

ABSTRACT
RG7667, a novel combination of two anticytomegalovirus (anti-CMV) monoclonal IgG1 antibodies (MCMV5322A and MCMV3068A), was designed to block CMV entry into host cells. It was developed as a potential therapy for preventing CMV infection and disease in transplant recipients. RG7667 was assessed for preventing CMV infection in a phase 2a trial with CMV-seronegative recipients of kidney transplants from CMV-seropositive donors. The patients received 4 intravenous doses of RG7667 (10 mg/kg of body weight of each antibody, n = 60) or placebo (n = 60) at the time of the transplant and at 1, 4, and 8 weeks after the transplant. Serum samples were collected for pharmacokinetic (PK) analysis and antidrug antibody (ADA) evaluation. To guide future dose selection, the relationships between RG7667 exposure and pharmacological activity were evaluated. MCMV5322A and MCMV3068A exposures were confirmed in all RG7667-treated patients. Mean clearances for MCMV5322A and MCMV3068A were 2.97 and 2.65 ml/day/kg, respectively, and the terminal half-lives of MCMV5322A and MCMV3068A were 26.9 and 27.4 days, respectively. The ADA incidence was low and was not associated with lower drug exposure. Patients with RG7667 or component antibody exposures greater than the respective median values had a lower incidence of viremia at 12 weeks and 24 weeks after transplantation and a longer delayed time to detectable CMV viremia than patients with exposures less than the median values. MCMV5322A and MCMV3068A exhibited expected IgG1 PK profiles in high-risk kidney transplant recipients, consistent with the earlier PK behavior of RG7667 in healthy subjects. Higher drug exposure was associated with better anti-CMV pharmacological activity. (This study has been registered at ClinicalTrials.gov under identifier NCT01753167.)
KEYWORDS: RG7667, cytomegalovirus, kidney transplantation, monoclonal antibodies, pharmacokinetics
INTRODUCTION
Cytomegalovirus (CMV)-associated disease is a significant cause of posttransplant complications, despite the administration of antiviral medications. In particular, CMV-seronegative recipients of a solid organ transplant (SOT) from a CMV-seropositive donor (donor-positive and recipient-negative [D+R−] transplants) are at high risk for CMV infection and subsequent CMV disease (1, 2). Although antivirals, such as acyclovir, ganciclovir, or valganciclovir, have dramatically decreased the incidence of CMV-associated disease during the early posttransplant period, they have significant adverse effects, such as neutropenia (3) and impaired cell-mediated immunity (4). Late-onset CMV disease, which occurs more than 3 months after transplantation, or very late disease, which occurs at any time later than 1 to 2 years following transplantation, may continue to occur and is also associated with graft failure and decreased patient survival (4–6).
Previous studies have demonstrated that CMV intravenous immunoglobulin (CMV-IVIG) can decrease the incidence of CMV infection and disease in renal transplant recipients in D+R− transplants, suggesting that antibody-based therapies may be effective for treating CMV infection (1, 7, 8). However, CMV-IVIG is expensive and not commonly used due to the availability of antiviral medications (9, 10). RG7667 is composed of two monoclonal antibodies, administered in a 1:1 ratio, that bind to distinct CMV antigens required for cellular entry and inhibit infection of relevant host cells (11). The first component, MCMV5322A, is a human immunoglobulin G1(κ) [IgG1(κ)] antibody that binds with single-digit nanomolar affinity to gH, a human CMV envelope protein that is present on the surface of the virus and that is required for entry into all cell types susceptible to CMV infection. The second component, MCMV3068A, a humanized IgG1(λ) antibody, binds with single-digit nanomolar affinity to a human CMV epitope that is formed by a complex of five CMV envelope proteins (11) and that is required for viral entry into epithelial cells, endothelial cells, and macrophages (12–16). Although MCMV3068A is better at neutralizing CMV than MCMV5322A (11), it is unable to prevent infection of fibroblasts (12–16). Therefore, the antibody combination should provide the optimal potency for blocking viral entry into all four key cell types that are targeted by CMV and suppressing viral resistance (11).
RG7667, initially tested in a phase 1 study in healthy volunteers, was well tolerated up to single intravenous (i.v.) doses of 10 mg/kg of body weight of each antibody (20 mg/kg total) (11). Terminal half-life (t1/2) estimates for MCMV5322A and MCMV3068A were similar and dose independent, with mean values ranging from 24.6 to 28.3 days for both antibodies following single doses of RG7667. The pharmacokinetic (PK) parameters obtained after the administration of multiple i.v. doses of MCMV5322A and MCMV3068A were consistent with data obtained after the administration of single doses (11). The PK of each antibody were comparable to those of a typical human IgG1 lacking an endogenous target in humans after monotherapy (17, 18).
In a phase 2a, randomized, double-blind, placebo-controlled study (ClinicalTrials.gov identifier NCT01753167), we examined the safety and pharmacological activity of multiple i.v. doses of RG7667 in preventing CMV infection in kidney transplant recipients in D+R− transplants (19). In addition to RG7667 treatment, the study used a preemptive approach instead of universal prophylaxis to prevent CMV disease: anti-CMV therapy with other antiviral agents was initiated only in patients with the early replication of CMV. Although RG7667 did not significantly reduce the proportion of patients with CMV viremia within 12 weeks posttransplant, it significantly reduced the proportion of patients with CMV viremia within 24 weeks posttransplant. RG7667 also significantly delayed the median time to detectable CMV viremia compared to that achieved with placebo (139 days versus 46 days). The serum concentrations of both antibodies exhibited a biphasic disposition, with an initial rapid distribution phase followed by a slow elimination phase. The mean terminal half-lives were 26.9 for MCMV5322A and 27.4 days for MCMV3068A, which were consistent with the terminal half-lives in healthy subjects. To guide dose selection for future clinical studies, this analysis of the phase 2a study further characterized the PK and immunogenicity of RG7667 and the relationship between drug exposure and pharmacological activity.
RESULTS
Patient demographics.
Details of the patient demographics, the randomization process, and the disposition of the patients have been previously reported (19). Briefly, 120 patients at 39 sites in 8 countries (the United States and the European Union) were enrolled from December 2012 to April 2014. Sixty patients were randomized to RG7667 (10 mg/kg of each antibody [20 mg/kg total] administered on days 1, 8, 29, and 57), and 60 patients were randomized to placebo. One patient in the RG7667 group and two patients in the placebo group did not receive either RG7667 or placebo because they tested CMV seropositive at screening. In addition, one patient in the placebo group withdrew before dosing. There were 116 patients in the modified intention-to-treat population (59 in the RG7667 group and 57 in the placebo group). The population in which PK were evaluable consisted of 59 patients who received at least 1 dose of RG7667. The dose groups were generally well balanced for demographic characteristics, including body weight, age, gender, and other demographic characteristics, and there were no imbalances that would have affected efficacy, safety, or PK (19).
Pharmacokinetics.
As previously described (19), serum MCMV5322A and MCMV3068A concentrations exhibited a biphasic disposition, with an initial rapid distribution phase followed by a slow elimination phase (Fig. 1), in kidney transplant recipients at high risk of CMV infection. Additionally, the serum concentrations observed from the phase 2a study were consistent with predicted concentrations obtained from a Monte Carlo simulation based on a model built using the human PK observed in the RG7667 phase 1 study (Fig. 1) (11). At week 12 (day 84), the primary efficacy time point, the observed concentrations of each antibody were well above the target trough concentrations of 5.45 μg/ml (MCMV5322A) and 3.88 μg/ml (MCMV3068A), which were based on in vitro activity and other factors (see Discussion for more details) for all subjects with available PK data (Fig. 1; see also Fig. S1 in the supplemental material). The mean values of the PK parameters for MCMV5322A and MCMV3068A following the administration of multiple i.v. doses of RG7667 are summarized in Table 1. For MCMV5322A, the observed mean clearance (CL) and t1/2 were 2.97 ml/day/kg and 26.9 days, respectively. The observed mean volume of distribution at steady state (Vss) was 110 ml/kg. The accumulation ratios based on the maximum observed serum concentration (Cmax), the minimum observed serum concentration (Cmin), and the area under the serum concentration-time curve (AUC) were 1.47, 1.20, and 1.60, respectively. For MCMV3068A, the observed mean CL and t1/2 were 2.65 ml/day/kg and 27.4 days, respectively. The observed mean Vss was 99.8 ml/kg. The accumulation ratios based on Cmax, Cmin, and AUC were 1.50, 1.28, and 1.74, respectively.
FIG 1.

Observed concentrations of MCMV5322A (A) and MCMV3068A (B) versus time in a phase 2a overlay with predictions from a simulation using a two-compartment population PK model based on phase 1 PK data. Black circles, observed phase 2a data; blue shaded areas, 95% confidence interval; gray dashed lines, median; blue dashed lines, target concentration for each antibody. The 95% confidence intervals and medians were derived on the basis of the simulated concentrations for 1,000 subjects.
TABLE 1.
PK parameters for MCMV5322A and MCMV3068A
| Parametera | MCMV5322A |
MCMV3068A |
||
|---|---|---|---|---|
| No. of patients | Mean ± SD value | No. of patients | Mean ± SD value | |
| Cmax_dose_1 (μg/ml) | 59 | 203 ± 40.2 | 59 | 213 ± 50.3 |
| Cmax_dose_4 (μg/ml) | 50 | 294 ± 69.8 | 50 | 306 ± 71.9 |
| AR_Cmax | 50 | 1.47 ± 0.270 | 50 | 1.50 ± 0.358 |
| Cmin_wk_1 (μg/ml) | 54 | 73.3 ± 24.7 | 55 | 88.0 ± 34.0 |
| Cmin_wk_12 (μg/ml) | 49 | 84.6 ± 30.3 | 49 | 104 ± 36.1 |
| AR_Cmin | 49 | 1.20 ± 0.326 | 49 | 1.28 ± 0.435 |
| AUC0–7 (μg · day/ml) | 56 | 740 ± 145 | 50 | 831 ± 158 |
| AUC56–63 (μg · day/ml) | 35 | 1,170 ± 508 | 34 | 1,350 ± 145 |
| AUC56–84 (μg · day/ml) | 35 | 3,180 ± 1,400 | 34 | 3,810 ± 1,590 |
| AUCinfb (μg · day/ml) | 36 | 7,830 ± 3,260 | 37 | 9,050 ± 3,790 |
| AR_AUC7 | 32 | 1.60 ± 0.607 | 28 | 1.74 ± 0.596 |
| Vssc (ml/kg) | 36 | 110 ± 30.3 | 37 | 99.8 ± 42.8 |
| CLc (ml/day/kg) | 36 | 2.97 ± 1.38 | 37 | 2.65 ± 1.49 |
| t1/2 (day) | 54 | 26.9 ± 9.42 | 54 | 27.4 ± 10.1 |
AR_Cmax, accumulation ratio at Cmax, where AR_Cmax = Cmax_dose_4/Cmax_dose_1; AR_Cmin accumulation ratio at Cmin, where AR_Cmin = Cmin_wk_1/Cmin_wk_12; AR_AUC7, accumulation ratio at the AUC at day 7, where AR_AUC7 = AUC56–63/AUC0–7; AUCinf, area under the serum concentration-time curve from day 0 extrapolated to infinity; AUC56–63, area under the serum concentration-time curve from days 56 to 63; AUC56–84, area under the serum concentration-time curve from days 56 to 84; AUC0–7, area under the serum concentration-time curve from days 0 to 7; CL, clearance; Cmax_dose_1, maximum observed serum concentration after the first dose; Cmax_dose_4, maximum observed serum concentration after the last (i.e., fourth) dose; Cmin_wk_1, minimum observed serum concentration at 1 week after the first dose; Cmin_wk_12, minimum observed serum concentration at 4 weeks after the last dose was administered at day 57; t1/2, terminal half-life; Vss, volume of distribution at steady state.
AUCinf was excluded if the percentage of AUC extrapolation exceeded 20%.
CL and Vss were excluded from the calculation if the last dose was missing.
ADA incidence.
Antidrug antibody (ADA)-positive patients were defined as those patients who tested negative at the baseline and positive after dosing during the study and had evaluable immunogenicity data (at least one postbaseline sample). Among the RG7667-treated patients, 3 of 56 developed ADA responses only to MCMV5322A and 4 of 56 developed ADA responses only to MCMV3068A. One patient developed an ADA response to both MCMV5322A and MCMV3068A (19). We also found that the MCMV5322A and MCMV3068A serum concentration-time profiles for ADA-positive patients and ADA-negative patients overlapped (Fig. 2), indicating that the presence of ADAs was not associated with lower drug exposure.
FIG 2.
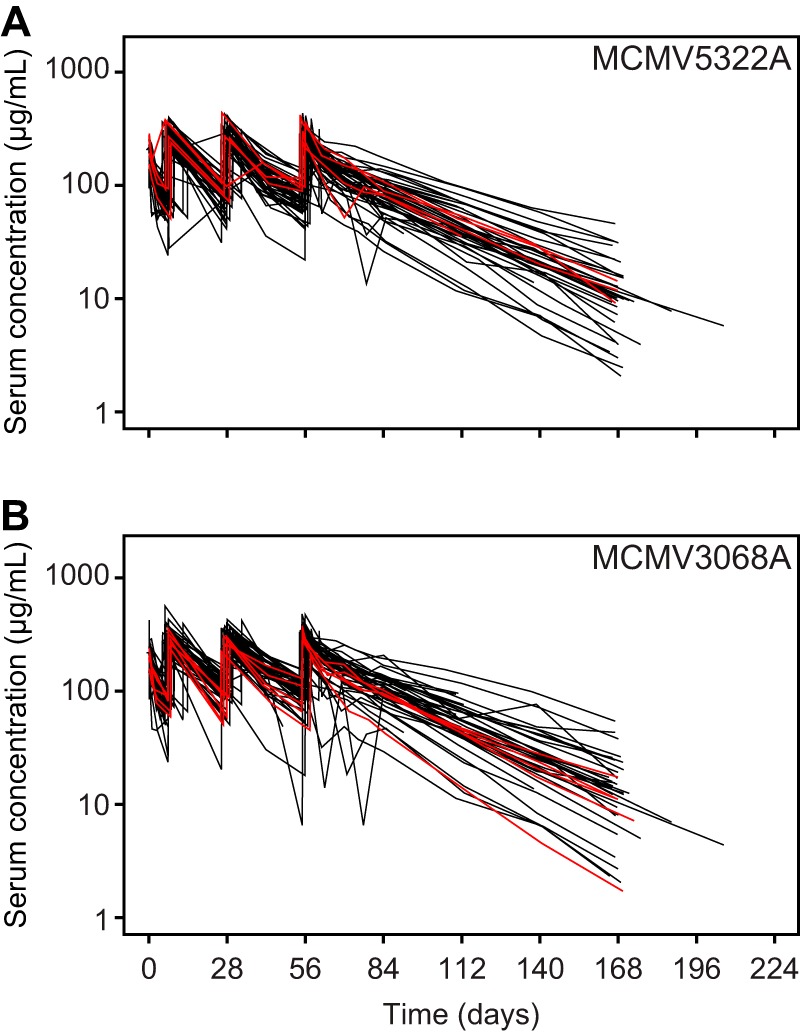
MCMV5322A (A) and MCMV3068A (B) serum concentration-time profiles for ADA-positive patients (red lines) and ADA-negative patients (black lines).
Pharmacological activity and exposure-response analysis.
The proportion of patients with detectable CMV viremia (viral load ≥ 150 copies/ml [137 IU/ml]) within 12 weeks after transplantation was the primary endpoint of the study. As previously reported (19), CMV viremia developed in 45.8% (27/59) of RG7667-treated patients and in 61.4% (35/57) of placebo-treated patients by the end of 12 weeks after transplantation, but this difference was not statistically significant. By the end of 24 weeks after transplantation, there were significantly fewer RG7667-treated patients (50.8% [30/59]) than placebo-treated patients (70.2% [40/57]) with CMV viremia (stratum-adjusted difference, 19.3%; P = 0.040). The median time to detectable CMV viremia, a secondary endpoint, was also significantly delayed (RG7667 versus placebo, 139 days versus 46 days; hazard ratio, 0.53; P = 0.009), and significantly fewer RG7667-treated patients than placebo-treated patients developed CMV disease (19).
To analyze the exposure-response relationship for each component antibody or the combination, we stratified patients by high exposure (Cmin-high; in which the Cmin at week 12 [Cmin_wk_12], 4 weeks after the last dose, was greater than or equal to the median Cmin) or low exposure (Cmin-low; in which Cmin_wk_12 was less than the median Cmin). We found that MCMV5322A Cmin-high patients (median Cmin, 84.6 μg/ml) had a lower incidence of viremia at 12 weeks after transplantation (36.0%) than patients who either had a low level of MCMV5322A exposure (54.2%) or were in the placebo group (61.4%) (Fig. 3A). Similarly, for MCMV3068A Cmin-high patients (median Cmin, 100 μg/ml), the incidence of viremia was reduced (32%) compared to the incidence in patients with Cmin-low values (62.5%). For patients with both high MCMV5322A exposures and high MCMV3068A exposures, the incidence of viremia was 29.4%, whereas it was 68.8% in those with Cmin-low values (Fig. 3A). At 24 weeks after transplantation, we also observed the same pattern: patients with high week 12 exposures for the component antibodies or the combination also had a reduced incidence of viremia (Fig. 3B).
FIG 3.
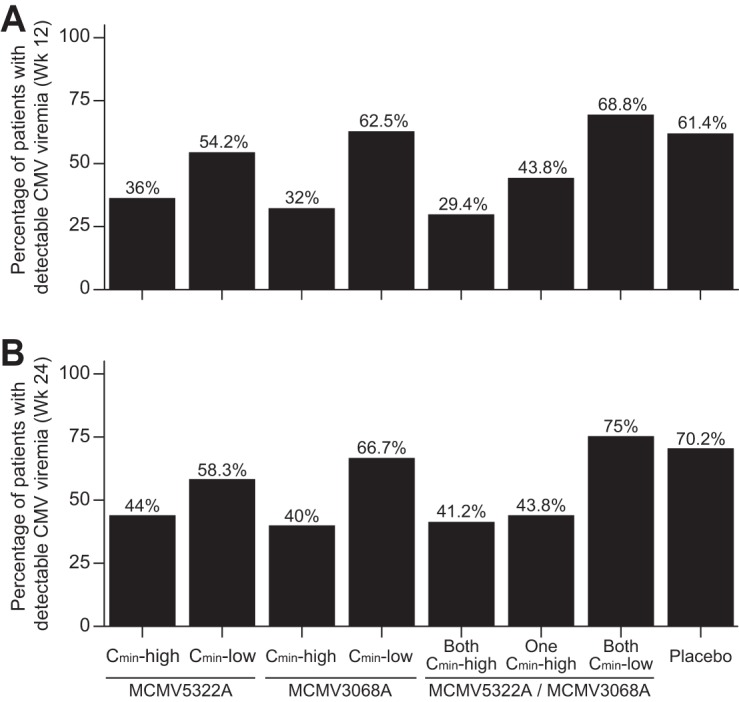
Incidence of viremia (viral load ≥ 150 copies/ml [137 IU/ml]) at 12 weeks (primary efficacy endpoint) (A) and 24 weeks (secondary efficacy endpoint) (B) posttransplantation stratified by the median Cmin_wk_12 of either or both antibodies.
MCMV5322A Cmin-high patients also had a longer median time to detectable CMV viremia than Cmin-low patients or placebo-treated patients (in which CMV viremia was not reached during the study [MCMV5322A Cmin-high patients] period or the median time to detectable CMV viremia was 75 days [Cmin-low patients] and 46 days [placebo-treated patients]) (Fig. 4A; Table 2). We observed similar results for MCMV3068A (Fig. 4C; Table 2) and the combination (Fig. 4E; Table 2).
FIG 4.

Time to detectable CMV viremia stratified by the median Cmin_wk_12 (A, C, E) and the median AUC56–84 (B, D, F). (A, B) MCMV5322A; (C, D) MCMV3068A; (E, F) RG7667.
TABLE 2.
Median time to detectable CMV viremiaa stratified by Cmin_wk_12 and AUC56–84
| Parameterb | MCMV5322A |
MCMV3068A |
MCMV5322A and MCMV3068A |
MCMV5322A or MCMV3068A |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of patients | Median time to detectable viremia (days) | P valuec | No. of patients | Median time to detectable viremia (days) | P value | No. of patients | Median time to detectable viremia (days) | P value | No. of patients | Median time to detectable viremia (days) | P value | |
| Cmin-high | 25 | Not reachedd | 0.00433 | 25 | Not reached | 0.00512 | 17 | Not reached | 0.0921 | 16 | Not reached | 0.0168 |
| Cmin-low | 24 | 75 | 0.0828 | 24 | 60.5 | 0.256 | 16 | 56.5 | 0.675 | |||
| AUC56–84-high | 18 | Not reached | 0.00543 | 17 | Not reached | 0.0189 | 14 | Not reached | 0.0566 | 7 | 55 | 0.793 |
| AUC56–84-low | 17 | 56 | 0.505 | 17 | 60.5 | 0.204 | 13 | 60 | 0.297 | |||
The median time to detectable CMV viremia for the placebo group (n = 57) was 46 days.
AUC56–84-high, an AUC56–84 greater than or equal to the median AUC; AUC56–84-low, an AUC56–84 less than the median AUC; Cmin-high, a Cmin_wk_12 greater than or equal to the median Cmin; Cmin-low, a Cmin_wk_12 less than the median Cmin.
P values are relative to the median time to viremia of the placebo group.
Not reached, the median time to viremia was not reached during the study period.
Patients with a high MCMV5322A exposure at steady state (defined as an AUC from days 56 to 84 [AUC56–84] that was greater than or equal to the median of 3,390 μg · day/ml) had a longer time to detectable CMV viremia than either patients with a low AUC56–84 value (a value below the median) or patients in the placebo group (in which CMV viremia was not reached during the study period [MCMV5322A AUC56–84-high patients] or the median time to detectable CMV viremia was 56 days [AUC56–84-low patients] or 46 days [placebo-treated patients]) (Fig. 4B; Table 2). We also observed comparable findings when exposure measurements were based on MCMV3068A alone (Fig. 4D; Table 2) or the combination (Fig. 4F; Table 2). Of note, the median time to detectable CMV viremia for the subgroup with a high AUC56–84 for either antibody (Table 2; Fig. 4F, green line) was not statistically significantly different from that for the placebo group, most likely due to the small sample size (n = 7).
The driver for RG7667 efficacy is not yet fully understood. Our previous work with a preclinical model that tested an anti-influenza A virus monoclonal antibody demonstrated that initial systemic exposure may be important for efficacy (20). Therefore, to determine whether early RG7667 exposure (1 week after transplantation) was linked to its pharmacological activity, we examined the incidence of viremia at weeks 12 and 24 and the time to detectable CMV viremia stratified by early exposure parameters (Cmin and AUC during the first week). We found no obvious differences in either endpoint between the two subgroups for either component antibody (data not shown).
We then assessed whether the lower level of efficacy observed in patients with lower exposures was due to lower dose intensities. A relative dose intensity of 1 indicates that RG7667 was administered at the dose planned per the protocol, without delay and without cancellations. Four of 59 RG7667-treated patients had a relative dose intensity that was less than 1 (i.e., the patients did not receive the planned dose) and were excluded from our exposure-response analysis due to the lack of observed exposure data. The remaining 55 patients had relative dose intensities of 1 regardless of their level of exposure, thus eliminating dose intensity as a factor affecting efficacy.
To better establish the true exposure-response relationship, we examined whether important risk factors, such as geographic region, use of induction immunosuppression, or a human leukocyte antigen mismatch between the donor and the recipient, were balanced between patients with lower exposures and patients with higher exposures. We found no major imbalances, suggesting that the efficacy difference was most likely related to low exposures (Table 3). We further confirmed this result using a Cox regression model and identified exposure (Cmin_wk_12) to be a significant covariate (MCMV5322A, P = 0.005; MCMV3068A, P = 0.006).
TABLE 3.
Summary of categorical covariates between high-exposure, low-exposure, and placebo groups in the modified intention-to-treat populationa
| Characteristic | No. (%) of patients |
||||
|---|---|---|---|---|---|
| MCMV5322A |
MCMV3068A |
Placebo |
|||
| Cmin-high | Cmin-low | Cmin-high | Cmin-low | ||
| Total | 25 | 24 | 25 | 24 | 57 |
| Residence in the United States | 13 (52) | 9 (37.5) | 13 (52) | 9 (37.5) | 27 (47.4) |
| Induction immunosuppression | 6 (24) | 4 (16.7) | 5 (20) | 5 (20.8) | 15 (26.3) |
| No. of HLA-A mismatches | |||||
| 0 | 9 (36.0) | 7 (29.2) | 7 (28.0) | 8 (33.3) | 15 (26.3) |
| 1 | 8 (32.0) | 14 (58.3) | 12 (48.0) | 10 (41.7) | 25 (43.9) |
| 2 | 5 (20.0) | 3 (12.5) | 5 (20.0) | 4 (16.7) | 13 (22.8) |
| Not done or unobtainable | 3 (12.0) | 1 (4.0) | 2 (8.3) | 4 (7.0) | |
| No. of HLA-B mismatches | |||||
| 0 | 7 (28.0) | 5 (20.8) | 7 (28.0) | 6 (25) | 18 (31.6) |
| 1 | 6 (24.0) | 11 (45.8) | 7 (28.0) | 9 (37.5) | 17 (29.8) |
| 2 | 9 (36.0) | 8 (33.3) | 10 (40.0) | 7 (29.2) | 17 (29.8) |
| Not done or unobtainable | 3 (12.0) | 1 (4.0) | 2 (8.3) | 5 (8.8) | |
| No. of HLA-DR mismatches | |||||
| 0 | 8 (32.0) | 5 (20.8) | 8 (32.0) | 5 (20.8) | 16 (28.1) |
| 1 | 9 (36.0) | 9 (37.5) | 10 (40.0) | 8 (33.3) | 27 (47.4) |
| 2 | 4 (16.0) | 8 (33.3) | 5 (20.0) | 7 (29.2) | 9 (15.8) |
| Not done or unobtainable | 4 (16.0) | 2 (8.3) | 2 (8.0) | 4 (16.7) | 5 (8.8) |
Cmin-high, a Cmin_wk_12 greater than or equal to the median Cmin; Cmin-low, a Cmin_wk_12 less than the median Cmin; HLA, human leukocyte antigen.
DISCUSSION
Previously, there had been limited data on the PK of antibody treatments in transplant patients and little consistency among studies. MSL-109, the parental antibody whose affinity maturation led to MCMV5322A, had typical IgG1 PK in patients receiving allogeneic bone marrow transplants (21). However, two other anti-CMV monoclonal antibodies, SDZ89-104 and 89-109, exhibited shorter terminal half-lives in bone marrow transplant patients than in healthy subjects (22). Other PK studies demonstrated that soon after transplantation, the clearance and the volume of distribution of therapeutic antibodies in transplant recipients may be increased compared to those in nontransplant patients (23). Additional reports indicate that the terminal half-life of an antibody in SOT patients is dependent upon the time that has elapsed after transplantation. It is possible that the intrinsic catabolism of IgG is initially increased due to surgery, inflammation, and other disease factors, but after surgery, it approaches that found in healthy subjects. For example, the terminal half-life of CMV-IVIG in renal transplant patients is approximately 7 days immediately after transplantation but increases to 15 days after 2 months (23, 24). In cardiac transplant patients receiving CMV-IVIG during the first 14 days after transplantation, the terminal half-life is approximately 4 days, but it increases to 14 days during the next 90 days (25). An anti-hepatitis C virus monoclonal antibody had a typical IgG1 PK profile in healthy volunteers (26) but had a clearance that was approximately 2- to 3-fold faster in liver transplant patients (27). Overall, we determined that the PK of RG7667 in SOT recipients were similar to those in healthy volunteers, were consistent with the typical PK for monoclonal antibodies, and were unaffected by immunogenicity. We also noted that the timing of the first RG7667 dose relative to the time of transplant did not affect MCMV5322A or MCMV3068A PK parameters, such as Cmin after the last dose at week 1 (Cmin_wk_1), the AUC from days 0 to 7 (AUC0–7), Cmax after the first dose (Cmax_dose_1), and Cmin_wk_12 (data not shown). These differences may have arisen due to the different roles of the transplanted organs in antibody clearance. For monoclonal antibodies such as rituximab, bevacizumab, and trastuzumab, renal function and hemodialysis have no reported impacts on their PK (28–33). In our study, the CMV viral load was unlikely to impact the monoclonal antibody PK as a consequence of target-mediated drug disposition because the amount of CMV particles was insignificant compared with the concentration of RG7667 immediately following dosing (19).
For this study in kidney transplant patients, we aimed to select a safe and efficacious dosing regimen to achieve and maintain circulating concentrations of both antibodies that were at or above the effective level, which was the level necessary to prevent CMV infection in this population. Because the receptors used by CMV to infect different cell types are different between humans and other species, there is no relevant in vivo model. Therefore, we selected the target serum trough concentrations for MCMV3068A and MCMV5322A by quantitatively linking in vitro and clinical data (see Fig. S1 in the supplemental material). Briefly, these serum target concentrations were based on the 90% effective concentrations (EC90s) of MCMV5322A, MCMV3068A, and CMV-IVIG established from in vitro neutralization assays (data not shown), as well as the ratio of the observed CMV-IVIG serum trough concentration in patients to the in vitro EC90 of CMV-IVIG (data not shown). Specifically, the ratio of each antibody's target serum trough concentration to its in vitro EC90 was set to be 3-fold greater than that for CMV-IVIG (Fig. S1A). We also considered several factors: the possibility of altered PK in transplant patients, the projected target concentrations, data from a PK simulation in healthy volunteers and SOT patients (data not shown), and nonclinical safety factors from a 4-week toxicology study in rats. An interim analysis during the phase 1 trial indicated that 5 mg/kg of each antibody (10 mg/kg total) could achieve the PK target while assuming that clearance and the volume of distribution were 3-fold faster in SOT patients than in healthy volunteers (Fig. S1B). Therefore, the phase 1 study in healthy volunteers utilized doses of up to 10 mg/kg of each antibody (20 mg/kg total) dosed monthly for 3 months (11) and included a factor of 2-fold over the projected efficacious dose. The nonclinical toxicology study in rats utilized doses of each antibody of up to 100 mg/kg (200 mg/kg total) dosed weekly for 5 weeks and showed a no-observed-adverse-effect level of 100 mg/kg for each antibody (data not shown). Both studies supported the safety of the dose and dose frequency used in the phase 2a study. In addition, the regimen chosen was expected to achieve the target serum trough concentrations in more than 90% of SOT patients in the Monte Carlo simulation (n = 1,000; Fig. S1). Due to the uncertainties of the RG7667 PK in SOT patients, the additional dose on day 8 was added to ensure that antibody concentrations were above the target serum concentrations both during and after transplantation.
Although we observed antiviral activity, RG7667 did not reduce the incidence of CMV viremia within 12 weeks. We observed a significant reduction at 24 weeks but could not determine if one or both antibodies was responsible because we did not administer the antibodies separately. Target serum concentrations based on CMV-IVIG and in vitro EC90s may have been underestimated because it was unclear how well in vitro efficacy measures would translate into clinically meaningful activities. Higher, but not early, exposures to RG7667 were associated with improved pharmacological responses, suggesting that a higher dose or more frequent dosing may provide greater concentrations and, thereby, greater pharmacological activity in target tissues, such as the kidneys. High exposures to both antibodies had better activity than a high exposure to only one antibody (Fig. 3A and 4F), which could have been due to the suboptimal dosing of one antibody. Therefore, optimization of the antibody ratio instead of the use of a 1:1 ratio may increase pharmacological activity. Although we eliminated induction immunosuppression, geographic region, and HLA mismatches as risk factors likely to affect efficacy, other unknown risk factors may have been involved. A future randomized trial based on exposure would better capture the exposure-response relationship. Finally, RG7667 may not neutralize CMV entry into other unidentified cell types. Greater activity may be achieved by using a larger number of monoclonal antibodies against other CMV epitopes. In summary, RG7667 had favorable pharmacokinetic and immunogenicity profiles in kidney transplant recipients at high risk for CMV infection. Future studies will be necessary to determine the optimal dosing regimen for maximum efficacy against CMV infection.
MATERIALS AND METHODS
Study population and design.
The study population and design have been described previously (19), and the study has been registered at ClinicalTrials.gov under identifier NCT01753167. Briefly, CMV-seronegative patients who were at least 18 years old and receiving a first or second kidney transplant from a CMV-seropositive donor (D+R− transplants), who are at particularly high risk for CMV infection and subsequent CMV disease (1), were eligible in this proof-of-activity study. This study was conducted in full conformance with the International Council for Harmonisation E6 guidelines for Good Clinical Practice and the principles of the Declaration of Helsinki or the laws and regulations of the country in which the research was conducted, whichever offered the greater protection to the individual. All patients provided written, informed consent.
One hundred twenty patients were randomized 1:1 to either RG7667 or placebo. The sample size selection has been described previously (19). Briefly, sample size was determined on the basis of an expected event rate in the placebo group, expected treatment effects, a 10% potential dropout rate in the primary endpoint, and the estimate that 10% of patients would become CMV seropositive (i.e., both the donor and the recipient would be CMV seropositive) at baseline because of seroconversion after the initial CMV serological evaluation. RG7667 (10 mg/kg of each antibody [20 mg/kg total], based on actual body weight at day 1) and placebo were administered i.v. in 4 doses on days 1 (between 1 day before and 2 days after transplantation), 8, 29, and 57. The dose for this proof-of-activity phase 2a study was chosen on the basis of a preclinical toxicity study in rats and safety, projected efficacious dose, and PK data from the phase 1 trial (11) (see Fig. S1 in the supplemental material). The primary endpoint was the incidence of CMV viremia, defined as ≥150 copies/ml (137 IU/ml), measured in patient plasma samples within 12 weeks of transplantation. Secondary endpoints were the incidence of CMV viremia within 24 weeks of transplantation, the time to detectable CMV viremia, and the characterization of the pharmacokinetic and immunogenicity profiles of RG7667. The CMV viral load in plasma was measured by quantitative PCR (qPCR) weekly (weeks 0 to 12) or every other week (weeks 13 to 24). If viral load measurements were clinically meaningful, site investigators could initiate preemptive antiviral therapy (e.g., valganciclovir, ganciclovir, foscarnet, or CMV-IVIG) at their discretion.
Pharmacokinetic assessments.
As previously described (19), serum samples for PK analysis were collected up to 24 h prior to the first dosing (day 1); at 1 h (±15 min), 4 h (±2 h), 24 h (day 2), and 72 h (day 4) after the first dosing; on day 8 before dosing and 1 h (±15 min) after dosing; on day 29 before dosing and 1 h (±15 min) after dosing; on day 57 before dosing and 1 h (±15 min) and 24 h (day 58) after dosing; on days 43, 64, 71, 78, 85, 113, and 141; and at study completion (on day 169 or at early termination). RG7667 levels in serum were measured using validated, quantitative, enzyme-linked immunosorbent assays (ELISA) (11). The levels of each molecule were analyzed independently. The lower limits of quantification for MCMV3068A and MCMV5322A were 350 ng/ml and 200 ng/ml, respectively.
Immunogenicity.
As described by Ishida et al. (19), serum samples for the detection of ADAs were collected on days 1 (predose), 29 (predose), 57 (predose), 85, 113, and 141 and at study completion (day 169) or at early termination. ADAs were detected using validated bridging immunoassays. Samples that tested positive in the screening assay were further tested in a confirmatory assay (11).
Statistical methods.
The statistical methods used to analyze patient demographic, efficacy, and safety data and perform sample size calculations have been described previously (19). For RG7667 PK, individual and mean serum concentration-time data for MCMV5322A and MCMV3068A were tabulated and plotted. PK data analysis was performed using standard noncompartmental analysis in the Phoenix WinNonlin (version 6.2) program (Certara, L.P., Princeton, NJ). Actual blood sampling times and actual doses were used. Predose data below the lower limit of quantification were set to zero. Postdose data below the lower limit of quantification were set to missing. The serum PK of MCMV5322A and MCMV3068A were summarized by estimating the following parameters: the area under the serum concentration-time curve (AUC), calculated using the log-linear trapezoidal rule (AUC from day 0 extrapolated to infinity [AUCinf], AUC from days 0 to 7 [AUC0–7], AUC from days 56 to 63 [AUC56–63], AUC from days 56 to 84 [AUC56–84]); the maximum observed serum concentration after the first and last (i.e., fourth) doses (Cmax_dose_1 and Cmax_dose_4, respectively); the minimum observed serum concentration (Cmin) at 1 week after the first dose (Cmin_wk_1) and 4 weeks after the last dose was administered at day 57 (Cmin_wk_12); and the serum terminal half-life (t1/2) after the last doses (as appropriate for the data collected). Total serum clearance (CL) and the volume of distribution at steady state (Vss) were also determined. Estimates for these parameters were tabulated and summarized (mean and standard deviation). Interpatient variability and drug accumulation based on Cmax, Cmin, and AUC following the first and last doses were evaluated. The MCMV5322A and MCMV3068A serum concentration-time data were compared to the data available from the phase 1 study of RG7667 (11).
A two-compartment PK model that was developed to describe the MCMV5322A and MCMV3068A PK data observed in phase 1 (data not shown) was used to simulate the expected concentrations in the phase 2a study of MCMV5322A and MCMV3068A. Monte Carlo simulations were performed for 1,000 subjects using NONMEM (version 7.2) software (ICON Development Solutions, Ellicott City, MD). The effects of covariates on PK parameters and covariance between clearance and the volume of distribution within the central compartment were not included in the simulation.
To analyze the exposure-response relationship, pharmacological activity (CMV viremia at week 12 and week 24 and the time to detectable CMV viremia on the basis of the number of viral copies in plasma samples) was correlated with the observed exposure (Cmin_wk_12, AUC56–84, Cmin, and AUC after the first week) to the single antibodies and/or the antibody combination by stratifying the exposures as greater than or equal to the medians and less than the medians. Kaplan-Meier estimates were used to compare virus-free status over time after treatment among the groups stratified by different exposure metrics. The median times to detectable CMV viremia for each group were calculated from Kaplan-Meier curves, and log-rank tests were performed for each subgroup versus the placebo group, with P values being shown in Table 2. Relative dose intensity was calculated as the total actual dose divided by the total planned dose during the study period before death/censoring occurred. A relative dose intensity of less than 1 indicates that patients did not receive the planned dose. All exposure-response analyses, including Cox regression, were performed using S-plus software (Tibco Software Inc., Palo Alto, CA).
Supplementary Material
ACKNOWLEDGMENTS
R. Deng, M. Maia, T. Burgess, and J. M. McBride designed the study and analyzed and interpreted the data. Y. Wang analyzed and interpreted the data. X. C. Liao and J. A. Tavel designed the study and interpreted the results. W. D. Hanley interpreted the data. All authors helped prepare the manuscript.
At the time of this study, all authors were employees of Genentech, Inc., a subsidiary of the Roche Group, and owned Roche stock. X. C. Liao is currently an employee of Immune-Onc Therapeutics, Inc. Genentech, Inc. was involved in the study design, data interpretation, and the decision to submit the work for publication in conjunction with the authors.
This study was supported by Genentech, Inc. Editing and writing assistance was provided by Deborah Solymar (Genentech, Inc.) and funded by Genentech, Inc.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01108-17.
REFERENCES
- 1.Snydman DR, Werner BG, Heinze-Lacey B, Berardi VP, Tilney NL, Kirkman RL, Milford EL, Cho SI, Bush HL, Levey AS, Strom TB, Carpenter CB, Levey RH, Harmon WE, Zimmerman CE II, Shapiro ME, Steinman T, LoGerfo F, Idelson B, Schröter GPJ, Levin MJ, McIver J, Leszczynski J, Grady GF. 1987. Use of cytomegalovirus immune globulin to prevent cytomegalovirus disease in renal-transplant recipients. N Engl J Med 317:1049–1054. doi: 10.1056/NEJM198710223171703. [DOI] [PubMed] [Google Scholar]
- 2.Ramanan P, Razonable RR. 2013. Cytomegalovirus infections in solid organ transplantation: a review. Infect Chemother 45:260–271. doi: 10.3947/ic.2013.45.3.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodson EM, Ladhani M, Webster AC, Strippoli GF, Craig JC. 2013. Antiviral medications for preventing cytomegalovirus disease in solid organ transplant recipients. Cochrane Database Syst Rev 2:CD003774. doi: 10.1002/14651858.CD003774.pub4. [DOI] [PubMed] [Google Scholar]
- 4.Paya C, Humar A, Dominguez E, Washburn K, Blumberg E, Alexander B, Freeman R, Heaton N, Pescovitz MD, Valganciclovir Solid Organ Transplant Study Group. 2004. Efficacy and safety of valganciclovir versus oral ganciclovir for prevention of cytomegalovirus disease in solid organ transplant recipients. Am J Transplant 4:611–620. doi: 10.1111/j.1600-6143.2004.00382.x. [DOI] [PubMed] [Google Scholar]
- 5.Razonable RR, Rivero A, Rodriguez A, Wilson J, Daniels J, Jenkins G, Larson T, Hellinger WC, Spivey JR, Paya CV. 2001. Allograft rejection predicts the occurrence of late-onset cytomegalovirus (CMV) disease among CMV-mismatched solid organ transplant patients receiving prophylaxis with oral ganciclovir. J Infect Dis 184:1461–1464. doi: 10.1086/324516. [DOI] [PubMed] [Google Scholar]
- 6.Husain S, Pietrangeli CE, Zeevi A. 2009. Delayed onset CMV disease in solid organ transplant recipients. Transpl Immunol 21:1–9. doi: 10.1016/j.trim.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Falagas ME, Snydman DR, Ruthazer R, Griffith J, Werner BG, Freeman R, Rohrer R. 1997. Cytomegalovirus immune globulin (CMVIG) prophylaxis is associated with increased survival after orthotopic liver transplantation. Clin Transplant 11:432–437. [PubMed] [Google Scholar]
- 8.Griffiths PD, Stanton A, McCarrell E, Smith C, Osman M, Harber M, Davenport A, Jones G, Wheeler DC, O'Beirne J, Thorburn D, Patch D, Atkinson CE, Pichon S, Sweny P, Lanzman M, Woodford E, Rothwell E, Old N, Kinyanjui R, Haque T, Atabani S, Luck S, Prideaux S, Milne RS, Emery VC, Burroughs AK. 2011. Cytomegalovirus glycoprotein B vaccine with MF59 adjuvant in transplant recipients: a phase 2 randomised placebo-controlled trial. Lancet 377:1256–1263. doi: 10.1016/S0140-6736(11)60136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saylor C, Dadachova E, Casadevall A. 2009. Monoclonal antibody-based therapies for microbial diseases. Vaccine 27(Suppl 6):G38–G46. doi: 10.1016/j.vaccine.2009.09.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Both L, Banyard AC, van Dolleweerd C, Wright E, Ma JK, Fooks AR. 2013. Monoclonal antibodies for prophylactic and therapeutic use against viral infections. Vaccine 31:1553–1559. doi: 10.1016/j.vaccine.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishida JH, Burgess T, Derby MA, Brown PA, Maia M, Deng R, Emu B, Feierbach B, Fouts AE, Liao XC, Tavel JA. 2015. Phase 1 randomized, double-blind, placebo-controlled study of RG7667, an anticytomegalovirus combination monoclonal antibody therapy, in healthy adults. Antimicrob Agents Chemother 59:4919–4929. doi: 10.1128/AAC.00523-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci U S A 102:18153–18158. doi: 10.1073/pnas.0509201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wille PT, Knoche AJ, Nelson JA, Jarvis MA, Johnson DC. 2010. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J Virol 84:2585–2596. doi: 10.1128/JVI.02249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fouts AE, Comps-Agrar L, Stengel KF, Ellerman D, Schoeffler AJ, Warming S, Eaton DL, Feierbach B. 2014. Mechanism for neutralizing activity by the anti-CMV gH/gL monoclonal antibody MSL-109. Proc Natl Acad Sci U S A 111:8209–8214. doi: 10.1073/pnas.1404653111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bai S, Jorga K, Xin Y, Jin D, Zheng Y, Damico-Beyer LA, Gupta M, Tang M, Allison DE, Lu D, Zhang Y, Joshi A, Dresser MJ. 2012. A guide to rational dosing of monoclonal antibodies. Clin Pharmacokinet 51:119–135. doi: 10.2165/11596370-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 18.Lim JJ, Deng R, Derby MA, Larouche R, Horn P, Anderson M, Maia M, Carrier S, Pelletier I, Burgess T, Kulkarni P, Newton E, Tavel JA. 2016. Two phase 1, randomized, double-blind, placebo-controlled, single-ascending-dose studies to investigate the safety, tolerability, and pharmacokinetics of an anti-influenza A virus monoclonal antibody, MHAA4549A, in healthy volunteers. Antimicrob Agents Chemother 60:5437–5444. doi: 10.1128/AAC.00607-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishida JH, Patel A, Mehta AK, Gatault P, McBride JM, Burgess T, Derby MA, Snydman DR, Emu B, Feierbach B, Fouts AE, Maia M, Deng R, Rosenberger CM, Gennaro LA, Striano NS, Liao XC, Tavel JA. 2017. Phase 2 randomized, double-blind, placebo-controlled trial of RG7667, a combination monoclonal antibody, for prevention of cytomegalovirus infection in high-risk kidney transplant recipients. Antimicrob Agents Chemother 61:e01794-16. doi: 10.1128/AAC.01794-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta P, Kamath AV, Park S, Chiu H, Lutman J, Maia M, Tan MW, Xu M, Swem L, Deng R. 2016. Preclinical pharmacokinetics of MHAA4549A, a human monoclonal antibody to influenza A virus, and the prediction of its efficacious clinical dose for the treatment of patients hospitalized with influenza A. MAbs 8:991–997. doi: 10.1080/19420862.2016.1167294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drobyski WR, Gottlieb M, Carrigan D, Ostberg L, Grebenau M, Schran H, Magid P, Ehrlich P, Nadler PI, Ash RC. 1991. Phase I study of safety and pharmacokinetics of a human anticytomegalovirus monoclonal antibody in allogeneic bone marrow transplant recipients. Transplantation 51:1190–1196. doi: 10.1097/00007890-199106000-00009. [DOI] [PubMed] [Google Scholar]
- 22.Aulitzky WE, Schulz TF, Tilg H, Niederwieser D, Larcher K, Ostberg L, Scriba M, Martindale J, Stern AC, Grass P, Mach M, Dierich MP, Huber C. 1991. Human monoclonal antibodies neutralizing cytomegalovirus (CMV) for prophylaxis of CMV disease: report of a phase I trial in bone marrow transplant recipients. J Infect Dis 163:1344–1347. doi: 10.1093/infdis/163.6.1344. [DOI] [PubMed] [Google Scholar]
- 23.Snydman DR. 2001. Historical overview of the use of cytomegalovirus hyperimmune globulin in organ transplantation. Transpl Infect Dis 3(Suppl 2):S6–S13. doi: 10.1034/j.1399-3062.2001.00002.x. [DOI] [PubMed] [Google Scholar]
- 24.Snydman DR, McIver J, Leszczynski J, Cho SI, Werner BG, Berardi VP, LoGerfo F, Heinze-Lacey B, Grady GF. 1984. A pilot trial of a novel cytomegalovirus immune globulin in renal transplant recipients. Transplantation 38:553–557. doi: 10.1097/00007890-198411000-00026. [DOI] [PubMed] [Google Scholar]
- 25.Metselaar J, Velzing PH, Rothbarth PH, Simoons ML, Bos E, Weimar W. 1987. A pharmacokinetic study of anti-cytomegalovirus hyperimmunoglobulins in cytomegalovirus seronegative cardiac transplant recipients. Transplant Proc 14:4063–4065. [PubMed] [Google Scholar]
- 26.Leav BA, Sloan S, Blair BM, Cheslock P, Knauber M, Ambrosino DM, Molrine DC. 2010. Safety and pharmacokinetics of a novel human monoclonal antibody directed against the E2 glycoprotein of hepatitis C virus (MBL-HCV1) in healthy volunteers. The 45th Annu Meeting of the European Association for the Study of the Liver. J Hepatol 52(Suppl 1):S118. [Google Scholar]
- 27.Chung RT, Gordon FD, Curry MP, Schiano TD, Emre S, Corey K, Markmann JF, Hertl M, Pomposelli JJ, Pomfret EA, Florman S, Schilsky M, Broering TJ, Finberg RW, Szabo G, Zamore PD, Khettry U, Babcock GJ, Ambrosino DM, Leav B, Leney M, Smith HL, Molrine DC. 2013. Human monoclonal antibody MBL-HCV1 delays HCV viral rebound following liver transplantation: a randomized controlled study. Am J Transplant 13:1047–1054. doi: 10.1111/ajt.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jillella AP, Dainer PM, Kallab AM, Ustun C. 2002. Treatment of a patient with end-stage renal disease with rituximab: pharmacokinetics evaluation suggests rituximab is not eliminated by hemodialysis. Am J Hematol 71:219–222. doi: 10.1002/ajh.10213. [DOI] [PubMed] [Google Scholar]
- 29.Castro M, Van Auken J, Sponzo R. 2001. Successful use of anti-CD20 monoclonal antibody, rituximab, in patients with non-Hodgkin's lymphoma and renal insufficiency. Proc Am Soc Clin Oncol 20:233b. [Google Scholar]
- 30.Gupta NK, El-Tarabily M, Bedient S, Michaels I, Gomez S, Lee D, Stilter M. 2003. Rituximab can be safely administered in patients with end-stage renal disease on hemodialysis without dose modification. Blood 102:4916. [Google Scholar]
- 31.Garnier-Viougeat N, Rixe O, Paintaud G, Ternant D, Degenne D, Mouawad R, Deray G, Izzedine H. 2007. Pharmacokinetics of bevacizumab in haemodialysis. Nephrol Dial Transplant 22:975. [DOI] [PubMed] [Google Scholar]
- 32.McKeage K, Perry CM. 2002. Trastuzumab: a review of its use in the treatment of metastatic breast cancer overexpressing HER2. Drugs 62:209–243. doi: 10.2165/00003495-200262010-00008. [DOI] [PubMed] [Google Scholar]
- 33.Thariat J, Azzopardi N, Peyrade F, Launay-Vacher V, Santini J, Lecomte T, Etienne-Grimaldi MC, Paintaud G, Milano G. 2008. Cetuximab pharmacokinetics in end-stage kidney disease under hemodialysis. J Clin Oncol 26:4223–4225. doi: 10.1200/JCO.2008.18.7674. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.