipeline (27, 28). The overlapping regions of the assembled genome sequences were trimmed, and the MiSeq sequence reads were then mapped to contigs using CLC Genomics Workbench 8.5.1 (CLC Bio, Qiagen, Aarhus, Denmark) to correct errors in reads obtained from the PacBio sequencing platform. The complete genome sequence was annotated using the RAST genome annotation server v. 2.0 (29). The genome of MRY12-0051 was also sequenced using the MiSeq system (2 × 300 paired-end run); the reads were then mapped onto the complete MRY08-1234 genome. A variant analysis was performed using CLC Genomics Workbench; variants occurring with a >97% frequency were defined as SNPs. All SNPs were confirmed by Sanger sequencing.
Analysis of PBP-encoding genes.
The full-length coding regions of the penicillin-binding protein (PBP)-encoding genes pbpA, pbp2, and ftsI were amplified from genomic DNA using the primer pairs F1 and R1 listed in Table S2. Sequencing of both strands of purified PCR products was performed using sequencing primers listed in Table S2, a BigDye Terminator v. 3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA), and an Applied Biosystems 3130 genetic analyzer. The obtained sequences were assembled and aligned using the sequence assembly software ATGC (Genetyx, Tokyo, Japan) and GENETYX v. 13 (Genetyx).
Construction of mutants.
Knockout mutants were generated by replacing the target gene with a cat cassette, as follows. Up- and downstream regions (ca. 1 kb) of the target gene were PCR amplified using the primer pairs Lf/Lr and Rf/Rr, respectively (primer sequences are listed in Table S3). Primers Lr and Rf contain a linker sequence that overlaps with the cat cassette. After the cat cassette was amplified from the pHel2 plasmid (30), the three PCR products were purified, mixed, and amplified using primers Lf and Rr to obtain a construct comprising the cat gene with flanking target gene sequences. The amplified constructs were cloned into pUC19, and each plasmid was used to create knockout mutants by electroporation, as previously described (31). The transformants were selected on blood agar plates containing chloramphenicol (10 μg/ml).
To generate pbpA, pbp2, and ftsI mutants, the amplified target genes from MRY12-0051 were cloned into pUC19, and each plasmid was used for electroporation to create transformants harboring the target mutations. Transformants were selected on blood agar plates containing CRO (16 μg/ml) or ampicillin (8 μg/ml).
PEN binding assay.
The affinities of PBPs for CRO and IPM were analyzed by a competition assay with Bocillin FL (BoFL) (Invitrogen-Molecular Probes, Eugene, OR) according to a previously described method (32). Whole bacterial cells were collected from 10 blood agar plates after 3 days of incubation. They were suspended in 9 ml of KPN buffer (20 mM potassium phosphate, 140 mM NaCl [pH 7.5]) and disrupted using an ultrasonicator. Bacterial lysates were centrifuged at 10,000 × g for 45 min at 4°C to remove unbroken cells. The supernatant was centrifuged at 300,000 × g for 60 min at 4°C. The pellet was washed briefly with KPN buffer and suspended in 200 ml of KPN buffer. The protein concentrations were measured using the Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Rockford, IL), and the membrane proteins were stored at −80°C until use. For the PEN binding assay, protein concentrations were adjusted to 50 μg in 14 μl of KPN buffer and increasing concentrations of CRO or IPM were added to membrane proteins. After incubation for 10 min at 30°C, Bocillin FL was added at a final concretion of 13.3 mM and the mixture was incubated for an additional 30 min. Sample buffer solution (Wako Pure Chemical Industries, Ltd., Osaka, Japan) supplemented with 100 mM dithiothreitol (DTT; Sigma, St. Louis, MO) was added to the mixture and the samples were boiled for 3 min at 98°C. The boiled samples were separated by SDS-PAGE using a 4% to 20% gradient gel (Wako Pure Chemical Industries, Ltd.), and the gels were scanned using an LAS-3000 instrument (Fuji Photo Film Co., Ltd., Tokyo, Japan).
Gene expression analyses.
RNA was isolated from bacteria using the TRIzol Plus RNA purification kit (Ambion, Austin, TX). Reverse transcription was performed using SuperScript IV VILO master mix with ezDNase (Invitrogen, Carlsbad, CA). Quantitative PCR (qPCR) was performed using Power SYBR green master mix (Applied Biosystems). Primers used for qPCR are listed in Table S4. All samples were assayed in duplicate, and threshold cycle (CT) values were averaged. Relative expression levels were calculated using the recA gene as a reference and are reported as the fold change relative to the levels in the CCUG18818 strain using the comparative CT method (33).
Accession number(s).
High-throughput sequences generated in the course of this study are available under the following GenBank accession numbers: AP017374 (complete genome of MRY08-1234) and DRA005701 (MRY12-0051 sequencing reads).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Grant-in-Aid for Young Scientists (B) from the Japan Society for Promotion of Science (grant no. 25860105), a grant from the Ministry of Health, Labor, and Welfare of Japan (grant no. H24-Shinkou-Ippan-010), and funds from the Research Program on Emerging and Re-emerging Infectious Diseases, from the Japan Agency for Medical Research and Development (AMED).
We thank Shunji Takahashi, Satoshi Yamamoto, and Masaya Mukai for providing strains used in this study. We also thank Satowa Suzuki and Mari Matsui for helpful discussions.
We have no conflicts of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02036-17.
REFERENCES
- 1.Totten PA, Fennell CL, Tenover FC, Wezenberg JM, Perine PL, Stamm WE, Holmes KK. 1985. Campylobacter cinaedi (sp. nov.) and Campylobacter fennelliae (sp. nov.): two new Campylobacter species associated with enteric disease in homosexual men. J Infect Dis 151:131–139. doi: 10.1093/infdis/151.1.131. [DOI] [PubMed] [Google Scholar]
- 2.Gebhart CJ, Fennell CL, Murtaugh MP, Stamm WE. 1989. Campylobacter cinaedi is normal intestinal flora in hamsters. J Clin Microbiol 27:1692–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez KR, Hansen LM, Vandamme P, Beaman BL, Solnick JV. 2002. Captive rhesus monkeys (Macaca mulatta) are commonly infected with Helicobacter cinaedi. J Clin Microbiol 40:1908–1912. doi: 10.1128/JCM.40.6.1908-1912.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burman WJ, Cohn DL, Reves RR, Wilson ML. 1995. Multifocal cellulitis and monoarticular arthritis as manifestations of Helicobacter cinaedi bacteremia. Clin Infect Dis 20:564–570. doi: 10.1093/clinids/20.3.564. [DOI] [PubMed] [Google Scholar]
- 5.Murakami H, Goto M, Ono E, Sawabe E, Iwata M, Okuzumi K, Yamaguchi K, Takahashi T. 2003. Isolation of Helicobacter cinaedi from blood of an immunocompromised patient in Japan. J Infect Chemother 9:344–347. doi: 10.1007/s10156-003-0265-3. [DOI] [PubMed] [Google Scholar]
- 6.Kitamura T, Kawamura Y, Ohkusu K, Masaki T, Iwashita H, Sawa T, Fujii S, Okamoto T, Akaike T. 2007. Helicobacter cinaedi cellulitis and bacteremia in immunocompetent hosts after orthopedic surgery. J Clin Microbiol 45:31–38. doi: 10.1128/JCM.01507-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lasry S, Simon J, Marais A, Pouchot J, Vinceneux P, Boussougant Y. 2000. Helicobacter cinaedi septic arthritis and bacteremia in an immunocompetent patient. Clin Infect Dis 31:201–202. doi: 10.1086/313930. [DOI] [PubMed] [Google Scholar]
- 8.Okubo H, Goto M, Sato M, Sugiyama T, Kawano M, Matsunaga T, Akaike T. 2014. Helicobacter cinaedi meningitis: a case report and review of previous cases. J Neurol Sci 347:396–397. doi: 10.1016/j.jns.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 9.Bartels H, Goldenberger D, Reuthebuch O, Vosbeck J, Weisser M, Frei R, Battig V. 2014. First case of infective endocarditis caused by Helicobacter cinaedi. BMC Infect Dis 14:586. doi: 10.1186/s12879-014-0586-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kakuta R, Yano H, Kanamori H, Shimizu T, Gu Y, Hatta M, Aoyagi T, Endo S, Inomata S, Oe C, Tokuda K, Ozawa D, Goto H, Katori Y, Kaku M. 2014. Helicobacter cinaedi infection of abdominal aortic aneurysm, Japan. Emerg Infect Dis 20:1942–1945. doi: 10.3201/eid2011.140440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holst H, Andresen K, Blom J, Hojlyng N, Kemp M, Krogfelt KA, Christensen JJ. 2008. A case of Helicobacter cinaedi bacteraemia in a previously healthy person with cellulitis. Open Microbiol J 2:29–31. doi: 10.2174/1874285800802010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyake N, Chong Y, Nishida R, Nagasaki Y, Kibe Y, Kiyosuke M, Shimomura T, Shimono N, Shimoda S, Akashi K. 2015. A dramatic increase in the positive blood culture rates of Helicobacter cinaedi: the evidence of differential detection abilities between the Bactec and BacT/Alert systems. Diagn Microbiol Infect Dis 83:232–233. doi: 10.1016/j.diagmicrobio.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 13.Murata S, Suzuki H, Sakamoto S, Miki T, Rimbara E, Shibayama K, Koyama S, Tamai K, Yaguchi Y, Tada M. 2015. Helicobacter cinaedi-associated vertebral osteomyelitis in an immunocompetent patient. Intern Med 54:3221–3224. doi: 10.2169/internalmedicine.54.4574. [DOI] [PubMed] [Google Scholar]
- 14.Akashi Y, Igarashi J, Suzuki H, Rimbara E, Shibayama K, Nin S, Tamai K, Yaguchi Y, Shiigai M, Oikawa T, Suzuki M. 2015. Pararenal lymphatic cyst infection caused by Helicobacter cinaedi. Intern Med 54:1437–1440. doi: 10.2169/internalmedicine.54.3991. [DOI] [PubMed] [Google Scholar]
- 15.Seto T, Takano T, Ichimura H, Fujii T, Komatsu K, Ohtsu Y, Terasaki T, Wada Y, Fukui D, Murata S, Amano J. 2014. Pericoronary pseudotumor caused by Helicobacter cinaedi. Int Heart J 55:463–465. doi: 10.1536/ihj.14-018. [DOI] [PubMed] [Google Scholar]
- 16.Uçkay I, Garbino J, Dietrich PY, Ninet B, Rohner P, Jacomo V. 2006. Recurrent bacteremia with Helicobacter cinaedi: case report and review of the literature. BMC Infect Dis 6:86. doi: 10.1186/1471-2334-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adachi Y, Moriya C, Fujisawa T, Shu E, Kanoh H, Nakayama A, Yonetamari J, Seishima M. 2016. Recurrent superficial cellulitis-like erythema associated with Helicobacter cinaedi bacteremia. J Dermatol 43:844–846. doi: 10.1111/1346-8138.13282. [DOI] [PubMed] [Google Scholar]
- 18.Kiehlbauch JA, Tauxe RV, Baker CN, Wachsmuth IK. 1994. Helicobacter cinaedi-associated bacteremia and cellulitis in immunocompromised patients. Ann Intern Med 121:90–93. doi: 10.7326/0003-4819-121-2-199407150-00002. [DOI] [PubMed] [Google Scholar]
- 19.Rimbara E, Mori S, Matsui M, Suzuki S, Wachino J, Kawamura Y, Shen Z, Fox JG, Shibayama K. 2012. Molecular epidemiologic analysis and antimicrobial resistance of Helicobacter cinaedi isolated from seven hospitals in Japan. J Clin Microbiol 50:2553–2560. doi: 10.1128/JCM.06810-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rimbara E, Noguchi N, Tanabe M, Kawai T, Matsumoto Y, Sasatsu M. 2005. Susceptibilities to clarithromycin, amoxycillin and metronidazole of Helicobacter pylori isolates from the antrum and corpus in Tokyo, Japan, 1995–2001. Clin Microbiol Infect 11:307–311. doi: 10.1111/j.1469-0691.2005.01099.x. [DOI] [PubMed] [Google Scholar]
- 21.Versalovic J, Shortridge D, Kibler K, Griffy MV, Beyer J, Flamm RK, Tanaka SK, Graham DY, Go MF. 1996. Mutations in 23S rRNA are associated with clarithromycin resistance in Helicobacter pylori. Antimicrob Agents Chemother 40:477–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morita Y, Tomida J, Kawamura Y. 2012. Multidrug efflux systems in Helicobacter cinaedi. Antibiotics 1:29–43. doi: 10.3390/antibiotics1010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rimbara E, Noguchi N, Kawai T, Sasatsu M. 2008. Mutations in penicillin-binding proteins 1, 2 and 3 are responsible for amoxicillin resistance in Helicobacter pylori. J Antimicrob Chemother 61:995–998. doi: 10.1093/jac/dkn051. [DOI] [PubMed] [Google Scholar]
- 24.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 25.Akiba M, Lin J, Barton YW, Zhang Q. 2006. Interaction of CmeABC and CmeDEF in conferring antimicrobial resistance and maintaining cell viability in Campylobacter jejuni. J Antimicrob Chemother 57:52–60. doi: 10.1093/jac/dki419. [DOI] [PubMed] [Google Scholar]
- 26.Rimbara E, Mori S, Kim H, Matsui M, Suzuki S, Takahashi S, Yamamoto S, Mukai M, Shibayama K. 2013. Helicobacter cinaedi and Helicobacter fennelliae transmission in a hospital from 2008 to 2012. J Clin Microbiol 51:2439–2442. doi: 10.1128/JCM.01035-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagasaki H, Mochizuki T, Kodama Y, Saruhashi S, Morizaki S, Sugawara H, Ohyanagi H, Kurata N, Okubo K, Takagi T, Kaminuma E, Nakamura Y. 2013. DDBJ read annotation pipeline: a cloud computing-based pipeline for high-throughput analysis of next-generation sequencing data. DNA Res 20:383–390. doi: 10.1093/dnares/dst017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaminuma E, Mashima J, Kodama Y, Gojobori T, Ogasawara O, Okubo K, Takagi T, Nakamura Y. 2010. DDBJ launches a new archive database with analytical tools for next-generation sequence data. Nucleic Acids Res 38:D33–D38. doi: 10.1093/nar/gkp847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heuermann D, Haas R. 1998. A stable shuttle vector system for efficient genetic complementation of Helicobacter pylori strains by transformation and conjugation. Mol Gen Genet 257:519–528. doi: 10.1007/s004380050677. [DOI] [PubMed] [Google Scholar]
- 31.Ge Z, Taylor DE. 1997. H. pylori DNA transformation by natural competence and electroporation, p 145–152. In Clayton CL, Mobley HLT (ed), Helicobacter pylori protocols. Humana Press Inc., Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 32.Asli A, Brouillette E, Krause KM, Nichols WW, Malouin F. 2016. Distinctive binding of avibactam to penicillin-binding proteins of gram-negative and gram-positive bacteria. Antimicrob Agents Chemother 60:752–756. doi: 10.1128/AAC.02102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.