

ABSTRACT
Bacteriophage therapy is a promising alternative treatment to antibiotics, as it has been documented to be efficacious against multidrug-resistant bacteria with minimal side effects. Several groups have demonstrated the efficacy of phage suspension in vivo to treat lung infections using intranasal delivery; however, phage dry-powder administration to the lungs has not yet been explored. Powder formulations provide potential advantages over a liquid formulation, including easy storage, transport, and administration. The purpose of this study was to assess the bactericidal activities of phage dry-powder formulations against multidrug-resistant (MDR) strain Pseudomonas aeruginosa FADDI-PA001 in a mouse lung infection model. Phage PEV20 spray dried with lactose and leucine produced an inhalable powder at a concentration of 2 × 107 PFU/mg. P. aeruginosa lung infection was established by intratracheal administration of the bacterial suspension to neutropenic mice. At 2 h after the bacterial challenge, the infected mice were treated with 2 mg of the phage powder using a dry-powder insufflator. At 24 h after the phage treatment, the bacterial load in the lungs was decreased by 5.3 log10 (P < 0.0005) in the phage-treated group compared with that in the nontreated group. Additionally, the phage concentration in the lungs was increased by 1 log10 at 24 h in the treated group. These results demonstrate the feasibility of a pulmonary delivery of phage PEV20 dry-powder formulation for the treatment of lung infection caused by antibiotic-resistant P. aeruginosa.
KEYWORDS: bacteriophage therapy, Pseudomonas aeruginosa, pulmonary infections, murine model, powder aerosols
INTRODUCTION
Respiratory infections have increasingly become a global medical challenge due to multidrug-resistant (MDR) bacteria. Infections caused by MDR Pseudomonas aeruginosa are difficult to treat and often lead to high rates of recurrence, mortality, and morbidity (1). Inhaled bacteriophage (phage) therapy is a promising alternative treatment option for the treatment of lung infections caused by MDR P. aeruginosa (2). Phages are naturally occurring antibacterial agents with low inherent toxicity and, importantly, are able to replicate inside the host (3, 4). Their highly specific nature enables the eradication of MDR bacteria without unnecessary damage to nontargeted bacteria.
Earlier mouse lung infection studies on phages used intranasal instillation for the initiation of the infection and treatment (5–7). These studies have provided strong support for inhaled phage therapy by successfully reducing the bacterial load and inflammation in the mouse lung infection model. However, the precise control of bacteria and phage dose is challenging via the intranasal route. Alemayehu et al. (7) intranasally administered bioluminescent P. aeruginosa to establish lung infection in mice. Luminescence imaging showed that bacteria had spread all over the body after 8 h, with heavy loads in the nose, lungs, and stomach. More recently, a number of groups have employed intratracheal instillation for bacteria and antibiotic administration to enable a direct and reproducible delivery to the lungs (8–10). Intratracheal administration using a MicroSprayer aerosolizer and a Dry powder insufflator enables noninvasive delivery; yet, this administration route is still underexplored.
The feasibility of producing a dry-powder phage formulation has been demonstrated in vitro. Our group and others have used spray drying (11–13), spray-freeze drying (12), and lyophilization (14) to generate stable phage powder with a minimal titer loss. Furthermore, under a suitable storage setting, spray-dried phage powders can retain bioactivity and aerosol performance even after 12 months (15). Although the in vitro efficacy of these phage powders has been established, in vivo investigations are limiting. The aim of the present study was to evaluate the efficacy and safety of dry-powder formulations of P. aeruginosa phages in a murine lung infection model. MDR P. aeruginosa was selected for this study because the Infectious Disease Society of America listed it as a pathogen that is difficult to treat and requires urgent attention for the discovery of novel treatments (16).
RESULTS AND DISCUSSION
In this study, we demonstrated that intratracheally delivered aerosolized phage powder can significantly reduce MDR P. aeruginosa load in mouse lungs. An inhalable dry-powder phage formulation was produced by spray drying highly purified PEV20 with lactose and leucine. Lactose stabilizes the phages against the stress exposed during spray drying, and leucine provides protection from moisture (12, 17, 18). After spray drying, the bioactivity of phage was retained with a 0.3-log10 titer reduction. Spray-dried phage powder formed spherical particles with slightly rough surfaces (Fig. 1). As expected, lactose and leucine in combination produced inhalable spray-dried particles with adequate in vitro aerosol performance. The fine particle fraction ([FPF] <5.3 μm) of the formulation was 51.6% ± 0.7% with a recovery of 90.8% ± 0.3%. Since the FPF values of most commercial dry-powder inhaler products are in the range of 20 to 30% (19), our spray-dried phage formulation has a much better aerosol performance. The median particle size (D50) was 2.11 ± 0.00 μm with a span of 1.62 ± 0.07 μm. The spray-dried phage formulation is suitable for inhalation delivery, as particles of less than 3 μm are considered inhalable. The phage content in the powder was 2 × 107 PFU/mg, which was suitable for assessing the antibacterial activity at a multiplicity of infection (MOI) of 100 in murine lungs, as the delivery device is designed to give 1 to 2 mg.
FIG 1.

SEM image of phage powder containing PEV20, lactose, and leucine. Particles formed were spherical with slightly rough surfaces.
The toxicity of the phage powder was evaluated in vitro with a resazurin cell viability assay. After 24 h of exposure, cell survival rates of human epithelial (A549 and HEK239) and macrophage (THP-1) cell lines were 97.3% ± 1.5%, 96.0% ± 1.0%, and 97.0% ± 1.7%, respectively (P < 0.05). As the phage powder was nontoxic to alveolar epithelial and macrophage cells in vitro, a further safety evaluation was performed in vivo by histological investigation (Fig. 2). The intratracheal administration of air resulted in no damage to the lung tissues in healthy mice, with semiquantitative scores (SQS) of 0 at 24 h posttreatment (Table 1 and Fig. 2). The phage powder resulted in minimal damage to the lung tissues, with an SQS of 1. As expected, the intratracheal inoculation of FADDI-PA001 (2.5 × 106 CFU/mouse) in neutropenic mice caused severe damage to the lung tissues, with SQS values of 5.0 in 3 of 4 mice and 2.0 in the other mouse. Treatment with phage powder moderately relieved the lung damage caused by the infection, with SQS values of 1, 2, 2, and 5 in each of the four mice. Phages alone, depending on the purification of the solution, do not stimulate inflammatory responses (5, 20, 21), and furthermore, phages can alleviate the lung damage caused by P. aeruginosa infection even in the absence of the host immune system.
FIG 2.

Lung histology in mice infected with P. aeruginosa. Lung sections from a healthy mouse with no treatment (a) and intratracheal delivery of air (b) showing normal lung structures. (c) Lung section from healthy mouse with intratracheal administration of phage powder, showing mild damage to the lung structure. Lung sections from P. aeruginosa-infected mice with no treatment showing severe damage (d), which was reduced by treatment with phage powder (e).
TABLE 1.
Histopathological examination of lung toxicity after intratracheal administration of phage powder in healthy mice and in neutropenic mice infected with P. aeruginosa
| Group | Treatment | SQSa |
|||
|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | ||
| Healthy mice | None | 0 | |||
| Air | 0 | 0 | |||
| Phage powder | 1 | 1 | 1 | 1 | |
| Neutropenic mice infected with P. aeruginosa | Air | 2 | 5 | 5 | 5 |
| Phage powder | 1 | 2 | 2 | 5 | |
SQS, semiquantitative score.
Inhaled phage therapy can be studied in vivo by two primary modes of a pulmonary route: intranasal and intratracheal. To date, the majority of the published work on the in vivo efficacy of phages has used intranasal delivery to treat lung infections, probably due to the ease of application (5–7). These studies have shown improved survival rates and reduced bacterial loads in the lungs after intranasal phage treatment or prophylactic treatment. One drawback to intranasal delivery is the difficulty of controlling and enumerating the number of bacteria and phages deposited in the lungs. In addition, the nasal route results in a substantial loss of the deliverables, with only 15% deposition in the lungs due to mucociliary clearance (22).
Intratracheal instillation enables the direct application of the bacteria and phages to the lungs with minimal loss in the nose, throat, and upper airways, which enables precise dosing (23). In our experiments, the method achieved a delivery of 97.0% ± 2.2% of the loaded sample, and the residual volume in the syringe after delivery was negligible. Recent studies by our group confirmed the suitability and consistencies in dose delivery using the intratracheal route, even for complex pharmacokinetic and pharmacodynamics studies (9, 10). In the present study, the bacterial loads after inoculation in four mice were log10 5.6, log10 5.6, log10 5.5, and log10 5.5 CFU/lung, reflecting the tight control of dose given via the intratracheal route. Despite the consistent dose given, some level of variation posttreatment seems inevitable due to variations in biological experiments. The intratracheal route also provides superior antibacterial activities over parenteral administration due to the direct delivery to the site of infection (9, 10). As humans use oral inhalation for lung delivery, the intratracheal route would naturally be the optimal administrative route for studying inhaled phage therapy.
Phage administration using a dry-powder inhaler is an attractive delivery mode over nebulizers due to the convenience in storage, transport, and administration. The in vitro efficacy of phage-powder formulations has been well documented (11–13), but the lack of in vivo studies highlights the importance of this proof-of-concept efficacy study. To the best of our knowledge, this is the first study to test the efficacy of dry-powder phage formulations for the treatment of lung infections. Neutropenic mice were used to establish lung infections in mice to assess the efficacy of phages in a dry-powder formulation without the influence of the immune system. The bacterial load recovered from the lung tissues immediately after the intratracheal instillation of the FADDI-PA001 suspension averaged 3.4 × 105 CFU/lung (range, 2.8 × 105 to 4.1 × 105 CFU/lung) (Fig. 3). After 4 h, no significant changes were seen in the nontreated group (average, 7.7 × 105 CFU/lung) and the phage-treated group (average, 6.9 × 104 CFU/lung) (Table 2). The actual dose of intratracheally delivered phage powder ranged from 1.4 × 107 to 1.2 × 108 PFU/lung (Fig. 4a). At 4 h after the phage administration, the phage dose given equaled the number of phages recovered in the lung homogenates in 50% of treated mice, and there was a 1-log10 lower recovery in the other 50% of treated mice. Phage infection, amplification, and subsequent bacteriolysis heavily rely on the kinetics of bacterial growth and replication (24). Bacteria may become stagnant while adjusting to the new environment in the lungs; consequently, phages are unable to amplify, as confirmed by the consistent numbers of bacteria (Fig. 3) and phages (Fig. 4a) at 0 h and 4 h. In addition, low concentrations of phages (range, 3.3 × 101 to 3 × 104 PFU/ml) were found in plasma samples (Fig. 4b). After 24 h, a significant reduction (−5.3 log10) of bacterial load was observed in the treated group compared with that in the nontreated group (P < 0.0005) (Fig. 3). The CFU/lung in the nontreated group averaged 1.3 × 1010 CFU/lung, and the treated group averaged 6.0 × 104 CFU/lung. In turn, the phage titers in lung homogenates were increased by 1 log10 in all the treated mice, confirming phage amplification in the lung tissue (Fig. 4a). In addition, phage concentrations in plasma samples were higher (Fig. 4b), with an average of 3.8 × 105 PFU/ml (range, 1.4 × 105 to 7.5 × 105 PFU/ml), which suggests phage proliferation in the lungs and a subsequent increase in systemic spread.
FIG 3.
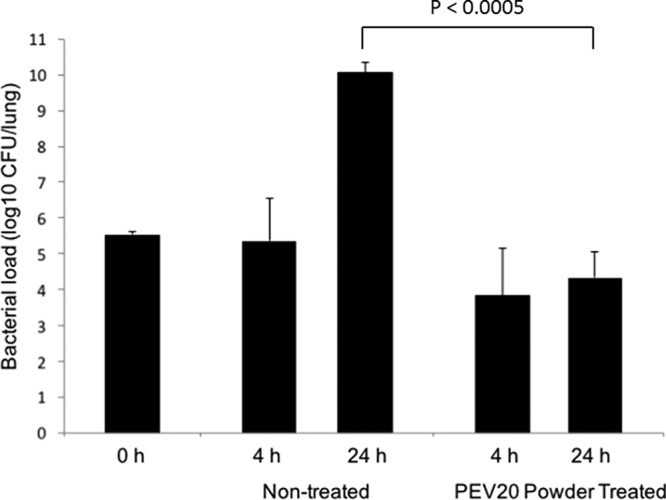
Killing activity of phage powder against P. aeruginosa FADDI-PA001 in mouse lung infection model. Bacterial burden in the lung was reduced by 5.3 log10 in the phage-treated group compared with that in the nontreated group at 24 h.
TABLE 2.
Bacterial loads in the lung homogenates at 4 h and 24 h after administration of phage powders
| Treatment | Bacterial load (CFU/lung) at: |
|||
|---|---|---|---|---|
| 4 h |
24 h |
|||
| Avg | Range | Avg | Range | |
| Nontreated | 7.7 × 105 | 4.4 × 103 to 1.8 × 106 | 1.3 × 1010 | 3.0 × 109 to 2.3 × 1010 |
| Phage treated | 6.9 × 104 | 4.4 × 102 to 2.5 × 105 | 6.0 × 104 | 3.0 × 103 to 1.9 × 105 |
FIG 4.

Phage concentrations in lung homogenates (a) and plasma samples (b) of mice infected with P. aeruginosa FADDI-PA001 at 4 h and 24 h posttreatment. Phage concentrations in the lung and plasma samples were increased at 24 h.
The pharmacology of antibacterial agents is affected by the presence of host immune system neutrophils. P. aeruginosa is an opportunistic pathogen, and healthy mice are capable of clearing the infection. Hence, we used neutropenic mice model to assess the efficacy of phages in a dry-powder formulation without the influence of the immune system (9, 10, 25). Indeed, the extent of bacterial infection was pronounced, achieving 1010 CFU/lung by 24 h in the nontreated group (Fig. 3), and a single dose of phage treatment was unable to completely clear the infection. Sample collection and investigation at 24 h may benefit from the observed further reduction of the infective burden by active therapy. Although we were unable to confirm the phage resistance of bacterial cells recovered at 24 h, it is suspected that these would be either phage-resistant mutants or persister cells. Now that we have validated the efficacy of spray-dried phage powders in this proof-of-concept study, in future studies, we plan to use nonneutropenic mice for establishing a lung infection model to investigate the effect of neutrophils on phage and bacteria interactions in the lung tissue. In this study, a stable spray-dried phage formulation was produced at a therapeutic dose for in vivo testing. Phage concentrations in the lungs can be increased to achieve >108 PFU by (i) increasing the amount of powder delivered, (ii) increasing the phage titer in the feed solution, or (iii) applying multiple dosing.
In conclusion, this is the first proof-of-concept study demonstrating the efficacy of the phage dry-powder formulation in a mouse lung infection model against a clinical MDR P. aeruginosa isolate. Moreover, the safety of the spray-dried phage formulation containing lactose and leucine has been confirmed in mouse lungs. Our findings suggest that dry-powder phage formulations can potentially be used for the treatment of clinical P. aeruginosa respiratory infections. Further pharmacokinetics, pharmacodynamics, and toxicity studies are urgently needed to expedite the commercialization of inhaled phage therapy to combat antibiotic-resistant bacterial infections.
MATERIALS AND METHODS
Bacterial strain.
A clinical P. aeruginosa strain (FADDI-PA001) was freshly subcultured from a −80°C stock prior to the experiment. This clinical isolate was resistant to most antibiotics, including amikacin, ciprofloxacin, doxycycline, and rifampin.
Bacteriophages.
Phage PEV20 active against P. aeruginosa was supplied by AmpliPhi Biosciences AU at a high titer of 1010 PFU/ml in phosphate-buffered saline (0.01 M phosphate buffer, 0.0027 M KCl, and 0.137 M NaCl; pH 7.5). This phage was originally isolated from the sewage treatment plant in Olympia (WA, USA) by the Kutter lab (Evergreen phage lab) using a P. aeruginosa strain from a cystic fibrosis patient at Children's Hospital in Seattle. PEV20 is a podovirus with a molecular mass of 91 kb and a capsid size of 60 nm. PEV20 is active against 56% of 90 clinical and multidrug-resistant strains collected from hospitals in Australia (17). This strain was employed as a reference bacterial strain to assess the phage titer with a plaque assay (12).
Spray drying.
The liquid feed was composed of 18 ml of excipient solution (17 mg/ml of lactose and 8 mg/ml of leucine) in ultrapure water and 2 ml of phage suspension (1010 PFU/ml) with the pH adjusted to 7.4. This feed suspension was spray dried using a Büchi 290 spray dryer coupled to a conventional two-fluid nozzle for atomization. The feed rate of 1.8 ml/min was used with an atomizing airflow of 742 liter/h and aspiration rate at 35 m3/h. The inlet temperature was set at 60°C with the outlet temperature ranging between 39 and 41°C. Dried powder was passed through the cyclone and collected in a vial. To determine phage content in the powder, phosphate-buffered saline (PBS) was added to a small amount of phage powder to give a concentration of 25 mg/ml before performing the plaque assay. Phage powder was stored with silica beads at room temperature until use.
Particle size distribution and scanning electron microscopy imaging.
To measure the particle size distribution of the phage powder, laser diffraction (Mastersizer 2000; Malvern Instruments Ltd., UK) was used. Phage powders were loaded onto a Scirocco 2000 dry-powder module (Malvern Instruments, UK), followed by dispersion compressed air at 4.0 bars. Volumetric diameters (D10, D50, and D90) and span ([D90 − D10]/D50) were reported. The particle size and morphology was analyzed using scanning electron microscopy ([SEM] Zeiss Ultra Plus; Carl Zeiss NTS GmbH, Oberkochen, Germany). Samples were mounted on a carbon tape and coated with 15 nm of gold with a K550X sputter coater (Quorum Emitech, Kent, UK).
Aerosol dispersion.
The in vitro aerosol performance of the phage powder was assessed using a multistage liquid impinger (MSLI) followed by a chemical assay using high-pressure liquid chromatography (HPLC) or a plaque assay. In brief, powder samples (30 mg each) were loaded to size 3 hydroxypropyl methylcellulose capsules and dispersed using an Osmohaler at 100 liter/min for 2.4 s. PBS and ultrapure water were used as the collecting solvents to determine the deposition profiles of the phage and the sugar excipient, respectively. The cutoff diameters of the MSLI stages 1 to 4 at 100 liter/min were 10.1, 5.3, 2.4, and 1.32 μm, respectively. The fine particle fraction (FPF) was defined as the mass fraction of particles <5.3 μm with respect to the loaded dose. The deposition of lactose in the inhaler, capsule, adaptor, throat, and each part of the MSLI was determined using an HPLC system with refractive index (RI) detection (model LC-20; Shimadzu, Japan). The HPLC system consisted of a LC-20AT pump, CBM-20A controller, RID-10A RI detector, an Agilent Hi-Plex Ca ligand exchange column (300 mm by 7.7 mm, 8 μm; Phenomenex, Torrance, CA), SIL-20A HT auto-sampler, and LCSolution software. The chromatographic conditions were as follows: injection volume, 50 μl; flow rate, 0.6 ml/min; oven temperature, 85°C; and mobile phase ultrapure water.
In vitro cell viability assay.
The toxicity of the phage powder was assessed in vitro with a resazurin cell viability assay. Three separate cell lines, A549, HEK239, and THP-1 (105 cells), were exposed to 2 mg of phage powder no. 1 for 24 h. Cells were incubated with resazurin for a further 2 h at 37°C and 5% CO2, and then the fluorescence was measured using FLUOstar Omega (excitation, 530 nm; emission, 590 nm; BMG LABTECH GmbH, Ortenberg, Germany).
Animal experiments.
All animal experiments were performed under the animal ethics approval by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee. Swiss mice (8 to 10 weeks) were obtained from Monash Animal Services (Clayton, Victoria, Australia). All mice were handled per criteria of the Australian code of practice for the care and use of animals for scientific purposes. A neutropenic mouse lung infection model was employed in this study (9). Briefly, neutropenia was induced by an intraperitoneal injection of cyclophosphamide (Baxter Healthcare Pty Ltd., New South Wales, Australia). Bacterial infection was established as follows. Mice were anesthetized by isoflurane and placed in the supine position against the restraining platform angled at 60 to 70° from the horizontal. A FADDI-PA001 suspension at early logarithmic phase (approximately 106 cells in 25 μl) was sprayed directly into the trachea using a MicroSprayer aerosolizer (model IA-1C; Penn-Century, Philadelphia, PA, USA). Mice were maintained in the upright position for 1 min and then placed on a warm pad for recovery. After 2 h, mice were anesthetized by isoflurane, and the phage powder (1 to 4 mg) was administered using a Dry powder insufflator (model DP-4M; Penn-Century). At 4 h and 24 h posttreatment, mice (n = 4) were humanely killed, and blood and lung samples were collected from each animal. Bacterial load and phage concentrations in the lung homogenates and plasma were determined. The statistical significance of the data was examined using the Student t test. The null hypothesis was rejected if the P value was <0.05.
Histopathology of lungs after pulmonary delivery of the phage powder.
Histopathological examinations were performed after treatment with inhaled phage powder no. 1 in the infected and healthy mice. After 24 h, mice were humanely killed, and lungs were harvested and fixed in 10% formalin. Fixed tissues were sent to Australian Phenomics Network Histopathology and Organ Pathology Service (Victoria, Australia) for histopathological examination. The following SQS were used to quantify the extent of lung damages: 0, no significant change; 1, mild damage; 2, mild to moderate damage; 3, moderate damage; 4, moderate to severe damage; and 5, severe damage (9).
ACKNOWLEDGMENTS
This study was supported by National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R21 AI121627 (H.-K.C. and J.L.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. J.L. is an Australian National Health and Medical Research Council (NHMRC) Senior Research Fellow. H.-K.C. thanks Stephen Abedon for helpful discussion and valuable advice.
REFERENCES
- 1.Geller DE. 2009. Aerosol antibiotics in cystic fibrosis. Respir Care 54:658–670. [DOI] [PubMed] [Google Scholar]
- 2.Burrowes B, Harper DR, Anderson J, McConville M, Enright MC. 2011. Bacteriophage therapy: potential uses in the control of antibiotic-resistant pathogens. Expert Rev Anti Infect Ther 9:775–785. doi: 10.1586/eri.11.90. [DOI] [PubMed] [Google Scholar]
- 3.Wittebole X, De Roock S, Opal SM. 2014. A historical overview of bacteriophage therapy as an alternative to antibiotics for the treatment of bacterial pathogens. Virulence 5:226–235. doi: 10.4161/viru.25991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loc-Carrillo C, Abedon ST. 2011. Pros and cons of phage therapy. Bacteriophage 1:111–114. doi: 10.4161/bact.1.2.14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Debarbieux L, Leduc D, Maura D, Morello E, Criscuolo A, Grossi O, Balloy V, Touqui L. 2010. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J Infect Dis 201:1096–1104. doi: 10.1086/651135. [DOI] [PubMed] [Google Scholar]
- 6.Morello E, Saussereau E, Maura D, Huerre M, Touqui L, Debarbieux L. 2011. Pulmonary bacteriophage therapy on Pseudomonas aeruginosa cystic fibrosis strains: first steps towards treatment and prevention. PLoS One 6:e16963. doi: 10.1371/journal.pone.0016963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alemayehu D, Casey PG, McAuliffe O, Guinane CM, Martin JG, Shanahan F, Coffey A, Ross RP, Hill C. 2012. Bacteriophages phiMR299-2 and phiNH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. mBio 3:e00029-. doi: 10.1128/mBio.00029-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheah SE, Wang J, Nguyen VT, Turnidge JD, Li J, Nation RL. 2015. New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: smaller response in lung infection. J Antimicrob Chemother 70:3291–3297. doi: 10.1093/jac/dkv267. [DOI] [PubMed] [Google Scholar]
- 9.Lin YW, Zhou QT, Cheah SE, Zhao J, Chen K, Wang J, Chan HK, Li J. 2017. Pharmacokinetics/pharmacodynamics of pulmonary delivery of colistin against Pseudomonas aeruginosa in a mouse lung infection model. Antimicrob Agents Chemother 61:e02025-. doi: 10.1128/AAC.02025-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin YW, Zhou QT, Onufrak NJ, Wirth V, Chen K, Wang J, Forrest A, Chan HK, Li J. 2017. Aerosolized polymyxin B for treatment of respiratory tract infections: determination of pharmacokinetic/pharmacodynamic indices for aerosolized polymyxin B against Pseudomonas aeruginosa in a mouse lung infection model. Antimicrob Agents Chemother 61:00211-17. doi: 10.1128/AAC.00211-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matinkhoo S, Lynch KH, Dennis JJ, Finlay WH, Vehring R. 2011. Spray-dried respirable powders containing bacteriophages for the treatment of pulmonary infections. J Pharm Sci 100:5197–5205. doi: 10.1002/jps.22715. [DOI] [PubMed] [Google Scholar]
- 12.Leung SS, Parumasivam T, Gao FG, Carrigy NB, Vehring R, Finlay WH, Morales S, Britton WJ, Kutter E, Chan HK. 2016. Production of inhalation phage powders using spray freeze drying and spray drying techniques for treatment of respiratory infections. Pharm Res 33:1486–1496. doi: 10.1007/s11095-016-1892-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vandenheuvel D, Singh A, Vandersteegen K, Klumpp J, Lavigne R, Van den Mooter G. 2013. Feasibility of spray drying bacteriophages into respirable powders to combat pulmonary bacterial infections. Eur J Pharm Biopharm 84:578–582. doi: 10.1016/j.ejpb.2012.12.022. [DOI] [PubMed] [Google Scholar]
- 14.Merabishvili M, Vervaet C, Pirnay JP, De Vos D, Verbeken G, Mast J, Chanishvili N, Vaneechoutte M. 2013. Stability of Staphylococcus aureus phage ISP after freeze-drying (lyophilization). PLoS One 8:e68797. doi: 10.1371/journal.pone.0068797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leung SS, Parumasivam T, Gao FG, Carter EA, Carrigy NB, Vehring R, Finlay WH, Morales S, Britton WJ, Kutter E, Chan HK. 2017. Effects of storage conditions on the stability of spray dried, inhalable bacteriophage powders. Int J Pharm 521:141–149. doi: 10.1016/j.ijpharm.2017.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 17.Chang RY, Wong J, Mathai A, Morales S, Kutter E, Britton W, Li J, Chan HK. 2017. Production of highly stable spray dried phage formulations for treatment of Pseudomonas aeruginosa lung infection. Eur J Pharm Biopharm 121:1–13. doi: 10.1016/j.ejpb.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L, Sun S, Parumasivam T, Denman JA, Gengenbach T, Tang P, Mao S, Chan HK. 2016. l-Leucine as an excipient against moisture on in vitro aerosolization performances of highly hygroscopic spray-dried powders. Eur J Pharm Biopharm 102:132–141. doi: 10.1016/j.ejpb.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Steckel H, Müller BW. 1997. In vitro evaluation of dry powder inhalers I: drug deposition of commonly used devices. Int J Pharm 154:19–29. doi: 10.1016/S0378-5173(97)00113-0. [DOI] [Google Scholar]
- 20.Carmody LA, Gill JJ, Summer EJ, Sajjan US, Gonzalez CF, Young RF, LiPuma JJ. 2010. Efficacy of bacteriophage therapy in a model of Burkholderia cenocepacia pulmonary infection. J Infect Dis 201:264–271. doi: 10.1086/649227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu KY, Yang WH, Dong XK, Cong LM, Li N, Li Y, Wen ZB, Yin Z, Lan ZJ, Li WP, Li JS. 2016. Inhalation study of mycobacteriophage D29 aerosol for mice by endotracheal route and nose-only exposure. J Aerosol Med Pulm Drug Deliv 29:393–405. doi: 10.1089/jamp.2015.1233. [DOI] [PubMed] [Google Scholar]
- 22.Patil JS, Sarasija S. 2012. Pulmonary drug delivery strategies: a concise, systematic review. Lung India 29:44–49. doi: 10.4103/0970-2113.92361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bivas-Benita M, Zwier R, Junginger HE, Borchard G. 2005. Non-invasive pulmonary aerosol delivery in mice by the endotracheal route. Eur J Pharm Biopharm 61:214–218. doi: 10.1016/j.ejpb.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Abedon ST. 2009. Kinetics of phage-mediated biocontrol of bacteria. Foodborne Pathog Dis 6:807–815. doi: 10.1089/fpd.2008.0242. [DOI] [PubMed] [Google Scholar]
- 25.Tiwari BR, Kim S, Rahman M, Kim J. 2011. Antibacterial efficacy of lytic Pseudomonas bacteriophage in normal and neutropenic mice models. J Microbiol 49:994–999. doi: 10.1007/s12275-011-1512-4. [DOI] [PubMed] [Google Scholar]