

ABSTRACT
Lopinavir-ritonavir forms the backbone of current first-line antiretroviral regimens in young HIV-infected children. As multidrug-resistant (MDR) tuberculosis (TB) frequently occurs in young children in high-burden TB settings, it is important to identify potential interactions between MDR-TB treatment and lopinavir-ritonavir. We describe the pharmacokinetics of and potential drug-drug interactions between lopinavir-ritonavir and drugs routinely used for MDR-TB treatment in HIV-infected children. A combined population pharmacokinetic model was developed to jointly describe the pharmacokinetics of lopinavir and ritonavir in 32 HIV-infected children (16 with MDR-TB receiving treatment with combinations of high-dose isoniazid, pyrazinamide, ethambutol, ethionamide, terizidone, a fluoroquinolone, and amikacin and 16 without TB) who were established on a lopinavir-ritonavir-containing antiretroviral regimen. One-compartment models with first-order absorption and elimination for both lopinavir and ritonavir were combined into an integrated model. The dynamic inhibitory effect of the ritonavir concentration on lopinavir clearance was described using a maximum inhibition model. Even after adjustment for the effect of body weight with allometric scaling, a large variability in lopinavir and ritonavir exposure, together with strong correlations between the pharmacokinetic parameters of lopinavir and ritonavir, was detected. MDR-TB treatment did not have a significant effect on the bioavailability, clearance, or absorption rate constants of lopinavir or ritonavir. Most children (81% of children with MDR-TB, 88% of controls) achieved therapeutic lopinavir trough concentrations (>1 mg/liter). The coadministration of lopinavir-ritonavir with drugs routinely used for the treatment of MDR-TB was found to have no significant effect on the key pharmacokinetic parameters of lopinavir or ritonavir. These findings should be considered in the context of the large interpatient variability found in the present study and the study's modest sample size.
KEYWORDS: Mycobacterium tuberculosis, antiretroviral agents, drug interactions, human immunodeficiency virus, multidrug resistance, pediatric drug therapy, pediatric infectious disease, pharmacokinetics, population pharmacokinetics
INTRODUCTION
In 2013, the World Health Organization (WHO) estimated that 3.2 million children were living with HIV infection (1). Tuberculosis (TB) is a leading cause of death among children living with HIV infection (2, 3), who now also face the emerging threat of multidrug-resistant TB (MDR-TB; which is caused by Mycobacterium tuberculosis strains resistant to at least both isoniazid and rifampin). WHO estimated that there were 480,000 new cases of MDR-TB in 2015 (2), with modeled estimates indicating that 25,000 to 32,000 of these new cases occur annually in children (4, 5). In settings with high burdens of HIV infection, a substantial proportion (20 to 53.9%) of these children are coinfected with M. tuberculosis and HIV (6–8). HIV infection is associated with rates of morbidity and mortality in children with MDR-TB (7, 9) higher than those in children with MDR-TB not infected with HIV. Cotreatment of MDR-TB and HIV in children is complex due to the high pill burden, limited child-friendly antituberculosis drug formulations, the potential for additive toxicities, possible drug-drug interactions (DDIs), immune reconstitution inflammatory syndrome, other comorbidities, and the potential for the acquisition of additional drug resistance (10, 11). Optimization of the treatments for both HIV infection and MDR-TB in coinfected children is critically important.
Current treatment for MDR-TB in children typically includes 4 effective second-line antituberculosis drugs plus pyrazinamide for between 9 and 18 months. According to the revised recent WHO classification of drugs for the treatment of MDR-TB, such regimens usually include one drug from group A (levofloxacin or moxifloxacin), one drug from group B (amikacin, kanamycin, or capreomycin), and at least two drugs from group C (ethionamide-prothionamide, cycloserine-terizidone, linezolid, or clofazimine). If a minimum of 4 effective antituberculosis drugs cannot be combined as required, drugs from groups D1 to D3 (add-on drugs), such as ethambutol, high-dose isoniazid, delamanid, para-aminosalicylic acid (PAS), and meropenem-clavulanate, are added (12) (bedaquiline is not yet recommended for use in children <12 years of age, given the absence of pediatric data for that drug). High-dose isoniazid (15 to 20 mg/kg of body weight) is used for the treatment of low-level isoniazid-resistant cases (13), typically associated with a mutation in the inhA promoter region, which is also associated with ethionamide cross-resistance (14). Pyrazinamide is usually added to the regimen, as it may have a synergistic effect with other drugs (15).
WHO advises a lopinavir-ritonavir (LPV/r)-based antiretroviral regimen to be a preferred treatment for HIV-infected children below 3 years of age. This regimen should include lamivudine and abacavir or zidovudine (16). Given the natural history of TB in young children with the highest risk of TB disease progression (17), many MDR-TB cases occur in children below 5 years of age. Therefore, data regarding the concomitant use of and potential DDIs between lopinavir-ritonavir and second-line antituberculosis drugs are highly relevant. DDIs between the first-line antituberculosis drugs and several antiretrovirals (ARVs) are well documented (3, 18–20). However, knowledge regarding the impact of the second-line antituberculosis drugs on the pharmacokinetics (PK) of ARVs is limited, and there are no data for children. Children might be less or more susceptible to DDIs due to development-related changes in drug disposition and the variable weight-adjusted doses of interacting drugs (21, 22). Lopinavir is a sensitive substrate of cytochrome P450 3A4 (CYP3A4), and P-glycoprotein and is highly protein bound in plasma (98 to 99%) (23–25). Absorption is dependent on gastric pH, intestinal CYP3A4 expression (23), and P-glycoprotein activity. Hepatic and intestinal CYP3A4 expression contributes to the systemic clearance of lopinavir (23). Ritonavir is highly protein bound (98 to 99%) (25) and a substrate of CYP3A4, P-glycoprotein, and, to a lesser extent, CYP2D6. These are all sites for possible DDIs with second-line antituberculosis drugs.
Of the second-line antituberculosis drugs, moxifloxacin undergoes partial hepatic metabolism (26), PAS and linezolid are thought to be metabolized by the liver (27–29), and renal mechanisms account for the elimination of levofloxacin, ofloxacin, amikacin, capreomycin, and terizidone (30). Clofazimine is a weak inhibitor of CYP3A4 (31), and ethionamide might have substrate specificities overlapping those of CYP P450 (32). It is possible that overlapping pathways, especially through CYP P450, might potentially impact the concentrations of ARVs. Isoniazid potentially inhibits CYP2C19 and CYP3A4 in a concentration-dependent manner (33). Preventive therapy with isoniazid, given at 5 mg/kg, increased the median lopinavir area under the concentration-time curve (AUC) at steady state by 5% (the AUC of lopinavir administered alone was 141.1 mg · h/liter, whereas it was 122.9 mg · h/liter when it was coadministered with isoniazid) in a study of 16 adults, but the difference was not statistically significant (P = 0.41) (34). The effect of high-dose isoniazid on lopinavir concentrations is unknown. As substrates of P-glycoprotein, linezolid and moxifloxacin (35–37) might interact with lopinavir through efflux pump transport mechanisms.
As pharmacokinetic data on second-line antituberculosis drugs are limited, potential DDIs with lopinavir-ritonavir are difficult to predict. A summary of the current knowledge on the pharmacokinetics of second-line antituberculosis drugs with a focus on possible DDI mechanisms is presented in Table S1 in the supplemental material. Any DDIs with second-line antituberculosis drugs which could reduce the concentrations of ARVs could potentially compromise the efficacies of the ARVs and may lead to the development of ARV resistance, while interactions increasing ARV concentrations could potentially result in increased toxicity.
We used a population pharmacokinetic model to describe the pharmacokinetics of lopinavir and ritonavir and to identify and quantify possible DDIs with second-line antituberculosis drugs. This technique provides a semimechanistic platform to interpret pharmacokinetic data able to adjust for the concomitant effect of multiple factors. The population pharmacokinetics of lopinavir and ritonavir in children (38–42) and in children cotreated with first-line antituberculosis drugs (18, 20) have been described in previous published reports. We aimed to characterize the effects of antituberculosis drugs routinely used for the treatment of MDR-TB on the pharmacokinetics of lopinavir and ritonavir in HIV-infected children.
RESULTS
Patients and data description.
Thirty-two children (16 HIV-infected children on MDR-TB treatment and 16 HIV-infected controls without TB) contributing a total of 191 samples (1 patient provided only one sample before receipt of the ARV dose) were included in the study. The lopinavir concentrations in three samples and the ritonavir concentrations in four samples were below the lower limit of quantitation (LLOQ).
A summary of the participants' characteristics is presented in Table 1. Patient characteristics were not statistically significantly different between the two groups. Twenty-nine of 32 (91%) children were on first-line combination antiretroviral therapy (cART) containing abacavir, lamivudine, and lopinavir-ritonavir, two controls received zidovudine, and one MDR-TB case received stavudine as a substitute for abacavir. The HIV viral load was taken at a median cART duration of 8 months for the MDR-TB group and 21 months for the control group, and CD4 counts were tested at 18 months for the MDR-TB group and 12 months for the control group. A single 12-year-old participant was excluded from the weight-for-age Z-score measurement, as the WHO chart includes scores only for individuals up to 10 years of age. WHO-derived Z-scores were used over other norms, as they are more applicable to the study population. Two children received rifampin preceding the sampling day because of inconclusive rifampin susceptibility results, and one of these children also received a dose of rifampin on the sampling day; both participants were also on superboosted ritonavir, which reduces the effect of rifampin on lopinavir bioavailability and clearance (20). Exclusion of the data for these two participants did not affect our findings.
TABLE 1.
Summary of characteristics of HIV-infected children with and without MDR-TB on a lopinavir-ritonavir-based antiretroviral treatment regimena
| Characteristic | Values for: |
P valueb | |
|---|---|---|---|
| MDR TB group (n = 16) | Control group (n = 16) | ||
| Median (IQR) age (yr) | 1.9 (1.0–2.7) | 2.2 (0.7–5.3) | 0.880 |
| No. (%) of children of black ethnicity | 13 (81.3) | 9 (56.3) | 0.252 |
| No. (%) of male children | 6 (37.5) | 6 (37.5) | 1.000 |
| Median (IQR) wt (kg) | 9.9 (7.7–13.0) | 11.8 (8.0–15.3) | 0.678 |
| Median (IQR) length/ht in cm | 78.8 (70.1–86.2) | 83.9 (67.8–103.4) | 0.821 |
| Median (IQR) body surface area (m2) | 0.5 (0.39–0.56) | 0.51 (0.39–0.67) | 0.678 |
| No. (%) of children with a Z-score of <−2c | |||
| Wt-for-age (n = 15) | 4 (26.7)d | 4 (25.0) | 1.000 |
| Length-for-age | 8 (50.0) | 4 (25.0) | 0.273 |
| Median (IQR) fat-free mass (kg) | 7.86 (6.28–10.08) | 8.88 (6.25–12.77) | 0.651 |
| Median (IQR) CD4+ T-cell count (cells/μl) | 1,347 (1,055–1,975) | 1,026 (491.5–1,512.5) | |
| Median (IQR) viral load (no. of copies/ml) | 804 (59.5–10,686.3) | LDL (LDL–897) | |
| No. (%) of children with the following WHO HIV staging: | |||
| 1 | 13 (81.3) | ||
| 2 | 1 (6.3) | ||
| 3 | 12 (75.0) | 1 (6.3) | |
| 4 | 4 (25.0) | 1 (6.3) | |
| No. (%) of children receiving the following formulation: | 0.481 | ||
| Suspension | 14 (87.5) | 11 (68.8) | |
| Whole tablet | 1 (6.3) | 4 (25.0) | |
| Crushed tablet | 1 (6.3) | 1 (6.3) | |
| No. (%) of children receiving drug by NGT administration | 13 (81.3) | 8 (50.0) | 0.135 |
| No. (%) of children with the following MDR-TB resistance pattern: | |||
| MDR-TB | 13 (81.3) | Not applicable | |
| Pre-XDR- or XDR-TB | 3 (18.8) | ||
Data are for 32 children. IQR, interquartile range; WHO, World Health Organization; LDL, lower than the detectable limit; NGT, nasogastric tube; MDR, multidrug resistant (resistant to isoniazid and rifampin); XDR, extensively drug resistant (resistant to isoniazid, rifampin, fluoroquinolones, and a second-line injectable agent); pre-XDR, resistant to isoniazid and rifampin and either the fluoroquinolones or a second-line injectable agent.
The following statistical tests were used: the Wilcoxon rank-sum test for continuous variables and either the chi-square test or Fisher's exact test for categorical variables.
Weight- and height-for-age Z-scores of <−2 include moderately underweight/stunted to severely underweight/stunted participants.
One 12-year-old participant in the MDR-TB group was excluded in the weight-for-age measurement.
One participant in the MDR-TB group received sodium valproate and mycophenolate. Sodium valproate is a CYP3A4 inhibitor which may increase lopinavir concentrations (43). As a precaution, we excluded the data for this participant when evaluating TB treatment as a covariate, but this made no difference, so the data for the patient were retained in the final model.
Pharmacokinetic model.
One-compartment models with first-order absorption and elimination were found to suitably describe the pharmacokinetics of both lopinavir and ritonavir. The data for the two drugs were then combined into a joint population model whose structure and final parameter estimates are presented in Fig. 1 and Table 2, respectively. A visual predictive check (VPC) plot stratified by MDR-TB treatment status is displayed in Fig. 2, showing that the 10th, 50th, and 90th percentiles of the observed data are in agreement with the respective 90% confidence intervals simulated by the model, thus supporting the adequacy of the model. The maximum inhibition (Imax) model was selected to characterize the ritonavir-lopinavir interaction because it provided the best goodness of fit among the alternatives tested. This inclusion improved the model fit in terms of the −2 log likelihood (−2LL) value (a 14-point decrease), the decreased variability for lopinavir (a 25% decrease in the interindividual variability [IIV] in clearance, a 23% decrease in the interoccasion variability [IOV] in bioavailability), and a general improvement in the goodness-of-fit plots and VPCs. The dynamic influence of the ritonavir concentration on the clearance of lopinavir is illustrated in Fig. 3. We attempted to reestimate the parameter values of the Imax model, but this did not improve the fit and made the model unstable, possibly due to the limited sample size, so we used the values from a previous report of a study with a comparable population (20).
FIG 1.
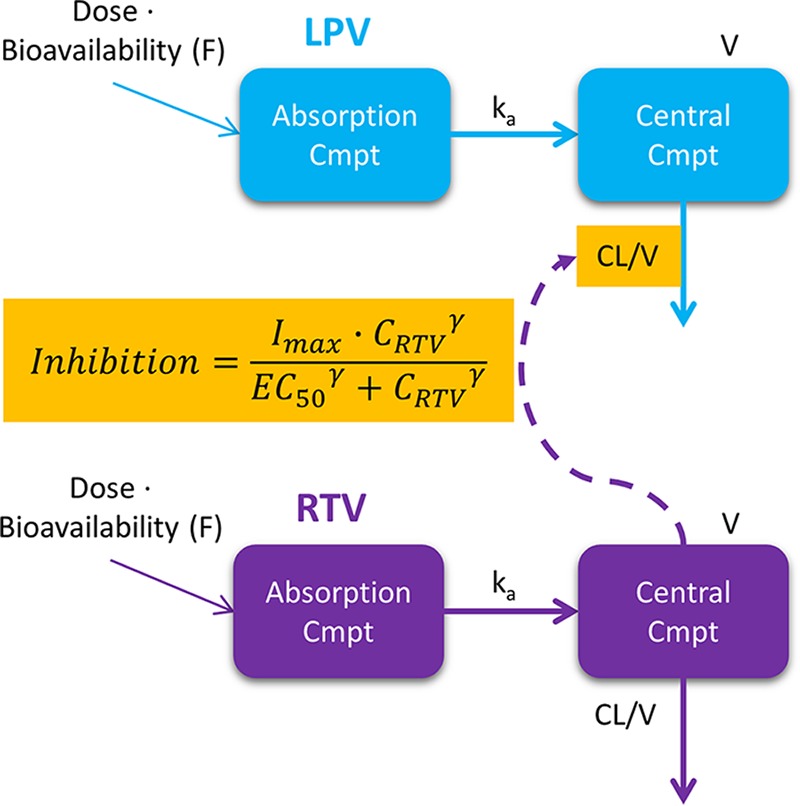
Structure of the final integrated lopinavir-ritonavir pharmacokinetic model. Cmpt, compartment; LPV, lopinavir; RTV, ritonavir; CL, clearance; V, apparent volume of distribution; ka, absorption rate constant; inhibition = (Imax · CRTVγ)/(EC50γ + CRTVγ), which describes the inhibitory effect of the concentration of ritonavir (CRTV) on the clearance of lopinavir described with an Imax relationship, where Imax is the maximum inhibitory effect of the ritonavir concentration on lopinavir clearance, EC50 is the concentration at which 50% of the maximal inhibitory effect is obtained, and γ, also known as the Hill coefficient, is the shape factor of the curve.
TABLE 2.
Parameter estimates from the final combined model for lopinavir and ritonavir in HIV-infected children with and without MDR-TB treatmenta
| Parameter | Lopinavir estimate (%RSE) | Ritonavir estimate (%RSE) | Lopinavir-ritonavir interaction estimate (%RSE) |
|---|---|---|---|
| CL (liters/h)b | 2.14c (3) | 16.8 (11) | |
| V (liters)b | 26.4 (16) | 74.1 (12) | |
| ka (1/h) | 0.424d (36) | 0.281d (4) | |
| F (unitless) | 1 (fixed) | 1 (fixed) | |
| Additive error (mg/liter) | 0.0039 (fixed)e | 0.00097 (fixed)e | |
| Proportional error (%) | 15 (7) | 29 (7) | |
| Change in CL at night (%) | −29 (3) | −38 (6) | |
| IIV in CL (%CV)f | 50 (19) | 69 (16) | |
| IOV in F (%CV)f | 41 (20) | 64 (15) | |
| IOV in ka (%CV)f | 61 (23) | ||
| Imax (unitless) | 0.82 (fixed)g | ||
| EC50 (mg/liter) | 0.098 (fixed)g | ||
| γ (unitless) | 2.8 (fixed)g | ||
| Correlation for IIV in CL lopinavir-ritonavir (%) | 64 (22) | ||
| Correlation for IOV in F lopinavir-ritonavir (%) | 99 (4) |
Data are for 32 children. RSE, relative standard error; CL, apparent oral clearance; V, apparent volume of distribution; ka, absorption rate constant; F, bioavailability; IIV, interindividual variability; IOV, interoccasion variability; CV, coefficient of variation; Imax, maximum inhibition; EC50, the concentration at which 50% of the maximal inhibitory effect is obtained; γ, the Hill coefficient, or the shape factor of the curve.
Clearance and volume of distribution were estimated using allometric scaling, and the values reported here are for a child of 11 kg.
The value for lopinavir clearance reported here was calculated for a median ritonavir concentration of 0.3 mg/liter to include the inhibitory effect of ritonavir. The model would have otherwise extrapolated a value for lopinavir clearance in the absence of ritonavir, which is not the case, since lopinavir was never administered alone.
A Bayesian prior with values from Zhang et al. (20) was used for the estimation of ka.
The estimate of additive error for lopinavir and ritonavir was not stable and tended to 0, so it was fixed to 20% of the LLOQ for each drug.
IIV and IOV were assumed to have a lognormal distribution, and their magnitudes are reported here as approximate coefficients of variation.
These values were fixed to the values previously reported for a similar population (20) to improve model stability.
FIG 2.

Visual predictive check of the combined pharmacokinetic model for lopinavir and ritonavir in HIV-infected children stratified by MDR-TB versus controls, using 1,000 simulations. The solid and dashed lines represent the 10th, 50th, and 90th percentiles of the observed data, while the shaded areas (pink and blue) are the model-predicted 90% confidence intervals for the same percentiles. Observed data are displayed as dots, with resimulated censored data points (points at the LLOQ) being indicated in red.
FIG 3.

Dynamic influence of the ritonavir concentration on lopinavir clearance in a typical 11-kg child. The dots represent the range (minimum and maximum) over which the ritonavir concentrations were observed.
The model generally detected a large IIV in clearance and a large IOV in bioavailability for both lopinavir and ritonavir. Correlations between the random effects on the pharmacokinetic parameters were found to be very strong: 99% for the IOV in bioavailability (a 34-point decrease in the −2LL value) and 64% for the IIV in clearance (a 9-point decrease in the −2LL value). In the integrated model, estimates of the typical values of the absorption rate constant (ka) for lopinavir and ritonavir were obtained using a Bayesian prior based on values reported in a comparable population by Zhang et al. (20) (ka, ∼0.385 1/h for both drugs) and 30% uncertainty. The final values obtained when the priors were included in the joint model were similar to those estimated in the separate models for lopinavir and ritonavir. The estimate of additive error for lopinavir and ritonavir was not stable and was therefore conservatively fixed to 20% of the LLOQ for each drug, since 20% was the level of error used by the laboratory technicians when defining the LLOQ of the assay.
The observed morning predose concentrations obtained in samples drawn approximately 12 h after the previous evening's dose were generally higher than the concentrations observed in the last samples in the schedule, which were drawn at 7 to 10 h after the morning dose. This highlights a difference between the exposures during nighttime and daytime, which was best captured in the model by an overnight decrease in clearance for both lopinavir (−29%, an improvement in the −2LL value of 11 points) and ritonavir (−38%, an improvement in the −2LL value of 11 points).
Covariate selection.
Allometric scaling was used to account for size differences, and its inclusion improved the model fit (a 17-point decrease in the −2LL value). Use of the fat-free mass instead of total body weight for allometric scaling did not provide any meaningful further benefit in terms of model fit. No effect of age (maturation) could be detected. The typical clearance for lopinavir and ritonavir for a child weighing 11 kg, the median weight in our study, was found to be 2.14 liters/h and 16.8 liters/h, respectively. The value for lopinavir clearance was calculated for the average ritonavir concentration observed in the study (0.3 mg/liter), to account for the typical inhibitory effect of ritonavir observed and allow comparison with the results of previous studies. The model would otherwise have estimated the value for lopinavir clearance in the absence of ritonavir, which would be an extrapolation, since lopinavir was never administered alone.
Treatment for MDR-TB was not found to significantly affect the pharmacokinetics of lopinavir or ritonavir. None of the models including effects on clearance, bioavailability, or ka achieved a significant improvement in the −2LL value. The VPC stratified by MDR-TB group versus the controls is shown in Fig. 2, showing that a model assuming no effect of treatment for MDR-TB was suitable for the data set: the observed data fell within the confidence intervals predicted by the model for both groups. When the effect was included in the model, there was a trend toward lower levels of exposures of lopinavir in the MDR-TB group as a result of either increased clearance or decreased bioavailability, but this did not reach statistical significance and did not provide convincing improvements in the diagnostic plots and VPCs. The a posteriori power calculations predicted that, at a significance level of a P value of <0.01, our study design is expected to be able to detect with 80% power a 70% increase in clearance or a 35% decrease in bioavailability in the MDR-TB group. This means that if a difference greater than these were present in the data, our model would have an 80% chance of detecting it at a P value of <0.01.
The individual morning trough concentrations for lopinavir predicted by our model are presented in Fig. 4. The median predicted morning trough concentrations for children with and without treatment for MDR-TB were 5.7 mg/liter and 6.5 mg/liter, respectively. Therapeutic lopinavir trough concentrations (morning trough concentration, >1 mg/liter) were achieved in 13/16 (81%) of the children with MDR-TB and in 14/16 (88%) of the controls.
FIG 4.
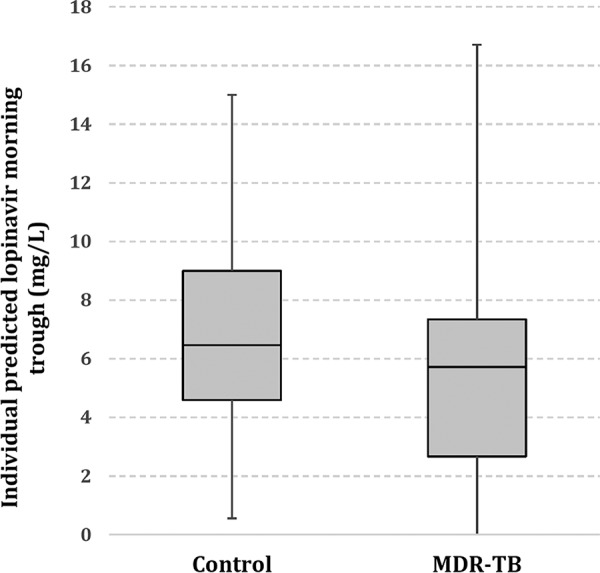
Model-based individual estimates of lopinavir morning trough concentrations (0 h) in HIV-infected children stratified by children with MDR-TB (cases) and children without MDR-TB (controls). These values were obtained on the basis of the model individual predictions (a posteriori mode).
No effect of sex, formulation, method of administration, or nutritional status on pharmacokinetics was detected, but this may have been due to the fact that most patients were female and received the suspension via a nasogastric tube and the fact that only eight patients were underweight for age, with only one case of underweight for age being severe.
DISCUSSION
In this first study to identify possible DDIs between treatments for MDR-TB including second-line antituberculosis drugs and ARVs in children, we report on the pharmacokinetics of lopinavir and ritonavir in HIV-infected children treated and not treated for MDR-TB. Our analysis did not detect any significant DDIs or effects of the MDR-TB treatment on the pharmacokinetic parameters of lopinavir and ritonavir. A slight trend toward lower exposures in the MDR-TB group was observed. This could have been due to the large variability between patients and the modest sample size.
Despite the modest numbers, our results are reassuring for the antituberculosis drugs commonly used in children, including high-dose isoniazid, ethionamide, ethambutol, pyrazinamide, amikacin, and terizidone. For other drugs, such as PAS, capreomycin, linezolid, moxifloxacin, levofloxacin, and ofloxacin, definitive conclusions were not possible, given our small numbers.
The estimated value for lopinavir clearance/bioavailability of 2.14 liters/h for a median ritonavir concentration of 0.3 mg/liter was similar to the values previously published in reports of studies performed in HIV-infected individuals (18, 20, 38–42, 44): the median weight-adjusted lopinavir clearance/bioavailability was 2.54 liters/h (range, 1.29 to 5.87 liters/h) for a total of eight studies (n = 920) with similar populations. Overall, the findings from these studies were compatible with our data. These studies included children with a wide age range (infants to 18 years), and two studies also included children receiving cotreatment with first-line antituberculosis drugs.
In our analysis, the interaction between lopinavir clearance and the ritonavir concentration was described by the inclusion of an Imax model, using parameter values from a previously published report of a study performed with a population similar to that used in the present study (20). The interaction model predicts that the inhibitory effect of ritonavir is already elicited at low concentrations and already reaches the maximum value at concentrations of about 1 mg/liter, as shown in Fig. 3.
A diurnal variation in ritonavir and lopinavir clearance was observed. A circadian phase dependency for the pharmacokinetics of ritonavir (20, 45) and lopinavir (20, 46) has been reported. CYP3A4 activity is thought to have some diurnal variation (47). The higher concentrations of lopinavir and ritonavir observed in the morning in our study, however, could be due to multiple factors. Delayed nocturnal absorption could describe the higher concentrations in the morning. Food is known to increase the bioavailability of ritonavir (48). It is possible that the children received the drug with food the evening before the pharmacokinetic study visit, which would enhance the bioavailability and increase the concentration. In our model, the best fit was provided when the effect on clearance was included, but due to the sparseness of our data, it was not possible to thoroughly investigate differences in bioavailability or other pharmacokinetic parameters. Our findings are probably due to a combination of these factors, with circadian variation having some effect.
We detected a large variability in lopinavir and ritonavir exposures. TB and HIV infection are known causes of hypoalbuminemia (49, 50), which could theoretically lead to an increase in the free fraction of highly protein bound drugs, such as lopinavir and ritonavir, thus enhancing drug metabolism and elimination. Another important consideration is that most (75%) of the children received lopinavir-ritonavir as the Kaletra oral suspension, which has poor palatability (it is very bitter). Administration of this poorly palatable suspension, together with other ARVs and MDR-TB treatment, is challenging, especially in younger children. Malnutrition has been described to be a possible cause for the lower lopinavir exposures (42). In our study, underweight for age did not affect lopinavir exposure; however, only one child was severely underweight for age, and our sample size was limited. A final important consideration is that 2 children received crushed lopinavir-ritonavir tablets. Crushing of the tablets is known to significantly reduce lopinavir-ritonavir exposures (51).
Our study has several limitations: lopinavir-ritonavir exposures are known to have a large IIV and IOV (52). This was also evident from the exposures observed in our cohort and was captured by the parameter estimates in our model. Because of the large variability, one would require a larger sample size to detect relatively small effects. Due to the success of vertical HIV transmission prevention programs, the prevalence of HIV infection among children with MDR-TB has decreased markedly in our study setting; therefore, recruitment of this study population was challenging. Despite our modest sample size, we believe that our findings are of value, as it is expected that large effects which would be clinically relevant would have been detected. A crossover design may have increased the power of the analysis and reduced the effect of the large IIV in clearance, but this was not feasible in our study. Another limitation is that this was an observational study in which children were on individualized MDR-TB treatment regimens with various combinations of antituberculosis drugs, which increased the possibility of the presence of unknown confounders. We explored the possibility of grouping the anti-MDR-TB drugs according to their suspected DDI potentials and testing each separately, but this could not be done due to the limited sample size.
In conclusion, we found no significant effect of antituberculosis drugs, including high-dose isoniazid, pyrazinamide, ethambutol, ethionamide, terizidone, and amikacin, on the key pharmacokinetic parameters of lopinavir and ritonavir when they were coadministered in HIV-infected children. While the size of our study was modest, our findings are clinically reassuring. The optimization of treatments for both HIV infection and MDR-TB in coinfected children is essential. Additional research is needed to evaluate DDIs between lopinavir, ritonavir, and other ARVs with increasingly used anti-TB drugs, including moxifloxacin, levofloxacin, clofazimine, and linezolid and the novel drugs bedaquiline and delamanid.
MATERIALS AND METHODS
Study population.
The study was conducted in Cape Town, Western Cape Province, South Africa. Data from 32 HIV-infected children (age, 0 to 15 years) on an ARV regimen routinely containing lopinavir-ritonavir were included: 16 HIV-infected children on an individualized MDR-TB treatment regimen (MDR-TB group) and 16 HIV-infected controls without TB (control group). All children were on combination antiretroviral therapy (cART) containing lamivudine and another nucleotide reverse transcriptase inhibitor (NRTI; abacavir, stavudine, or zidovudine), in addition to lopinavir-ritonavir. The antituberculosis drugs and doses used as part of the routine MDR-TB treatment regimen are summarized in Table 3.
TABLE 3.
Routine antituberculosis drugs and the administered and recommended doses used in HIV-infected children with MDR-TB on a lopinavir-ritonavir-based antiretroviral treatment regimena
| Antituberculosis drug (source) | No. (%) of children | Median (range) daily dose (mg/kg) | Daily recommended dose (mg/kg) |
|---|---|---|---|
| High-dose isoniazid (Sanofi) | 14 (87.5) | 19.3 (13.8–20) | 15–20 |
| Ethambutol (Sandoz) | 14 (87.5) | 22.3 (16.7–24.7) | 15–25 |
| Pyrazinamide (Sandoz) | 16 (100) | 34.7 (27.9–41.3) | 30–40 |
| Ethionamide (Sanofi) | 14 (87.5) | 20 (19.9–20) | 15–20 |
| Amikacin (Fresenius) | 15 (93.8) | 17.1 (14.9–20) | 15–20 |
| Capreomycin (Pharmacare) | 2 (12.5) | 15 (14.6–15.4) | 15–20 |
| Ofloxacin (Sanofi) | 5 (31.3) | 20 (19.9–20) | 15–20 |
| Levofloxacin (Sandoz) | 7 (43.8) | 20 (14.9–20) | 15–20 |
| Moxifloxacin (Dr. Reddy's) | 2 (12.5) | 10 (9.9–10) | 7.5–10 |
| Terizidone (Sanofi) | 15 (93.8) | 20 (19.9–20) | 15–20 |
| para-Aminosalicylic acid (Jacobus) | 3 (18.8) | 75 (74.9–83.1) twice daily | 150–200 in two divided doses |
| Linezolid (Pfizer) | 2 (12.5) | 10 (10) twice daily | 10 (twice daily if younger than age 10 yr) |
| Clofazimine (Sangrose) | 2 (12.5) | 5 (4.6–5.3) | 3–5c |
| Rifampinb (Sandoz) | 1 (6.25) | 16.7 | 10–20 |
Data are for 16 children. MDR-TB, multidrug-resistant tuberculosis.
Two children were on rifampin preceding the pharmacokinetic sampling day, but only one of them received rifampin on the sampling day.
Up to a maximum of 100 mg daily. In younger children, doses may be given on alternate days because of the milligram size of the gel capsules.
All children had been receiving a first-line cART including lopinavir-ritonavir for at least 2 weeks prior to enrollment. Children in the MDR-TB group underwent pharmacokinetic sampling following routinely initiated MDR-TB treatment for a minimum of 2 weeks and for up to 16 weeks. Exclusion criteria were laboratory-documented anemia (hemoglobin concentration, <8 g/dl) or a weight below 5 kg. Pharmacokinetic sampling in children with severe acute illness was deferred until the children were clinically stable.
Standard of care for treatment of MDR-TB and HIV infection.
The majority of children with MDR-TB in the study setting initially received inpatient care mainly due to the use of a second-line injectable antituberculosis agent. Children with MDR-TB received individualized treatment (Table 3) informed by the drug susceptibility test result for the child's or the known source case's isolate, TB disease severity, and consideration of the toxicity of the anti-TB drugs. During 2012, levofloxacin and moxifloxacin replaced ofloxacin as the fluoroquinolones of choice for MDR-TB treatment in South Africa. All children with TB were routinely tested for HIV; HIV-infected children not yet established on cART were initiated on cART within 2 weeks of the start of MDR-TB treatment.
Study procedures.
Data on demographic and clinical characteristics, including WHO HIV clinical staging, HIV viral load, CD4+ T-cell count (most recent routine result), TB disease status, and type of TB (the MDR-TB group), were collected for all children at the time of pharmacokinetic sampling. Anthropometric measurements, WHO-derived Z-scores, gestational age if it was <2 years, concomitant medication, and concurrent illnesses were documented. Adherence to antituberculosis drugs and ARVs was assessed by a visual analogue score and 3-day recall.
Pharmacokinetic design.
The lopinavir-ritonavir products used were the Kaletra 80/20-mg/ml oral suspension and Aluvia 100/25-mg tablets (Abbott, Chicago, IL, USA).
All children received a 300/75-mg/m2 dose of lopinavir-ritonavir twice daily; children on rifampin as part of their MDR-TB treatment regimen received superboosted lopinavir-ritonavir with an additional 226 mg/m2 ritonavir twice daily per South African guidelines (53). For the MDR-TB group, the evening dose was observed by routine ward personnel, as the children in this group were inpatients. The control group's evening dose was documented by the parent/caregiver. Mealtimes in relation to the timing of the evening dose were not recorded. On the day of pharmacokinetic sampling, the exact doses of each drug were calculated for each child. Suspensions were appropriately inverted several times and measured to the nearest 0.1 ml. Tablets were cut and weighed to the nearest 0.1 mg and given either whole or crushed and suspended in 5 to 10 ml water, according to patient tolerance. All antituberculosis drugs were administered after a minimum of a 4-h fast; ARVs, including lopinavir-ritonavir, were dosed 1 h later, and afterwards a standard breakfast was given. Some children below 2 years of age were dosed using a nasogastric tube to improve drug administration on the sampling day on the basis of drug administration challenges.
Pharmacokinetic data were collected using intensive pharmacokinetic sampling. Blood samples were collected at 6 time points: 2 samples were collected before dosing of the ARVs (1 h before the ARV dose [time −1 h] and immediately before [time zero]), and one sample each was collected at 1, 3, 7, and either 5 or 10 h after the observed dosing. The samples were collected in EDTA-containing Vacutainer tubes, and the tubes were centrifuged. Plasma was separated within 30 min and frozen at −80°C. As the study was nested in a larger pharmacokinetic study on drugs for the treatment of MDR-TB, the sampling schedule was optimized around the time of dosing of the antituberculosis drugs, resulting in two samples being collected prior to lopinavir-ritonavir dosing.
Analytical method.
Lopinavir and ritonavir concentrations were determined using a validated liquid chromatography-tandem mass spectrometry assay at the Division of Clinical Pharmacology, University of Cape Town. The lower limits of quantification (LLOQ) were 0.0195 mg/liter for lopinavir and 0.00488 mg/liter for ritonavir. The intraday and interday coefficients of variation were below 11%. The laboratory subscribes to the AIDS Clinical Pharmacology Quality Assurance Antiretroviral Proficiency Testing Program of the National Institute of Allergy and Infectious Diseases (NIAID) Division of AIDS (DAIDS).
The study was approved by Stellenbosch University (N11/03/059) and the University of Cape Town (397/2011) Health Research Ethics Committees. Written informed consent was obtained from the parents/legal guardians, and assent was obtained from the participants when appropriate.
Population pharmacokinetic modeling.
Population pharmacokinetic analyses were completed using Monolix software (version 2016R1, 2016; Lixoft SAS Antony, France) and the stochastic approximation expectation-maximization (SAEM) (54) for parameter estimation. Computations were performed using facilities provided by the University of Cape Town ICTS high-performance computing team (http://hpc.uct.ac.za).
Several structural models for lopinavir and ritonavir were tested, including models with one- and two-compartment dispositions with first-order elimination and first-order absorption with and without absorption lag time or transit compartments (55).
A sequential approach was used to develop the model. First, two separate independent models for lopinavir and ritonavir were developed, and then the models for the two drugs were integrated into a joint population model in order to estimate the correlation between the random effects of lopinavir and ritonavir and to characterize the effect of ritonavir concentrations on the pharmacokinetics of lopinavir. Several models previously proposed were tested to describe the inhibitory effect of the ritonavir concentration on lopinavir clearance, including models with linear or exponential relationships (18, 41) and a maximum inhibition (Imax) model (20, 56).
The Imax model is a dynamic interaction model that determines how the real-time model-predicted ritonavir concentration affects lopinavir clearance using the relationship shown below:
| (1) |
where CLLPV is the clearance of lopinavir, CL0 is the lopinavir clearance when the ritonavir concentration (CRTV) is 0, Imax is the maximum inhibitory effect of the ritonavir concentration on lopinavir clearance, EC50 is the concentration at which 50% of the maximal inhibitory effect is obtained, and γ, also known as the Hill coefficient, is the shape factor of the curve.
The relative bioavailability was fixed to 1 for a typical patient. The pharmacokinetic samples collected predose were treated as a separate occasion in the model to allow estimation of both interindividual variability (IIV) and interoccasion variability (IOV). A lognormal distribution was assumed for these random effects, and the correlation between them was investigated at both the IIV level and the IOV level. The residual unexplained variability (RUV) was evaluated using a combined additive and proportional model. Censored data below the lower limit of quantification (LLOQ) were handled with the Monolix implementation of the M3 method (57). Instead of minimizing the distance between the model prediction and the observed concentration, which is the standard procedure used for all noncensored values, the M3 method maximizes the likelihood that the observation is lower than the LLOQ.
Allometric scaling using fixed coefficients was applied to the apparent clearance and volume of distribution (V) to account for differences in body size (58). Besides total body weight, fat-free mass (59) and body surface area were also tested as alternative descriptors of body size.
After the inclusion of allometric scaling, covariate selection was performed by first narrowing the search to factors that were either known to affect or physiologically plausibly affected a certain pharmacokinetic parameter. Then, the plots of individual random effects (empirical Bayes estimates) (60) versus covariates were used to screen possibly significant trends in the data. Finally, the candidate covariate effects were tested and included in the model using a stepwise procedure with forward inclusion (P < 0.05, based on a drop in the −2 log likelihood [−2LL] value) and backward elimination (P < 0.01). Additionally, the improvement in the goodness of fit (including the VPCs), the reduction in unexplained variability, and the stability of the model parameter estimates were considered to retain the effects in the model.
Age was tested using a sigmoid maximum-effect maturation model (61, 62). MDR-TB treatment, as a single combined variable, was used as a categorical covariate to test whether parameter estimates (oral clearance, bioavailability, and the absorption rate constant [ka]) were different between the MDR-TB group and the control group both in the final integrated model and in the separate ritonavir and lopinavir models.
Other covariates tested for significance in the model were sex, the method of drug administration on the sampling day (via a nasogastric tube versus by the oral route), the drug formulation (suspension or whole tablets), and nutritional status (WHO-derived Z-scores for weight for age [WAZ] and height for age [HAZ]). According to WHO growth standards (2010), a value lower than −2 was used to classify low WAZ (underweight) and low HAZ (stunted) (63).
Model development was guided by changes in the −2LL value (with drops of more than 6.63 points being considered significant at a P value of <0.01 for the inclusion of 1 additional parameter in the model), precision in parameter estimates (relative standard error [RSE], in percent), graphical analysis of goodness-of-fit plots (including VPC) (64), and scientific plausibility.
Statistical power calculations.
The analysis presented in this work was opportunistic and built into a larger study of children with MDR-TB, and no formal calculation of statistical power was prospectively performed. To assess the statistical power of our data to detect significant differences in clearance and bioavailability between children with MDR-TB and controls, we performed an a posteriori power analysis based on simulations (65). Since no automatic procedure is available in Monolix, the model was reimplemented in the NONMEM (version 7.4) program (66) and the simulation analysis was implemented using the stochastic simulation and estimation (SSE) tool of Perl-speaks-NONMEM (67). Briefly, the final PK model was used to resimulate (n = 200) the current trial (thus assuming the same patient covariates, doses, and sampling times), but with postulation of a known difference in clearance or bioavailability between the MDR-TB and control groups. Then, alternative models with or without this MDR-TB covariate effect were fit to each simulated data set and compared to evaluate whether the effect was statistically significant in terms of improvement of the −2LL value. The percentage of simulated data in which the effect could be detected as significant provided the statistical power.
Supplementary Material
ACKNOWLEDGMENTS
We thank the children and parents for participating in this study. We also thank the clinical team, including Heather R. Draper, Marianne Willemse, Adelaide Carelse, Primrose Puling, Zibelezenkosi Hlebani, Nickey Jass, Klassina Zimri, and Zingiwe Mramba, for their assistance in study implementation. Special recognition goes to the University of Cape Town ICTS high-performance computing team (http://hpc.uct.ac.za) for the use of its facilities and to Norvatis for sponsoring the Monolix workshop that L. E. van der Laan attended.
Funding was provided by an R01 grant from The Eunice Kennedy Shriver National Institute of Child Health and Human Development (grant NICHD-069169-01; principal investigator, A. C. Hesseling) and the South African National Research Foundation (SArCHI chair, A. C. Hesseling). H. McIlleron received a National Research Foundation grant for rated researchers 90729. Drug assays at the UCT pharmacokinetic laboratory were supported in part by NIAID (UM1 AI068634, UM1 AI068636, UM1AI106701, U01 AI068632), NICHD, and the National Institute of Mental Health (AI068632).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00420-17.
REFERENCES
- 1.World Health Organization. 2013. Treatment of children living with HIV. World Health Organization, Geneva, Switzerland: http://www.who.int/hiv/topics/paediatric/en/. [Google Scholar]
- 2.World Health Organization. 2015. Tuberculosis. World Health Organization, Geneva, Switzerland. http://www.who.int/mediacentre/factsheets/fs104/en/. [Google Scholar]
- 3.McIlleron H, Meintjes G, Burman WJ, Maartens G. 2007. Complications of antiretroviral therapy in patients with tuberculosis: drug interactions, toxicity, and immune reconstitution inflammatory syndrome. J Infect Dis 196:S63–S75. doi: 10.1086/518655. [DOI] [PubMed] [Google Scholar]
- 4.Dodd PJ, Sismanidis C, Seddon JA. 2016. Global burden of drug-resistant tuberculosis in children: a mathematical modelling study. Lancet Infect Dis 16:1193–1201. doi: 10.1016/S1473-3099(16)30132-3. [DOI] [PubMed] [Google Scholar]
- 5.Jenkins HE, Tolman AW, Yuen CM, Parr JB, Keshavjee S, Pérez-Vélez CM, Pagano M, Becerra MC, Cohen T. 2014. Incidence of multidrug-resistant tuberculosis disease in children: systematic review and global estimates. Lancet 383:1572–1579. doi: 10.1016/S0140-6736(14)60195-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fairlie L, Beylis NC, Reubenson G, Moore DP, Madhi SA. 2011. High prevalence of childhood multi-drug resistant tuberculosis in Johannesburg, South Africa: a cross sectional study. BMC Infect Dis 11:28. doi: 10.1186/1471-2334-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seddon JA, Hesseling AC, Willemse M, Donald PR, Schaaf HS. 2012. Culture-confirmed multidrug-resistant tuberculosis in children: clinical features, treatment, and outcome. Clin Infect Dis 54:157–166. doi: 10.1093/cid/cir772. [DOI] [PubMed] [Google Scholar]
- 8.Hesseling AC, Kim S, Madhi S, Nachman S, Schaaf HS, Violari A, Victor TC, McSherry G, Mitchell C, Cotton MF. 2012. High prevalence of drug resistance amongst HIV-exposed and -infected children in a tuberculosis prevention trial. Int J Tuberc Lung Dis 16:192–195. doi: 10.5588/ijtld.10.0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seddon JA, Hesseling AC, Godfrey-Faussett P, Schaaf HS. 2014. High treatment success in children treated for multidrug-resistant tuberculosis: an observational cohort study. Thorax 69:458–464. doi: 10.1136/thoraxjnl-2013-203900. [DOI] [PubMed] [Google Scholar]
- 10.Seddon JA, Hesseling AC, Marais BJ, Jordaan A, Victor T, Schaaf HS. 2012. The evolving epidemic of drug-resistant tuberculosis among children in Cape Town, South Africa. Int J Tuberc Lung Dis 16:928–933. doi: 10.5588/ijtld.11.0679. [DOI] [PubMed] [Google Scholar]
- 11.Seddon JA, Hesseling AC, Marais BJ, McIlleron H, Peloquin CA, Donald PR, Schaaf HS. 2012. Paediatric use of second-line anti-tuberculosis agents: a review. Tuberculosis 92:9–17. doi: 10.1016/j.tube.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 12.World Health Organization. 2016. Treatment guidelines for drug-resistant tuberculosis. World Health Organization, Geneva, Switzerland: http://apps.who.int/iris/bitstream/10665/250125/1/9789241549639-eng.pdf?ua=1. [Google Scholar]
- 13.Schaaf HS, Victor TC, Engelke E, Brittle W, Marais BJ, Hesseling AC, Van Helden PD, Donald PR. 2007. Minimal inhibitory concentration of isoniazid in isoniazid-resistant Mycobacterium tuberculosis isolates from children. Eur J Clin Microbiol Infect Dis 26:203–205. doi: 10.1007/s10096-007-0257-9. [DOI] [PubMed] [Google Scholar]
- 14.Schaaf HS, Victor TC, Venter A, Brittle W, Jordaan AM, Hesseling AC, Marais BJ, Van Helden PD, Donald PR. 2009. Ethionamide cross- and co-resistance in children with isoniazid-resistant tuberculosis. Int J Tuberc Lung Dis 13:1355–1359. [PubMed] [Google Scholar]
- 15.Chang K-C, Leung C-C, Yew W-W, Leung EC-C, Leung W-M, Tam C-M, Zhang Y. 2012. Pyrazinamide may improve fluoroquinolone-based treatment of multidrug-resistant tuberculosis. Antimicrob Agents Chemother 56:5465–5475. doi: 10.1128/AAC.01300-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.World Health Organization. 2016. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach. World Health Organization, Geneva, Switzerland. [PubMed] [Google Scholar]
- 17.Marais BJ, Gie RP, Schaaf HS, Hesseling AC, Obihara CC, Starke JJ, Enarson DA, Donald PR, Beyers N. 2004. The natural history of childhood intra-thoracic tuberculosis: a critical review of literature from the pre-chemotherapy era. Int J Tuberc Lung Dis 8:392–402. [PubMed] [Google Scholar]
- 18.Elsherbiny D, Ren Y, McIlleron H, Maartens G, Simonsson USH. 2010. Population pharmacokinetics of lopinavir in combination with rifampicin-based antitubercular treatment in HIV-infected South African children. Eur J Clin Pharmacol 66:1017–1023. doi: 10.1007/s00228-010-0847-9. [DOI] [PubMed] [Google Scholar]
- 19.Reference deleted.
- 20.Zhang C, Denti P, Decloedt EH, Ren Y, Karlsson MO, McIlleron H. 2013. Model-based evaluation of the pharmacokinetic differences between adults and children for lopinavir and ritonavir in combination with rifampicin. Br J Clin Pharmacol 76:741–751. doi: 10.1111/bcp.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salem F, Rostami-Hodjegan A, Johnson TN. 2013. Do children have the same vulnerability to metabolic drug-drug interactions as adults?. A critical analysis of the literature J Clin Pharmacol 53:559–566. [DOI] [PubMed] [Google Scholar]
- 22.Salem F, Johnson TN, Barter ZE, Leeder JS, Rostami-Hodjegan A. 2013. Age related changes in fractional elimination pathways for drugs: assessing the impact of variable ontogeny on metabolic drug-drug interactions. J Clin Pharmacol 53:857–865. doi: 10.1002/jcph.100. [DOI] [PubMed] [Google Scholar]
- 23.ter Heine R, Van Waterschoot RAB, Keizer RJ, Beijnen JH, Schinkel AH, Huitema ADR. 2011. An integrated pharmacokinetic model for the influence of CYP3A4 expression on the in vivo disposition of lopinavir and its modulation by ritonavir. J Pharm Sci 100:2508–2515. doi: 10.1002/jps.22457. [DOI] [PubMed] [Google Scholar]
- 24.Vishnuvardhan D, Moltke LL, Richert C, Greenblatt DJ. 2003. Lopinavir: acute exposure inhibits P-glycoprotein; extended exposure induces P-glycoprotein. AIDS 17:1092–1094. doi: 10.1097/00002030-200305020-00023. [DOI] [PubMed] [Google Scholar]
- 25.Patterson KB, Dumond JB, Prince HA, Jenkins AJ, Scarsi KK, Wang R, Malone S, Hudgens MG, Kashuba ADM. 2013. Protein binding of lopinavir and ritonavir during four phases of pregnancy: implications for treatment guidelines. J Acquir Immune Defic Syndr 63:51. doi: 10.1097/QAI.0b013e31827fd47e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anonymous. 2008. Moxifloxacin. Tuberculosis 88:127–131. doi: 10.1016/S1472-9792(08)70016-7. [DOI] [PubMed] [Google Scholar]
- 27.Lehmann J. 1969. The role of the metabolism of p-aminosalicylic acid (PAS) in the treatment of tuberculosis. Interaction with the metabolism of isonicotinic acid hydrazide (INH) and the synthesis of cholesterol. Scand J Respir Dis 50:169. [PubMed] [Google Scholar]
- 28.MacGowan AP. 2003. Pharmacokinetic and pharmacodynamic profile of linezolid in healthy volunteers and patients with Gram-positive infections. J Antimicrob Chemother 51:ii17–ii25. doi: 10.1093/jac/dkg248. [DOI] [PubMed] [Google Scholar]
- 29.Wynalda MA, Hauer MJ, Wienkers LC. 2000. Oxidation of the novel oxazolidinone antibiotic linezolid in human liver microsomes. Drug Metab Dispos 28:1014–1017. [PubMed] [Google Scholar]
- 30.Garcia-Prats Donald PR, Hesseling AC, Schaaf HSAJ. 2013. Second-line antituberculosis drugs in children: a commissioned review for the World Health Organization 19th Expert Committee on the Selection and Use of Essential Medicines. World Health Organization, Geneva, Switzerland: http://www.who.int/selection_medicines/committees/expert/19/applications/TB_624_C_R.pdf. [Google Scholar]
- 31.Cholo MC, Steel HC, Fourie PB, Germishuizen WA, Anderson R. 2012. Clofazimine: current status and future prospects. J Antimicrob Chemother 67:290–298. doi: 10.1093/jac/dkr444. [DOI] [PubMed] [Google Scholar]
- 32.Henderson MC, Siddens LK, Morré JT, Krueger SK, Williams DE. 2008. Metabolism of the anti-tuberculosis drug ethionamide by mouse and human FMO1, FMO2 and FMO3 and mouse and human lung microsomes. Toxicol Appl Pharmacol 233:420–427. doi: 10.1016/j.taap.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desta Z, Soukhova NV, Flockhart DA. 2001. Inhibition of cytochrome P450 (CYP450) isoforms by isoniazid: potent inhibition of CYP2C19 and CYP3A. Antimicrob Agents Chemother 45:382–392. doi: 10.1128/AAC.45.2.382-392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Decloedt EH, van der Walt JS, McIlleron H, Wiesner L, Maartens G. 2015. The pharmacokinetics of lopinavir-ritonavir when given with isoniazid in South African HIV-infected individuals. Int J Tuberc Lung Dis 19:1194–1196. doi: 10.5588/ijtld.15.0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Escribano I, Rodriguez JC, Llorca B, Garcia-Pachon E, Ruiz M, Royo G. 2007. Importance of the efflux pump systems in the resistance of Mycobacterium tuberculosis to fluoroquinolones and linezolid. Chemotherapy 53:397–401. doi: 10.1159/000109769. [DOI] [PubMed] [Google Scholar]
- 36.Bolhuis MS, van Altena R, van Soolingen D, de Lange WCM, Uges DRA, van der Werf TS, Kosterink JGW, Alffenaar J-WC. 2013. Clarithromycin increases linezolid exposure in multidrug-resistant tuberculosis patients. Eur Respir J 42:1614–1621. doi: 10.1183/09031936.00001913. [DOI] [PubMed] [Google Scholar]
- 37.Brillault J, De Castro WV, Harnois T, Kitzis A, Olivier J-C, Couet W. 2009. P-glycoprotein-mediated transport of moxifloxacin in a Calu-3 lung epithelial cell model. Antimicrob Agents Chemother 53:1457–1462. doi: 10.1128/AAC.01253-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bouazza N, Urien S, Blanche S, Hirt D, Foissac F, Benaboud S, Tréluyer J-M, Frange P. 2014. Concentration-response model of lopinavir/ritonavir in HIV-1-infected pediatric patients. Pediatr Infect Dis J 33:e213–e218. doi: 10.1097/INF.0000000000000298. [DOI] [PubMed] [Google Scholar]
- 39.Bouazza N, Foissac F, Fauchet F, Burger D, Kiechel J-R, Treluyer J-M, Capparelli EV, Lallemant M, Urien S. 2015. Lopinavir/ritonavir plus lamivudine and abacavir or zidovudine dose ratios for paediatric fixed-dose combinations. Antivir Ther 20:225–233. doi: 10.3851/IMP3016. [DOI] [PubMed] [Google Scholar]
- 40.Jullien V, Urien S, Hirt D, Delaugerre C, Rey E, Teglas J-P, Vaz P, Rouzioux C, Chaix M-L, Macassa E. 2006. Population analysis of weight-, age-, and sex-related differences in the pharmacokinetics of lopinavir in children from birth to 18 years. Antimicrob Agents Chemother 50:3548–3555. doi: 10.1128/AAC.00943-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikanjam M, Chadwick EG, Robbins B, Alvero C, Palumbo P, Yogev R, Pinto J, Hazra R, Hughes ML, Heckman BE. 2012. Assessment of lopinavir pharmacokinetics with respect to developmental changes in infants and the impact on weight band-based dosing. Clin Pharmacol Ther 91:243–249. doi: 10.1038/clpt.2011.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartelink IH, Savic RM, Dorsey G, Ruel T, Gingrich D, Scherpbier HJ, Capparelli E, Jullien V, Young SL, Achan J. 2015. The effect of malnutrition on the pharmacokinetics and virologic outcomes of lopinavir, efavirenz and nevirapine in food insecure HIV-infected children in Tororo, Uganda. Pediatr Infect Dis J 34:e63. doi: 10.1097/INF.0000000000000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DiCenzo R, Peterson D, Cruttenden K, Morse G, Riggs G, Gelbard H, Schifitto G. 2004. Effects of valproic acid coadministration on plasma efavirenz and lopinavir concentrations in human immunodeficiency virus-infected adults. Antimicrob Agents Chemother 48:4328–4331. doi: 10.1128/AAC.48.11.4328-4331.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urien S, Firtion G, Anderson ST, Hirt D, Solas C, Peytavin G, Faye A, Thuret I, Leprevost M, Giraud C. 2011. Lopinavir/ritonavir population pharmacokinetics in neonates and infants. Br J Clin Pharmacol 71:956–960. doi: 10.1111/j.1365-2125.2011.03926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu A, Granneman GR, Witt G, Locke C, Denissen J, Molla A, Valdes J, Smith J, Erdman K, Lyons N. 1997. Multiple-dose pharmacokinetics of ritonavir in human immunodeficiency virus-infected subjects. Antimicrob Agents Chemother 41:898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Justesen US. 2008. Protease inhibitor plasma concentrations in HIV antiretroviral therapy. Dan Med Bull 55:165–185. [PubMed] [Google Scholar]
- 47.Ohno M, Yamaguchi I, Ito T, Saiki K, Yamamoto I, Azuma J. 2000. Circadian variation of the urinary 6β-hydroxycortisol to cortisol ratio that would reflect hepatic CYP3A activity. Eur J Clin Pharmacol 55:861–865. doi: 10.1007/s002280050708. [DOI] [PubMed] [Google Scholar]
- 48.Awni W, Chiu YL, Zhu T, Braun N, Klein C, Heuser R, Breitenbach J, Morris J, Doan T, Brun S. 2005. Significantly reduced food effect and pharmacokinetic variability with a novel lopinavir-ritonavir tablet formulation. Age 35:19–55. http://www.medadvocates.org/resources/conferences/3rd%20_ias/3rdiasawni.pdf. [Google Scholar]
- 49.Ramakrishnan K, Shenbagarathai R, Kavitha K, Uma A, Balasubramaniam R, Thirumalaikolundusubramanian P. 2008. Serum zinc and albumin levels in pulmonary tuberculosis patients with and without HIV. Jpn J Infect Dis 61:202–204. [PubMed] [Google Scholar]
- 50.Swaminathan S, Padmapriyadarsini C, Sukumar B, Iliayas S, Kumar SR, Triveni C, Gomathy P, Thomas B, Mathew M, Narayanan PR. 2008. Nutritional status of persons with HIV infection, persons with HIV infection and tuberculosis, and HIV-negative individuals from southern India. Clin Infect Dis 46:946–949. doi: 10.1086/528860. [DOI] [PubMed] [Google Scholar]
- 51.Best BM, Capparelli EV, Diep H, Rossi SS, Farrell MJ, Williams E, Lee G, van den Anker JN, Rakhmanina N. 2011. Pharmacokinetics of lopinavir-ritonavir crushed versus whole tablets in children. J Acquir Immune Defic Syndr 58:385. doi: 10.1097/QAI.0b013e318232b057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guiard-Schmid J-B, Poirier J-M, Meynard J-L, Bonnard P, Gbadoe AH, Amiel C, Calligaris F, Abraham B, Pialoux G, Girard P-M. 2003. High variability of plasma drug concentrations in dual protease inhibitor regimens. Antimicrob Agents Chemother 47:986–990. doi: 10.1128/AAC.47.3.986-990.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Department of Health of the Republic of South Africa. 2015. National consolidated guidelines for the prevention of mother-to-child transmission of HIV (PMTCT) and the management of HIV in children, adolescents and adults. Department of Health of the Republic of South Africa, Pretoria, South Africa. [Google Scholar]
- 54.Kuhn E, Lavielle M. 2005. Maximum likelihood estimation in nonlinear mixed effects models. Comput Stat Data Anal 49:1020–1038. doi: 10.1016/j.csda.2004.07.002. [DOI] [Google Scholar]
- 55.Savic RM, Jonker DM, Kerbusch T, Karlsson MO. 2007. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn 34:711–726. doi: 10.1007/s10928-007-9066-0. [DOI] [PubMed] [Google Scholar]
- 56.Moltó J, Barbanoj MJ, Miranda C, Blanco A, Santos JR, Negredo E, Costa J, Domingo P, Clotet B, Valle M. 2008. Simultaneous population pharmacokinetic model for lopinavir and ritonavir in HIV-infected adults. Clin Pharmacokinet 47:681–692. doi: 10.2165/00003088-200847100-00005. [DOI] [PubMed] [Google Scholar]
- 57.Beal SL. 2001. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 28:481–504. doi: 10.1023/A:1012299115260. [DOI] [PubMed] [Google Scholar]
- 58.Anderson BJ, Holford NHG. 2008. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol 48:303–332. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 59.Al-Sallami HS, Goulding A, Grant A, Taylor R, Holford N, Duffull SB. 2015. Prediction of fat-free mass in children. Clin Pharmacokinet 54:1169–1178. doi: 10.1007/s40262-015-0277-z. [DOI] [PubMed] [Google Scholar]
- 60.Lavielle M, Ribba B. 2016. Enhanced method for diagnosing pharmacometric models: random sampling from conditional distributions. Pharm Res 33:2979–2988. doi: 10.1007/s11095-016-2020-3. [DOI] [PubMed] [Google Scholar]
- 61.Anderson BJ, Holford NHG. 2009. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 24:25–36. doi: 10.2133/dmpk.24.25. [DOI] [PubMed] [Google Scholar]
- 62.Holford N, Heo Y, Anderson B. 2013. A pharmacokinetic standard for babies and adults. J Pharm Sci 102:2941. doi: 10.1002/jps.23574. [DOI] [PubMed] [Google Scholar]
- 63.World Health Organization. Moderate malnutrition. World Health Organization, Geneva, Switzerland: http://www.who.int/nutrition/topics/moderate_malnutrition/en/. [Google Scholar]
- 64.Karlsson MO, Holford N. 2008. A tutorial on visual predictive checks. Annu Meet Population Approach Group in Europe, Marseille, France. [Google Scholar]
- 65.Lee PI. 2001. Design and power of a population pharmacokinetic study. Pharm Res 18:75–82. doi: 10.1023/A:1011030827847. [DOI] [PubMed] [Google Scholar]
- 66.Beal S, Sheiner L, Boeckmann A, Bauer R. 2009. NONMEM user's guides 1989-2009. Icon Development Solutions, Ellicott City, MD. [Google Scholar]
- 67.Lindbom L, Ribbing J, Jonsson E. 2004. Perl-speaks-NONMEM (PsN)—a Perl module for NONMEM related programming. Comput Methods Programs Biomed 75:85–94. doi: 10.1016/j.cmpb.2003.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.