

Abstract
Electrochemistry represents one of the most intimate ways of interacting with molecules. This review discusses advances in synthetic organic electrochemistry since 2000. Enabling methods and synthetic applications are analyzed alongside innate advantages as well as future challenges of electroorganic chemistry.
Graphical abstract
1. INTRODUCTION
Electrochemistry represents one of the most intimate and visceral ways of interacting with molecules: electrostatic attractions between electrons and nuclei constitute the most fundamental forces in chemistry—electrochemistry entails the addition or removal of electrons from such interactions through the direct application of an electrical potential. Thus, it is raw redox-chemistry and, as such, is one of the oldest forms of reaction setups explored in a laboratory. The storied history of electroorganic chemistry (illustrated in Figure 1A)1 can be traced back to 1800 when the invention of the Volta Pile, the first electric battery, allowed the continual movement of electrons through a circuit.2
Figure 1.
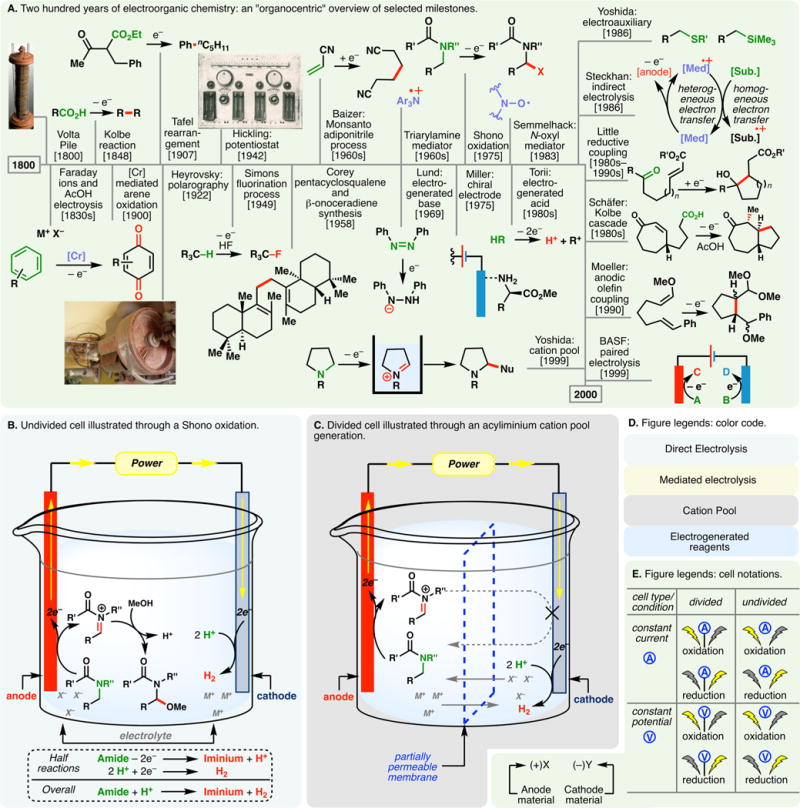
(A) Two hundred years of electroorganic chemistry: an “organocentric” view of selected milestones; basic principles of the undivided cell (B) and divided cell (C) explained through amide oxidations; color code (D) and cell notations (E) used in the review. Image credits: the image of “Volta Pile”, Copyright Wellcome Images, adapted under CC BY 4.0; the image of “Heyrovsky’s Polarograph”, Copyright Lukáš Mižoch, adapted under CC BY-SA 3.0; the image of Janke & Kunkel “Electrolytical Work Table”, Copyright IKA, adapted with permission.
However, it was not until the 1830s when Faraday’s pioneering efforts sparked the interest in utilizing this current to drive nonspontaneous organic reactions.1,3 Faraday’s systematic studies galvanized the foundational principles and lexicon of electrochemistry. To be sure, terms including electrolysis, anode, and cathode were introduced during this formative period. Faraday keenly observed that the electrochemical potential induced the movement of ions through a solution, thereby spawning the modern-day usage of ionic salts as electrolytes to increase the conductivity of organic systems. His electrolysis of acetic acid represents the first preparative organic electrochemical experiment, inspiring the invention of the venerable Kolbe electrolysis in 1847 where electrochemical oxidation (anodic oxidation) of ubiquitous carboxylic acids provides a convenient means to access alkyl radicals.4 Schoebein’s5 dehalogenation of trichloromethanesulfonic acid appears to be the first electrochemical reduction (cathodic reduction) of an organic compound, while the Tafel rearrangement, a well-known cathodic reduction, was developed in 1907 to enable preparations of hydrocarbons.6
These early forays gave rise to prototypes of today’s electrochemical setup: a power source is connected to a reaction mixture through an electrode where electron transfer with the substrate molecule occurs to generate a reactive intermediate for downstream functionalizations (illustrated with an anodic oxidation in Figure 1B). This electrode is known as the working electrode. To complete the circuit, another electrode (known as the counter electrode) is needed to conjoin the reaction with the other end of the power source. Depending on where reaction with the substrate takes place, either an anode or cathode can be the working electrode (with paired electrolysis being an exception, vide infra). Thus, in an anodic oxidation, oxidation at the working electrode is balanced by reduction at the cathode as electrons donated by the substrate at the anode move around the circuit to reduce solvent molecules, protons, or other species cathodically. Each electrochemical reaction can therefore be construed as a combination of two half-reactions. In the simplest setup, working and counter electrodes reside in the same chamber (an undivided cell); however, scenarios can arise where high energy intermediates generated at the working anode are prematurely reduced at the cathode and vice versa. This can be overcome with divided cells where anodic and cathodic chambers are segregated by a partially permeable membrane or a salt bridge (Figure 1C, illustrated with the generation of an acyliminium cation pool, vide infra). Maigrot and Stabates’s work in 1889 represents an early example of membrane electrolysis.7 For cathodic reductions, a sacrificial anode consisting of readily oxidizable materials (e.g., Mg, Zn, or Fe) can be used to forestall the undesired oxidation of reactive intermediates in undivided cells. In this case, the oxidative dissolution of the anode occurs preferentially.
Electrosynthesis in the 19th century exclusively relied on galvanostatic conditions where the reaction mixture is subjected to a constant stream of current and the potential increases over time. To this end, Hickling’s invention of the potentiostat in 1942 opened a new dimension in electrosynthesis: reactions could be conducted under constant potential, and the current decreases over time (shown in Figure 1A is an electrochemical power source from Janke & Kunkel (now IKA) manufactured in the same era).8,9 This was further aided by the advent of voltammetry techniques such as Heyrovsky’s polarography (1922),10,11 a forerunner to modern cyclic voltammetry (the first cyclic voltammetry experiment was described by Randles in 1948),12 where voltage and current are correlated. Developments and perfection of such analytical prowess allowed the electrochemical potentials of individual functionalities to be accurately determined—selective manipulation of functional groups is thus made possible by “dialing in” these measured properties under constant potential (potentiostatic) electrolysis. In potentiostatic setups, a standard electrode (commonly Ag/Ag+ or saturated calomel electrode) is used to accurately gauge the potential at the working electrode. Notwithstanding the many benefits of divided cells and constant potential electrolysis, the use of undivided galvanostatic setups remains strategic, owing to the operational simplicity.
Inventions of enabling equipment (vide supra), coupled with foundational advances in the 1800s, heralded awe-inspiring applications of electroorganic chemistry in the mid-1900s. The Simons fluorination process13 and the Monsanto adiponitrile process,14 both conducted on industrial scales, showcased the innate scalability of electrosynthesis. Corey’s elegant synthesis of pentacyclosqualene and onoceradiene testified to the potential of electroorganic chemistry to furnish simplifying disconnections.15
Electrodes are critical to organic electrosynthesis—under direct electrolysis, substrate molecules undergo electron transfers with the electrode surface. Such heterogeneous processes can impose a high kinetic barrier. Besides, reactions in the space near the electrode surface (known as the double layer)16 lead to accumulation of high energy species (e.g., radical cations and anions). Some of these reactive intermediates can diffuse back into the bulk solution for downstream functionalizations while others decompose to trigger electrode deactivation (known as passivation), impeding further reactions. To circumvent these problems, a mediator (or redox catalyst) can be used that undergoes heterogeneous electron transfer with the electrode surface to form a stabilized intermediate. This reactive species can then oxidize or reduce a substrate molecule homogeneously in an indirect electrolytic process. While indirect electrolysis using inorganic mediators found applications in as early as 1900 when chromium salts were harnessed to facilitate the anodic synthesis of quinones,17 powerful organic redox mediators such as triaryl amines18 and nitroxyl radicals19 were popularized during the 1970s and 1980s; the principles of indirect electrolysis were formalized by Steckhan in the 1980s.20,21 Transition metal complexes and ionic halides represent two other types of common mediators—the latter can undergo anodic oxidation to generate molecular halogen, hypohalite, or halogen cation species.
Other notable achievements in this era include synthetic applications of electrogenerated base22 and acid,23 where electromotive force is enlisted to effect proton transfers in an environmentally friendly fashion. Miller’s invention of the chiral electrode in 1975 pointed to a new direction in asymmetric catalysis that is perhaps worth revisiting.24 In the same year, the development of the Shono oxidation allowed the α-functionalization of alkyl amides (Figure 1B, illustrated with an undivided cell);25,26 this versatile reaction represents one of the most widely studied and utilized electroorganic transformations to this day.
The last quarter of the 20th century witnessed path-pointing achievements which shaped a considerable portion of synthetic electrochemistry discussed in this review. Yoshida introduced the concept of electroauxiliaries where sulfur- and silicon-containing functional groups are incorporated into substrate molecules to lower their electrochemical potentials (vide infra), allowing controls over regio- and chemoselectivities.27 Steckhan’s contribution to indirect electrolysis, as discussed briefly earlier, brought forth numerous mediated processes in recent history. Little’s efforts established cathodic reduction as a robust means to accomplish empowering ring-forming reactions that would otherwise require sensitive single-electron metal reductants.28–30 Studies by Schäfer culminated in a series of cascade reactions initiated by Kolbe electrolysis, substantially expanding the scope of the classical reaction.31,32 Moeller’s rigorous analysis of anodic olefin coupling furnished fundamental insights on the polarity of radical cations; it also led to many spectacular synthetic applications in the 21st century (vide infra).33 BASF’s utilization of paired electrolysis where synthetically useful processes take place simultaneously at both the anode and the cathode highlight the possibility of bolstering the energy efficiency of electro-organic chemistry in an industrial setting.34,35 Nucleophilic trapping of anodically generated cationic species constitutes a significant portion of organic electrochemical reactions. However, nucleophiles present in the electrolytic setup can undergo competitive oxidation, limiting the possible types of reactions. Yoshida’s concept of cation pool circumvented such longstanding problems: anodic oxidation is carried out under cryogenic conditions in the absence of trapping nucleophiles, allowing the accumulation of reactive cations which can engage various nucleophilic species in downstream functionalizations.36,37 As cation pools may be prone to cathodic reduction, they are usually generated in a divided cell as shown in Figure 1C.
These monumental achievements have been summarized in various review articles. Anodic oxidations have been comprehensively surveyed by Moeller in 200033,38 while Schäfer and Wright recounted advances in both anodic and cathodic processes in 198139 and 2006,16 respectively. Developments in mediated electrolysis have been analyzed by Little40 while Yoshida’s 2008 report dissected the emerging trends,42 enabling techniques and reaction engineering aspects of organic electrochemistry.41 The environmental impact of electrochemistry as pertaining to sustainability and greenness has been addressed by Frontana-Uribe/Little43 and Schäfer44 recently. Thus, the aim of this review is to examine reactions that have been published after 2000, and the focus will be placed exclusively on the synthetic applications of each transformation.45 Topics including electrogenerated acid,46 electrogenerated base,47 electrochemical enzymatic reactions,48 and electrocatalytic hydrogenation will not be detailed.49
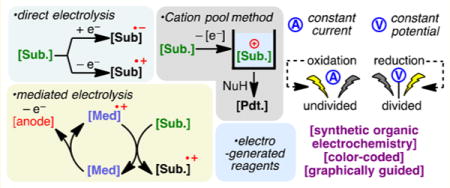
This overview of the field is organized by reaction type: oxidations (anodic), reductions (cathodic), and paired electrolysis (both occurring in the same vessel). To aid the reader, each section is subdivided by categories of functional group transformations and color-coded by the electrochemical technique (Figure 1D). Thus, seafoam green, cream yellow, gray, and powder blue backgrounds are utilized for direct electrolysis, mediated processes, cation pool, and electrogenerated reagents, respectively. A set of “cell notations” has also been devised to graphically represent the electrochemical parameters of each reaction, including cell type (divided vs undivided), electrolytic conditions (constant current vs constant potential), and electrode compositions (Figure 1E). Electrochemical potential values quoted in this review represent reported values against designated standards. To ease discussions, comparisons of potentials are made on the basis of their absolute values. For example, if a compound has a more negative reduction potential, it is described to have a higher reduction potential (absolute value), making it more difficult to reduce. Electrochemical potentials of common organic functional groups have been summarized elsewhere and will not be reiterated.50,51 It is hoped that this color coded and annotated format will aid the reader to rapidly identify classes of reactions and setups without needing to refer to the text.
2. ANODIC OXIDATION
2.1. Oxidation of carboxylates: The Kolbe reaction and related processes
Perhaps the best known electrochemical transformation, the Kolbe reaction, commences with the anodic oxidation of alkyl carboxylates whereupon the resulting radical undergoes facile decarboxylation. The ensuing alkyl radical then dimerizes to forge a C–C bond. Development and applications of Kolbe electrolysis before 2000 have been reviewed;31,33,52 more recently, this classical process has been utilized to synthesize benzathine,53 construct bis-phosphine oxide ligands,54 and dimerize silylacetic acids55–57 and fatty acids.58 The mixed-Kolbe reaction, wherein a heterodimerization between two different alkyl carboxylic acids occurs, is less common—an excess of one acid component is often necessary to favor the cross-coupling product.59 Renaud and co-workers applied a mixed-Kolbe electrolysis to synthesize nephromopsinic, phaseolinic, and dihydropertusaric acids (Figure 2A).60 In the third case, the ketal containing carboxylic acid was used in eight equivalents.
Figure 2.
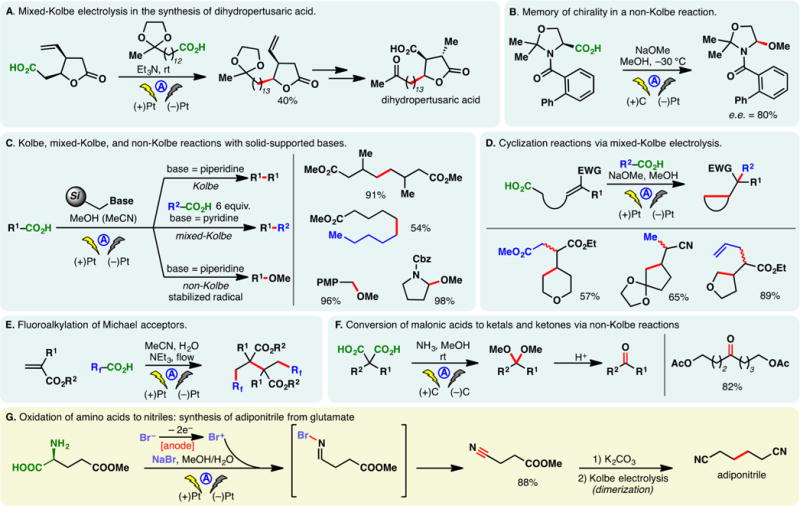
Oxidation of carboxylates: the Kolbe reaction and related processes.
Should the electrochemical decarboxylation process afford a radical that is stabilized by an α-substituent (e.g., an α-amino group in an amino acid), further electrochemical oxidation can occur—the resulting cation (e.g., an acyliminium cation) may be trapped with an external nucleophile in a non-Kolbe reaction, as is the case in Figure 2B.61 In this example, Matsumura and co-workers reported that the electrochemical oxidation of an N-acylated serine derivative exhibited “memory of chirality”—the methoxylated product was afforded in 80% enantiomeric excess (e.e.) (Figure 2B). The bulky o-phenyl benzoyl protecting group was believed to be the main factor for the stereoselectivitiy, as lower e.e. values were observed with a simple benzoyl group.62,63
In the Kolbe electrolysis, a base is added to a solution of carboxylic acids, forming carboxylate salts which could serve as substrates and electrolytes. Tajima, Fuchigami, and co-workers developed protocols to conduct Kolbe,64 mixed-Kolbe,65 and non-Kolbe66,67 reactions with solid-supported bases (piperidine or pyridine) (Figure 2C). Upon completion of the electrolysis, the base could be removed through filtration, rendering the process operationally simple. Chiba has reported an efficient protocol for the Kolbe reaction using cycloalkane-based thermomorphic systems;68 Compton and co-workers devised an aqueous protocol for the Kolbe reaction of water-immiscible aliphatic acids through ultrasonication and emulsion formation—it was observed that the yields of products obtained from this biphasic system are independent of the electrode material used, on the contrary to homogeneous systems.69
A variant of the Kolbe reaction is the conjunctive cyclization cascade pioneered by Schafer.31, 70 In this process, a carboxylic acid is tethered to an olefin which can engage the electrogenerated alkyl radical in a cyclization; the resulting radical can combine, intermolecularly, with an alkyl radical derived from the anodic decarboxylation of an exogenous carboxylic acid. Earlier examples of this process have been detailed in review articles.33 More recently, Márko and co-workers reported a method to prepare five- and six-membered rings based on this cascade (Figure 2D).71 The use of an electron-deficient olefin is critical to ensure high yields, owing to the nucleophilic character of alkyl radicals. Taking advantage of an intermolecular version of this conjunctive coupling, Wirth and co-workers reported a method for the “dimerizative” fluoroalkylation of Michael acceptors (Figure 2E) in flow reactors.72 The analogous batch reaction was reported by Uneyama in 1988.73
Recently, Maŕko disclosed a method to convert malonic acids into ketals and subsequently ketones via two tandem anodic decarboxylation reactions (Figure 2F).74
Electrolysis of a glutamic acid derivative in the presence of a bromide mediator produced the corresponding nitrile after decarboxylation (Figure 2G).75 Putatively, a bromo-imine or equivalent thereof is involved.76 This process was harnessed by Fu and co-workers to synthesize adiponitrile from glutamic acid—a classical Kolbe dimerization was used in a later step.
2.2. Oxidation of sulfinic acid salts
Sulfinates, the sulfur congeners of carboxylates, can also be readily oxidized into the corresponding sulfonyl radicals under electrochemical conditions. Aryl sulfonyl radicals may be trapped by olefins prior to SO2 extrusion. This reactivity allowed Wang and co-workers to develop a synthesis for E-vinyl sulfones from cinnamic acids.77 The reaction is believed to proceed through the addition of an aryl sulfonyl radical onto the double bond, followed by electrochemical decarboxylation (Figure 3A, left). A similar example in 2015 showcased that the same type of product could be accessed via the reaction of electrogenerated sulfonyl radicals and styrenes—NaI was used as the mediator, allowing the formation of a β-iodosulfone intermediate (Figure 3A, right).78 Halide mediated oxidations of aryl sulfinates have also found applications in the preparations of oxindole (Figure 3B, top),79 indenones (Figure 3B, bottom),80 and sulfonamides81 (Figure 3C) through sulfonyl radical initiated reactions.
Figure 3.
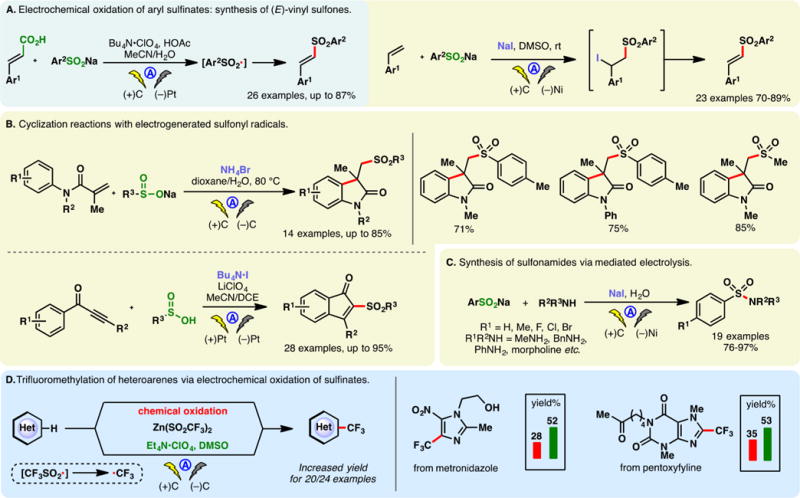
Oxidation of sulfinic acid salts.
Baran, Blackmond, and co-workers reported that the direct electrolysis of zinc bis-trifluoromethylsulfinate (TFMS) led to the formation of trifluoromethyl radicals which could chemoselectively engage a broad selection of heteroarenes including drug molecules such as metronidazole and pentoxyfyline (Figure 3D).82 Although this transformation can alternatively be achieved with a chemical oxidant (TBHP), the electrochemical protocol demonstrated enhanced yields for many substrates while conferring additional scalability. Tommasino and co-workers have used potassium trifluoromethylsulfinate to effect electrochemical trifluoromethylation of dimethoxybenzenes, durene, and several olefins.83
2.3. Oxidation of amines and amides: formation of N-centered radicals and nitrenes
2.3.1. Formation of N-centered radicals
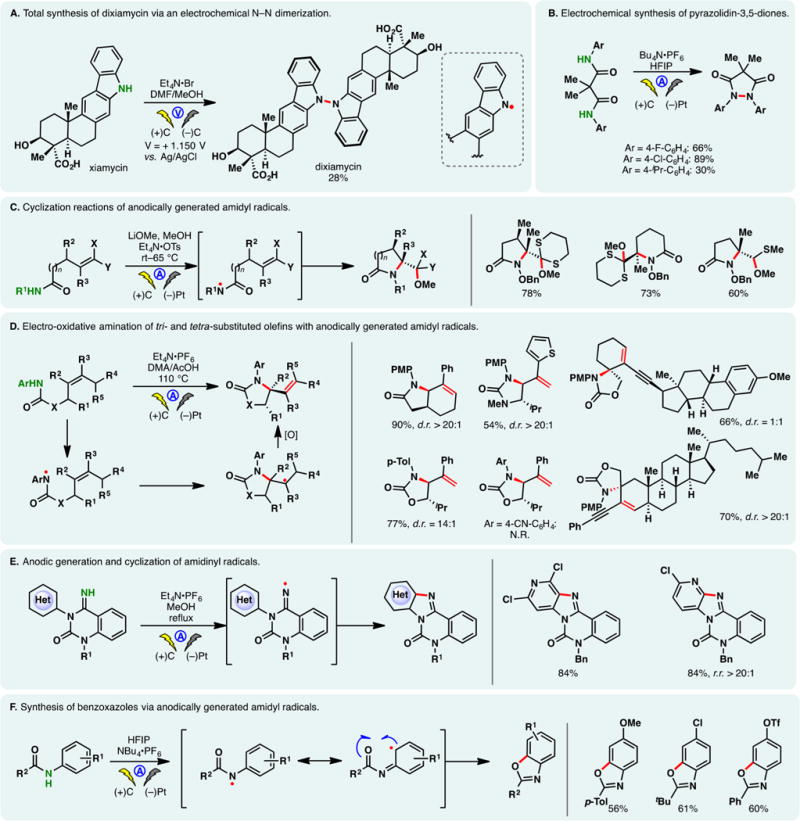
Anodic oxidation offers a convenient avenue to access N-centered radicals directly from amines and amides. Taking advantage of constant potential electrolysis, Baran and co-workers reported a synthesis of dixiamycin through the direct dimerization of xiamycin (Figure 4A).84 Controlling the potential at +1.150 V (vs Ag/AgCl) allowed the chemoselective oxidation of the carbazole nitrogen while leaving other reactive functionalities (e.g., a free alcohol and a carboxylic acid) intact. Forays to achieve this N–N coupling chemically were unsuccessful; meanwhile, the hydrazine-based strategy to construct this natural product would likely require a lengthy sequence to prepare the carbazole core. Anodic N,N-dimerization of amidyl radicals derived from aryl amides is also possible—Moeller, Waldvogel, and co-workers developed a galvanostatic protocol to synthesize pyrazolidin-3,5-diones, which are important motifs in heterocyclic chemistry (Figure 4B).85,86 This reaction can be carried out in a simple undivided cell.
Figure 4.
Generation of N-centered radicals through direct electrolysis.
Moeller reported that electrophilic amidyl radicals, generated through anodic oxidation of O-bezylhydroxamates or N-phenyl amides, can undergo facile cyclization onto electro-rich olefins (e.g., enol ethers or dithioketene acetals) (Figure 4C).87 Five- and six-membered rings could be effectively forged through 5- and 6-exo attacks, respectively. The benzyloxy group on the amide lowers the oxidation potential of the amide nitrogen while stabilizing the ensuing radical species. The cyclized carbon-centered radical could undergo further electrochemical oxidation wherein the carbocation may be trapped by methanol. The radical nature of the cyclizations was supported by cyclic voltammetry studies and DFT calculations.
Building on this precedent, Xu and co-workers developed an electrochemical method for the oxidative amination of tri- and tetra-substituted olefins (Figure 4D).88 Amides, carbamates, and ureas substituted with electron-rich aryl groups can be used as the radical precursor under galvanostatic conditions. In the absence of an alcoholic solvent, the cyclized radical intermediate can be oxidized further to provide an olefin. A broad substrate scope was presented, including complex natural product derivatives. The same group also reported the use of direct anodic oxidation to generate amidinyl radicals (Figure 4E).89 These reactive species were found to cyclize onto electron-deficient (hetero)arenes, thereby allowing the construction of polycyclic benzimidazoles and pyridoimidazoles.
During their investigations on N, N-dimerization (vide supra), Waldvogel and co-workers observed that amidyl radicals of aryl-amides exhibited considerable radical character within the aromatic ring. By leveraging this reactivity, they developed an efficient method for the construction of benzoxazoles from anilides (Figure 4F).90 The use of HFIP is critical as it putatively prolongs the lifetime of the radical intermediate.
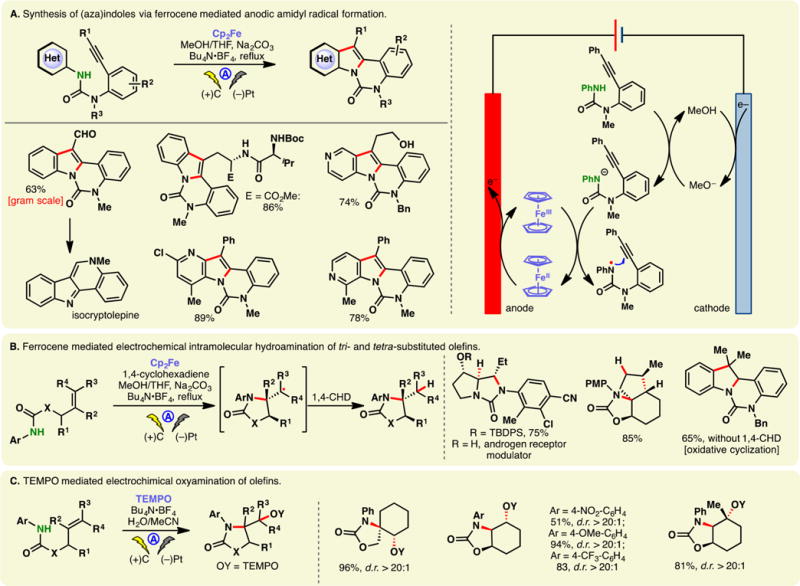
N-Aryl-amidyl radicals could also be accessed via indirect electrolysis using redox mediators. Xu has conducted extensive studies using ferrocene mediators to generate amidyl radicals electrochemically. These radicals can undergo intramolecular addition onto hetero(arenes) (Figure 5A)91 or olefins (Figure 5B).92 The former case allows access to polycyclic (aza)-indoles (Figure 5A) while the addition onto olefins enabled hydroamination reactions in the presence of a hydrogen atom donor (e.g., cyclohexadiene (1,4-CHD), Figure 5B). The choice of solvent was crucial: the addition of THF to MeOH was found to lower the oxidation potential of N-aryl-amides while raising that of ferrocene. Therefore, electron transfer between the oxidized form of the mediator and the substrate molecule is possible. No cyclization products were detected in the absence of THF. Natural product derivatives and drug analogs have been synthesized through these processes, attesting to their utilities. An analogous reaction using TEMPO as the mediator has also been detailed by Xu and co-workers, in which the coupling between the intermediary C-centered radical and TEMPO gave rise to aminooxygenation products (Figure 5C).93 Thus, TEMPO serves the dual-purpose as the mediator and the oxygen source.
Figure 5.
Generation of N-centered radicals through indirect electrolysis.
2.3.2. Formation of nitrenes and nitreniums
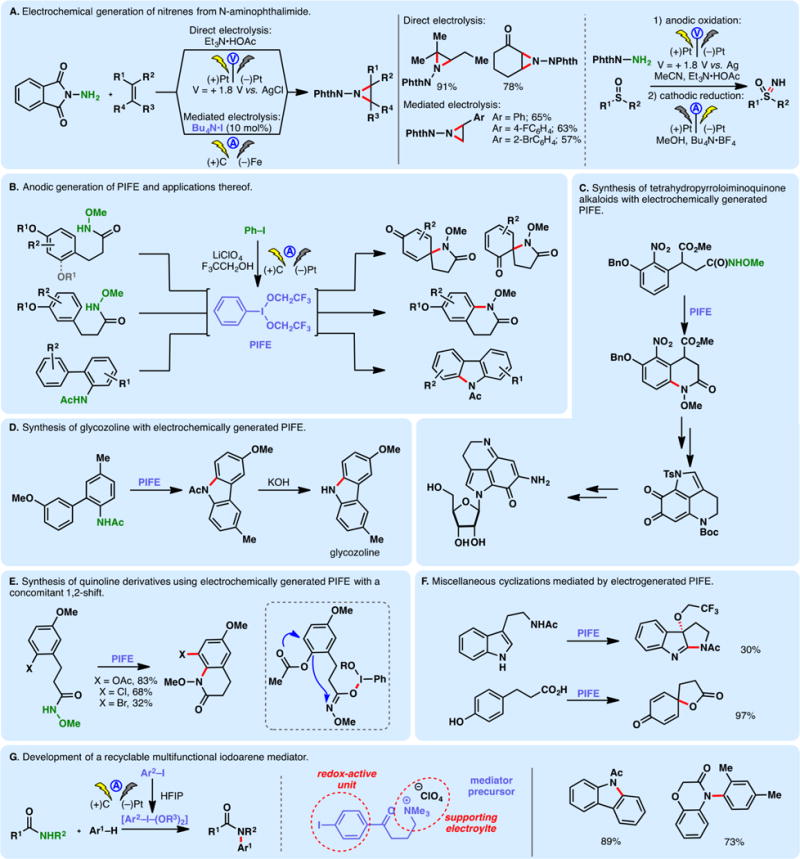
N-Aminophthalimide can serve as a nitrene precursor wherein the terminal nitrogen can be oxidized by a chemical oxidant to furnish a nitrene. Nevertheless, this oxidation commonly requires stoichiometric quantities of harsh reagents (e.g., Pb(OAc)4). Yudin and co-workers discovered that the N-phthalimido-nitrene can be accessed through direct electrolysis at +1.8 V (vs Ag/AgCl) in an environmentally friendly fashion (Figure 6A).94,95 Under the potentiostatic conditions, both electron-rich and electron-deficient olefins are aziridinated effectively. Although some electron-rich substrates exhibit similar oxidation potentials compared to N-aminophthalimide, the latter’s lower overpotential (additional potential beyond the thermodynamic requirement needed to drive a reaction at a certain rate) allowed its selective oxidation. Moreover, this electrochemical protocol could be used to effect imination of sulfoxides (Figure 6A, right).96 Little, Zeng, and co-workers developed a mediatory system to carry out the aziridination reaction under simple constant current conditions with inexpensive electrode materials; tetrabutylammonium iodide was found to be the optimal mediator (Figure 6A, bottom).97
Figure 6.
Electrochemical generation of nitrene and nitrenium species.
Anodic oxidation of iodoarenes leads to the formation of hypervalent iodine species which can serve as redox mediators for further functional group oxidations.98 The oxidation of iodobenzene in the presence of trifluoroethanol furnishes phenyliodine bis(trifluoroethoxide) (PIFE); Nishiyama and co-workers have extensively utilized this electrogenerated reagent to effect the oxidation of amides or hydroxamic acid derivatives into the corresponding nitreniums (Figure 6B).99 The nitreniums thus generated can undergo cyclization with electron-rich arenes to afford quinoline scaffolds,100 carbazoles,101 or spirocycles99 depending on the substitution patterns of the arene. This method has found applications in the synthesis of tetrahydropyrroloiminoquinone alkaloids (Figure 6C) as well as glycozoline (Figure 6D). When an arene is substituted with a (pseudo)halide at the ortho-position, concomitant substituent migration was observed alongside oxidative cyclization (Figure 6E).102 Additional applications of electrogenerated PIFE include oxidative cyclization of tryptamine and dihydrocinnamic acid derivatives (Figure 6F).99,103 Francke and co-workers developed a multifunctional electrochemical mediator for nitrenium generation by merging an iodoarene scaffold with an ionic side chain.104 Pre-electrolysis of this aryl iodide in HFIP led to the formation of a hypervalent iodine species which could trigger the oxidative arylation of anilides. Because of the ionic motif, no external electrolyte is necessary. The presence of the keto group at the benzylic position of the mediator prevents oxidative degradation—the mediator precursor can be recycled at the end of the reaction (Figure 6G). From a pragmatic perspective, it is worth noting that the above examples (Figure 6B–G) involve an electrochemical reagent generation step followed by addition of the substrate (with no electricity).
2.4. Oxidation of amines and amides: Shono-type oxidation
2.4.1. Variation of nitrogen substituents
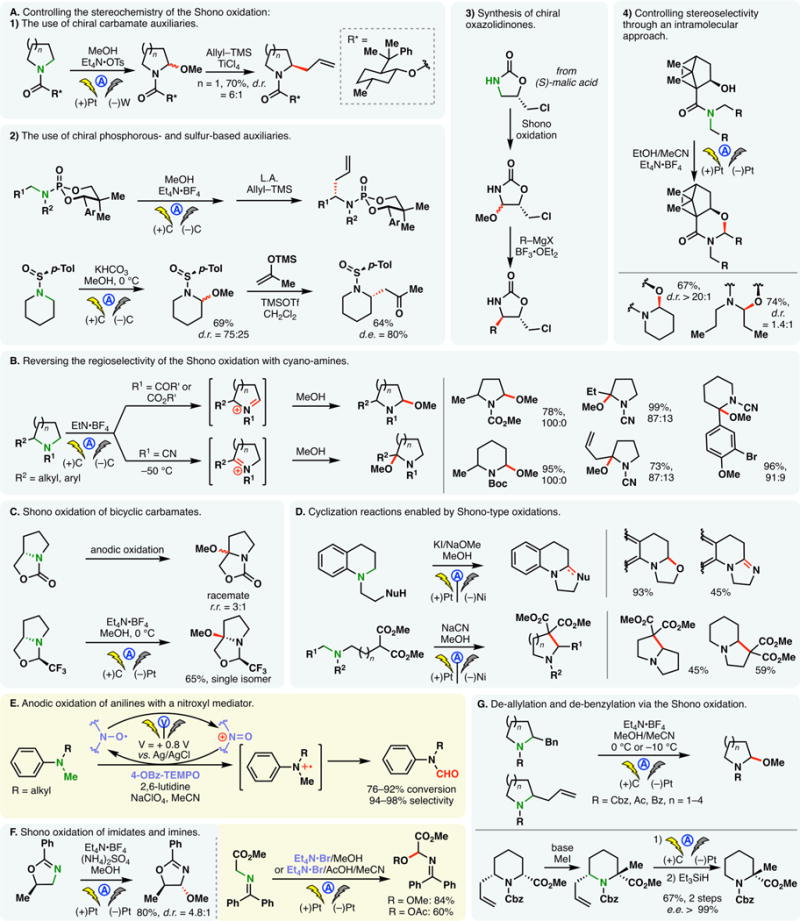
Similar to aryl amides, the anodic oxidation of alkyl amides or carbamates gives rise to N-centered radical cations. However, these reactive intermediates readily undergo fragmentation to afford N-acyl or N-carbamoyl iminium ions in what is known as the Shono oxidation.105,106 This classic reaction is customarily carried out in an alcoholic solvent with the reactive iminium trapped as an isolable N,O-acetal. This species can revert to an acyliminium upon treatment with acids to allow further functionalizations. Efforts have been expended to render this sequence asymmetric with chiral auxiliaries (Figure 7A). For example, Shono methoxylation of chiral carbamates,107–110 phosphoramides,111,112 and sulfinamides113 has been surveyed (Figure 7A(1)–(3)). Generally, while poor diastereoselectivity was observed in the methoxylation step, the introduction of auxiliaries imparts stereocontrol in the downstream functionalizations, allowing nucleophiles such as allyl silane to be added at the α-position stereoselectively. Good d.r. values of the initial electrochemical oxidation were noted when the alkoxy nucleophile was tethered with chiral cyclic amides; nevertheless, the electrolysis of an analogous acyclic amides led to virtually no selectivity (Figure 7A(4)).114 Shono oxidation has also been combined with the Evans oxazolidinone chemistry to afford enantiomerically enriched Mannich adducts.115
Figure 7.
Shono oxidation: variation of nitrogen substituents.
When an unsymmetrical secondary amide or carbamate is used in the Shono oxidation, oxidations occur preferentially at the less substituted position as illustrated in Figure 7B. However, efforts by Onomura revealed that the regiochemical outcome may be reversed using cyanoamines wherein the methoxylation at the more substituted position is favored.116 Computational studies suggested that the cyano group substantially stabilized the more substituted iminium ion, thereby favoring methoxylation at the more substituted position. Conversely, for carbamates, the iminium intermediates are of similar stability; steric effects thus predominate.
The Shono oxidation of bicyclic carbamates takes place mainly at the ring junction (the more substituted position), albeit with little stereocontrol. As shown in Figure 7C, the oxidation of a proline-derived chiral carbamate led to racemic products.117 The use of a 1-alkoxy-2,2,2-trifluoroethyl group in place of the carbamate was found to enhance the regio- and stereoselectivity of the process as the methoxylation product was predominantly afforded as a single regio- and stereoisomer (Figure 7C).
Despite the low redox potentials of amines, anodic oxidation of alkyl amines is utilized to a lesser extent compared to amides, presumably owing to the instability of the aminyl radical cations and imines. Gallardo and co-workers have exploited the anodic oxidation of alkyl amines to synthesize substituted imidazolinium, tetrahydropyrimidinium,118 and hindered alkyl diamines.119 When a nucleophile is embedded within the amine substrates, the reactive intermediates can be trapped in various modalities of cyclizations (Figure 7D).120,121 Additionally, the Shono-type oxidation is more useful with anilines or benzyl amines which form more stabilized radical cation intermediates.122 Electrochemical oxidation of dialkyl anilines was achieved with a stabilized nitroxyl mediator (4-OBz-TEMPO) (Figure 7E).123 The regioselectivity of this process resembles that of the analogous amide/carbamate oxidation—the reaction takes place at the less substituted position; the N-methyl group is preferentially oxidized, furnishing a formamide. Additionally, the Shono-type oxidation has also been successfully performed on imidates and imines (Figure 7F).124 In the oxidation of cyclic imidates, the addition of ammonium sulfate was found to improve the reaction yield; although its precise role is unclear, ammonium sulfate presumably maintains a neutral pH in the electrochemical cell, suppressing the oxidation of cathodically generated methoxide. α-Methoxylation/acetoxylation of imines can be achieved with the aid of tetraethylammonium bromide as a mediator (Figure 7F, right).
Aside from oxidizing the C–H bonds on the α-carbon of amides and carbamates, Onomura showed that the Shono oxidation may be extended to cleave allyl and benzyl groups affixed at that position (Figure 7G).125 Preliminary mechanistic studies on the deallylation reaction revealed that the process involves allyl cationic species rather than the analogous radicals. This method was successfully applied to the synthesis of optically active N-acylated α-alkyl-α-amino acid esters (Figure 7G, bottom).
2.4.2. Variation of trapping nucleophiles
Alcohols are ideal nucleophiles in Shono oxidations, due to their relatively high oxidation potential compared to amides/carbamates. Trapping the intermediary iminium with carbon-centered nucleophiles such as cyanides or enol ethers can conceivably be complicated by the competing anodic oxidations of these nucleophiles. Instead, a two-step process is used wherein an N,O-acetal is isolated and subjected to further manipulations. For example, the N,O-acetals obtained in Shono oxidations may be treated with triphenylphosphonium salts to afford 1-(N-acylamino)alkyltriphenylphosphonium wherein triphenylphosphine formally displaces the alkoxy group.126 These α-amidoalkylating agents can in turn be converted into amidosulfones which are commonly used surrogates for acyl iminiums.127 N-[1-(Benzotriazol-1-yl)alkyl]amides may be obtained in a similar fashion.128
Tajima and co-workers devised a one-step anodic α-cyanation reaction based on the principle of “site isolation” (Figure 8A).129 Bu4N·BF4 and the cyanide salt of a solid-supported quaternary ammonium cation (PS-NMe3·CN in Figure 8A) were introduced concurrently into the reaction—the former served as the supporting electrolyte while the latter reduced the effective concentration of cyanide in the solution phase, keeping CN– in vicinity to the polymer support. Cyanide oxidation is thus suppressed (see Figure 8A inset cyclic voltammogram).
Figure 8.
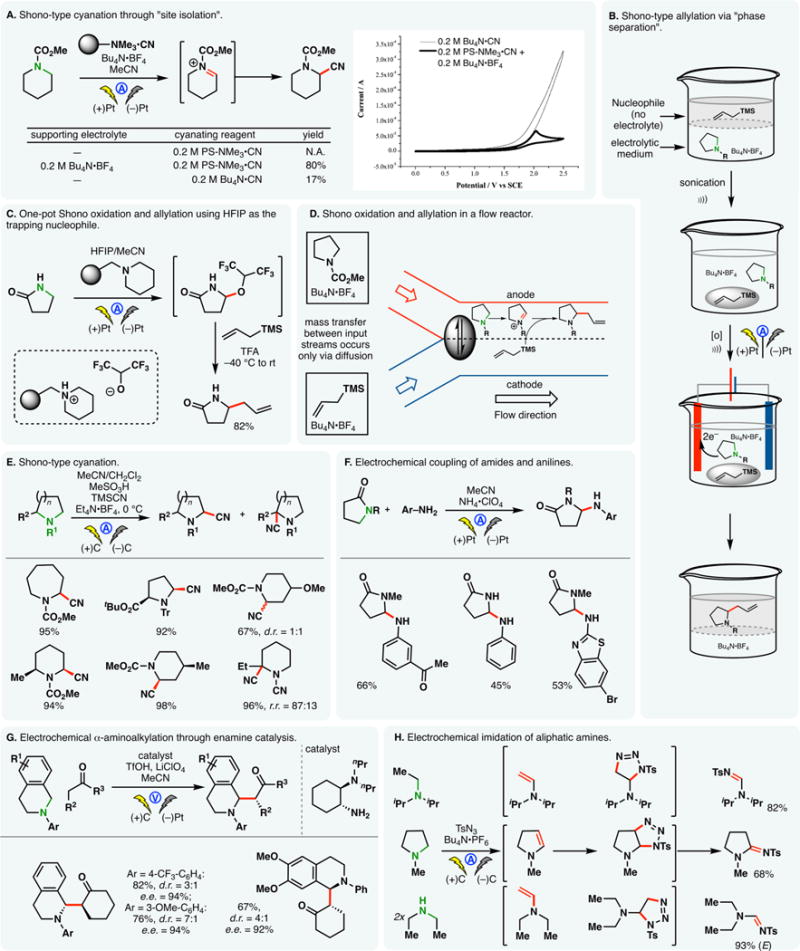
Shono oxidation: variation of trapping nucleophiles. Figure 8A inset: cyclic voltammogram adapted with permission from J. Am. Chem. Soc. 2008, 130, 10496–10497. Copyright (2008) American Chemical Society.
Leveraging similar principles, Atobe reported an example of direct Shono allylation (Figure 8B).130 In this case, an ionic liquid131 (EMM·BF4) was chosen as the reaction medium wherein the nucleophile (allyl–TMS) has minimal solubility. While sufficient interaction between the nucleophile and the anodically generated iminium can be attained through acoustic emulsification (sonication), oxidation of the nucleophile is thwarted by its poor solubility and conductivity. Besides, even though the allylated product had similar oxidation potential compared to the starting carbamate, its lower solubility in EMM·BF4 prevented its oxidation.
Tajima also reported an alternative approach for the one-pot allylation of lactams (Figure 8C).132 This strategy utilizes a Shono oxidation with HFIP as the trapping electrophile in the presence of a solid-supported base. The base allows for the in situ generation of electrolytes; it may be removed through a filtration, and the hexafluoroisopropoxy group in the Shono product can be readily displaced by carbon nucleophiles (e.g., allyl-TMS). The use of solid-supported bases in MeOH has allowed the same group to develop a series of electrochemical methoxylation reactions wherein methoxide anions are generated in situ.133–135
Atobe and co-workers demonstrated that a one-step α-allylation of amides may be achieved with the aid of parallel laminar flow in microflow reactors (Figure 8D).136,137 When the carbamate and the nucleophile are introduced in different streams, mass transfer between these separate streams can only occur via diffusion between the liquid–liquid contact area. Due to the small size of the flow channel, this area will remain stable and laminar. As the nucleophiles are spatially removed from the anode, undesired oxidation is minimized.
Onomura showcased that a one-step anodic cyanation of amides/carbamates is also possible using TMS–CN as the nucleophile (Figure 8E); the addition of methanesulfonic acid (MeSO3H) was found to be crucial for this reaction.138 Using cyanoamines as substrates, more substituted amino nitriles were obtained (vide supra).
Huang and co-workers demonstrated that anilines could be introduced into the α-position of lactams (such as NMP) through direct anodic oxidation when the latter was used in excess (Figure 8F).139 Maintaining a high current density at the anode was found to be critical: even though the oxidation potential of aniline coupling partners is lower than that of lactams, the more concentrated lactam could undergo preferential oxidation at high current densities. The use of a thin Pt wire as an anode and a Pt foil as the cathode was thus favorable for this purpose. This setup is often referred to as a “quasi-divided cell” wherein the surface areas of the working and counter electrodes differ significantly, creating different current densities. It is also possible that the acid electrolyte, ammonium perchlorate, has partially protonated the anilines, thereby disfavoring their oxidations.
Luo and co-workers developed an anodic coupling between tetrahydroisoquinoline derivatives and alkyl ketones through the combination of Shono-type oxidation and enamine catalysis.140 The reaction proceeded with moderate to good enantioselectivity (Figure 8G). In another variant of anodic amine α-oxidation, Wang and co-workers developed an electrochemical protocol for the imidation of aliphatic amines with tosyl azide.141 Tertiary, secondary, and primary amines could be converted into N-tosyl amidines in this process (Figure 8H). Although the imidation of primary amines was not shown in Figure 8H, the reactions followed a similar course compared to those of secondary amines. An oxidative dimerization occurs to furnish N-alkyl imines/enamines. Putatively, the anodically generated imine/iminium tautomerizes into the corresponding enamine which can engage tosyl azides in a cycloaddition whereupon fragmentation of the cycloadduct affords the amidine products.
2.4.3. The use of electroauxiliaries
The chemo- and regioselectivities of the Shono oxidation can be controlled with electroauxiliaries. As discussed in the Introduction, electroauxiliaries are structural moieties introduced to lower the electrochemical potential of substrates. For example, the introduction of TMS or tributylstannane motifs at the α-position of a carbamate lowers the redox potential substantially (Figure 9A).41,142 Through the use of silyl containing amino acids, Moeller and co-workers achieved the chemoselective introduction of acyliminiums into complex peptides under anodic oxidation.142,143 The silyl side-chain may be oxidized into an N,O-acetal (Figure 9B, bottom); alternatively, cyclic peptidomimetics can be synthesized through intramolecular ring closures (Figure 9B, top).144
Figure 9.
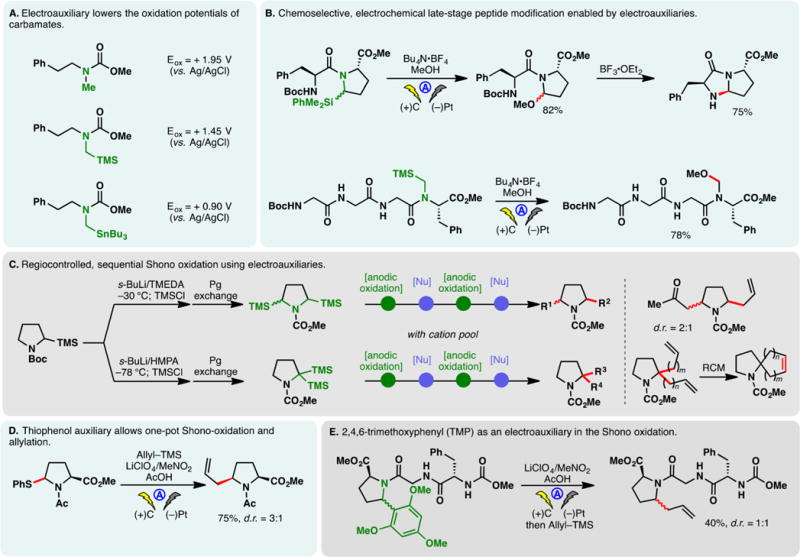
Use of electroauxiliary in the Shono oxidation.
Suga, Yoshida, and co-workers synthesized a 2,5-disilylated pyrrolidine derivative along with a 2,2-disilylated analog through directed metalation.145,146 These compounds could each be subjected to sequential Shono oxidations—the differential placement of silyl auxiliaries led to complementary regiochemical outcomes (Figure 9C). While the former afforded 2,5-disubstituted pyrrolidines, the latter delivered pyrrolidine products bearing two substituents on the same α-carbon. 2,2-Diallyl pyrrolidine thus obtained may be elaborated further to access spirocycles.
Efforts by Chiba established thiophenyl (Figure 9D)147 and 2,4,6-trimethoxybenzene (TMP) (Figure 9E)148 as viable electroauxiliaries in the Shono oxidation. The thiophenyl auxiliary allowed the direct anodic α-allylation of amides without oxidizing the allyl-TMS nucleophile; the TMP auxiliary facilitated the anodic modification of a tripeptide using a cation pool approach (vide infra). The use of LiClO4 in MeNO2 was deemed advantageous in both cases; indeed, this solvent system was believed to stabilize the intermediary acyliminium cation—this cation may be accumulated in MeNO2/LiClO4 at 0 °C in an undivided cell.147–149
2.4.4. Electrochemical generation of the acyl iminium cation pool
The scope of the Shono oxidation has been substantially expanded with the advent of the “cation pool” method wherein the anodic oxidation is carried out under cryogenic temperature, allowing for the accumulation of iminium species in a cation pool.41 These highly reactive intermediates can subsequently be intercepted by various nucleophiles in different transformations. As no nucleophilic species is present during the initial electrolysis, competing nucleophile oxidation is not a concern. The synthetic applications of the cation pool strategy has been reviewed by Yoshida and co-workers;41 Figure 10A provides a brief summary of iminium reaction manifolds enabled by this approach.
Figure 10.
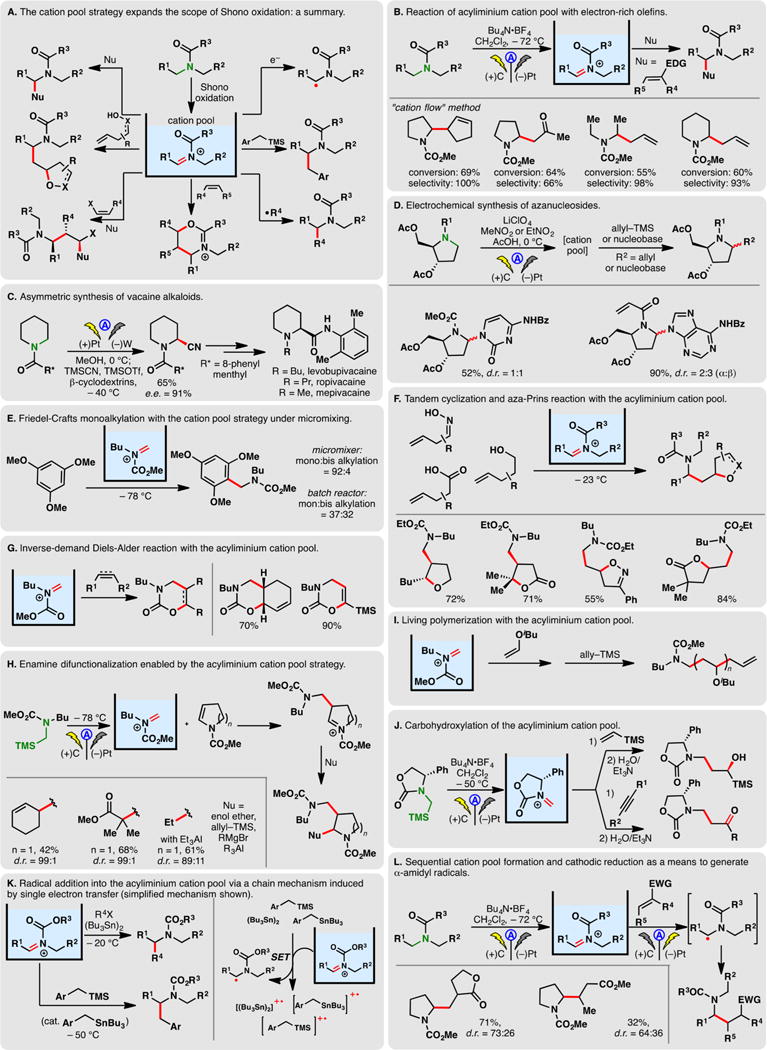
Electrochemical generation of acyl iminium cation pools.
First and foremost, the acyliminium cation pool can readily engage carbon-centered nucleophiles such as allyl silane, enol ethers, or organometallic reagents to furnish C–C bonds (Figure 10B)—this may be accomplished in a flow system through the “cation flow” method.150,151 Yoshida and co-workers have reported the synergistic applications cation pool and microflow systems for electrochemical combinatorial organic syntheses.152,142 The addition of cyanide onto the electrogenerated iminium cation pool allowed expedient asymmetric syntheses of vacaine alkaloids (Figure 10C).153 Chiba and co-workers prepared a series of aza-nucleosides by treating acyliminium cation pools with different nucleobases (Figure 10D).154,155 Reactive functionalities such as acetyl and the acryloyl groups were tolerated.156 Iminium cation pools have also found uses in other nucleophilic processes such as the Friedel–Crafts reaction (Figure 10E),157 the aza-Prins reaction (Figure 10F),158 and the inverse electron demand Diels–Alder reaction (Figure 10G).159 In the case of Friedel–Crafts alkylation, Yoshida and co-workers demonstrated that the use of micromixing effectively suppressed bis-aminoalkylation.
Reactions of the acyl/carbamoyl iminium cation pools with enamines led to the formation of another iminium ion which can be subjected to another iteration of nucleophilic trapping—Yoshida and co-workers have developed a tandem three-component reaction based on this reactivity (Figure 10H).160,161 In a similar vein, the Yoshida group demonstrated the feasibility of using the electrogenerated carbamoyl iminium cation pools in living polymerization162 (Figure 10I) as well as carbohydroxylation163 reactions (Figure 10J).
Yoshida has also reported radical addition into electrogenerated iminium ions with benzyl silanes164,165 or alkyl halides166,167 serving as radical progenitors alongside catalytic amounts of organotin species (tributyl-benzyl-stannane for alkyl halides or hexabutyl-di-tin for benzyl silanes) (Figure 10K). It was believed that a single electron transfer (SET) between acyl iminium cation and the organotin initiates a chain process. In the absence of catalytic benzyl-stannane, benzyl silanes could also participate in the SET process. When arylthiomethylsilanes and aryloxymethylsilanes are used as the nucleophiles, this type of SET-based reaction may also be initiated with electricity.165
Additionally, Yoshida showcased that, upon the completion of the anodic oxidation, the iminium cation pool can be reduced to α-amidyl radicals in a cathodic process.168 The nucleophilic radicals thus generated were found to readily add onto Michael acceptors in Giese reactions (Figure 10L).
2.4.5. Applications of Shono-type oxidations
The Shono oxidation has found numerous applications in the synthesis of natural products or other compounds of biomedical interest. For example, Ley and co-workers utilized sequential Shono oxidation and Pictet–Spengler reactions to access nazlinine and its structural analogs in a rapid fashion (Figure 11A).169 This process was conducted with a flow electrochemical cell which helped reduce the amount of requisite electrolyte.
Figure 11.
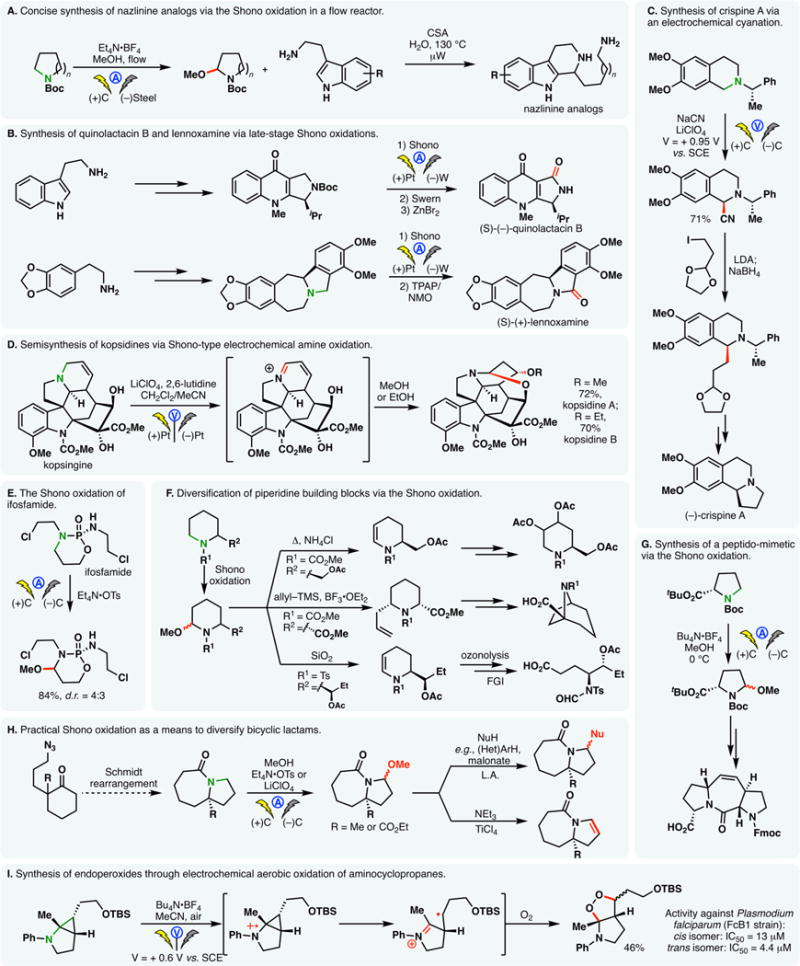
Synthetic applications of Shono-type anodic oxidations.
When water is used as the trapping nucleophile in the Shono oxidation, the resulting hemiaminal can be readily subjected to Swern or Ley oxidations, furnishing the corresponding lactams. Santos and co-workers exploited this sequence in their syntheses of quinolactacin B170 and lennoxamine171 (Figure 11B). Notably, the anodic oxidation was carried out at a late stage in each case, underscoring the functional group compatibility of the electrochemical reaction.
Anodic oxidation of stabilized amines has also found applications. Hurvois and co-workers prepared a series of tetrahydroisoquinoline alkaloids through the electrochemical cyanation of tetrahydroisoquinoline derivatives (e.g., crispine A, Figure 11C).172,173 The reaction was shown to proceed with good stereoselectivity in the presence of a chiral auxiliary. The same group capitalized on similar transformations to synthesize pumiliotoxin C.174 Kam showcased that kopsine, kopsidine, kopsinitrarine, and bisindole derivatives can be obtained through the anodic Shono-type oxidation of aspidofractinine-type alkaloids (Figure 11D).175
The Shono oxidation could also be used in metabolic studies of drug molecules. For example, applications of the Shono oxidation to ifosfamide and cyclophosphamide allowed Royer and co-workers to obtain the methoxylated analogs of these oxazaphosphorinane anticancer drugs in high yields (Figure 11E).176 Under galvanostatic conditions, the oxidation took place in a chemoselective fashion at the α-position of the tertiary nitrogen. The Shono oxidation has also been applied to synthesize 3-oxadiazolyl/triazolyl morpholines which are novel scaffolds for drug discovery.177
Owing to the economic viability of the electrochemical process, the N,O-acetal obtained from the Shono methoxylation of piperidine and pyrrolidine derivatives are versatile building blocks in synthesis. These intermediates not only allow the introduction of α-substitutions; conversion to the corresponding enamine facilitates functionalizations of the β- and γ-carbons. Shono oxidation of piperidine derivatives enabled the preparations of aza-sugar derivatives,178 bicyclic structures,179 and γ-amino acids180 (Figure 11F). A poison frog alkaloid, 195C, was synthesized in a similar fashion through the Shono oxidation of a chiral piperidine derivative.181 Additionally, the Shono oxidation of a proline derivative was utilized to synthesize a Pro-Pro dipeptide mimetic on gram scales (Figure 11G).182 Combinations of Shono allylation and ring-closing metathesis allowed Moeller183 and Steckhan184 to synthesize a series of bicyclic lactams.
Recent efforts by Moeller, Aube and co-workers demonstrated that bicyclic lactams derived from sequential Diels–Alder and Schmidt reactions are also viable substrates for Shono oxidation (Figure 11H). By combining a practical oxidation protocol with various downstream functionalization reactions, they were able to access various complex scaffolds.185 The Shono oxidation of azetidine derivatives was exploited by Wanner and co-workers to synthesize a series of GABA-uptake inhibitors.186
Buriez and co-workers explored the electrochemical oxidation of aminocyclopropanes under aerobic conditions.187 The radical cation intermediate was found to undergo fragmentation readily, whereupon reaction of the resulting radical species with oxygen gave rise to endoperoxides with antimalarial properties (Figure 11I). The potentiostatic electrolysis was conducted in a divided cell; the cathodic reduction of the labile endoperoxide motif was thus averted.
2.5. Oxidation of alcohols
Semmelhack’s seminal report in 1983 spawned continual interest in the indirect anodic oxidation of alcohols employing TEMPO or other nitroxyl radicals as mediators.19 To date, various other nitroxyl mediators have been developed (Figure 12A) for electrochemical alcohol oxidations, offering complementarity to existing chemical methods. Under electrochemical conditions, nitroxyl radicals can be oxidized into the corresponding oxoammonium species which are the reactive oxidants. Nitroxyl mediators bearing ionic tags have been synthesized (e.g., C, Figure 12A), substantially increasing their solubility in aqueous electrolyte solutions.188 Bicyclic mediators such as D were found to be superior in the anodic oxidation of hindered secondary alcohols compared to TEMPO (Figure 12B)—their enhanced reactivity was ascribed to their smaller sizes.189
Figure 12.
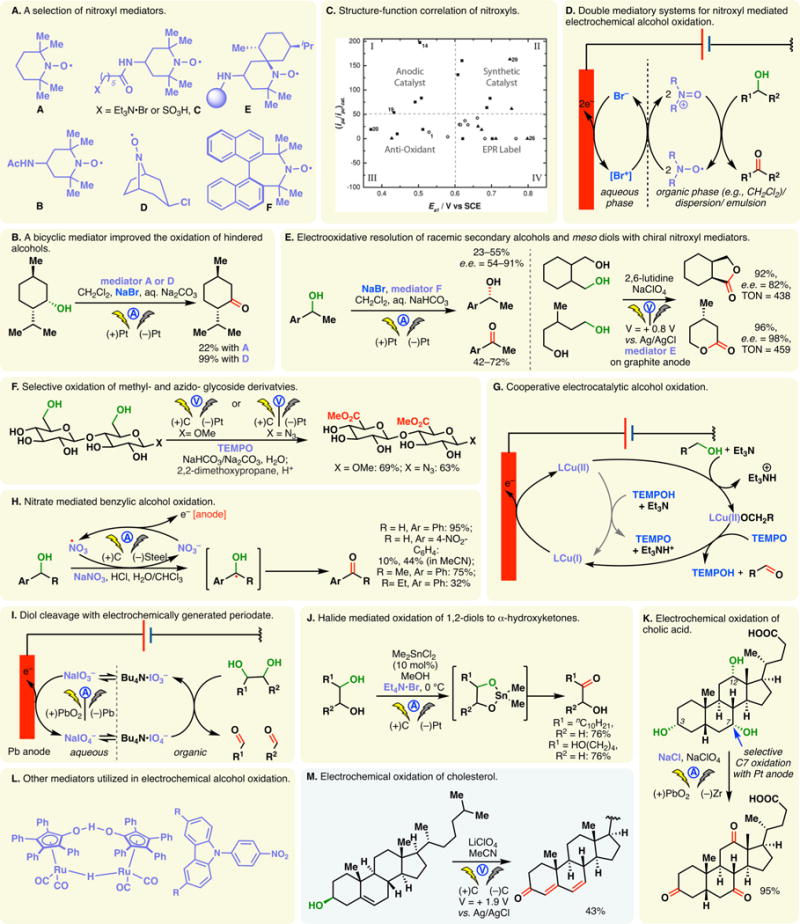
Electrochemical oxidation of alcohols. Reprinted from J. Am. Chem. Soc. 2015, 137, 16179–16186. Copyright (2015) American Chemical Society.
Stahl and co-workers later reported that the catalytic activity of nitroxyl mediators is more strongly influenced by the nitroxyl/oxoammonium redox potential than steric effects.190 Their study led to the identification of B as an effective and inexpensive mediator (Figure 12A); the high reactivity of B is even more pronounced at high pHs. Minteer, Sigman, and co-workers have conducted extensive studies on the structure–functional relationships of nitroxyl radicals, culminating in a computational model to accurately predict their electrochemical potentials and catalytic activities (Figure 12C).191
TEMPO and many other nitroxyl mediators are sparingly soluble in polar electrolytic media, limiting their synthetic utilities. This problem can be circumvented through a double mediatory system (Figure 12D), involving halides and nitroxyls.192 A biphasic solvent mixture is usually employed for such a system wherein the halide undergoes oxidation in the aqueous phase. The ensuing dihalogen or hypohalite can then effect the oxidation of nitroxyl at the aqueous/organic interface. For instance, the reaction showcased in Figure 12B (vide supra) utilized such a biphasic double mediator system. A method to resolve secondary benzyl alcohols was reported using a double mediatory system with NaBr and the axially chiral mediator, F (Figure 12E, left).193 While nitroxyl mediators are customarily dissolved in an organic solvent such as dichloromethane, they could be dispersed onto a polymer support or an emulsion.194–199 TEMPO mediated electro-oxidation of primary and secondary alcohols can also be achieved in a microfluidic electrolytic cell.197 Another means to address this solubility issue entails anchoring nitroxyl mediators onto the surface of an electrode.200–202 For example, by attaching a chiral spirocyclic N-oxyl mediator (E, Figure 12A) onto a graphite anode, Kashiwagi and co-workers developed an electrocatalytic method to desymmetrize meso-diols (Figure 12E, right).203 High enantioselectivity and catalyst turnover were achieved concurrently under constant potential conditions. In another notable example, Stahl and co-workers developed a pyrene-tethered TEMPO derivative which undergoes in situ noncovalent immobilization onto a carbon anode.204 This electrode system was found to demonstrate high catalytic activities in the oxidation of alcohols, exhibiting turnover numbers and frequencies of close to 2000 and 4000 h–1. Sigman, Minteer, and co-workers reported a method to covalently immobilize TEMPO onto linear poly-(ethylenimine) which is then cross-linked onto the surface of a glassy carbon electrode.205 This modified electrode exhibited substantially enhanced catalytic current density compared to the analogous homogeneous systems employing TEMPO as the redox catalyst. Brown and co-workers have developed an efficient protocol for TEMPO-mediated electrochemical oxidation of primary and secondary alcohols in a microfluidic electrolyte cell.
The TEMPO mediated electro-oxidation has been applied by Schäfer and co-workers to convert methyl-glycosides and disaccharides into the corresponding uronic acid derivatives in a chemoselective fashion—the primary alcohols were oxidized selectively under potentiostatic conditions, leaving secondary alcohols and other functional groups intact (Figure 12F).206 Uronic acid esters of azido-disaccharides can be obtained in an analogous fashion; however, the use of a divided cell is necessary to prevent the cathodic reduction of the azide motif.
The alcohol oxidation methods described above invoke the interconversion between nitroxyl and oxoammonium which occurs at a relatively high potential compared to that between the hydroxylamine and nitroxyl. Stahl and co-workers reported a cooperative electrocatalytic system using Cu(II) and TEMPO which exploit the low-potential conversion between TEMPO and its hydroxylamine congener (TEMPOH) (Figure 12G).207 This system allows alcohol oxidations to occur at higher rates and at a significantly lower potential.
Nitrate salts have been employed as electrochemical mediators to oxidize secondary benzyl alcohols wherein the anodically generated nitrate radical serves as the reactive oxidant (Figure 12H).208,209 Halides and hypohalite anions could also serve as mediators in the anodic oxidation of alcohols (Figure 12I–K). For example, electrochemical methods for the oxidative cleavage of diols have been reported wherein sodium periodate is anodically generated in a biphasic system (Figure 12I);210 unmediated diol cleavage has also been described.211 Onomura and co-workers demonstrated that 1,2-diols can be efficiently oxidized into α-hydroxyketones under electrochemical conditions using Et4N·Br as the mediator (Figure 12J).212 Me2SnCl2 is the essential additive for the reaction as it converts the vicinal diol to the corresponding stannylene acetal whereupon the reversible cleavage of a Sn–O bond allows halide mediated alcohol oxidation. Only a catalytic amount of the organotin reagent is required under the galvanostatic protocol, whereas the analogous chemical protocol requires the preformation of the stannylene acetal in a separate step. For 1,2-diols containing a primary and a secondary alcohol groups, anodic oxidation of the secondary alcohol was found to occur preferentially. The reaction system also discriminates 1,2-diols from 1,3-diols or isolated hydroxyl groups effectively. The organotin reagent used in this reaction may be replaced by a copper salt wherein the addition of a chiral ligand allowed an asymmetric electrochemical oxidation of 1,2-diols, aminoalcohols, and aminoaldehydes into α-hydroxyketone or α-aminoesters in moderate enantioselectivity; racemic substrates may be resolved in this fashion.213
A NaCl mediated oxidation of cholic acid into dehydrocholic acid (Figure 12K) was reported.214 The nature of the anode was found to play an important role—with a PbO2 anode,215 anodic oxidations occurred sequentially at the C7, C12, and C3 alcohols; the use of Pt electrode led to the selective oxidation of C7, without reactions at C12 or C3.
Other mediators for alcohol oxidation include the Shvo’s catalyst216 and N-aryl carbazole (Figure 12L).217 Oxidation of cholesterol, through direct electrolysis at a carbon anode, led to the formation of cholesta-4,6,-diene-3-one under constant potential conditions in a four-electron, four-proton process (Figure 12M).218 The electrolysis conditions were optimized on a laboratory synthetic scale with a flow cell.219
2.6. Electrochemical formation of oxocarbenium and thionium ions
Anodic oxidation of alkyl ethers can give rise to oxocarbenium ions in an analogous fashion to the Shono oxidation. For example, Markó and co-workers developed a synthesis of simple spiroketals through the electrochemical cyclization of a pendant alcohol onto the α-carbon of a cyclic ether.220 Nevertheless, as ethers generally have high redox potentials, competing oxidation of solvent molecules can take place. The use of non-nucleophilic solvents such as trifluoroethanol is beneficial;221 direct electrolysis of ethers was sparsely used.
Instead, to access oxocarbenium ions through anodic oxidation, the introduction of auxiliary groups such as trialkylsilyl222 and SAr223 groups at the α-carbon is oftentimes necessary. Aided by these electroauxiliaries, Yoshida achieved the electrochemical generation of oxocarbenium ion pools at cryogenic temperatures (Figure 13A, left).222,223 These reactive cations, stable below −50 °C, can then be trapped with various carbon and heteroatom-centered nucleophiles. Oxocarbenium ion pools may also be accessed through the anodic oxidative scission of C–C bonds (Figure 13A, right).224 For example, the electrolysis of 1,2-dimethoxy-1,2-diphenylethane leads to the formation of an alkoxybenzyl cation pool. This approach is amenable to generate dication pools as is depicted in Figure 13A. Alternatively, oxocarbenium cation pools may be obtained from thioacetals using electrogenerated ArS+ cation equivalents, and this will be discussed in section 2.12.3 along with olefin functionalization reactions using these electrochemically generated electrophiles.
Figure 13.
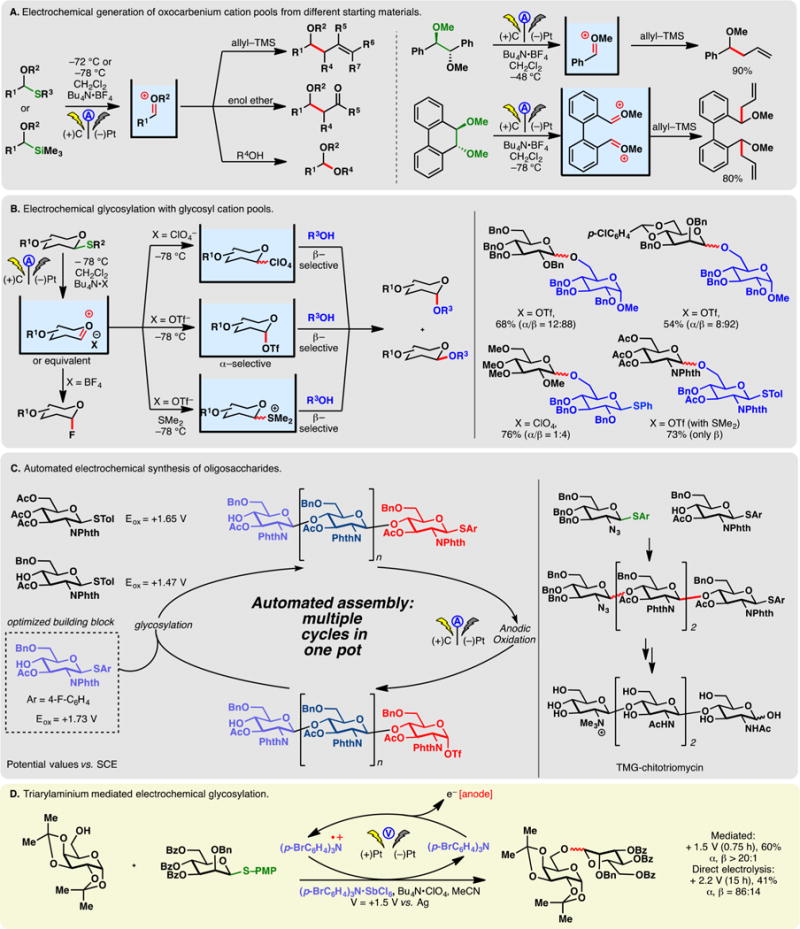
Generation of oxocarbenium cation pool and applications in glycosylation.
An important application of the oxocarbenium ion pool is glycosylation (Figure 13B). Electrochemical glycosylation can be accomplished by judiciously matching the relative potentials of glycosyl donors and acceptors;225–227 alternatively, the cation pool strategy can be used (vide infra). Toward this end, the choice of electrolyte is critical. The use of Bu4N·BF4 led to the glycosyl fluoride.223 Meanwhile, the presence of triflate anions allowed the selective formation of α-glycosyl triflates which could be characterized by low-temperature NMR.228 The OTf group can be readily displaced by an alcohol to forge a glycosyl linkage with β-selectivity. The use of ClO4– counterion was also effective, although the intermediate was less well-defined. The treatment of electrogenerated glycosyl triflates allowed the formation of storable sulfonium cations which was found to be viable intermediates for β-selective glycosylation reactions.229,230 The β-selectivity in electrochemical glycosylation was also noted by Tanaka in their synthesis of 2′,3′-dideoxynucleosides through electro-oxidative glycosylation via the cation pool method.231 As the glycosyl linkage is forged under nonoxidative conditions, readily oxidizable thioglycosides can be used as glycosyl acceptors (Figure 13B, bottom right), thereby allowing iterative glycosylations.
The Yoshida group has streamlined the electrochemical glycosylation into an automated process to synthesize oligosaccharides (Figure 13C).232–234 Finding an optimal monomeric building block is critical—as the oxidation potentials of thioacetals were found to correlate inversely with the efficiency of the glycosylation reaction, the monomer in Figure 13C was deemed ideal due to its high electrochemical potential. Iterative cation pool formation and glycosylation then enabled the synthesis of complex oligosaccharides, including a potential N,N,N-trimethyl-D-glucosaminylchitotriomycin precursor. It is noteworthy that glycosyl linkages between the optimized monomer were all forged with exclusive β-selectivity.
Electrochemical glycosylation can also be carried out in the presence of a triarylaminium radical cation mediator which allows oxidation to occur at a lower potential than the measured values for the thioglycoside. The trapping nucleophile can thus be present during the electrolysis (Figure 13D).235 In the case depicted in Figure 13D, a higher yield and α-selectivity were obtained using the mediated protocol.
Another approach toward iterative electrochemical glycosylation reactions involves the use of different electroauxiliaries.236 In the case depicted in Figure 14A, monosaccharide building blocks bearing SePh and STol auxiliaries were subjected to electrolysis in the same pot.236 At 1.7 V (vs Ag wire), only the selenoacetal was oxidized, leading to the selective formation of a β-glycosyl linkage. A second glycosylation was triggered when a potential of .0 V was applied, through the oxidation of the STol group. In accordance with the relative oxidizability between sulfur- and selenium-based auxiliaries, stannane auxiliaries are more prone to oxidation than silane-based congeners.237 When both TMS and tributylstannane groups were affixed at the same α-ethereal carbon, the latter was selectively cleaved under anodic oxidation (Figure 14B).
Figure 14.
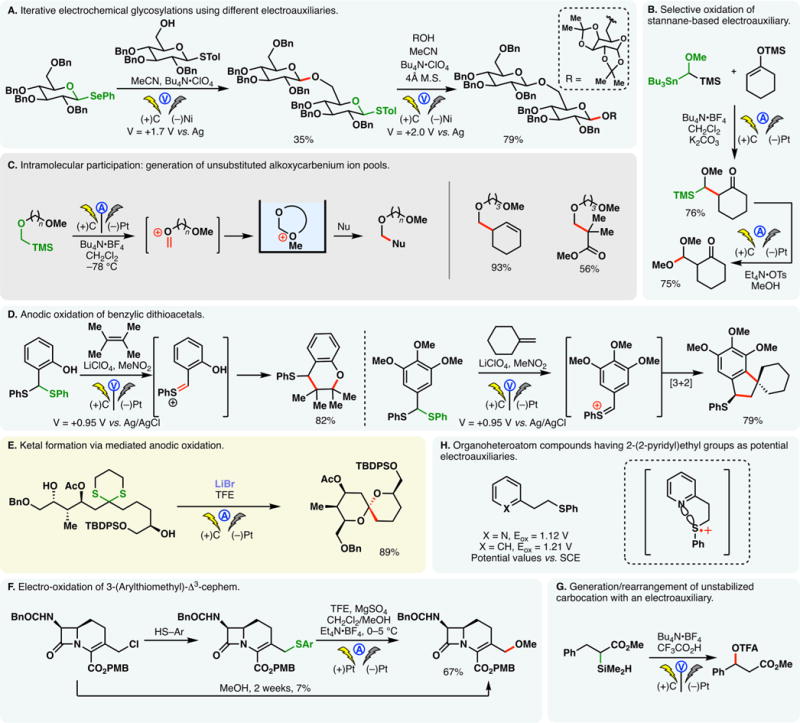
Generation of oxocarbenium, thionium, and other carbocations with electroauxiliaries.
While substituted alkoxycarbenium cations can be accumulated below −50 °C, generation of unsubstituted oxocarbenium ions is more challenging owing to their lower stabilities. Yoshida developed a method to stabilize these fleeting intermediates intramolecularly with an alkoxy group.238 The stabilized cation pool can then be used for nucleophilic functionalizations with allyl silanes, silyl enol ethers, or silyl ketene acetals (Figure 14C).
Anodic oxidation of dithioacetals or ketals offers a convenient means to access reactive thionium cations. Chiba and co-workers demonstrated that the thionium species derived through the anodic oxidation of benzylic dithioacetals could engage olefins in formal [4 + 2] as well as [3 + 2] cycloadditions (Figure 14D).239 The intermediary thionium cations can also be trapped with activated olefins such as allyl silanes. The use of LiClO4/MeNO2 electrolyte system was found to be critical. Nishiyama exploited the bromide mediated anodic oxidation of dithianes to access the spirocyclic ketal motif of antitumor antibiotic ossamycin in a high yield (Figure 14E).240 Electrochemical oxidation of dithiane motifs has also been applied by Kutateladze and co-workers to achieve the deprotection of carboxylic acids from their 2-(hydroxymethyl)-1,3-dithiane (Dim) esters.241
Aside from oxocarbenium and thionium cations, sulfur- or silicon-based electroauxiliaries also offer a convenient means to access other stabilized carbocations such as allylic cations. For example, electro-oxidation of 3-(arylthiomethyl)-Δ3-cephem was utilized to synthesize a 3-methoxy-cephem analog when attempted displacement of the analogous allyl chloride did not yield the product in satisfactory yields (Figure 14F).242 Barba and co-workers reported an efficient method to convert xanthates into thiocarbonates through anodic oxidation.243
Fry has demonstrated that electrochemical oxidation using a silane auxiliary can lead to an unstabilized α-carbonyl cation which readily rearranges to the lower energy benzyl cation (Figure 14G).244 In this example, the phenyl group at the β-position was believed to play an important role—it stabilizes the initial radical cation intermediate, lowing the oxidation potential of the substrate.
Oxidation potentials of electroauxiliaries may be lowered further through the incorporation of heteroaromatic moieties—for example, compounds bearing the 2-(2-pyridyl)-ethyl groups exhibited lower redox potential compared to the phenyl congeners as the pyridine nitrogen provides stabilization to the radical cation intermediate arising from anodic oxidation (Figure 14H).245
2.7. Oxidation of aldehydes
Relatively few examples of anodic aldehyde oxidation have been reported within the period covered by this review. Boydston devised an electrochemical method to convert aldehydes into esters through N-heterocyclic carbene (NHC) catalysis (Figure 15A).246 Under this system, a Breslow intermediate formed between the aldehyde and NHC is oxidized under a low potential (+0.1 V vs Ag/AgNO3) whereupon interception of the oxidized product by an alcohol leads to the ester product. The reaction may be conducted using various types of batteries as the power source. This ester synthesis was adapted by Brown and co-workers in microflow cells under constant current conditions.247 The low potential required for this process was found to be compatible with other nucleophilic species, allowing the groups of Boydston and Brown to develop methods of synthesizing amides and thioesters directly from aldehydes.248,249 The electrochemical conversion of aldehydes into nitriles has also been reported wherein ammonium iodide served as the source of nitrogen as well as the mediator (Figure 15B).250
Figure 15.
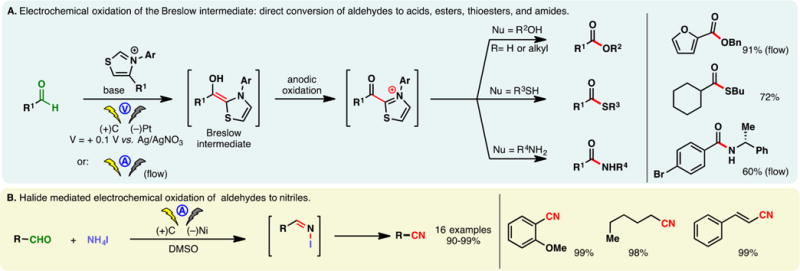
Electrochemical oxidation of aldehydes.
2.8. α-Oxidation of carbonyls
Oxidation at the α-position of carbonyls may be accomplished through the oxidation of the corresponding enol ethers—this will be detailed in section 2.12.1. Alternatively, halide mediated anodic processes could be used to oxidize the more enolizable systems such as aryl-alkyl ketones and malonate derivatives.
Ionic halides such as ammonium iodide can undergo facile anodic oxidation whereupon the incipient molecular iodine can engage enolizable carbonyls, forming α-halo compounds (e.g., an α-iodoketone, Figure 16A, right) possibly through the intermediacy of an α-radical. These intermediates are susceptible to nucleophilic substitution, thereby allowing the introduction of different functionalities. For example, the inclusion of secondary amines in the electrolytic system allowed the direct α-amination (Figure 16A).251 The iodide mediated α-oxidation of aryl ketones has also found applications to cleave the β-O-4 lignin model compounds.252 Aryl ketones could also be elaborated into α-hydroxy ketals using a NaI/NaOH system under electrolysis (Figure 16B).253–255 In a similar vein, Zeng, Little, and co-workers developed an electrosynthesis of 3-amino-2-thiocyanato-α,β-unsaturated carbonyl derivatives through a bromide mediated α-oxidation of 1,3-dicarbonyls where the amino and thiocyanato moieties either originate from a single reagent or a combination of ammonium acetate and potassium isocyanate (Figure 16C).256 Similarly, Yuan and co-workers reported an iodide mediated electrosynthesis of β-keto sulfones.257 In the absence of a strong nucleophile, electrogenerated α-chloro 1,3-dicarbonyls may be isolated as noted by Kakiuchi (Figure 16D).258 Halide mediated anodic α-oxidation has also been combined with Aldol and Michael reactions to furnish spirobenzofuran derivatives (Figure 16E).259
Figure 16.
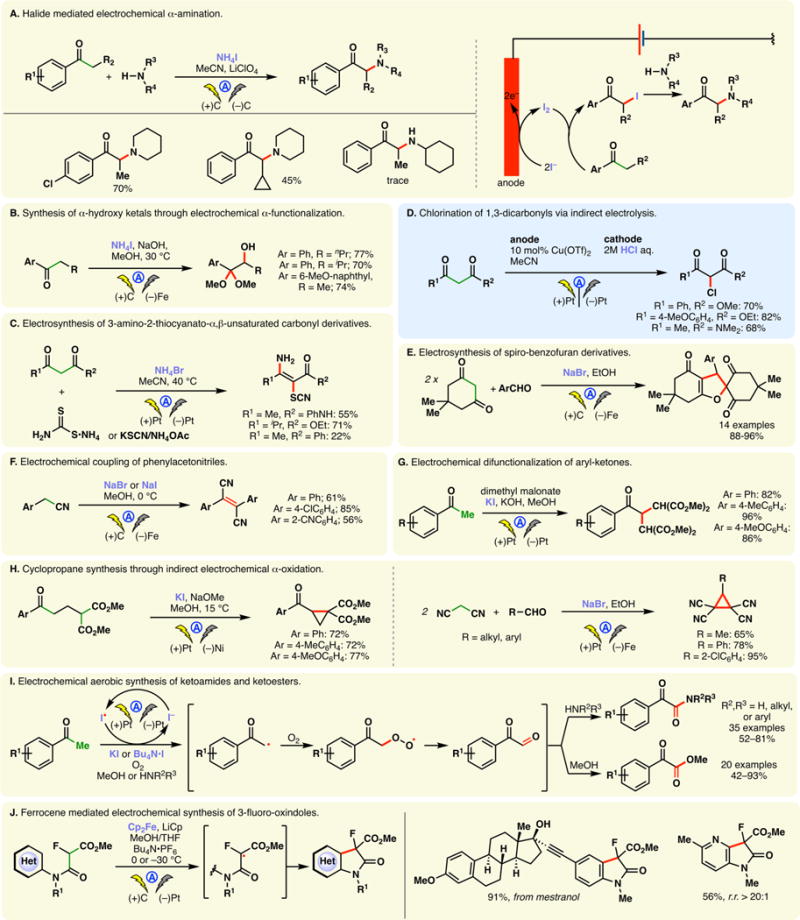
Indirect electrochemical α-functionalization of carbonyls.
Additionally, electrochemically generated α-halo-carbonyl compounds may be intercepted with an enol or equivalents thereof to construct 1,4-dicarbonyls. For example, under bromide or iodide mediated electrolysis, phenylacetonitriles could be effectively homocoupled to afford trans-α,β-dicyanostilbenes (Figure 16F).260 Wang and co-workers also reported that using KI as a mediator, two equivalents of malonates can be appended at the α-position of an aryl alkyl ketone under basic conditions through two consecutive electrochemical α-iodinations (Figure 16G).261 Radical trapping experiment suggested the intermediacy of an iodine radical. Intramolecular variants of such processes can be used to construct rings;262,263 for example, halide mediated α-coupling between malonates and aryl ketones have been utilized to prepare cyclopropane derivatives (Figure 16H).264–272
In the iodide meditated oxidation of methyl aryl ketones, Wang and co-workers noted that the intermediary α-radical may be intercepted with triplet oxygen—the fragmentation of the ensuing peroxo species furnishes a 2-oxo-aryl-acetaldehyde (Figure 16I).273 This intermediate was found to react with alcohol or amine nucleophiles whereupon further electrochemical oxidation led to α-keto esters and amides respectively (Figure 16I).274 A similar aerobic reaction was reported by the same group wherein methyl aryl ketones were transformed into the corresponding vinylogous amides through iodide mediated anodic oxidation.275 An electrochemical method to synthesize isatins has also been developed based on iodide mediated α-oxidation under aerobic conditions.276
The ferrocene-based mediator system, commonly used to generate amidyl radicals, is also amenable to oxidize the α-position of malonates as shown in Figure 16J. The resulting radical cyclizes onto a (hetero)arene to furnish 3-fluorooxindole derivatives.277 Under the electrochemical conditions, the oxidant and base (EGB) are generated in a continuous fashion, allowing the synthesis of base- and heat-sensitive products while showing compatibility with various functional groups; it can be used to modify complex substrates such as mestranol.
2.9. Anodic benzylic functionalization
Electron-rich and electron-neutral arenes exhibit relatively low redox potentials; they can therefore undergo anodic oxidation to form the corresponding radical cations. When an arene is substituted with an alkyl side chain, the aryl radical cation oftentimes undergoes further oxidation to a benzyl cation after losing a proton. This offers a convenient gateway to benzylic functionalization. An application of this electrochemical process is the oxidative cleavage of para-methoxybenzyl (PMB) ethers.278 Brown and co-workers recently detailed an electrochemical protocol for the removal PMB protecting group in flow reactors (Figure 17A). The reaction mass efficiency and atom economy scores of this deprotection process compare favorably with common chemical methods involving strong oxidants such as CAN; other common alcohol protecting groups were tolerated.278 Wang and co-workers demonstrated that the benzyl cation arising from direct anodic oxidations could be intercepted with water whereupon further oxidation of the resulting benzyl alcohol furnishes the ketone (Figure 17B).279 This process is most effective for methylene units substituted with two arenes; lower yields were noted otherwise.
Figure 17.
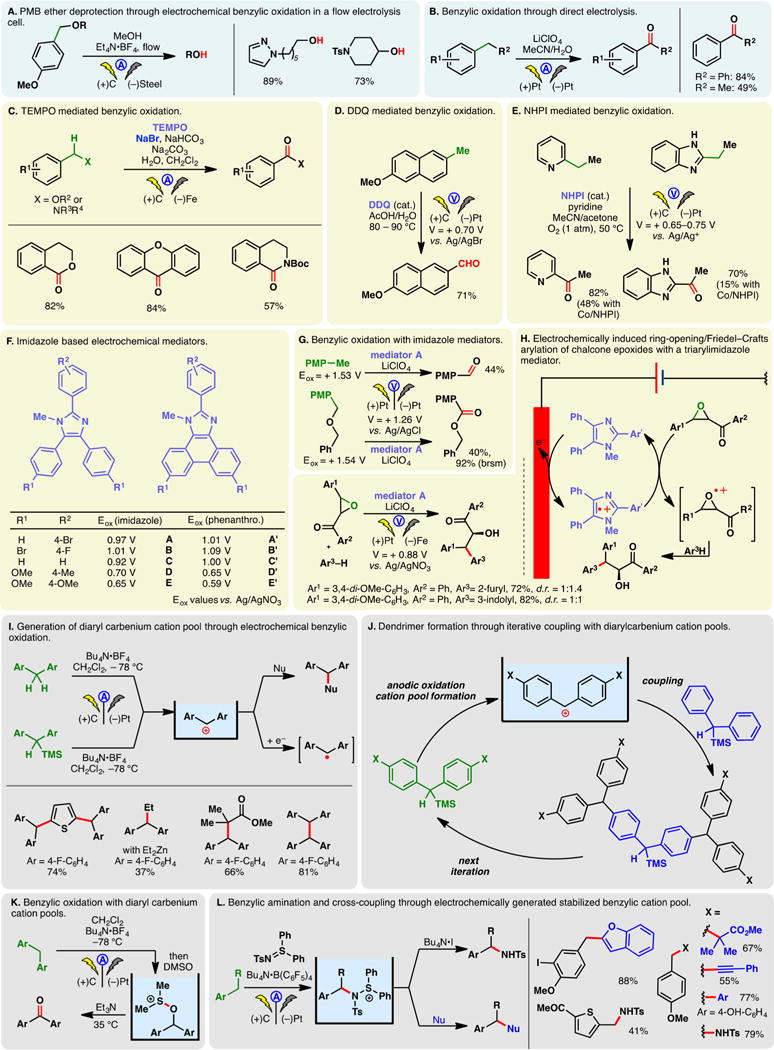
Electrochemical benzylic oxidation.
Benzylic electrochemical oxidation could also be achieved with the aid of different mediators. For example, Zeng, Little, and co-workers developed an electrochemical method to access dihydroisoquinolinones, isochromanones, and xanthenones (Figure 17C), through benzylic C–H oxidations with a dual mediator system comprising TEMPO and sodium bromide.280 While DDQ is a competent oxidant in benzylic oxidation reactions, the use of this reagent in stoichiometric quantities undermines the atom economy while rendering the reaction purification challenging. Through anodic oxidations, catalytic amounts of DDQ can be used to mediate benzylic oxidation as the resulting DDHQ is reoxidized (Figure 17D).281 Recently, Stahl and co-workers reported that N-hydroxy-phthalimide (NHPI) could mediate benzylic oxidation reactions under aerobic and electrochemical conditions (Figure 17E). Although the electrolytic conditions could be substituted by a cobalt catalyst, the electrochemical protocol is advantageous for heterocyclic substrates where the heteroatoms may coordinate to cobalt, triggering catalyst poisoning. Examples of such substrates are depicted in Figure 17E.282
Little and co-workers have developed a series of triarylimidazoles as redox mediators in electrochemical oxidation reactions (Figure 17F). These compounds were found to undergo quasi-reversible anodic oxidation. The presence of three arene rings allows the oxidation potential of the mediators to be modulated through the introduction of electron-donating or electron-withdrawing substituents.283,284 Electrochemical oxidation of these conjugated compounds gives rise to stabilized radical cations which could facilitate the benzylic oxidation reactions under mild conditions (Figure 17G).283 Mediator A was later used to induce the ring-opening and Friedel–Crafts arylation of chalcone epoxides wherein the anodically generated imidazole radical cation undergoes electron transfer with the epoxide to accelerate the arylation (Figure 17H).285 The use of a mediator circumvented the overoxidation of product even though it has a lower oxidation potential than the starting material; formation of the mediator radical cation likely triggered a chain mechanism as only catalytic amount of electricity is necessary. This reaction can also be conducted using a polymeric ionic liquid and carbon black composite as the supporting electrolyte.286 The use of this electrolyte allows the in situ modification of electrode surface, improving the reaction kinetics. Further optimization of the arylimidazole mediators led to the identification of 2-aryl-1-methylphenanthro[9,10-d]imidazoles which demonstrated higher radical cation stability and a broader range of accessible electrochemical potentials (Figure 17F).287
Although their stabilities strongly depend on the substituents on the arene rings, diaryl carbenium cations obtained through benzylic electrochemical oxidation may, in some cases, be accumulated under cryogenic temperatures in a cation pool (Figure 17I). These cations may be trapped with carbon-centered nucleophiles such as heteroarenes, organozinc reagents, or silyl ketene acetals (Figure 17I).288,289 Alternatively, they may be subjected to cathodic reduction where the ensuing radical was found to undergo homodimerization. Generation of the diarylcarbenium cation pool could be facilitated with silane electroaxuiliaries—this strategy also allowed the synthesis of dendritic molecules through iterative cation pool formation and nucleophile trapping (Figure 17J).290–293 Yoshida reported that diaryl carbenium cations may be trapped with DMSO; the resulting sulfonium can be treated with a base to furnish the ketone, much akin to a Swern oxidation (Figure 17K).294 An in situ protocol was also reported wherein the oxidation of an arene is carried out in the presence of DMSO. Anodic oxidations of toluene derivatives and aryl-substituted olefins into benzaldehydes and 1,2-diketones could be accomplished in this manner. Moreover, the oxidation of unsaturated compounds bearing a nucleophilic substituent gives rise to cyclized carbonyl compounds (vide infra, olefin oxidation).
Unlike diaryl carbeniums, other benzyl cations are generally unstable even at −78 °C—direct generation of cation pools are therefore not feasible. Instead, Yoshida showed that these fleeting cations may be generated in the presence of a sulfilimine, leading to the formation of a stabilized benzylaminosulfonium cation pool (Figure 17L).295 The aminosulfonium group can be readily displaced by a carbon-based nucleophile to effect net benzylic substitution. The reported scope of nucleophiles includes electron-rich aromatics, allyl silanes, silyl ketene acetals, etc. Alternatively, treatment of the stabilized benzyl cation pool with iodide was found to cleave the N–S bond, resulting in the formation of benzylic amination products.296 Due to the high oxidation potential of tosyl sulfilimine (decomposition potential: 2.01 V vs SCE), a wide variety of arenes can be oxidized in its presence. This two-step benzylic amination method is advantageous in that the presence of the stabilized aminosulfonium cation circumvents overoxidation of the amination product.
2.10. Oxidation of the arene nucleus
2.10.1. Oxidation of electron-rich and -neutral arenes
As alluded to in the preceding section, electron-rich arenes have relatively low oxidation potentials and may be readily oxidized into the corresponding radical cations under electrochemical conditions—the reaction of these species with nucleophiles can enable the direct functionalization of arene C–H bonds. However, a caveat exists for this approach—nucleophiles may be susceptible toward competing oxidations; this problem is especially pronounced when oxidations of less electron-rich arenes are sought. For example, Tajima and co-workers reported a simple electrochemical protocol for arene acetoxylation using a solid-supported base in AcOH; the electrolyte is generated in situ through an acid–base reaction, and it can be removed through filtration (Figure 18A).297 Under galvanostatic conditions, 1,4-dimethoxybenzene can be satisfactorily acetoxylated. Nevertheless, attempts to functionalize benzene were unsuccessful as the high requisite potential triggered acetate oxidation. Instead, switching to trifluoroacetic acid as the solvent broadened the permissible potential window—benzene and even unactivated C–H bonds in norbornane could be functionalized. Alternatively, selective arene oxidation can be achieved under constant potential electrolysis, through the judicious control of oxidation potential. Royer and co-workers demonstrated that PMP-anilines can be selectively deprotected in the presence of other oxidizable functionalities such as dithiane and phenols through potentiostatic electrolysis (Figure 18B).298 An analogous electrochemical protocol to cleave PMP-ethers has also been reported by the same group.299
Figure 18.
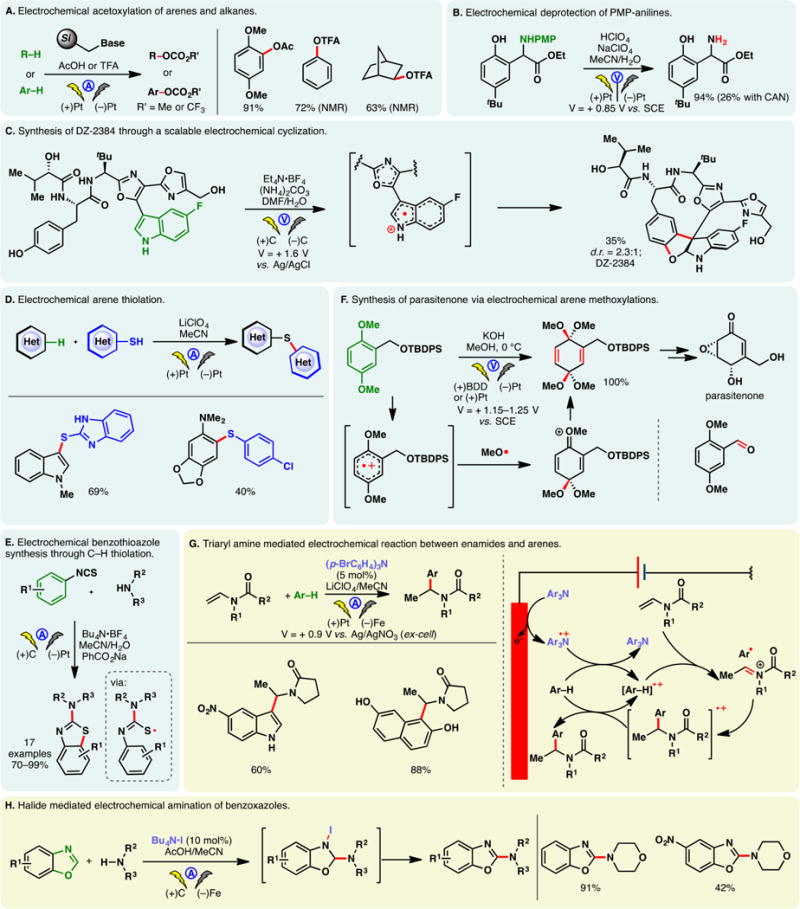
Arene functionalization through electrochemical oxidation.
As shown in Figure 18C, constant potential electrolysis also allowed Harran and co-workers to selectively oxidize an indole motif in a complex synthetic intermediate, in the presence of an electron-rich phenol and free alcohols.300 Nucleophilic attack of the phenol onto the indolyl radical cation elicited a macrocyclization, furnishing DZ-2384, a diazonamide analog of therapeutic potential against various types of cancers. Notably, the reaction has been reliably performed above 60 g scale on a similar substrate, attesting the utility of anodic arene oxidation.
Lei and co-workers recently described an electrochemical method for arene arylthiolation wherein an electron-rich arene such as indole is electrolyzed alongside a thiophenol derivative in an undivided cell (Figure 18D).301 Although the thiophenol can also be oxidized to the corresponding disulfide under the reaction conditions, it was believed that the aryl radical cation could engage the disulfide or the intermediary sulfur-centered radical to afford the thiolation product. An intramolecular thiolation process has been devised by Lei to synthesize benzothiazoles from aryl-isothiocyanate electrochemically (Figure 18E)—this reaction presumably occurs through the intermediacy of a sulfur-centered radical;302,303
Sometimes, concomitant oxidation of solvent and substrates is strategic as an electrogenerated aryl radical cation may be intercepted by a radical species arising from solvent oxidation. For example, Nishiyama and co-workers reported that the radical cation arising from the electrochemical oxidation of a 1,4-dimethoxybenzene derivative could react with anodically generated methoxy radicals to afford a bis-ketalization product (Figure 18F).304 The formation of methoxy radical was confirmed through trapping experiments and ESR while the bis-ketal product could be expediently elaborated to furnish parasitenone. The choice of anode is important for what has been termed an “EECrCp” reaction:305 the broad potential window of boron-doped diamond (BDD) or platinum electrode allowed the generation of methoxy radicals, leading to the formation of bis-ketal. Conversely, the use of graphite electrode led to exclusively benzylic oxidation as is the case with chemical oxidants. It was reasoned that with graphite anode, the concentration of methoxy radicals was lower; consequently, the aryl radical cation could undergo elimination to afford the corresponding aldehyde. A similar electrochemical arene methoxylation using the BDD anode was utilized by the same group to synthesize cyclohexadienones carrying an α-D-glucopyranosyl moiety.306 An analogous electrochemical methoxylation of N-protected-4-methoxy anilines has also been reported wherein quinone imine acetals are formed as products.307
Electro-oxidation of electron-rich arenes could also be accomplished with redox mediators when arenes donate an electron to the oxidized form of the mediator. For example, Zeng, Little, and co-workers disclosed a triarylamine mediated method for enamide α-arylation where the anodically generated aminium radical cation triggered the oxidation of electron-rich arenes (Figure 18G) in an ex-cell protocol.308 The resulting aryl radical cation could undergo electron transfer with the enamine, generating a highly electrophilic acyliminium ion and enabling a Friedel–Crafts reaction. This cationic chain mechanism, through the single-electron oxidation of aromatics, is supported by CV analysis and control experiments.
Zeng and Little have also described an electrochemical process for the 2-amination of benzoxazole using tetraalkylammonium iodides or bromides as redox mediators (Figure 18H). The proposed mechanism involved a benzoxazoline intermediate where electrogenerated X+ species facilitates rearomatization.309 The reaction is carried out under galvanostatic conditions in a simple undivided cell, allowing simple product isolation.
Electrochemical oxidation of furans has been widely utilized in organic synthesis. Such a process was employed by Frontana-Uribe and co-workers in the electro-oxidation of hispanolone.310 Anodic oxidation of furan has also found applications in the synthesis of dideoxynucleoside analogs311 and furosemide.312 Additionally, Breinbauer and co-workers demonstrated the electro-oxidation of furans on solid support.313,314 Anodic oxidation of 5-substituted uracils in aqueous solution was found to yield N(1)–C(5′)-linked dimers.315
2.10.2. Arene functionalization through the cation pool strategy
Although aryl radical cations are commonly perceived as unstable species, Yoshida and co-workers demonstrated that radical cation pools of fused aromatic systems (e.g., naphthalene or anthracene) can be generated through electrolysis at −78 °C whereupon addition of an electron-rich (hetero)arene nucleophile enabled the formation of biaryl systems at −90 °C in the absence of metal catalysts (Figure 19A).316 As the C–C bond formation takes place under nonoxidative conditions, this process circumvents nonselective oxidation of starting materials and overoxidation of coupling products. Atobe and co-workers reported a similar aryl–aryl coupling reaction using microflow reactor wherein the undesired oxidation of the aromatic nucleophile was effectively prevented with laminar parallel flows.317
Figure 19.
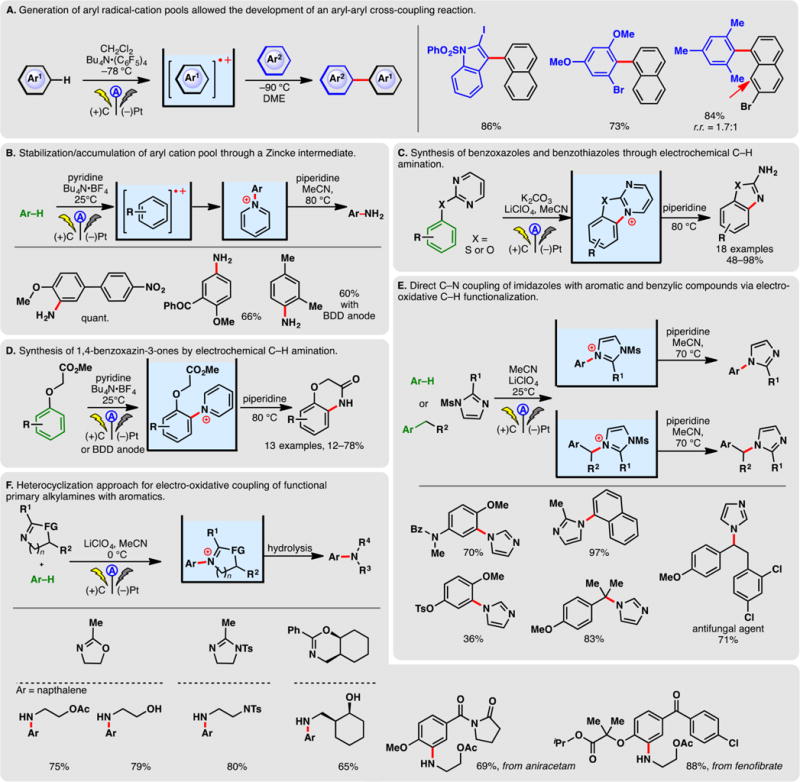
Arene oxidation with the cation pool strategy.
Less stable aryl radical cations may be accumulated in stabilized cation pools through in situ trapping with a judiciously chosen nucleophile. This is illustrated in a series of electrochemical arene amination methods pioneered by Yoshida and co-workers (Figure 19B–F).318 Constant current electrolysis of arenes in the presence of pyridines led to the formation of Zincke-type pyridinium cation which can then be treated with an dialkyl amine (e.g., piperidine) under heat to afford unprotected aniline products through net C–H amination (Figure 19B). The use of pyridine is strategic for several reasons. First, the π-deficient nature of pyridine disfavors its competing oxidation under anodic conditions; second, the strong nucleophilicity of the azine nitrogen allows facile reactions with fleeting aryl radical cations; third, formation of Zincke intermediate precludes overoxidation as the positively charged pyridinium motif withdraws electron density from the arene ring. While Yoshida’s original condition works most effectively for electron-rich arenes such as anisole derivatives, Waldvogel expanded the scope to include electron-neutral substrates with the use of a BDD anode—even diamination has been demonstrated on some substrates.319,320 In Waldvogel’s study, it was also noted that amination of the arene nucleus trumps benzylic functionalization for substrates substituted with alkyl appendages. An intramolecular variant of this method allowed access to 2-aminobenzoxazoles and benzothiazoles (Figure 19C).321 The electrochemical formation of Zincke intermediates also allowed Yoshida, Waldvogel, and co-workers to devise an expeditious syntheses of 1,4-benzoxazin-3-ones (Figure 19D).322
The Yoshida group later reported that methanesulfonyl imidazoles could also trap aryl radical cations under constant current electrolysis—in the absence of alkyl appendages, reactions occur at the arene nucleus; otherwise, the free imidazole nitrogen was found to add onto the benzylic position (Figure 19E).323 Heating the stabilized imidazolium cations in the presence of piperidine can cleave the sulfonyl protecting group, leading to an array of N-aryl imidazole products, including bioactive molecules. For example, an antifungal agent was prepared concisely. As with the Zincke salt, the positively charged imidazolium intermediate forestalls further electrochemical oxidation, allowing products to be obtained in good yields.
The scope of the C–H amination technology was expanded further when Yoshida and co-workers introduced various heterocyclic scaffolds as compatible nucleophiles in arene electrolysis (Figure 19F).324 These nonaromatic heterocycles, derived through the reaction of primary alkyl amines bearing a functional group with nitriles or equivalents thereof, can be readily cleaved upon formation of the stabilized ion pool, giving rise to a range of N-alkyl anilines containing functional group handles (e.g., a hydroxyl group or an amino group) on the alkyl chain for further elaborations. This chemoselective process allowed modification of drug molecules such as aniracetam and fenofibrate.
2.10.3. Oxidation of phenols
Electrochemical oxidation of phenols has been a subject of continual research interest—such processes have found applications in the synthesis of natural products.325,326 Anodic oxidation of phenols can lead to complex and variegated scaffolds which are valuable in the realm of organic synthesis. For example, Waldvogel and co-workers reported that the anodic oxidation of 2,4-dimethylphenol in an undivided cell afforded a dehydro-tetramer product in moderate yields.327–330 This robust process has been scaled beyond 20 g to furnish ample amounts of products which could be diverged in different ways to access various complex constructs (Figure 20).327
Figure 20.
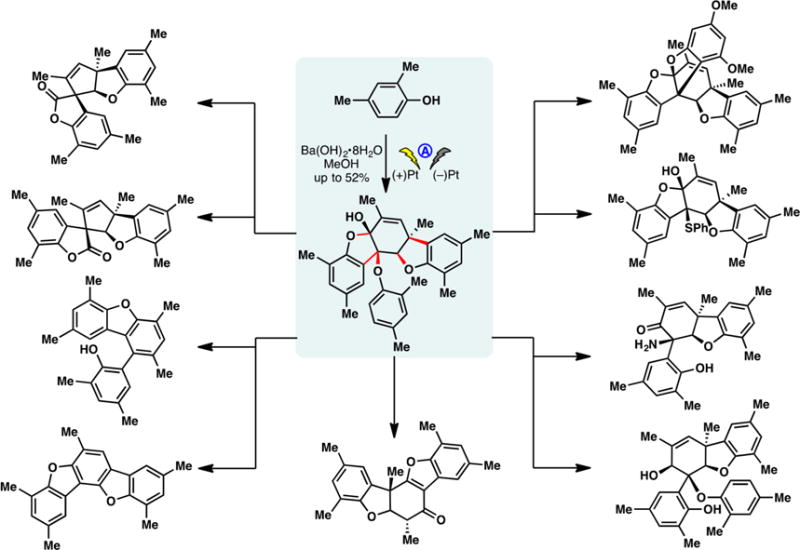
Diversity-oriented synthesis of polycyclic scaffolds through the modification of an anodic product derived from 2,4-dimethylphenol.
2.10.3.1. C–C biaryl coupling
Ortho-ortho biaryl coupling of phenols is an important process in organic synthesis that commonly employs stoichiometric amounts of strong chemical oxidants—an electrochemical method would thus represent an environmentally friendly and atom economical alternative.331 Nonetheless, unprotected phenols have a tendency to undergo dehydro-oligomerization through a series of C,C and C,O bond-forming reactions (vide supra)—the electrolysis of 2,4-dimethylphenol with a Pt anode in MeOH led to minimal formation of the biaryl coupling product (Figure 21A, top of right column). Toward this end, Waldvogel devised a template-directed strategy wherein phenols are converted into the corresponding tetraphenoxyborate complexes.332 Anodic oxidation of these complexes favored ortho-ortho coupling exclusively, even on kilogram scales.
Figure 21.
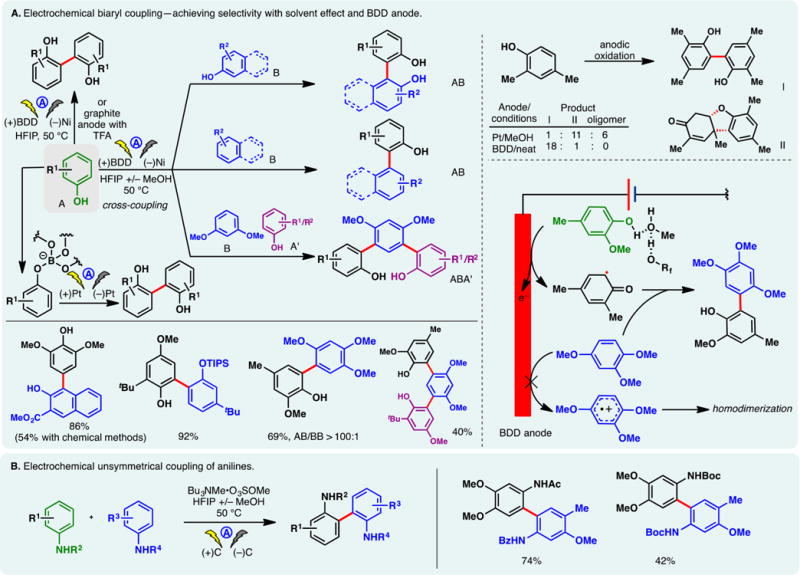
Biaryl synthesis through electrochemical phenol oxidation.
Alternatively, Waldvogel and co-workers discovered that in the anodic oxidation of 2,4-dimethylphenol, the ortho-coupling product could be preferentially afforded with a BDD anode.333,334 Because of its high overpotential for oxygen evolution in protic media, the BDD anode enabled the formation of oxyl radicals which could putatively serve as mediators for the reaction. Fluorinated solvents such as HFIP have beneficial effects as well, presumably because they could stabilize the oxygen spin centers. These findings allowed the development of an ortho-selective phenol homocoupling reaction (Figure 21A).333 Anodic coupling of guaiacol derivatives has also been reported based on these principles.335
This method could be extended to achieve cross-couplings of two phenols or a phenol and a naphthol.331,336 Similarly, solvents with high hydrogen-bonding capacities such as HFIP are used. While cross-coupling products were still afforded using formic acid instead of HFIP, yields of the biaryl products were found to be lower—this reduced efficiency was attributed to the significantly lower anodic stability of formic acid. The phenol with lower oxidation potential (A) undergoes preferential anodic oxidation; homocoupling of the resulting radical species can be suppressed when the coupling partner (B) is employed in higher molar quantities. Yields of electro-oxidative phenol cross-coupling reactions were found to be comparable with chemical methods.337 In addition, a phenol could be effectively coupled with TIPS-protected phenol in this manner.338 The implementation of O-silyl-protected phenols allowed enhanced yields and selectivity as the protecting group sterically suppresses the oxidation of coupling products.
The Waldvogel group also developed methods to achieve the cross-coupling between a phenol (A) and an electron-rich arene (B) (Figure 21A).339,340 Even when B has a lower oxidation potential compared to A, high selectivity of cross-coupling (AB) was observed relative to homodimerization (AA or BB). Addition of water or MeOH to HFIP was critical to this phenomenon.340 It was believed that methanol can act as a base in HFIP to form a hydrogen bonding network with the phenol substrate, lowering the oxidation potential of phenols while disrupting the shell of solvation to facilitate preferential oxidation at the electrode.341 The solvation shell serves to suppress the oxidation of the arene. In the case illustrated in Figure 21A (bottom right), even though trimethoxybenzene has a lower oxidation potential than 2-methoxy-4-methylphenol, this solvent effect allowed the “decoupling” of nucleophilicity and redox potential, thereby favoring cross-coupling. While the formation and influence of these solvates are substrate dependent, they could shift the oxidation potential of individual substrates to create matching pairs for cross-couplings.
Analogous coupling of two protected anilines has also been reported (Figure 21B);342 in a similar vein, a three-component reaction to synthesize meta-terphenyl-2,2″-diols was invented (Figure 21A).343 Practically, all these phenol coupling reactions could be conducted in simple undivided cells under galvanostatic conditions. The fluorinated solvents may be recycled at the end of the reaction.
Waldvogel and co-workers have also demonstrated that the homocoupling of phenols could be rendered more economical using a graphite anode and TFA as the essential additive.344
2.10.3.2. Aryl ether formation
Nishiyama and co-workers have investigated the anodic oxidation of ortho-halo-phenols extensively.325 Although the oxidation of monohalogenated phenols led exclusively to C–C coupling,345 anodic oxidation of 2,5-dibromo- or 2,5-dichloro-phenol derivatives followed by cathodic cleavage of a C–X bond led to the formation of diaryl ethers as shown in Figure 22A.346 This reaction proceeds through the C–O dimerization of a phenoxy radical where one of the halogen atoms serves as an auxiliary to guide the regiochemical course. This method was the key step in Nishiyama’s synthesis of O-methylthalibrine.346,347 It is note-worthy that additional electrochemical transformations are enlisted in the overall sequence, including an electrochemical halogenation. The same group accomplished the syntheses of verbenachalcone348 and isodityrosine349 using this strategy (Figure 22A, right column).
Figure 22.
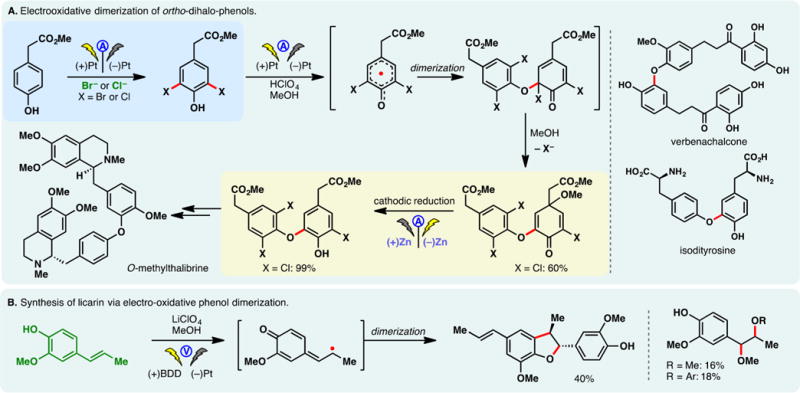
Aryl ether formation through anodic phenol dimerization.
Nishiyama also studied the anodic oxidation of isoeugenol where the intermediary phenoxy radical species was found to dimerize to deliver licarin in a formal [3 + 2] cycloaddition (Figure 22B).304 The use of BDD anode provided the product in the highest yield, presumably as it enabled the generation of methoxy radical as evidenced in the formation of a vicinal dimethoxylation byproduct.
2.10.3.3. Formation of phenonium cations
Phenols could also undergo two-electron anodic oxidation to yield the corresponding phenonium cations. This highly electrophilic species could engage nucleophiles intramolecularly, forming spirodienone products.350,351 Cyclization of the oxime oxygen onto an electrogenerated phenonium species constitutes the key step in Nishiyama’s synthesis of aeroplysinin (Figure 23A).352,353 The same group also used a similar strategy in their synthesis of heliannuol E where cyclization of an alcohol onto a phenonium intermediate furnished a spirodienone which was poised to undergo ring expansion upon treatment with a Lewis acid (Figure 23B).354,355
Figure 23.
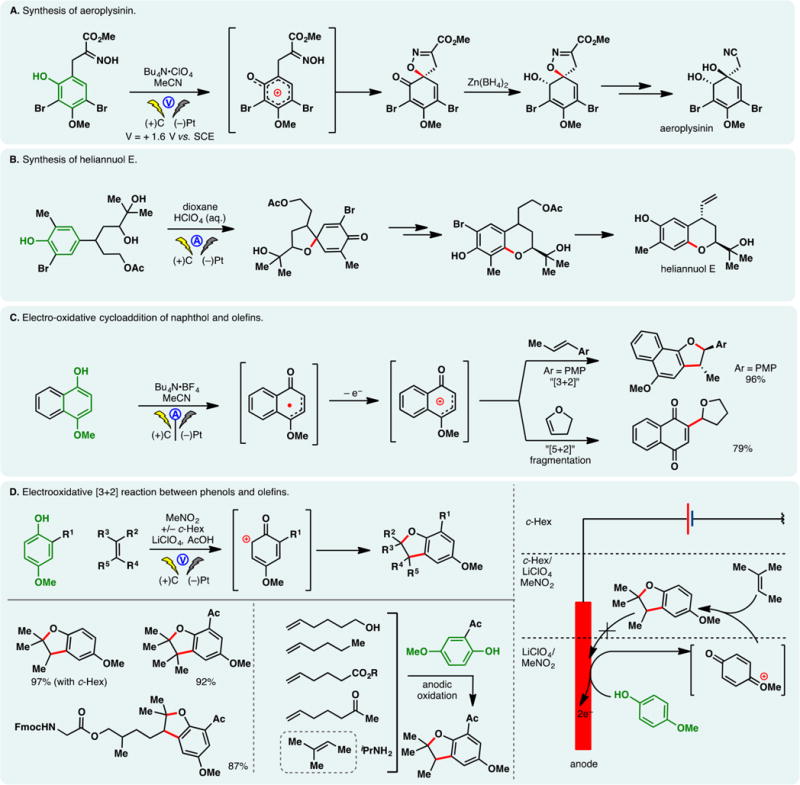
Electrochemical generation of phenonium cations.
Alternatively, phenonium cations can readily engage electron-rich olefins in various modalities of cycloadditions. For example, Nishiyama and co-workers showed that the naphthol cation could undergo [3 + 2] as well as [5 + 2] cycloadditions in this manner (Figure 23C).356
Chiba and co-workers developed an electrochemical [3 + 2] reaction between phenols and olefins through the intermediacy of a phenonium ion (Figure 23D).357,358 This reaction can be conducted in a temperature controlled multiphase solvent system wherein the phenol resides in the polar MeNO2 layer while the cycloadduct congregates in a less polar thermomorphic middle layer comprising MeNO2 and cyclohexane. Formation of this layer enhanced interactions between the phenonium intermediate and the nonpolar olefin while impeding overoxidation of the cycloadduct. This reaction exhibited selectivity for electron-rich tri- and tetra- substituted olefins over monosubstituted ones in competition experiments; primary amines were also tolerated.
The anodic oxidation of catechols readily gives rise to 1,2-benzoquinones. As catechol derivatives typically possess low oxidation potentials, various nucleophilic species can be included in the electrolytic setup without getting oxidized, allowing their interactions with the intermediary 1,2-benzoquinones in situ. Some examples of such transformations are summarized in Figure 24A. For example, reactions with thiols359–363 or sulfinates364 deliver sulfides or sulfones, respectively. Electrochemical reactions of catechol with 2-thiouracil365 and aminopyrimidine366 derivatives afford tricyclic heteroaromatic scaffolds after two iterations of sequential electrochemical oxidation and nucleophilic trapping processes (denoted an ECEC sequence where “E” stands for an electron transfer while “C” implies an ensuing chemical transformation). 3-aryl-oxindole can be obtained by electrolyzing catechol derivatives in the presence of an oxindole;367 anodic oxidation of catechol in the presence of benzylamine allows synthesis of 2-aryl-benzoxazoles.368 Electro-oxidation of a mixture of catechols and N,O-ketene acetals led to the formation of indoles;369,370 nevertheless, replacing the N,O-ketene acetal with ketene aminals371,372 or vinylogous amides373 led to a different outcome (Figure 24A, bottom right). Additionally, electrolysis of catechol derivatives in the presence of 1,3-dicarbonyl compounds was shown to afford benzofuran derivatives as products.374,375
Figure 24.
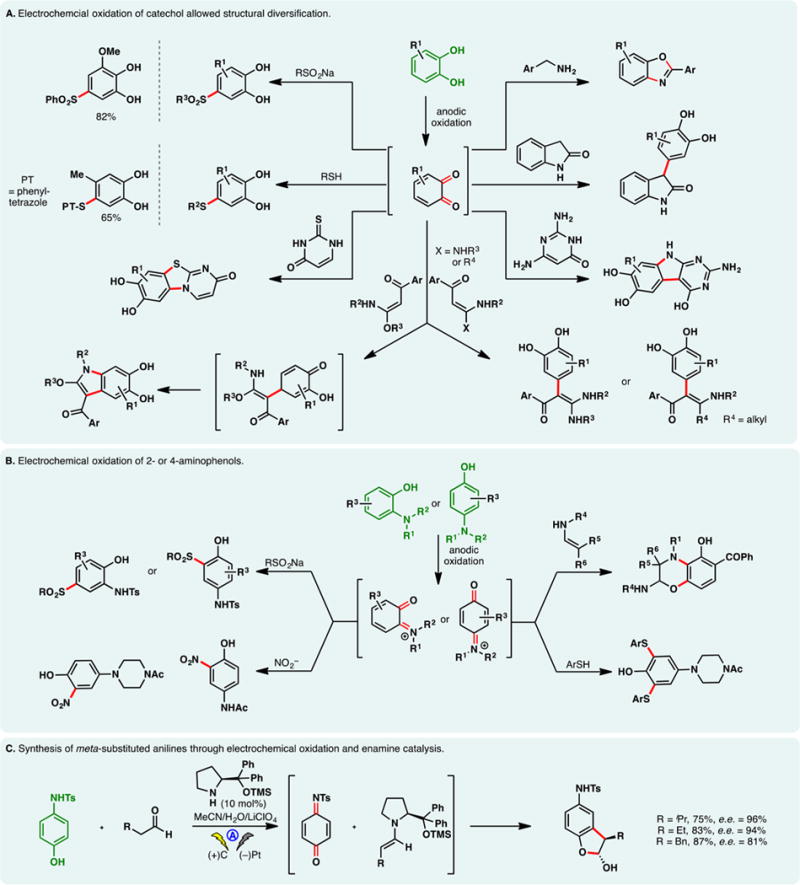
Electrochemical oxidation of catechol and aminophenols.
Similarly, electrophilic quinone imine species can be generated through the anodic oxidation of 2- or 4-aminophenol derivatives at relatively low potentials. Various transformations of these intermediates with nucleophiles such as sulfinates have been detailed (Figure 24B, top left).376 For example, Nematollahi and co-workers reported that electrochemical oxidation of N-(2-hydroxyphenyl)acetamide in the presence of a nitrite anion led to the formation of ortho-nitration products (Figure 24B, bottom left);377 the same group also achieved a di-thiolation of 4-aminophenol derivatives (Figure 24B, bottom right);378 Electrogenerated quinone-imines may also be captured by an electrogenerated imine in a Diels–Alder reaction as shown by Blattes et. al (vide infra).379 Likewise, 1,4-benzodioxin derivatives can be prepared from pyrogallols in such type of electrochemically induced Diels–Alder reaction.380
Jørgensen and co-workers have combined the anodic oxidation of 4-aminophenol derivatives with chiral enamine catalysis, allowing the preparation of meta-substituted anilines through formal α-arylation of aldehydes (Figure 24C). Excellent enantioselectivities were obtained for this process.381 Electro-oxidation of 2- or 4-aminoaniline produces quinone di-imines which were found to exhibit similar reactivities as Michael acceptors, compared to 1,2-benzoquinones or its monoimino congener.382–389
2.10.4. Oxidation of Meisenheimer complexes
Direct anodic oxidation of electron-deficient arenes is challenging due to their high oxidation potentials. However, since these substrates (e.g., nitroarenes) are prone to form σ-complexes with strong nucleophiles, electrochemical oxidation of these Meisenheimer intermediates can induce rearomatization, thereby enabling the indirect anodic functionalization of electron-poor arenes (Figure 25A).390–392 Two SNAr reaction modes are possible here—first, oxidation of a σH–Meisenheimer complex could trigger the elimination of a proton (denoted by Gallardo et al. as an SNH (or NASH) process as the nucleophile formally replaces a hydrogen);392 alternatively, when the arene is substituted with a leaving group (e.g., a halide), nucleophilic attack may occur ipso to this group, leading to the formation of a σX–complex where extrusion of X gives rise to the substitution product (denoted by Gallardo et al. as an SNX (or NASX) reaction where the nucleophile formally displaces a leaving group). It is noteworthy that for nitroarenes with two or more nitro groups, the nitro could serve as the leaving group in SNX processes. While SNH reactions proceed through a two-electron anodic oxidation step, mechanistic studies reveal that a one-electron oxidation is operative in the SNX reaction wherein homolysis of the C–X bond occurs to eliminate radical species. This differs from classical SNAr reactions. From a practical point of view, these electrochemical SNAr reactions are usually carried out under potentiostatic conditions, based on first oxidation peak of in situ generated Meisenheimer complexes.
Figure 25.
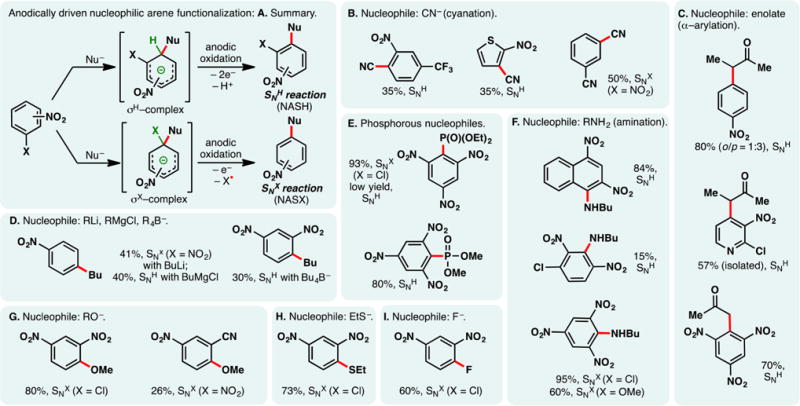
Electrochemically promoted nucleophilic aromatic substitution (GC/NMR yields listed in this figure).
Gallardo showed that cyanide (Figure 25B),393,394 alkyl metal reagents (Figure 25D) (e.g., alkyl lithium or Grignard),395,396 organophosphorous nucleophiles (Figure 25E),397 and amines (Figure 25F)398,394 could serve as nucleophiles in both SNX and SNH processes, while enolates399 and tetraalkylborate salts396 have been used as nucleophiles in SNH reactions (Figure 25C). Conversely, σH-complexes between nitroarenes and alkoxides,394 thiolates,394 or fluoride394 tend to undergo one-electron electrochemical oxidation, extruding the heteroatom nucleophile to regenerate the parent arene. These nucleophiles can instead engage in the so-called SNX reactions with nitroarenes containing a second leaving group (Figure 25G–I).394 The reactions may be conducted in ionic liquids.400 A recent report indicated that acridine derivatives can also participate in these transformations.401
2.10.5. SEAr reactions with electrogenerated electrophiles
In a different approach to electrochemical arene functionalizations, a halide or pseudohalide is oxidized anodically in the presence of an arene.402 The resulting electrophilic (pseudo)halogen cations or di(pseudo)halogen can engage the arene in SEAr reactions (Figure 26A). As most (pseudo)halides have relatively low oxidation potentials, their selective oxidation in the presence of an electron-neutral arene is possible. Electrochemical arene chlorination,403–406 bromination,407,408 iodination (vide infra), and thiocyanation409–411 have been reported within the time frame of this review. Applications of electrochemical arene bromination using aqueous NaBr have been surveyed on complex late-stage intermediates and drug molecules.412
Figure 26.
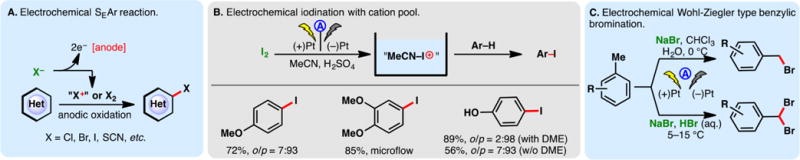
Arene functionalization with electrogenerated electrophiles and radicals.
A cation pool strategy may also be used where electrogenerated halogen cations are accumulated at low temperatures. For example, Yoshida devised a practical method for arene iodination wherein an “I+” ion pool was generated through stabilization by acetonitrile (Figure 26B).413,414 Subsequent addition of arene led to iodination products in good yields while circumventing competing arene anodic oxidation; overiodination may be suppressed with the use of microflow reactors.414 The para-selectivity of this reaction can be improved using DME as a cosolvent in the second step.
Halogen radical species can also be accessed anodically—this enabled electrochemical Wohl-Ziegler type benzylic halogenations (Figure 26C). Mono-415 and di-416 benzylic bromination of toluene derivatives can be achieved depending on the reaction conditions. The formation of HOBr was invoked in each case; bromination at the nucleus of more electron-rich arenes is the major competing reaction.
2.11. Fluorination
While anodic oxidation of chlorides, bromides, and iodides can be utilized to generate the electrophilic species, an analogous process with fluoride is conceivably difficult due to fluoride’s extremely high oxidation potential. Instead, the high potential of fluoride allowed various other motifs to be oxidized in its presence—electrogenerated cation or radical cations can then engage fluoride anions in nucleophilic fluorination processes. The most well-known example of such a transformation is perhaps the Simons process (Figure 1) which produces perfluorinated hydrocarbons on large scales. Electrochemical fluorination has been reviewed;417–419 some recent advances are summarized herein.
Taking advantage of arylsulfide-based electroauxiliaries, Fuchigami has developed a series of methods to achieve the selective anodic fluorination of organic molecules. For example, Fuchigami and co-workers showed that electrolysis of 4-arylthio-1,3-dioxolan-2-ones in Et3N·nHF can lead to the formation of fluorodesulfurization product A (Figure 27A) through the intermediacy of an oxocarbenium cation (vide supra).420,421 A major competing pathway is a Pummerer type fluorination (B) wherein the sulfur-centered radical cation arising from anodic oxidation is converted into a thionium cation instead. Fuchigami noted that the choice of solvent and fluoride source profoundly impacts the selectivity between these two pathways. The use of DME/Et3N·4HF exclusively produced B, while A was favored with CH2Cl2/Et3N·5HF. It was reasoned that DME stabilizes cationic intermediates and enhances the nucleophilicity of fluoride, thereby allowing the formation of a thionium cation; conversely, CH2Cl2 solvated cationic species poorly, and desulfurization occurred preferentially. This solvent effect was also observed in the anodic fluorination of 3-phenylthiophthalide wherein selective fluorodesulfurization took place selectively using CH2Cl2 or [emin][OTf] as the solvent.422,423 The Pummerer-type electrochemical fluorination may be rendered stereoselective through the use of a chiral auxiliary (Figure 27B).424
Figure 27.
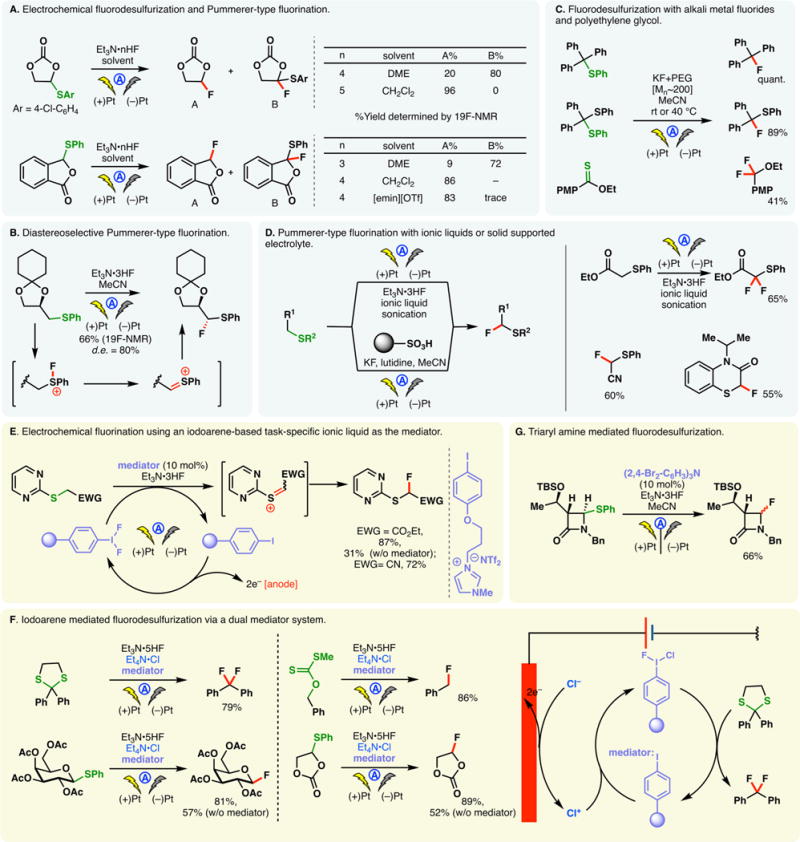
Electrochemical fluorodesulfurization and Pummerer-type fluorination.
HF complexes used in the reactions may be replaced with a simple alkali metal fluoride (e.g., KF) in the presence poly(ethylene glycol) (PEG) which coordinates with the cations to increase the nucleophilicity and solubility of the fluoride source (Figure 27C).425 Fluorodesulfurization was demonstrated with this system under constant current electrolysis. Thiocarbonyl groups can also be converted into geminal difluorides.
Pummerer-type electrochemical fluorination at the α-position of electron-withdrawing groups have been probed extensively (Figure 27D). An efficient protocol has been developed in Et3N·3HF ionic liquid; this process was found to be promoted by ultrasonication—as the viscosity of ionic liquid fluoride salts is higher than that of ordinary molecular solvents, sonication presumably facilitates the mass transport of substrate (Figure 27D, top).426 Difluorination is possible with this system, allowing the preparation of α,α-difluoro-esters. Minimal electrode passivation was observed using this protocol. An alternative system was also described which takes advantage of cation exchange between alkali-metals and solid-supported acids, allowing simple alkali metal fluorides to be used as the source of F atom (Figure 27D, bottom).427 The exchange reaction promotes the dissociation of alkali-metal fluoride salt in MeCN; in the presence of lutidine, lutidine·HF is generated in situ.
Direct electrochemical fluorination is often plagued by severe electrode passivation due to substrate decomposition under the reaction conditions—mediated processes can alleviate such problems. Fuchigami showed that, under electrolytic conditions and in the presence of a fluoride source, iodoarenes can be oxidized into hypervalent difluoro-iodoarene species. By attaching an iodoarene to an ionic tag, Fuchigami and co-workers developed a task specific ionic liquid which could be readily converted into ArIF2 in the presence of Et3N·HF under galvanostatic conditions;428 these hypervalent iodine species can then mediate Pummerer-type fluorinations. Yields of this mediated protocol compare favorably with direct electrolysis (Figure 27E). The mediator was also shown to remain intact after the reaction—it can thus be reused in subsequent runs.
As with other types of reactions involving the electrogeneration of thionium or oxocarbenium from the thioacetals (vide supra), fluorodesulfurization can be achieved through indirect electrolysis with the use of redox mediators. In the example depicted in Figure 27F, a double mediatory system was devised employing Et4N·Cl and a polystyrene-supported iodoarene to facilitate fluorination and difluorination of various sulfur-containing structural motifs.429 Triaryl amines could also be used to mediate fluorodesulfurization process—for example, a fluorinated β-lactam was obtained through constant current electrolysis with 10 mol % of (2,4-Br2–C6H3)3N as the mediator (Figure 27G).430
Trifluoroborate salts have also found utility in electrochemical fluorination (Figure 28A). When the pinacol ester of phenylthiomethyl boronic acid was subjected to electrolysis in the presence of Et3N·HF, the C–B bond was ruptured and replaced by a fluoride.431,432 A BF3 salt is believed to be the reactive species as the BF3 motif putatively serves as an electroauxiliary by exerting a β-effect. Indeed, conversion of a thiophene-based boronic acid to the corresponding BF3 salt lowered its oxidation potential substantially, allowing fluorodeborylation to occur (Figure 28A, right).432 However, since the oxidation potential of the monofluorination product is close to that of the trifluoroborate salt, further fluorination ensues. The use of BF3 salt as an electroauxiliary was also demonstrated in allylation reactions (Figure 28A, bottom left).
Figure 28.
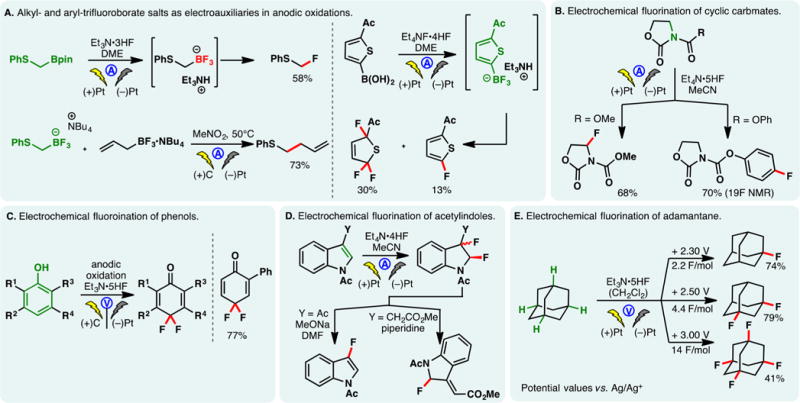
Electrochemical fluorination of miscellaneous functional groups.
Carbamates can also be fluorinated at the α-position under electrochemical conditions through a Shono-type process (Figure 28B).433 However, when the substrate contains an electron-rich aromatic ring, arene fluorination dominates. Likewise, electron-rich arenes such as phenol (Figure 28C)434 or acetyl-indole (Figure 28D)435 could be fluorinated under electrolytic conditions with HF complexes. However, achieving monofluorination is challenging. For example, difluorocyclohexadienones were obtained from phenols while acetylindoles were converted to 2,3-difluoroindolines. In the latter case, the fluoride group at C2 or C3 can be eliminated selectively depending on the substituent on C3. Benzylic positions of arenes can be fluorinated as well using similar systems.436
Aliphatic C–H bonds in adamantane can also be fluorinated under anodic conditions (Figure 28E).437,438 By adjusting the applied potential, mono-, di-, tri-, and tetra-fluorination products can be furnished selectively. Some other functional groups that can be fluorinated under electrolytic conditions include ethers,439 lactones,439 carbonates,439 conjugated dienes,440 pyrrole,441 benzofuran,442 benzothiophene,443 and thiazolidine.444 These processes have been reviewed by Fuchigami and co-workers.417,418
2.12. Olefin oxidation
2.12.1. Oxidation of enol ethers and ketene acetals or equivalents thereof
Anodic oxidation of enol ethers gives rise to radical cations with considerable radical character. This reactive species can undergo facile cyclization with an electron-rich olefin. An example is provided in Figure 29A which shows that this intramolecular reaction proceeds through a chair like transition state via kinetic control.445 The resulting disjointed radical cation could be oxidized and trapped with the solvent molecule (MeOH), affording an acetal product. Intriguingly, premature reaction between MeOH and the uncyclized enol ether radical cation was found to be minimal. This may be ascribed to the fact that the electrochemical oxidation occurs in a double-layer adjacent to the anode instead of in the bulk solution phase—movement of solvent molecules is thus restricted. Commonly denoted as anodic olefin couplings, these processes enable umpolung disconnections that are broadly applicable in the synthesis of complex molecules; they have been rigorously examined by Moeller and Wright with earlier findings summarized in review articles.33,446,447 This section focuses on recent developments within the designated time frame of this article.
Figure 29.

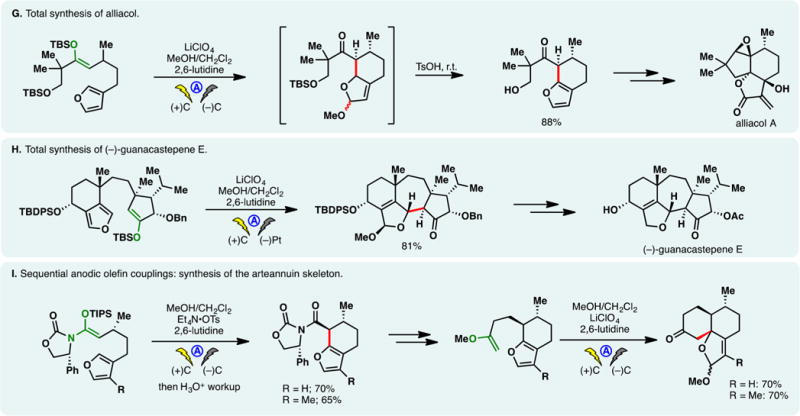
Anodic olefin coupling reactions of enol ethers and ketene acetal equivalents.
While dithioketene acetals were found to be viable anodic coupling partners with allyl silanes (Figure 29B, top), the process was less effective for silanes with lower reactivities (e.g., R = Me).448,449 Conversely, the use of N,O-ketene acetals under the same reaction conditions led to efficient cyclizations (Figure 29B, middle).450,451 This difference could be attributed to the polarity of radical cation intermediates: whereas the N,O-ketene acetal radical cation exhibits considerable radical character on the α-carbon, this position is more cationic for the dithioketene acetal congener (Figure 29B, bottom). As a result, N,O-ketene acetal radical cations preferentially undergo radical cyclization with olefins while dithioketene acetal radical cations are more prone to trapping by alcoholic solvents at the α-position.
Enol ether radical cations generated through anodic oxidation may be disposed to fragmentation pathways. For example, in the reaction presented in Figure 29C, the use of a simple methyl enol ether led to no desired product formation. Moeller and co-workers demonstrated that the intermediary radical cation can be stabilized through intramolecular trapping with a second alcohol nucleophile.452 The cyclization product was obtained in good yields as a result (Figure 29C).
Ketene acetal and its equivalents (e.g., thioketene acetals and N,O-ketene acetals) can be effectively coupled with enol ethers under anodic conditions (Figure 29D).449,453,454 In competition experiments, enol ethers were found to engage radical cations of dithioketene acetals more efficiently than allyl silanes.455 The bottom example in Figure 29D represents a cross-coupling reaction between enol ethers en route to ineleganolide.454,456 Functional group arrangements in the natural product necessitated the introduction of an oxygen atom at the α-position of the cyclic enol ether. While the introduction of OMe at that position was found to shut down the reaction, less electron-donating carbonate groups were tolerated, albeit furnishing the product in a modest yield. It is posited that an α-donating group alters the polarization of the radical cation.
Enol ether radical cations can also be trapped by electron-rich aromatic nucleophiles. For example, an anodic coupling reaction between a TMS-enol ether and a dimethoxybenzene derivative was utilized by Wright in the preparation of the hamigeran skeleton (Figure 29E).457 The coupling reactions between enol ethers and furans have attracted considerable attention (Figure 29F). Wright has developed expedient methods of building polycyclic scaffolds with such a transformation.458–463 For example seven-memebered rings can be constructed through such electrochemical annulations (Figure 29F, bottom). Intriguingly, the presence of an alkyl group at the ring junction was found to be critical: in the absence of this β-group, no cyclization product was observed. Cyclic voltammetry analysis revealed that presence of a gem-dialkyl group in the tether lowers the oxidation potential of the substrate. The cyclization precursors may be conveniently prepared through conjugate addition with enones.
Syntheses of complex natural products have been devised based on the coupling between enol ethers and furans (Figure 29G–I). For example, Moeller and co-workers developed a concise synthesis of alliacol A, wherein the bicyclic core was constructed using an anodic coupling (Figure 29G).464 While the silyl enol ether and the furan in this case are both electron-rich moieties, the former exhibited a lower oxidation potential and was thus preferentially oxidized under electrochemical conditions. A similar cyclization was utilized in Trauner’s synthesis of guanacastepene E—the anodic enol ether-furan coupling enabled the construction of the challenging seven-membered ring (Figure 29H).465,466 Anodic coupling with furan does not alter the heteroarene’s oxidation state—as a result, it could be readily aromatized as in the case with alliacol. This opens the possibility of sequential couplings. In their efforts toward arteannuin, Moeller and co-workers employed two anodic olefin-furan couplings to construct the tricyclic core of the complex natural product (Figure 29I).448 The first of these reactions makes use of a chiral N,O-acetal, allowing access to enantiomerically enriched materials while the second coupling forges a challenging tertiary center.
Radical cations arising from the anodic oxidation of enol ethers or ketene acetal equivalents can also be intercepted with a heteroatom-based nucleophile to forge C–heteroatom bonds. For example, substituted tetrahydrofuran and tetrahydropyran rings can be constructed through the intramolecular addition of alcohol nucleophiles onto anodically generated enol ether radical cations (Figure 30A).467 This type of reaction has been performed using a photovoltaic cell as the power source in an environmentally friendly fashion.468,469
Figure 30.
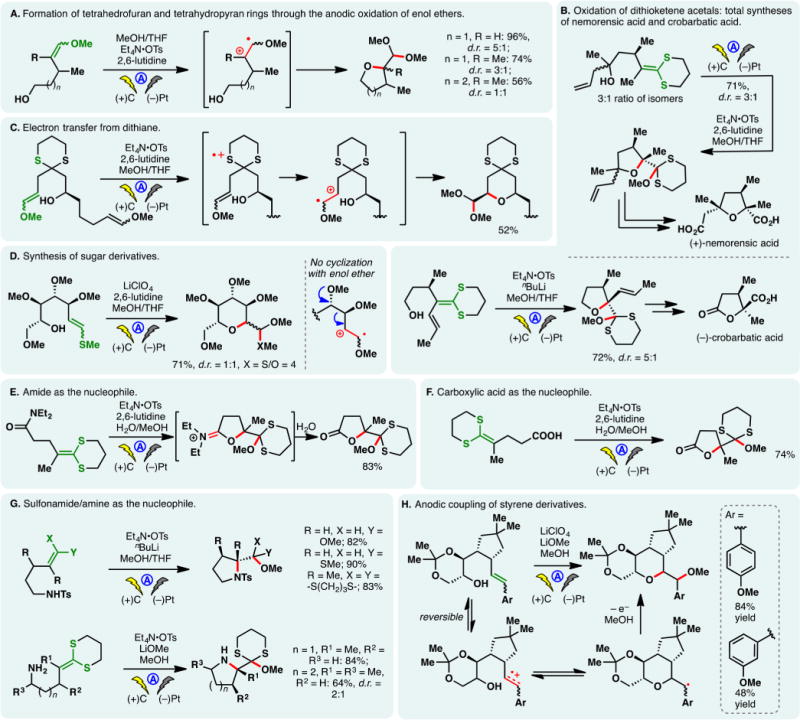
Anodic olefin coupling with heteronucleophiles.
As discussed in the preceding section, the polarity of the radical cations strongly influences their reactivity toward heteronucleophiles. Radical cations of dithioketene acetals, which possess considerable cationic character on the α-carbon, were found to undergo facile cyclization with alcohols (Figure 30B).449 This efficient mode of cyclization found use in Moeller’s syntheses of nemorensic acid470 and crobarbatic acid.471
The example in Figure 30C illustrates that enol ether radical cations can be generated through an intramolecular electron transfer via a Curtin–Hammet pathway.472 In this case, the dithiane motif was more readily oxidized under anodic conditions, affording a sulfur-centered radical cation which can accept an electron from the proximal enol ether motif. Meanwhile, the distal enol ether is left unscathed, despite exhibiting a similar electrochemical potential.
Anodic addition of oxygen nucleophiles onto electron-rich olefins also allowed Moeller to devise a synthesis of C-glycosides (Figure 30D).473 Again, the polarity of intermediary radical cations is critical to the success of such reactions. In the example presented in Figure 30D, the use of a methyl enol ether led to no cyclization product as a competing fragmentation ensued. However, by utilizing a less polarized vinyl sulfide instead, the undesired pathway can be effectively suppressed, delivering the pyranose-type product in good yields. Furanose derivatives, conversely, may be obtained from both enol ether and vinyl sulfide starting materials.
Amides474 and carboxylic acids475 can each function as nucleophiles toward anodically generated enol ether or ketene acetal radical cations. γ-Substituted valerolactones were afforded as shown in Figure 30E and F. In the case of amides, addition of water exerted a beneficial effect on the reaction presumably because water facilitated the hydrolysis of the cyclic imidate cation intermediate to the lactone product. In the absence of water, this intermediate was prone to elimination and further oxidation. When carboxylic acids are used as trapping nucleophiles, no decarboxylation product was observed. Additionally, sulfonamides are competent nucleophiles in anodic olefin coupling reactions, resulting in the formation of pyrrolidine rings (Figure 30G).476,477 This C–N cyclization is favored by less polarized radical cation intermediates as is the case with analogous C–O cyclizations; it is also promoted by the presence of stronger bases (e.g., LiOMe). Mechanistic studies suggested that the coupling of a sulfonamide to an electron-rich olefin exhibited a radical-like mechanism with the initial oxidation occurring at the deprotonated sulfonamide; however, intramolecular electron transfer between the resulting N-centered radical and the electron-rich olefin can occur, yielding an olefinic radical cation.478,479 The presence of a strong base favors the formation of sulfonamide anion. Later, it was discovered that radical cations derived from dithioketene acetals can be trapped with an unprotected amine under electrolytic conditions (Figure 30G, bottom). Even though the potential of the cyclization product (a secondary amine) is conceivably lower than that of the starting material, the high rate of the cyclization kinetically lowers the potential of the starting material.480
While studies on anodic olefin coupling focus on enol ethers and ketene acetal equivalents, electron-rich styrenes can also participate in this type of process. In the example presented in Figure 30H, an alcohol was shown to add onto a styrenyl radical cation in a reversible fashion.481 Thus, the success of this reaction is dependent on the ease with which the resulting benzylic radical is oxidized—the substrate with a para-methoxy phenyl group therefore gave higher yields compared to that containing a meta-methoxy phenyl.
2.12.2. Electrocatalytic cycloadditions of olefins
When the anodic oxidation of an enol ether is carried out in the presence of another olefin, the intermediary radical cation can react with the olefin, giving rise to a “cyclobutane radical cation” (Figure 31A, right column). This process is reversible as the cyclobutane radical cation is prone to fragmentation. However, Chiba and co-workers observed that if either the enol ether or olefin component contains an electron-rich arene ring—the aryl group could serve as a “redox-tag”, intramolecularly donating an electron to the cyclobutane radical cation.482–484 As a result, [2 + 2] cycloadducts can be obtained. Since the reaction follows a chain mechanism, only a catalytic amount of electricity is necessary to initiate the reaction.
Figure 31.
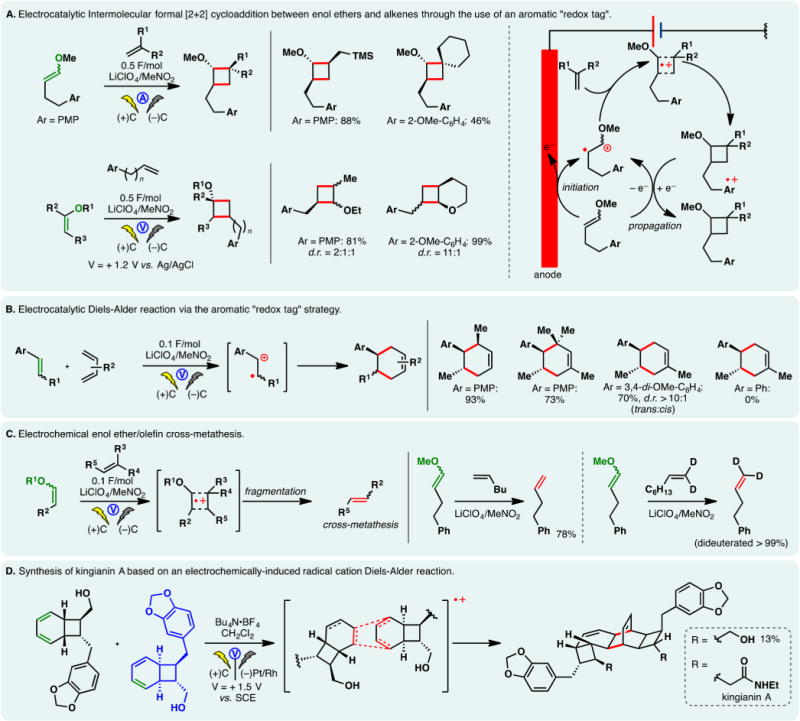
Electrocatalytic cycloadditions.
Chiba extended this method to the Diels–Alder reactions between electron-rich styrenes and dienes wherein the anodic oxidation of the former triggers off the chain process.485 The arene group serves as the “redox-tag”; the use of electron-neutral or electron-deficient styrenes led to no cycloadduct (Figure 31B).
As described above, the cyclobutane radical cation readily undergoes retro-[2 + 2] reactions in the absence of an arene electron donor. Toward this end, two pathways are possible—fragmentation could either lead to the regeneration of the starting olefin and enol ether radical cation; alternatively, a cross-metathesis product could be yielded. By leveraging this process, Chiba and co-workers developed an electrochemical method for the cross-metathesis of enol ethers and olefins (Figure 31C).486,487
Anodic oxidation has also been used to initiate an electronically mismatched Diels–Alder reaction en route to kingianin A. In this case, the dimerization of “pre-kingianin” monomer under potentiostatic electrolysis led to the desired cycloadduct in a modest yield (Figure 31D).488
Additionally, electrochemical [2 + 2] cycloaddition of cyclooctene and [2 + 2+2] reaction of cyclopentene have been achieved by Geiger and co-workers using rhenium-based or triaryl amine-based mediators.489,490
2.12.3. Oxidation of other electron-rich olefins
Enamines derived from the condensation between aldehydes and secondary amines were found to exhibit low oxidation potentials; they could therefore be oxidized to radical cations relatively easily under anodic conditions. Jang and co-workers demonstrated that such radical cations could be trapped with TEMPO to furnish α-oxyl-aldehydes following spontaneous hydrolysis of the iminium intermediate (Figure 32A, top).491 This allowed the development of an asymmetric protocol with substoichiometric amounts of chiral secondary amines, furnishing products in moderate e.e. values. Analogously, Jang also reported a method for α-alkylation of xanthene and cycloheptatriene through anodic enamine oxidation (Figure 32A, bottom).492
Figure 32.
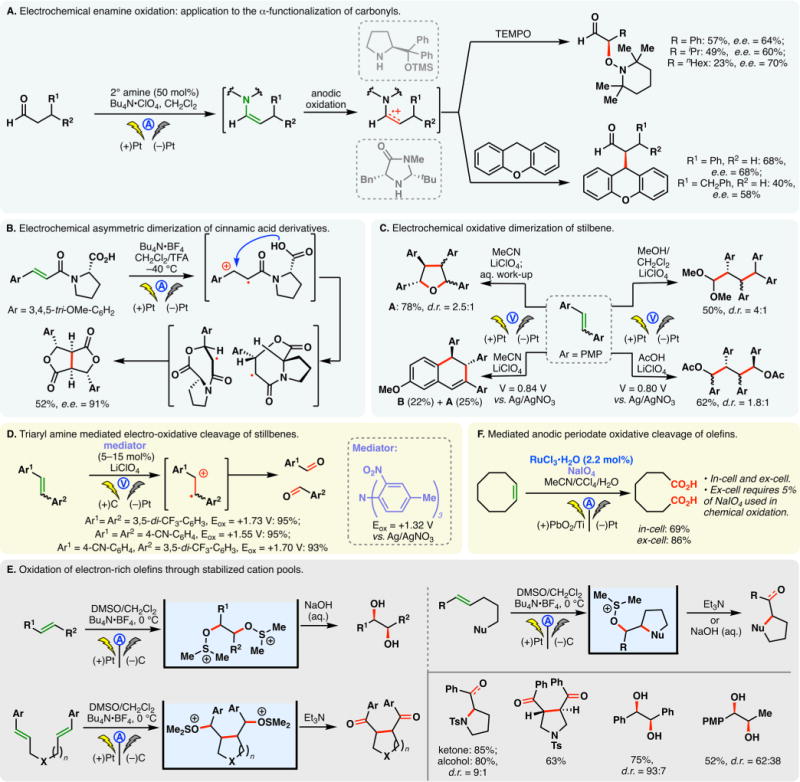
Anodic oxidation of other electron-rich olefins.
Watanabe described a synthesis of furofuran-lignans through the electrochemical oxidative dimerization of a cinnamic acid derivative (Figure 32B).493 Anodic oxidation of the olefin generates a radical cation, triggering the cyclization of the side-chain carboxylate group onto the benzylic position. This process furnishes an α-radical which could undergo spontaneous dimerization. The presence of a chiral proline auxiliary allowed the product to be obtained in good e.e. values.
Preparative anodic oxidation of stilbene and styrene derivatives has also been pursued. Kam and co-workers carried out extensive studies on the oxidative dimerization of stilbenes under constant potential conditions.494,495 The reaction outcomes were strongly influenced by the choice of solvent (Figure 32C). [3 + 2] and [4 + 2] adducts were obtained in MeCN with the former favored after an aqueous workup. Meanwhile, the introduction of MeOH or acetic acid led to the formation of different products. Effects of aromatic substitution on product distribution were probed in detail.
Fry and co-workers showed that electron-deficient stilbenes could undergo oxidative cleavage to provide aryl aldehydes under constant potential conditions (Figure 32D).496 The use of an electron-deficient triarylamine mediator is critical for this electrochemical equivalent of ozonolysis—the mediator has an unusually high oxidation potential of +1.32 V (vs Ag/AgCl). The reaction involves the formation of the corresponding 1,2-diols. Both symmetrical and nonsymmetrical stilbenes are viable substrates, affording aldehydes in good yields.
Radical cations derived from the anodic oxidation of electron-rich olefins can also be trapped with DMSO to generate stabilized cation pools. For example, by carrying out the electrolysis of stilbenes in the presence DMSO, Yoshida and co-workers generated a dication pool shown in Figure 32E (top left).497 Subsequent treatment with sodium hydroxide facilitated the cleavage of the S–O bond, affording a dihydroxylation product. Alternatively, when a styrene is tethered with a nucleophile, the latter can cyclize onto anodically generated radical cations. In the presence of DMSO, the cyclized radical cation could be trapped; subsequent treatment with NaOH or trimethylamine gives rise to a ketone or an alcohol respectively (Figure 32E, top right and bottom left).294,498
The Sharpless olefin oxidation, employing ruthenium(III) and periodate, represents one of the most commonly used methods for oxidative olefin cleavage. However, this venerable reaction is sometimes plagued by the need for large amounts of periodate salts. Schäfer and co-workers devised electrochemical means to regenerate sodium periodate during the reaction (Figure 32F).499 Electrochemical regeneration of periodate and olefin cleavage may take place in the same vessel, using an in-cell protocol. On the other hand, an ex-cell system has also been developed wherein the olefin cleavage is conducted in a biphasic solvent system. The aqueous phase can be channeled back and forth to a separate electrochemical reactor where the periodate regeneration takes place under electrolytic conditions. This excell protocol has successfully reduced the amount of requisite sodium periodate by 95%. The use of a lead oxide/titanium composite electrode was found to be optimal for this process. Additionally, Shäfer has also reported an electrochemical method to generate ozone using a lead oxide anode—ozonolysis of olefins could be accomplished in good yields, albeit in modest current efficiencies.500
Anodic oxidation of cyclooctatetraene has been probed by Fry and co-workers—it was found to undergo multiple C–C bond-forming and -cleavage events under electrolysis.501,502
2.12.4. Olefin oxidation with anodically generated electrophiles
While direct anodic oxidation provides a simple and practical means to functionalize electron-rich olefins such as styrenes and enol ethers, oxidizing other olefins is more challenging, owing to their higher oxidation potentials. However, anodic processes can still be used to functionalize these structural moieties by virtue of electrogenerated electrophiles. For example, halogen or chalcogen anions can be oxidized anodically where the resulting electrophilic species can engage olefins.
Yoshida and co-workers demonstrated that the electrolysis of halogen or chalcogen anions in the presence of DMSO led to the formation of stabilized halogen or chalcogen cation pools through coordination with the sulfoxide oxygen (Figure 33A).503,504 The stability of these cation pools increases in the order of Br+ < I+ < ArS+ < ArSe+ in accordance with the relative electronegativity of X. These stabilized cations were found to react readily with a wide variety of olefins, forming β-X-substituted alkoxysulfonium ions which are versatile synthetic intermediates that can be readily diverged to different functionalities. For example, treatment of the alkoxysulfonium with trimethylamine was shown to induce Swern-like oxidation, furnishing α-haloketones (Figure 33A, top); reaction with aqueous sodium hydroxide yielded vincinal halo/chalcohydrins (Figure 33A, middle); sodium methoxide was found to induce epoxide formation (Figure 33A, bottom). These processes are amenable for isotopic labeling when DMS18O is used. Application to 1,6-dienes led to the formation of cyclized products (Figure 33A).
Figure 33.
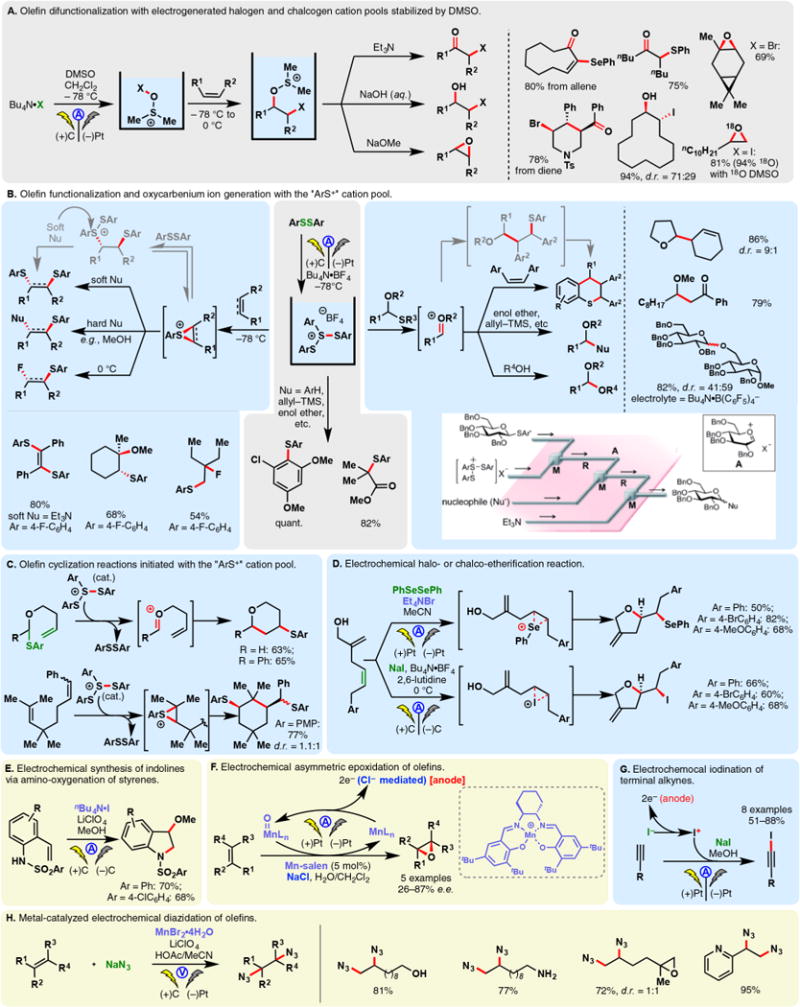
Olefin functionalization with electrogenerated electrophiles. Inset photo in Figure 33B reprinted from Angew. Chem. Int. Ed. 2011, 50, 5153–5156. Copyright (2011) John Wiley & Sons.
Similarly, the anodic oxidation of aryl disulfides at low temperatures allowed the accumulation of highly electrophilic ArS+ equivalent which could form episulfonium intermediates upon reaction with olefins or alkynes (Figure 33B, left).505–510 The thiirane could be opened directly with hard nucleophiles such as fluorides or alkoxides. Nevertheless, due to the presence of disulfides in solution, treatment of the episulfonium with soft nucleophiles led to the formation of vicinal disulfides instead. Catalytic protocols have also been developed wherein substoichiometric amounts of the ArS+ equivalent initiates a cation chain mechanism.507,508 This “ArS+ cation pool” also found utility in the functionalization of arenes, allyl silanes, enol ethers, and ketene acetals (Figure 33B, middle);511 moreover, it could be used to induce oxocarbenium formation through reaction with thioacetals (Figure 33B, right).512–514 This process has found application in glycosylation chemistry where efficient protocols with flow reactors have been detailed.515,516 Furthermore, the “ArS+ cation pool” was utilized (in catalytic amounts) to initiate olefin cyclization reactions, forming both heterocyclic and carbocyclic rings (Figure 33C).517,518
As with disulfides, the anodic oxidation of diselenides generates +SeAr cations which can be used to induce chalco-etherification type processes (Figure 33D).519 An analogous reaction employing an electrogenerated “I+ ion” has also been reported (Figure 33D, bottom).519 Zeng, Little, and co-workers took advantage of the electrochemical halocyclization reaction to develop a synthesis of indolines from the corresponding 2-aminostyrenes. The intermediary organoiodide can be displaced by methanol to afford the final product while regenerating iodide anion for anodic oxidation (Figure 33E).520 The electrochemical formation of cyclic iodonium intermediates was also exploited by Little, Zeng, and co-workers to achieve the regioselective azidoiodination of olefins.521
An electrochemical variant of the Jacobsen epoxidation was also reported wherein a halide mediated anodic oxidation generated a reactive chiral manganese-oxo species which could convert a variety of olefins into epoxides in moderate e.e.s (Figure 33F).522
Electrolysis of terminal alkynes in the presence of sodium iodide was found to yield iodo-alkynes through the intermediacy of “I+” ions (Figure 33G).523
Electrogenerated radical species can also be harnessed for olefin functionalizations. Very recently, Lin and co-workers detailed an electrochemical method for the diazidation of olefins (Figure 33H).524 An inexpensive manganese catalyst was used to facilitate the reaction. Under potentiostatic conditions, the ability to dial the potential at the minimum level required for the desired redox transformation conferred the reaction high degrees of chemoselectivity, allowing the functionalization of complex synthetic intermediates.
2.13. Dehydrogenation and heteroarene synthesis
Oxidative aromatization involving the net elimination of H2 is an important process in heteroarene synthesis which is commonly accomplished with stoichiometric amounts of chemical oxidants, such as CAN, DDQ, or KMnO4. Electrochemical dehydrogenation wherein H2 is removed anodically offers an alternative. Electrochemical syntheses of 2-substituted benzoxazoles have been developed through both direct477 and mediator facilitated525–527 processes from the Schiff bases of 2-aminophenols (Figure 34A). A system using NaI as the mediator has been developed by Zeng, Little, and co-workers, utilizing inexpensive electrode materials and mild carbonate bases (Figure 34A, right column).525 Alternatively, DDHQ can serve as the mediator for this transformation where electrogenerated DDQ is the reactive oxidant (Figure 34A, left column).527
Figure 34.
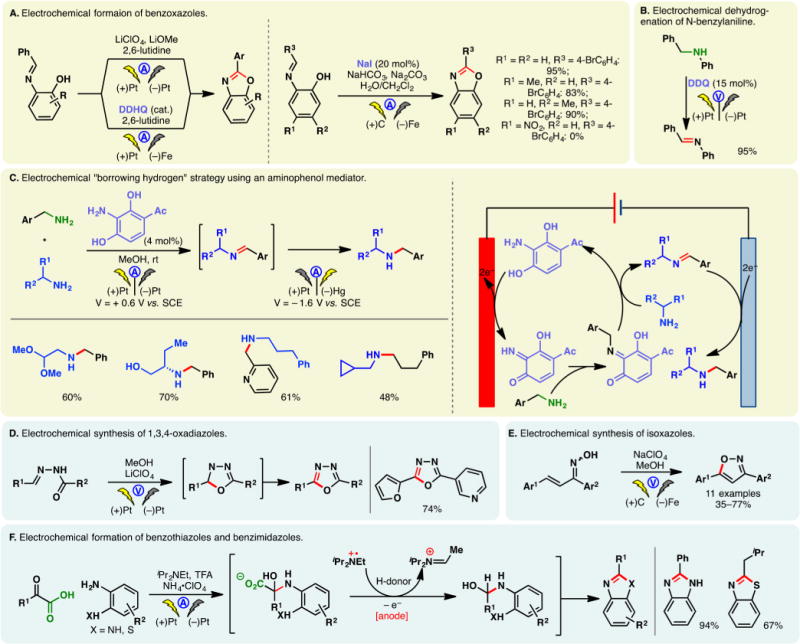
Electrochemical dehydrogenation and heteroarene synthesis.
DDQ has also found use as a mediator in the dehydrogenation of N-benzylaniline to the corresponding imine—the use of electrochemical conditions allowed the DDQ to be employed in catalytic quantities (Figure 34B).528
Largeron and Fleury devised an electrochemical “borrowing hydrogen” protocol to synthesize secondary amines from two sterically differentiated primary amines, through formal N-alkylation (Figure 34C). This one-pot procedure commences with the anodic dehydrogenation of a primary amine where an aminophenol is used as the mediator.529–531 Electrochemical oxidation of the mediator furnishes an iminoquinone which can oxidize the amine through condensation and imine isomerization (Figure 34C, right column). The resulting N-aryl-imine can engage another primary amine, giving rise to an N-alkyl-imine. This imine can then be reduced to the corresponding secondary amine under cathodic conditions in the same pot. Alternatively, conditions have also been developed which allow the intermediary iminoquinone to engage electrogenerated alkyl imines in a Diels–Alder reactions.532,533
Electrochemical dehydrogenations have been employed to synthesize 1,3,4-oxadiazoles (Figure 34D)534 and isoxazoles (Figure 34E).535 A recent report by Huang and co-workers detailed an electrochemical synthesis of benzothiazoles or benzimidazoles through the coupling between α-keto acids and an α-functionalized aniline (Figure 34F).536 Putatively, electrochemical decarboxylation and dehydrogenation take place sequentially in this reaction. Hünig’s base is believed to serve as a hydrogen donor for the radical intermediate formed in the decarboxylation step.
2.14. Oxidation of aliphatic C–H bonds
Aliphatic C–H bonds in both allylic and unactivated methylene systems have very high oxidation potentials. As a result, there are few examples on the direct anodic oxidation of these C–H bonds. In 2005, Sobkowiak reported that the allylic position of cholesterol can be acetoxylated under direct electrolysis with a platinum anode in modest yields (Figure 35A).537
Figure 35.
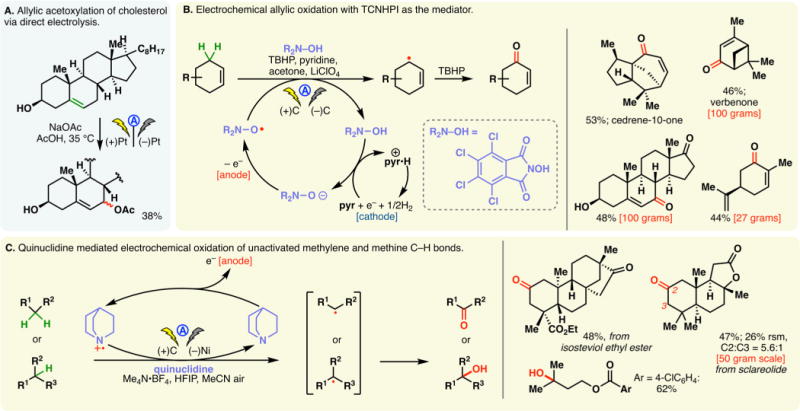
Electrochemical oxidation of allylic and unactivated C–H bonds.
The challenge of oxidizing aliphatic C–H bonds can instead be surmounted through indirect electrolysis employing an appropriate mediator. For example, Baran and co-workers reported a scalable means for allylic oxidation using tetrachloro-N-hydroxyphthalimide (TCNHPI) as the mediator (Figure 35B).538 Under basic conditions, it was believed that the anion of the mediator molecule can be oxidized anodically, giving rise to a reactive N-oxyl radical which can homolyze allylic C–H bonds. The electron-withdrawing chloride groups confers the nitroxyl species additional reactivity—the use of nonchlorinated NHPI led to inferior results. This process utilizes inexpensive electrode materials while tolerating functional groups such as ketones and alcohols. Complex steroids, monoterpenoids, and trieterpenoids can be oxidized on up to 100 g scales.
Attempted adaptation of the TCNHPI mediatory system to oxidize unactivated methylene and methine C–H bonds were unsuccessful, presumably because the nitroxyl radical species is not strong enough to cleave these bonds. Instead, Baran and co-workers developed a quinuclidine-based mediator system that allowed the functionalization of “deep-seated” methylene and methine units (Figure 35C).539 Selectivity for methylene oxidation is dependent on both steric and electronic factors; homolysis of the electron-rich C–H bond distal to electronegative functionalities is usually preferred. The scalability of this method is demonstrated through the oxidation of sclareolide on a 50-g scale, affording 2-oxo-product selectively. The anodically generated quinuclidine radical cation is believed to be the reactive species in this reaction.
2.15. Oxidation of miscellaneous functional groups
Aside from the examples presented in the preceding sections, anodic oxidations of various other functional groups have been reported recently. Chiba and co-workers reported a method to synthesize macrocyclic disulfides through anodic oxidation of acetamidomethyl cysteines in soluble solid-bound peptides (Figure 36A).540,541 The homogeneous reaction takes place under potentiostatic conditions in organic solvent mixtures; the solid-supported product could be precipitated out through dilution with counter solvents. Complex molecules such as somatostatin could be chemoselectively accessed.
Figure 36.
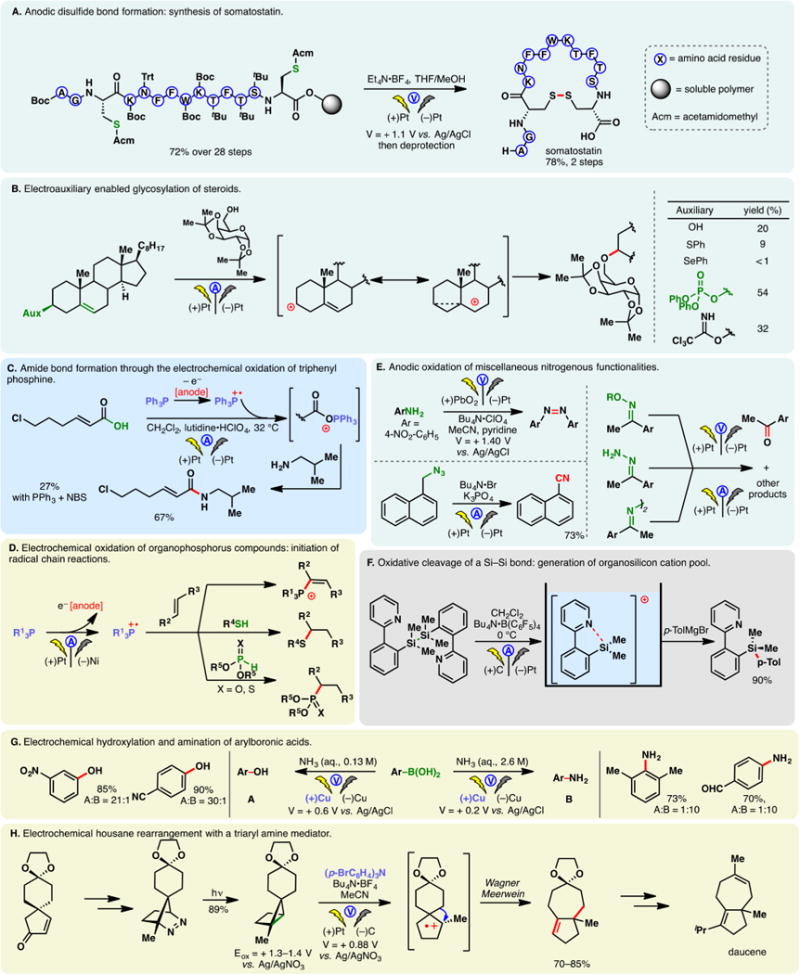
Oxidation of miscellaneous functional groups.
Anodic oxidation of phosphorus functionalities can also enable useful reactions. For example, in an effort to develop a homoallylic glycosylation reaction of steroids, Morzycki and co-workers examined various electroauxiliaries to facilitate the generation of a homoallyl cation (Figure 36B).542,543 In one study, while commonly used auxiliary groups such as SPh and SePh gave low yields, the diphenylphosphonate group was found to be most effective; the oxidation of the diphenylphosphonate group allowed fragmentation of the C–O bond, giving rise to a nonclassical cation to allow stereoselective glycosylation.543
Ohmori’s work in the 1990s demonstrated that triphenylphosphine could undergo anodic oxidation to furnish a radical cation species that could facilitate various reaction manifolds.544 In a more recent application of this chemistry, Frontana-Uribe used the electrogenerated phosphine radical cation to activate carboxylic acids for amide bond formation (Figure 36C).192 The reaction took place under galvanostatic conditions in the presence of the amine coupling partner; the yield of the amide compares favorably with analogous chemical processes employing triphenylphosphine and NBS. Nikitin and co-workers discovered that electrochemically generated organophosphorus radical cations can serve as a hydrogen atom acceptor, initiating hydrofunctionalization reactions of olefins in the presence of a hydrogen atom donor (Figure 36D).545 Thiols, dialkylthiophosphites as well as dialkylphosphites can all serve as donors in this case. A major competing pathway involves the direct reaction of the phosphorppcentered radical cation with alkenes, leading to the formation of vinylphosphonium salts.
Anodic oxidations of miscellaneous nitrogenous functionalities have also been examined. For example, the oxidation of arylamines to azoarenes has been reported under potentiostatic conditions with a lead(IV) oxide or nickel oxyhydroxide anode (Figure 36E, top left);546,547 Oxidation of benzyl azides was shown to afford aryl nitriles after extrusion of nitrogen (Figure 36E, bottom left).548 Becker has studied anodic oxidation of compounds containing C=N bonds, including oxime (and ethers thereof), hydrazones, and azines (Figure 36E, right).549
Yoshida and co-workers exploited the ability of pyridine to stabilize cationic intermediates to generate an organosilicon cation pool through the electrochemical oxidation of Si–Si bonds (Figure 36F).550 The reaction of this cation pool with a Grignard reagent was found to deliver a substituted silane.
Huang and co-workers explored the electrochemical transformation of aryl boronic acids (Figure 36G). Selective methods to convert boronic acids into phenols and anilines have been devised using copper electrodes under potentiostatic conditions.551 Electrolysis of aryl boronic acids in aqueous ammonia (0.13 M) at 0.6 V led to the exclusive formation of the corresponding phenols; conversely, when a potential of 0.2 V was applied in the presence of aqueous ammonia of higher concentrations (2.61 M), amination product predominated. It is postulated that electrochemical conditions allowed the generation of active catalysts for this reaction.
Little and co-workers utilized triarylamine mediated electrochemical conditions to oxidize the strained bicyclic housane framework (Figure 36H).552,553 The resulting radical cation was found to undergo Wagner-Meerwein rearrangement, allowing a concise synthesis of a sesquiterpene, daucene. The process was carried out under constant potential electrolysis at +0.88 V vs Ag/AgNO3 where the mediator was oxidized into the corresponding aminium radical cation. The redox potential of the substrate is approximately +1.4 V under these conditions. Although the electron transfer from housane to the aminium radical cation is not thermodynamically favorable, the irreversible Wagner-Meerwein rearrangement shifts the equilibrium, allowing the reaction to proceed.
2.16. Anodic oxidation in palladium catalysis
Palladium catalyzed reactions have played a pivotal role in all facets of synthetic chemistry over the past few decades. Many such processes require stoichiometric oxidants to regenerate Pd(II) catalysts from Pd(0). Electrochemical oxidation provides an atom economical alternative. The use of anodic oxidation to regenerate transition metal catalysts has been detailed in previous reviews.21,554,555 The ensuing discussion thus focuses on recent developments in this area. As the active metal catalysts generated in anodic processes are prone to reduction, divided cells are normally used.
The Wacker oxidation offers a classical example where an external oxidant (a copper salt) is used to reoxidize Pd(0) to Pd(II). Due to a lack of homogeneity, large amounts of copper and palladium salts may be necessary; moreover, the use of copper chloride oftentimes triggers the formation of chlorination side-products. Although protocols which are catalytic in copper have been developed using oxygen as the terminal oxidant, such processes can raise safety concerns on large scales. An electrochemical Wacker reaction using a benzoquinone mediator to turn over palladium was reported by Tsuji and co-workers.556 More recently, Mitsudo, Tanaka, and co-workers reported anodic conditions to effect the transformation of Pd(0) to Pd(II) using TEMPO as the mediator (Figure 37A).557 Simple Pd(OAc)2 serves as the palladium source; it is believed that a highly reactive cationic Pd(II) species is generated at the outset of the reaction when Pd(OAc)2 undergoes Kolbe decarboxylation (Figure 37A, right). A different protocol using a PEG/MeCN thermomorphic biphasic system was also developed which homogenizes upon heating to 60 °C; upon the completion of the reaction, the palladium catalyst can be recycled in the PEG phase.558 Tanaka showed that this Wacker oxidation method can be extended to enable oxidative cyclization559 and aryl boronic acid homocoupling.560,561 Around the same time, Jutand and co-workers reported a similar electrochemical approach to boronic acid dimerization with benzoquinone as the mediator.562
Figure 37.
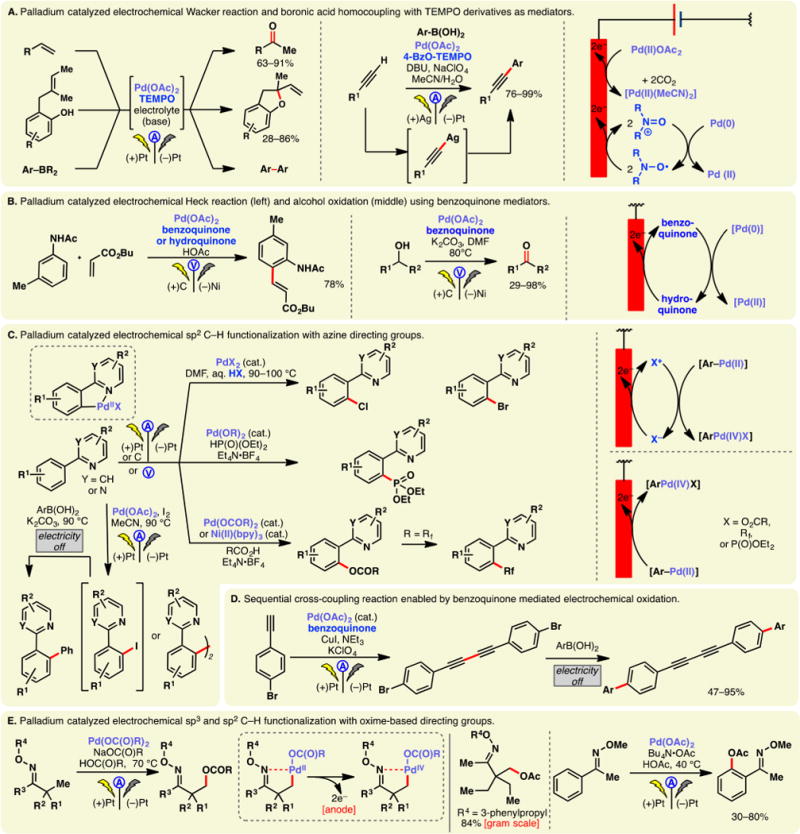
Anodic oxidation in palladium catalysis.
Another recent application of the Pd(OAc)2/TEMPO system is the cross coupling between terminal alkynes and aryl boronic acids (Figure 37A, middle).563 In this case, a silver anode is used which allows the formation of silver acetylides; coupling was also observed using a copper anode albeit in lower yields, while the use of Pt anode led to little product formation (≤5%). 4-BzO-TEMPO was found to be the optimal mediator.
Amatore, Jutand, and co-workers reported an electrochemical Heck-type reaction of N-acetylaniline with benzoquinone or hydroquinone as the redox mediator (Figure 37B).564 Unlike a traditional Heck reaction, this process does not commence with a C–X insertion. Instead, an electrophilic palladation (formal C–H palladation) takes place, followed by addition to olefin and β-hydride elimination. Thus, a Pd(II) catalyst is converted to Pd(0) at the end of this C–H functionalization reaction, and a benzoquinone oxidant is required to reinitiate the catalytic cycle (Figure 37B, right). To this end, electrochemistry furnished products in high yields with 10 mol % of benzoquinone or hydroquinone. The same group extended this system to effect palladium catalyzed electrochemical oxidation of primary and secondary alcohols to aldehydes and ketones (Figure 37B, middle).565 The analogous chemical reaction was developed by Larock in 1998 and involved the use of molecular oxygen; the anodic protocol was operative under anaerobic conditions.566
Anodic oxidation has also been utilized to aid palladium catalyzed C–H activation, exemplified by the ortho-C–H bromination and chlorination reactions of arylpyridines developed by Kakiuchi and co-workers (Figure 37C).567 Coordination of the Pd(II) catalyst onto the pyridine nitrogen led to the formation of a palladacycle (Figure 37C, dashed box). It was believed that halide mediated anodic oxidation of this intermediate facilitated reductive elimination, thereby affording an ortho-halogenation product. This reaction manifold was adapted by Budnikova and co-workers to achieve the ortho-phosphorylation568 and acetoxylation569 of arylpyridines (Figure 37C).570 In the acetoxylation process, C–H fluoroalkylation products were afforded with a perfluorinated alkyl carboxylic acid after longer electrolysis time.571 In the absence of halides as potential mediators, direct anodic oxidations of Pd(II) or Ni(II) are putatively operative (Figure 37C, bottom right). These electrochemical C–H functionalization reactions invoke a high valent palladium or nickel species (e.g., Pd(III), Pd(IV), Ni(III), etc.). Thus, electrolysis was carried out in divided cells to prevent premature cathodic reduction of metal catalysts.
In a similar vein as their C–H chlorination and bromination reactions, Kakiuchi developed a protocol for electrochemical C–H iodination of arylpyridines with I2 (Figure 37C, bottom).572 By increasing substrate concentration while reducing the amount of I2 and electricity, ortho-dimerization of arylpyridine could be realized.573 For substrates bearing meta-substituents in the arene ring, selective reaction at the less hindered ortho position took place—this differed from analogous chemical processes employing oxone as the terminal oxidant.574,575 Additionally, the iodination reaction can be combined in the same pot with a Suzuki coupling when the electric current is turned off, thereby allowing the net ortho-arylation of arylpyridines.
Mitsudo, Suga, and co-workers reported another example where palladium-catalyzed couplings under electrochemical and chemical conditions are combined in the same pot (Figure 37D).576 In this case, dimerization of terminal alkynes was achieved with the aid of anodic oxidation; when electricity was switched off, instead of getting oxidized at the anode, Pd(0) present in the reaction could undergo oxidative addition with the aryl bromide, resulting in a Suzuki coupling.
In a very recent example, Mei and co-workers demonstrated the application of anodic oxidation to facilitate Pd(II) catalyzed sp3 C–H functionalization (acetoxylation) for the first time (Figure 37E).577 This transformation utilizes an oxime-ether-based directing group to furnish a palladacycle through C–H activation with Pd(OAc)2. Direct anodic oxidation of this Pd(II) complex can lead to a Pd(IV) intermediate that is prone to reductive elimination, affording C–H acetoxylation products while regenerating Pd(II) catalyst. By exchanging the acetate group on the palladium catalyst, other oxygenated functionalities such as OTFA, OCOC3H7, OTs, or OMe can be introduced. The reaction has also been demonstrated on gram scales. An analogous process for the acetoxylation of sp2 C–H bonds was reported shortly thereafter (Figure 37E, right).578
3. CATHODIC REDUCTION
3.1. Reduction of aldehydes and ketones
Electrochemical reduction of aldehydes and ketones affords the corresponding ketyl radicals, much akin to dissolving metal reductions. Homodimerization of cathodically generated ketyl radicals through pinacol couplings has been known for more than half a century.579 Capitalizing on the conductivity of ionic liquids, this classic reaction could be carried out at room temperature in ionic liquids to avoid the use of extraneous electrolytes (Figure 38A).580–582 For example, Manchanayakage and co-workers reported an efficient reductive dimerization of ketones and aldehydes using 1-butyl-3-methylimidazolium tetrafluoroborate [BMIM][BF4] as the solvent.580 To avoid the oxidation of intermediary ketyl radicals, a tin electrode was used as the sacrificial anode.
Figure 38.
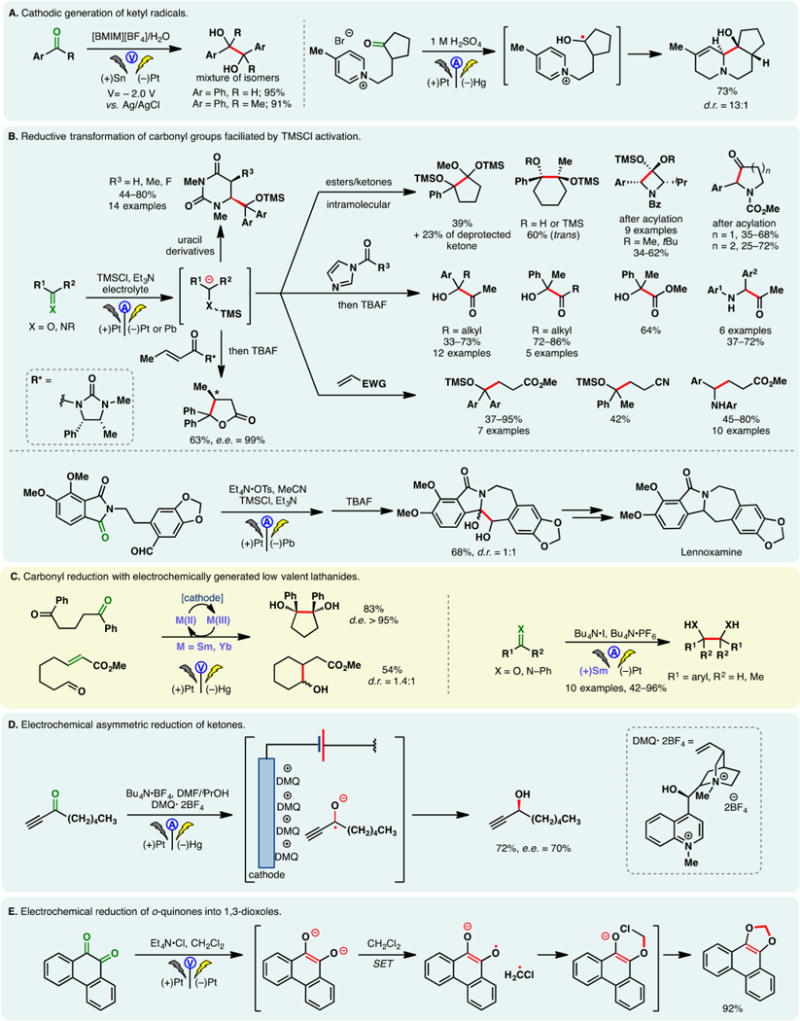
Reduction of aldehydes and ketones.
The reduction potential (absolute value) of carbonyls could be effectively lowered under acidic conditions when the carbonyl oxygen is protonated. For example, in dilute sulfuric acid, Schäfer and co-workers demonstrated the selective reduction of a ketone moiety in the presence of a pyridinium (Figure 38A, right).583 The intermediary hydroxyalkyl radical could cyclize onto the pyridinium to forge a tricyclic product in high yields and good diastereoselectivity. Cyclic voltammetry studies indicated that ketones are more reducible under the reaction conditions than pyridiniums.
Similarly, Kise and co-workers established that electrochemical reduction of carbonyls could be facilitated through activation with trimethylsilyl chloride (TMSCl) (Figure 38B) where interaction between TMSCl and the carbonyl oxygen significantly lowers the reduction potential (absolute value) of carbonyl compounds.584 Cathodic reduction of aromatic ketones in the presence of TMSCl furnishes TMS-protected ketyl radicals which could be reduced further to the analogous anions. These anionic species were shown to exhibit high reactivity toward an assortment of electrophilic species, enabling reductive cyclization onto esters,585 aliphatic ketones or aldehydes.586,587 In the latter case, a trans-product was preferentially obtained, in contrast to conventional intramolecular pinacol reactions employing low valent titanium or samarium reductants. Intermolecular variants of this technology have been devised wherein electrogenerated ketyl anion could couple with aliphatic aldehydes/ketones,587 N-acylimidazole,584,587 achiral588/chiral Michael acceptors589 and uracil derivatives.590,591 It was also demonstrated that TMSCl could be used to facilitate the electrochemical reduction of imines, allowing reductive formation of nitrogenous heterocycles592–595 as well as enabling reductive coupling with N-acylimidazole.596 Moreover, 3-methoxycarbonylindoles597,598 could be coupled with carbonyl compounds in a similar manner. Imides could also be reduced cathodically in the presence of TMSCl to the corresponding anion which was found to engage carbonyl compounds or Michael acceptors.599–602 As an application of the TMSCl promoted electroreductive coupling, Kise and co-workers utilized an intramolecular coupling between an aldehyde and a phthalimide to synthesize lennoxamine in a concise fashion (Figure 38B, bottom).603
Ketyl radicals could alternatively be generated with the aid of a redox mediator. For example, Little and co-workers reported intramolecular pinacol coupling using electrochemically generated low-valent lanthanide (Figure 38C, top left).604 This process entails the in situ cathodic reduction of Sm(III) or Yb(III) salts to Sm(II) and Yb(II). Redox potentials of lanthanide salts vary with the choice of anions—toward this end, the use of iodide electrolyte for samarium and the use of bromide electrolyte for ytterbium were found to be optimal. This protocol can be adapted for the reductive coupling between carbonyls and Michael acceptors (Figure 38C, bottom left). While unmediated version of such reactions known as the electroreductive cyclization (ERC, vide infra) have been rigorously examined by Little prior to 2000, several more recent reports have also emerged.583,605–607 The scope and limitations of ytterbium mediated reductions have been extensively studied by Pletcher and co-workers.608 Recently, reductive coupling of imines using a soluble samarium anode was reported (Figure 38C, right).609,610 This electrode not only serves as the sacrificial anode to forestall the premature oxidation of radical anion intermediates; it also provides samarium salts that serve as mediators in the reaction.
Aside from homocoupling or cyclization, electrogenerated ketyl radicals could receive another electron and proton to afford the corresponding alcohol. When the protonation takes place in a chiral environment, induction of enantioselectivity is possible. For instance, Yadav et al. reported that direct cathodic reduction of ketones in the presence of dimethylquininium tetrafluoroborate (DMQ·2BF4) led to moderate enantioselectivity (Figure 38D).611,612 It is proposed that the adsorption of DMQ cations onto the mercury cathode is critical for chiral induction.
Electrochemical reduction of 1,2-dicarbonyl compounds has also found applications. Barba and co-workers reported the syntheses of 1,3-dioxoles via reduction of o-quinones (Figure 38E).613 A mechanism comprising electron transfer from the hydroquinone dianion to dichloromethane was proposed instead of simple nucleophilic displacement. Several related studies have also been reported by the same group.614–618
3.2. Reduction of esters and amides
Alkyl esters exhibit high reduction potentials (absolute value); their cathodic reductions are hardly practical until Shono showcased in 1992 that these functional groups can be efficiently reduced with Mg electrodes.619 Some applications of this process are discussed in the paired electrolysis section (section 4.3). Electrochemical reduction of benzoate esters, nonetheless, requires lower reduction potentials (absolute value); these processes have been studied by Markó and co-workers in detail (Figure 39A). Cathodic reduction of toluate esters at elevated temperatures was found to trigger radical deoxygenation (known as the Markó-Lam deoxygenation), analogous to the Barton-McCombie reaction.620,621 Secondary and tertiary toluates gave deoxygenated products in good yields whereas lower efficiencies were observed for primary toluates (Figure 39A(1)). In the same report, it was also demonstrated that the intermediate alkyl radical could be trapped by a pendant alkyne in an intramolecular fashion (Figure 39A(1) middle). A few years later, the same group reported a similar radical deoxygenation reaction using diphenylphosphinates as a radical precursor (vide infra).622 An improved procedure for the deoxygenation of primary alcohols was also reported using a transesterification strategy where an in situ generated toluate ester undergoes cathodic deoxygenation (Figure 39A(1), bottom).623 When reductions of aryl esters were performed at lower temperatures and in the presence of a protic source, hydrolysis products (alcohols) were afforded in lieu of deoxygenation (Figure 39A).624 This observation was exploited to allow the selective hydrolysis of different aryl esters by adjusting the reaction potentials under potentiostatic conditions (Figure 39A(2)). The most electron-deficient di-CF3-phenyl ester was selectively hydrolyzed at −1.6 V while all three ester groups were ruptured at −2.5 V.
Figure 39.
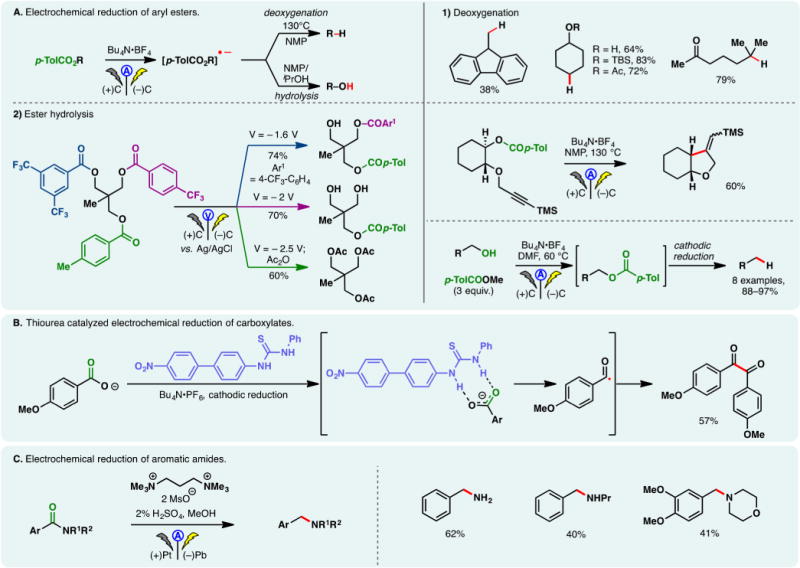
Reduction of esters and amides.
Ozaki and co-workers have reported an electrochemical method to reduce S-(2-methoxycarbonyl)phenyl thioesters. This process was shown to generate an acyl radical which could cyclize onto pendant olefins to forge cyclic ketones.625
Reduction of aryl carboxylates can also be accomplished with the aid of a hydrogen bond donor. In the case illustrated in Figure 39B, hydrogen bonding interactions between the carboxylate and a biphenyl thiourea presumably lowered its reduction potential (absolute value).626 Cathodic reduction then triggered acyl radical formation, followed by dimerization.
As with esters, amides typically exhibit high reduction potentials (absolute value); their electrochemical reduction could only be accomplished in strongly acidic media (e.g., concentrated sulfuric acid).627 Building on a system developed for oxime reduction (vide infra), Waldvogel and co-workers reported a method for the direct cathodic reduction of aromatic amides to amines (Figure 39C).628 Under moderately acidic conditions (2% H2SO4), various primary, secondary, and tertiary amides were efficiently reduced to the corresponding amine products. A lead cathode was used in combination with quaternary ammonium salt additives—these additives suppress the corrosion of the lead cathode as the ammonium cations form a compact layer to prevent interactions between protons and the electrode surface (vide infra).629 In addition to amide reduction, electrochemical reductions of phthalimide630 and sulfoxide628 have also been reported.
3.3. Reduction of C=C double bonds
In general, the electrochemical reduction of unactivated C=C double bonds is difficult due to their high reduction potentials (absolute value). Nevertheless, when double bonds are substituted with electron-withdrawing groups, cathodic reductions can become more practical.631 In the presence of a hydrogen donor, formal hydrogenation of electron-deficient olefins can thus be accomplished.632 For example, an electrochemical method of reducing Michael acceptors has been reported by Tajima and co-workers using a polymer-supported acid (Figure 40A).633,634 Although the acid is constrained to the solid support, its interaction with water confers sufficient conductivity to the reaction media—additional supporting electrolyte is unnecessary. Alternatively, such a reduction can be achieved with hydrogen adsorbed on the cathode surface.635
Figure 40.
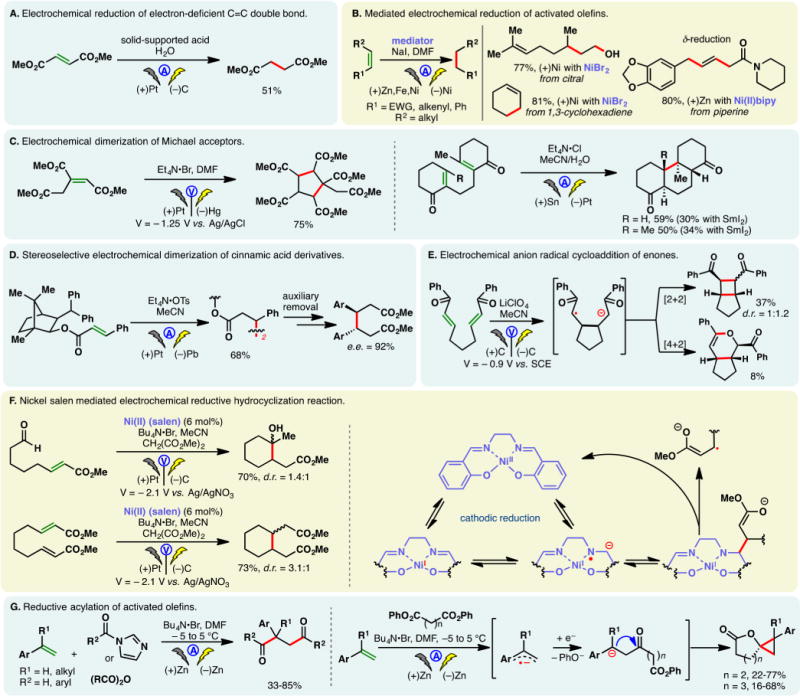
Reduction of activated olefins.
Indirect cathodic reduction of C=C double bonds has also been reported (Figure 40B).636,637 Navarro and co-workers investigated mediated cathodic reductions of cyclohexanone—it was observed that the use of Ni2+ mediators and a Ni anode led to the formation of cyclohexanone whereas employing a Fe2+ mediator in combination with a Fe sacrificial anode afforded cyclohexanol exclusively. Electrochemical reduction of other activated olefins such as Michael acceptors and dienes with nickel- or iron-based mediators have been detailed in a subsequent study by the same group.637 Figure 40B shows that partially hydrogenated products from cyclohexadiene, citral, and piperine were obtained in good yields and selectivities using Ni2+ mediators. Nonconjugated olefins are generally unreactive under such homogeneous electro-mediated reduction (HEMR) systems with the exception of allylic alcohols.
The cathodic reduction of activated olefins involves the formation of radical anions. These intermediates could dimerize (or formally engage another Michael acceptor) to furnish 1,6-dicarbonyls in the same vein as the adiponitrile process (known as the electrohydrodimerization or EHD).638 Electrochemical dimerizations of trimethyl aconitate (Figure 40C),639 methyl cinnamate,640 flavone,641 α,β-unsaturated ketones,642–645 and nitroolefins646 have been accomplished in this fashion. In the case of an intramolecular enone dimerization (Figure 40C, right), electrochemical conditions gave higher yields of electrohydrocyclization (EHC) products compared to the analogous chemical reactions employing SmI2 as the reductant.644 In the reductive dimerization of enones, pinacol couping can compete with the EHD pathway. Handy and co-workers reported a case wherein the use of samarium iodide as a reductant favored the EHD reaction while electrochemical conditions led to exclusively pinacol products.647 Kise and co-workers reported a stereoselective dimerization of cinnamic acid esters (Figure 40D).648 Although the initial efforts with menthol- and borneol-derived chiral auxiliaries led to little stereoselectivity, a bulky chiral auxiliary readily synthesized from (1R)-(+)-camphor was found to be effective; the dimerized product was obtained in a good yield with 92% e.e. upon cleavage of the auxiliary.
Bauld, Krische, and co-workers reported a radical anionic cycloaddition of enones under electrochemical conditions (Figure 40E).649–652 Both [2 + 2] and [4 + 2] products were obtained through constant potential electrolysis. A stepwise anion radical chain mechanism was invoked, involving single-electron reduction of the enones followed by cyclization—a distonic anion radical intermediate was invoked. Similar dimerizations of vinylsulfone have also been reported.653,654
Electrochemical reductions of Michael acceptors can also enable intramolecular reductive coupling with aldehydes or ketones through electroreductive cyclization (ERC) reactions. Little and co-workers described an indirect ERC using a Ni(II) salen complex as the mediator (Figure 40F, top left).655 A Ni(I) species is putatively generated at the cathode; the electron transfer from this Ni(I) complex to the C=C double bond is believed to occur via an inner sphere mechanism based on the experimental observation that substitution at the iminyl carbon of the salen ligand shut down the cyclization (Figure 40F, right). Under this mechanism, a covalent bond is putatively formed between the Michael acceptor and the reduced form of the mediator; homolysis of this bond affords a β-radical anion that can undergo protonation whereupon the resulting β-radical participates in cyclization. Alternative mechanistic possibilities have been discussed as well. Intramolecular electroreductive dimerization of Michael acceptors can also be realized using this system (Figure 40F, bottom left).
Nishiguchi and co-workers reported the reductive carboxyalkylation of activated olefins such as styrenes or Michael acceptors using magnesium as the reductant.656,657 Alternatively, the same group demonstrated that the electron-deficient olefins can be reduced cathodically to the radical anions whereupon reactions with two equivalents of acyl chlorides/acid anhydrides or N-acylimidazoles (Figure 40G, left) afford vicinal bis-acylation products after subsequent reductions.658,659 An analogous geminal double carboalkoxylation of imine derivatives has been achieved in a similar fashion.659 In the reduction of styrenes, the styrenyl radical anion could also react with phenyl diesters where the nucleophilic terminal carbon could add into a carbonyl. The resulting homoenolate was found to undergo cyclization to eventually afford cyclopropyl spirolactones after the loss of a phenol (Figure 40G, right).660
Olefins containing a leaving group at the allylic position can also be subjected to cathodic reduction. For example, Duñach and co-workers reported an electrochemical method to achieve the reductive deprotection of allyl carbamates with a nickel-based mediator.661 The electrogenerated low valent nickel species enabled smooth removal of the allyl group (Figure 41A). Deprotection of allyl carbonates using this system has also been described.662 Hudlicky and co-workers found that cinnamyl group exhibited lower reduction potentials (absolute value) compared to simple allyl groups. Cinnamyl ethers could, therefore, be reduced without a nickel mediator.663 Nevertheless, the high reduction potential (absolute value) necessitated the use of a mercury cathode. As described in Figure 41B, the cinnamyl group was selectively cleaved under electrochemical conditions, while an allyl group in the same molecule remained untouched. Conversely, chemical conditions employing sodium metal ruptured both protecting groups. The electrochemical reduction of cinnamyl esters and carbamates has also been investigated.664,665
Figure 41.
Reduction of allylic systems and aromatics.
The electrochemical reduction of an allyl acetate has been performed on a pilot plant scale en route to Ceftibuten (Figure 41C). The use of a tin cathode was found to be optimal for this reaction; various reducible functionalities such as carboxylates, a β-lactam, and a sulfoxide were left unscathed.666
The direct electrochemical reduction of aromatic systems is conceivably difficult owing to arenes’ electron-rich nature. Nevertheless, early efforts demonstrated that electrochemical conditions could be utilized to generate solvated electrons which could, in turn, enable the Birch reduction of aromatic systems.667–669 To date, conditions have been devised which allow the reduction of arenes in aqueous solutions through similar mechanisms.670–672 Ishifune and co-workers developed an electrochemical Birch-type reduction in tBuOH (Figure 41D, left).673 The use of Mg electrodes was critical. It was postulated that anodically generated Mg(II) mediates electron transfer. Electron-deficient pyridines could also be reduced cathodically. For example, dimethyl dipicolinate could undergo 2-electron or 4-electron reductions under direct electrolysis, depending on the choice of solvent, the amount of electricity, and the temperature of the reaction (Figure 41D, right).674 Electroreductive dimerization of coumarin and its analogs has been studied by Peters and co-workers.675
3.4. Reduction of alkyl halides
While the reduction of alkyl halides could be achieved with low valent metals, electrochemical methods offer a scalable and inexpensive alternative. For example, Waldvogel and co-workers detailed a highly practical method to reductively cleave the C–Br bonds in dibromocyclopropane motifs of complex substrates (Figure 42A).676 A leaded bronze cathode was used, owing to its higher resistance toward corrosion than pure lead. This method found applications in the reduction of a proline-derived dibromocyclopropane, an important intermediate in the synthesis of HCV NS5A inhibitors. Notably, attempts to reduce this substrate through chemical means led to racemization and ring opening. The debromination product was afforded on large scales (>45g) electrochemically; selective monodehalogenation is also possible. The cost-efficiency and functional group compatibility of this method make it amenable for applications in process settings.677 The reaction conditions were also applied in the debromination of a cyclosporin A analog where the product was afforded in a 98% yield.
Figure 42.
Reduction of alkyl halides.
The transformation of vicinal (pseudo)-dihalides to olefins could also be accomplished electrochemically (Figure 42B). For example, the cathodic reduction of alkyl vincinal dibromides using a cobalt salen complex as the mediator in ionic liquid was shown to afford the corresponding olefins (Figure 42B).678 Little and co-workers described a protocol to expediently reduce glycosyl bromides to glycals, using undivided cells and galvanostatic conditions (Figure 42C).679 Various glycals were obtained in good yields using a zinc sacrificial anode and an RVC cathode. This scalable method exhibited compatibility with acid sensitive functionalities (e.g., acetal) which might not have survived chemical reductive conditions using Zn/AcOH. While the reaction was believed to proceed through the direct electrochemical reduction of the C–Br bond followed by the elimination of the neighboring acetate, the involvement of zinc deposits at the cathode cannot be ruled out.
Budnikova and co-workers reported that perfluoroakyl halides could be reduced to fluoroalkyl radicals using nickel mediators under electrochemical conditions (Figure 42D).680 These radicals could engage electron-deficient olefins to afford monomeric or dimeric products.
Electrochemical reductions of benzyl halides have also found synthetic utilities. Utley and co-workers showcased that o-quinodimethane can be conveniently accessed in an aqueous electrolyte solution through the electrochemical reduction of α,α′-dibromo-o-xylene precursor (Figure 42E).681 This reactive intermediate could be used in Diels–Alder reactions with maleimide,681 maleate,682 and naphthoquinone.683 In some cases (e.g., when maleimide is used as the dienophile), the dienophile can also serve as the mediator which undergoes cathodic reduction to afford a stabilized radical anion that can in turn trigger the reduction of the benzylic bromide. The polymerization of p-quinodimethane has also been reported.684 Various mediatory systems have been described for the cathodic reduction of benzyl halides. For example, carborane685 and titanocene686 have been used to effect such transformations. A practical means to conduct the cathodic reduction of benzyl halides using microflow reactors has also been reported.687
Owing to the synthetic utility of allylation reactions, electrochemical reduction of allyl halides has received considerable interest (Figure 42F). Hilt reported a Barbier carbonyl allylation using an indium(III) salt as the mediator.688 An aluminum sacrificial anode was used; intriguingly, it was proposed that the aluminum metal on the electrochemically activated anodic surface promoted the reduction of In(III) to indium metal whereupon an anodic oxidation furnishes a highly reactive In(I) aluminum halide species. In other words, this reductive reaction mainly occurs at the anode. This In(I) can reduce allyl halides, thereby enabling a Barbier reaction. Aldehydes and ketones can both be allylated under the reaction conditions; additionally, esters were shown to undergo bis-allylation. Several years later, an aqueous-phase tin-mediated process was developed which utilizes graphite electrodes.689 Electrogenerated zinc690–692 and samarium(II)609,693 could also facilitate such transformations. Additionally, Durandetti and co-workers reported an iron-catalyzed electrochemical allylation of carbonyl compounds by allylic acetates.694 Navarro and co-workers developed electrochemical conditions for the prenylation of benzaldehyde wherein prenyl halides and benzaldehyde were adsorbed on a graphite powder cathode and reduced in a cavity cell.695 Intriguingly, high α-regioselectivity was observed with a graphite cathode and 3% tetrabutylammonium tetrafluoroborate, whereas the use of 2% silver-doped graphite led to exclusively γ-prenylation. Regioselectivity of electrochemical allylation processes can also be controlled with flow techniques.696,697
The indium-mediated electrochemical Barbier reaction (vide supra) can be extended to achieve imine allylation.698 Analogously, Huang and co-workers developed an efficient electrosynthesis of homoallylic amines from imines and allyl halides (Figure 42G).699 The reaction takes place readily in aqueous solutions, using zinc electrodes under galvanostatic conditions. Benzyl and alkyl halides could also be reduced using this protocol, allowing the benzylation/alkylation of imines. The cathodic zinc deposit formed during the reaction was found to have a significant effect on the reaction efficiency.
Allylic borylation represents another application of cathodic processes involving allyl halides (Figure 42H).700 Duñach and co-workers reported the preparation of allyl boronic esters from allyl halides through direct electrolysis in the presence of HBpin. When unsymmetrical allyl halides are used, the terminal allyl boronic ester was afforded. Borylation of aryl halide has also been reported and will be discussed in the section on aryl halide reduction.701–703 Additionally, electrochemical reduction of allyl halides has also found applications in conjugate additions with α,β-unsaturated esters.704,705
α-Halocarbonyls constitute another class of activated alkyl halides that have been extensively studied in cathodic processes. For example, an iron-mediated electrochemical Reformatsky reaction was described by Durandetti in 2003 using an iron sacrificial anode (Figure 42I).706 A wide variety of aldehydes and ketones are viable electrophiles in this reaction. A cathodically generated Fe(I) species presumably acts as the reductant for α-chloroesters—the cathode potential (−1.1 V vs SCE) corresponds to the reduction of bipyridyl complex of Fe(II). Electrochemical Reformatsky reactions can also be performed in water using a zinc anode;707 analogous processes with graphite cathodes have been reported as well.708,709 Electrochemical reductions of α-halocarbonyls were also utilized to effect the dimerization of phenacyl bromide derivatives710 and to synthesize coumarin.711
Similar to α-halocarbonyls, α,α,α-trichlorocarbonyl compounds could be reduced electrochemically, affording a stabilized dichlorocarbanion that could readily react with carbonyls. Quintanilla recently applied this methodology to the electrosynthesis of halogenated δ-lactones (Figure 42J, top).712 Thus, one of the C–Cl bond was cathodically cleaved to generate a gem-dichloro carbanion species whereupon an ensuing aldol condensation gave the products. Similar processes were exploited in the preparation of 4-amino-2-aryl-2-oxazolines,713 quinolinones,714 and coumarins.715 In another variant, the electrochemical reduction of trichloroacetates was applied to synthesize α-chloroepoxides (Figure 42J, bottom).716 In this case, the gem-dichlorocarbanions generated by direct cathodic reduction of C–Cl bond can undergo a Darzens reaction to give α-chloroepoxides. Chelation of Zn(II) ion in the transition state presumably allowed for high stereoselectivity. Médebielle and co-workers reported an electrochemical method to homolyze the C–Cl bond in chlorodifluoromethyl ketones, enabling the synthesis of heteroarene compounds containing difluoroacyl moieties.717
Vitamin B12 derivatives have been harnessed as redox mediators in the cathodic reduction of trichloromethyl groups. For example, Shimakoshi and Hisaeda reported the conversion of trichlorotoluene to esters and amides using a B12 model complex (Figure 42K).718 From a series of experiments, it was proposed that the reaction was initiated by a single electron transfer from the anodically generated Co(I) species to the substrate. Trapping of the resulting radical with oxygen allows further reactions, giving rise to esters and amides as final products.719
Trichloromethyl and gem-dichloro functionalities can formally serve as carbene equivalents under cathodic conditions. An electrochemical cyclopropanation protocol for electron-deficient olefins was reported by Paugam and co-workers, using copper and iron mediators (Figure 42L).720,721 Pre-electrolysis of CuBr with Fe sacrificial anode produces reactive Cu(0) and Fe(0) deposits on the cathode which were believed to be critical for the reaction. A follow-up study applied this method to synthesize 1-acyl-2,2-diphenylcycloproanes;722 detailed mechanistic study revealed that the reaction likely proceeds via a stepwise mechanism rather than a concerted pathway commonly observed with metal carbenoid species.723 Putatively, a C–Cl bond is initially reduced to generate a carbanion, which undergoes conjugate addition to activated double bond whereupon nucleophilic displacement of a remaining chloride affords the product. It was also found that Fe(II) ion plays an important role in the process, whereas copper may not be as essential. Epoxide synthesis is also possible by using aldehydes or ketones instead of active olefins.724 Cyclopropanation using a B12 model as the mediator was also reported.725 Feasson and co-workers have detailed a synthesis of cyclopropylphosphonates through electrochemical reduction of α,α-dichlorobenzylphosphonate in the presence of Michael acceptors using a magnesium sacrificial anode; a phosphonic analog of minalcipran was prepared in a similar fashion.726 Electrogenerated α-chlorocarbanion was also shown to readily react with triakylboranes to give alkylated products in a one-step reaction.727 Additional, a similar method of cyclopropane formation through nickel-catalyzed electroreductive coupling of unactivated gem-dibromo compounds and electron-deficient olefins has also been reported.728
3.5. Reduction of aryl and alkenyl halides
Similar to alkyl halides, aryl and alkenyl halides could also be reduced electrochemically—the resulting radical anion readily undergoes fragmentation to afford aryl radicals. In the absence of other electrophiles, the direct electrochemical reduction of aryl halides and vinyl halides thus results in the formation of biaryls.729,730 Proto-dehalogenation products are afforded in the presence of a hydrogen atom donor.731,732 Analogous to the allyl halide borylation reaction discussed in the preceding section, borylation of aryl halides can be accomplished through cathodic reduction of aryl halides in the presence of a boron electrophile. Pinacol borane634,702 and trialkyl borates701,703 have both been employed for such transformations. The use of the former allows the preparation of potassium trifluoroborates in one pot (Figure 43A).733 Unprotected phenol and heteroarenes were tolerated under electrochemical conditions, indicating that the reaction is insensitive to the electronic nature of aryl halides.
Figure 43.
Direct and indirect reduction of aryl halides.
Huang and co-workers developed a method for electrochemical hydrodefluoroination of fluoroaromatic compounds based on the cathodic cleavage of C–F bonds.833
Mitsudo and Suga observed the preferential electrochemical methylation of aryl fluorides during their study on halogen-deuterium exchange (Figure 43B, top).734 MeCN putatively provides the methyl group under electrolytic conditions while the deuterated product was formed in a smaller proportion. Intriguingly, when an aryl chloride was used, the reductive dehalogenation pathway predominated. On the other hand, electrochemical halogen deuterium exchange could be achieved efficiently using 9-fluoroenone as a redox mediator (Figure 43B, bottom).
Fluorene derivatives have also been utilized as mediators in the electrochemical reductive cyclization of aryl halides (Figure 43C).735 Aryl chlorides generally gave higher yields than aryl bromides and iodides. Mechanistic studies support the role of 9,9-diethylfluorene as a mediator. MeCN serves as the hydrogen atom donor to terminate this radical cyclization. Similar reductive cyclizations using organohalides as radical precursors have been achieved with phenanthrene736,737 or Ni(II)738–745 mediators instead.
Perylene bisimides have attracted considerable attention in materials science, culminating in applications to organic dyes, electronics, and fluorescent materials. This class of compounds is also known to form stable radical anions after single-electron reduction. This property was utilized by Zhu and co-workers (Figure 43D) to effect the electrochemical dehalogenative-arylation of pyrroles with a perylene-based mediator.746 It is postulated that the radical anion arising from the cathodic reduction of the perylene bisimide could reduce aryl halides to aryl radicals. Similar to aryl halides, aryl diazonium can also be reduced cathodically—this was utilized by Murphy and co-workers to prepare indolines and indoles through radical cyclizations.747 Barba, Batanero, and co-workers have reported the electrochemical conversion of diazonium salts into diaryl sulfides.748
Alternatively, cathodic reduction allows the generation of low valent transition metal species (e.g., Co(I), Ni(I), or Pd(0)) which could insert into aryl halides through oxidative addition, thereby indirectly accomplishing the reduction of C–X bonds. This mode of reactivity is detailed in the following sections.
3.6. Cathodic generation of low valent nickel and cobalt species: reduction of aryl and vinyl halides
The cathodic reduction of Ni(II) complexes offers a convenient means to access Ni(0) species that demonstrate a high propensity toward oxidative addition with aryl halides. This mode of reactivity has been reviewed by Nédélec, Périchon, and Troupel in 1997749 as well as by Duñach in 2003;750 some recent advances will be recounted herein. Condon and co-workers reported a nickel-catalyzed electroreductive conjugate addition wherein an electrogenerated Ni(0) species undergoes oxidative insertion into aryl halides to generate an aryl-nickel intermediate (Figure 44A).751 The reaction between this aryl-nickel complex and an activated olefin can then afford 1,4-addition products in a reductive Heck-type process. Both aryl bromides and aryl chlorides were found to be suitable substrates, although a higher temperature was required for the latter (100 °C) compared to the former (70 °C). In addition, sequential reactions between p-dibromobenzene and two different Michael acceptors were demonstrated. An iron sacrificial anode was used, providing iron ions that putatively cooperated with the coupling process to improve the yields. This type of electrochemical conjugate addition may be carried out in ionic liquids;752 the use of heteroaryl halides753 as substrates has also been reported. Additionally, formal 1,4-addition of aryl halides to acrolein can be achieved electrochemically via a nickel catalyzed reductive coupling between aryl halides and acrolein diethyl acetal.754 Hydrolysis of the resulting enol ether affords β-arylated aldehydes. Nickel-catalyzed electroreductive conjugate addition of aryl bromides has also been applied to synthesize medium-ring aromatic lactones.755
Figure 44.
Indirect reduction of aryl halides with transition-metal mediators.
Unsurprisingly, aryl-nickel species generated in this fashion could be harnessed in many chemical transformations besides conjugate additions. For example, alkyl aryl ketones can be afforded when the aryl-nickel intermediate is generated in the presence of Fe(CO)5 and an alkyl halide (Figure 44B).756 Alternatively, the aryl-nickel complex could undergo transmetalation with chromium salts to enable the Nozaki–Hiyama–Kishi (NHK) reaction (Figure 44C).757 A practical protocol for this reaction was reported where the transition metal catalysts were generated in situ through the pre-electrolysis of a stainless steel anode.
As with aryl halides, alkenyl halides could undergo similar electrochemical transformations to afford alkenyl nickel complexes—these reactive intermediates may be used in cross-electrophile couplings of various modalities (Figure 44D).758 Mechanistically, the oxidative insertion of an electrogenerated Ni(0) catalyst into C–X bonds gives rise to a Ni(II) complex which could undergo cathodic reduction to yield an aryl-Ni(I) intermediate. This intermediate can participate in a second iteration of oxidative addition whereupon a reductive elimination affords the coupling product (see Figure 44E for a simplified mechanistic picture).554 By judiciously choosing halides with different reactivies and through the slow addition of one component, cross-coupling products can be favored over homodimerization. As depicted in Figure 44D, alkenyl halides could be coupled with activated alkyl halides and heteroaryl halides. In the absence of an additional coupling partner, dimerization of alkenyl halides could occur. Electrochemical homocoupling of (hetero)aryl halides (such as 2-halopyridines) through this mechanism has been reported.759,760
Cross-couplings between aryl halides and heteroaryl halides through similar mechanisms have also been reported.761–764 Various nitrogen-rich heterocycles, which are challenging targets for conventional Suzuki coupling reaction, were shown to undergo electroreductive couplings in good yields (Figure 44F).762 The success of the electrochemical conditions was explained by the presence of Fe ions (from the pre-electrolysis using an iron sacrificial anode). Coordination of the nitrogen atom to Fe ion ensures efficient regeneration of nickel catalyst. Very recently, Hansen and co-workers reported an electrochemical method for the cross-electrophile coupling between aryl halides and unactivated alkyl halides.765 As another application of nickel catalyzed electroreductive transformations of aryl halides, silylation of aryl bromides was reported.766
Complexes of cobalt and pyridine exhibit similar reactivity with nickel in that they readily undergo cathodic reduction to generate low valent Co species which are capable of oxidative insertion into aryl C–X bonds. The resulting organocobalt complex can undergo transmetalation with zinc salts, offering an efficient means to prepare organozinc reagents (Figure 44G).767 A practical protocol using catalytic amounts of cobalt was reported. This electrochemical method obviated the need for organolithium or organomagnesium reagents; this is particularly beneficial for aryl halides bearing reactive functional groups such as esters and ketones which showed compatibility with the electrochemical protocol. Arylzinc reagents thus obtained can engage electrophiles such as acyl-pyridiniums768 or acyl chlorides.769 Alternatively, they could be used in Negishi couplings.767 Similar to their nickel counterparts, cobalt(II) pyridine complexes could facilitate the cross-electrophile coupling of aryl halides with allyl acetates/carbonates,770 alkenyl (pseudo)halides,771–773 and (hetero)aryl halides774,775 under cathodic conditions (Figure 44H). A similar mechanism comprising two oxidative addition events is most likely operative.
3.7. Cathodic reduction in palladium catalysis
As is the case with anodic oxidations (Section 2.16), cathodic reductions have also been exploited to facilitate palladium catalyzed reactions. Pd(0) can undergo oxidative addition with organohalides; under cathodic conditions, the resulting Pd(II) complex can be readily reduced to Pd(0), allowing a second iteration of oxidative addition and thereby enabling the coupling between two organohalides. The electroreductive homocoupling of aryl halides has been reported by Torii in the 1980s,776 and its mechanism was elucidated by Jutand to comprise two oxidative additions as briefly described above.777 Recently, Rothenberg and co-workers reported an electroreductive homocoupling of aryl halides which proceeds at room temperature in an ionic liquid (Figure 45A).778 Using a palladium anode in an undivided cell, palladium nanoparticles, putatively the reactive catalytic species in this reaction, can be generated under galvanostatic conditions.
Figure 45.
Cathodic reduction in palladium catalysis.
Tanaka and co-workers have developed pyridinium-based mediatory systems to effect the reduction of Pd(II) to Pd(0) in the cathodic homocoupling of aryl halides (Figure 45B).779,780 In the initial report, an N-octyl-viologen dicationic salt ([OctV2+]) was chosen as the mediator; it could be readily converted to the neutral form [OctV0] under electrolysis. Two equivalents of [OctV0] could then reduce Pd(II) to Pd(0), priming it for oxidative addition while generating two equivalents of [OctV+]. Although a different redox cycle ([OctV2+]/[OctV+]) might be possible, mechanistic studies revealed that the [OctV0]/[OctV+] cycle is primarily responsible for the reduction of palladium. Premature reoxidation of the palladium catalyst can be prevented with a sacrificial zinc anode. Electron-neutral and electron-deficient aryl bromides were dimerized in high yields; nevertheless, the protocol is less efficient for electron-rich substrates. Later, N-alkyl-4-alkoxycarbonylpyridinium781 and diquat derivatives782 were shown to be competent mediators for this electroreductive homocoupling reaction.
Recently, Huang and co-workers reported an electrochemical cross-coupling between alkyl halides (benzyl bromides and alkyl iodides) and allyl halides under palladium catalysis using a sacrificial zinc anode (Figure 45C).783 The reaction is conducted in an aqueous solvent under air. Although the mechanism is not entirely clear, it is proposed that anodically generated Zn2+ undergoes cathodic reduction, generating Zn metal which can reduce the alkyl halide to an organozinc species. Despite the proposed intermediacy of the organozinc nucleophile, the reaction was found to be compatible with aqueous solvent systems. Functional groups such as carboxylic acids, alcohols, and esters were also tolerated.
Moeller and co-workers have developed a series of site-selective palladium mediated reactions on electrochemically addressable, semiconducting chips.784 In these processes, Pd(OAc)2 is reduced on preselected electrodes of the chip, generating Pd(0) species which initiates cross-couplings such as the Heck reaction. The region surrounding the chosen electrode is submerged with a solution containing allyl methyl carbonate—any Pd(0) diffusing away from the electrode would be converted to inactive Pd(II). Thus, the Pd(0) species is spatially confined. Although the chip-based reactions per se are not in the scope of this review, while studying these processes, Moeller and co-workers discovered that preparative solution-phase Heck reactions could be substantially accelerated through the passage of electric current (Figure 45D).785 The electrochemically assisted Heck reaction can be conveniently carried out at room temperature under simple galvanostatic conditions in undivided cells. The use of strong ligands is not necessary. Control experiments indicated that the analogous chemical processes occur at much slower rates and oftentimes in lower yields. It is believed that the circulation of electric current constantly regenerates catalytically active Pd(0) species. Polymerization of Pd(0) to form “palladium black”, a common problem in palladium catalysis, is thus ameliorated.
3.8. Cathodic carboxylation reactions
Electrochemical “CO2 fixation” reactions have attracted considerable interest as cathodic reduction enables practical means to introduce CO2 onto olefins, alkynes, and carbonyls; electrochemical protocols have also been devised to transform organohalides into the corresponding carboxylates. Recent advances on this front has been reviewed by De Vos and co-workers.786 Electrochemical carboxylation usually involves the reduction of substrates and/or carbon dioxide, leading to the formation of carboxylate anions. Such reactions are commonly carried out using sacrificial anodes wherein the oxidative dissolution of anode materials provides the counterion while preventing the undesired oxidation of substrates or products.
3.8.1. Cathodic carboxylation of olefins, alkynes, and carbonyls
Senboku and co-workers reported a method to synthesize succinic acid derivatives through the dicarboxylation of styrenes (Figure 46A).787 Generally, dicarboxylated products were obtained in good to excellent yields using a Pt cathode and a Mg sacrificial anode. Cyclic voltammetry studies indicate that CO2 and styrenes have similar reduction potentials: thus, two reaction pathways are possible. For styrenes substituted with electron neutral or donating groups, the reduction of CO2 (−2.53 V vs Ag/Ag+) occurs preferentially whereupon the resulting CO2 radical anion can add onto the olefin.788 Conversely, for styrenes with lower reduction potentials (absolute value) than CO2, olefin reduction predominates and the resulting carbanion can be trapped by CO2. Both processes can be operative at the same time. The effects of electrodes, electrolytes, temperature, and other factors have been examined in follow-up studies.789,790
Figure 46.
Electrochemical carboxylation of alkenes, alkynes, and carbonyls.
Similar to styrenes, the cathodic reduction of 1,3-butadiene derivatives also affords dicarboxylation products.791–793 For example, 1,3-butadiene could be cleanly converted to 3-hexenedioic acid, an adipic acid precursor (Figure 46B).791 Although the precise mechanism is unclear, it is probable that cathodic reductions of both CO2 and the diene are occurring. Electrochemical carboxylation of butadiene can also be accomplished with the aid of a nickel-based mediator which can facilitate the one electron reduction of CO2.794
In addition to styrenes and dienes, the electrochemical carboxylation of Michael acceptors has also been examined. These electron-deficient olefins typically exhibit reduction potentials (absolute value) lower than that of CO2. Cathodic carboxylation of these substrates, therefore, proceeds primarily through the reduction of the activated olefin followed by trapping with CO2. The reductive carboxylation of ethyl cinnamate provides a representative example (Figure 46C).795–797 Efforts have been expended to render similar processes stereoselective.798 Moderate diastereoselectivity was observed using a chiral oxazolidinone auxiliary containing a bulky diphenylmethyl group. The carboxylation of bicyclo[N.1.0]alkylidene derivatives has also been reported.799
Alkynes undergo different modes of cathodic carboxylations depending on the reaction conditions. For example, Duñach reported the carboxylation of terminal alkynes to afford propionic acid derivatives (Figure 46D, top).800 The use of a silver cathode was critical for this transformation. It was suggested that the reaction proceeded via the deprotonation of terminal alkyne by an electrogenerated base (EGB) whereupon the ensuing acetylide was trapped with CO2—internal alkynes were not carboxylated under the reaction conditions. The strong interaction between the silver cathode and electrogenerated acetylides was invoked to account for the selectivity. An earlier report posited that hydrocarboxylation products were furnished with Markovnikov selectivity when terminal alkynes were subjected under electrolytic conditions in the presence of a Ni(II) mediator, a magnesium sacrificial anode, and a carbon cathode.801 Cathodically generated Ni(0) species can presumably interact with alkynes and CO2 to form a metallocycle. Dicarboxylation of terminal alkynes can be achieved under different reaction conditions as described by Jiang and co-workers. Maleic anhydride derivatives are obtained as products (Figure 46D, bottom).802 However, when catalytic amounts of CuI were introduced into the system (with higher CO2 pressure), a tricarboxylation product was furnished instead.803 Cyclic voltammetry studies suggested that the reductions of alkynes and CO2 occur at similar potentials. Therefore, CO2 radical anions are likely to be present under the reaction conditions. It is thus postulated that the copper salt can coordinate to the double bond of the dicarboxylate intermediate, facilitating the attack of CO2 radical anions to the double bond. CuI can also coordinate to CO2 to render its reduction potential (absolute value) lower.
Carboxylate groups can also be appended onto aromatic rings under electrochemical conditions.804 Yuan and co-workers reported a method to achieve dearomative carboxylation of polycyclic aromatic compounds such as naphthalene and anthracene (Figure 46E).805 Arene and CO2 likely undergo reductions simultaneously. The ensuing CO2 radical anion can react with arenes; likewise, arene radical anions can engage CO2. In the case of naphthalene, only a trans-dicarboxylic acid product was afforded. Reductive carboxylation of flavones is another example of electrochemical arene carboxylation (Figure 46E, bottom)—flavanone-2-carboxylic acids were afforded regioselectively.806,807
Reductive carboxylation of ketones is perhaps the most efficient way to synthesize α-hydroxycarboxylic acids. These structural moieties can be accessed from the corresponding cyanohydrins whose preparations require hazardous cyanide nucleophiles. A recent report by Yuan and co-workers detailed a protocol for the reductive carboxylation of ketones (Figure 46F, top).808 Although the scope of substrates is limited to aromatic ketones, α-hydroxycarboxylic acids were generally obtained in good yields together with small amounts of pinacol byproducts. Since the reduction potentials of ketones are close to that of CO2, it is proposed that the reduction of both CO2 and ketones could occur under galvanostatic conditions. Protocols using silver electrodes809 or ionic liquids810 have also been reported. Stereoselective electrochemical carboxylation of ketones could be achieved using a catalytic amount of cinchona alkaloids, and phenol—moderate enantioselectivities were observed (Figure 46F, right).811
In addition to reductive C–C bond formation at the carbonyl C=O, reductive carboxylation at the α-position of carbonyl functionalities was also reported (Figure 46F, bottom).812 Cyclic and acyclic aliphatic ketones were converted to the corresponding β-ketoacids in moderate to good yields. It is believed that CO2 is reduced to its radical anion under the reaction conditions; this radical anion species can facilitate enolate formation.
3.8.2. Cathodic carboxylation of halogenated compounds
As discussed in section 3.4, the direct electrochemical reduction of a carbon–halogen bond gives rise to a radical anion which readily undergoes C–X bond fragmentation. Reactions between the resulting radical or anion with carbon dioxide enable access to carboxylation products. In this context, Genarro and co-workers reported a reductive carboxylation of benzyl chlorides using a silver cathode (Figure 47A, left).813 It was found that, compared to other cathode materials, silver effectively lowered the reduction potentials (absolute value) of benzyl chlorides by approximately 0.6 V while the reduction potential of CO2 remains relatively unchanged. This enabled selective reduction of benzyl halides in the presence of CO2, minimizing side reactions caused by the reduction of CO2 while bolstering current efficiency. Several other studies on electrochemical carboxylations of benzyl halides using silver cathode have been published.814–816 An analogous cathodic carboxylation of alkyl halides has also been reported.815 Racemic817,818 and enantioselective819 syntheses of aryl-2-propionic acid derivatives, which are important drug motifs, could be accomplished via the reductive carboxylation reactions. Moreover, supercritical CO2820 and microemulsions821 could be utilized for such processes. Aside from halides, it has been reported that benzyl alcohols822 and benzyl carbonates823 could also serve as substrates in electrochemical carboxylation reactions.
Figure 47.
Electrochemical carboxylation of organohalides.
Like benzyl chlorides, benzylic flourides could undergo cathodic carboxylation with CO2. For example, galvanostatic electrolysis of α,α-difluorotoluene derivatives in the presence of CO2 gives α-fluorinated arylacetic acid derivatives, including fluorinated analogs of nonsteroidal anti-inflammatory drugs (Figure 47A, right).824 These compounds were previously synthesized using hazardous fluorinating agents such as diethylaminosulfur trifluoride (DAST), perchloryl fluoride (FClO3), and acetyl hypofluorite (AcOF).
Cathodic carboxylation of α-halocarbonyl compounds allows access to 1,3-dicarbonyl products. This process may be rendered stereoselective with chiral auxiliaries, as shown by Feroci and Inesi (Figure 47B).825,826 Stereoselective dehalogenative carboxylation protocols employing β-cyclodextrin supramolecular assemblies827 and chiral cobalt salen-mediators have also been reported.828
Furthermore, the reductive carboxylation of alkenyl and aryl (pseudo)halides may be achieved electrochemically. Senboku studied the cathodic carboxylation of vinyl triflates and bromides (Figure 47C, left).829 When triflates derived from 1,3-dicarbonyl compounds or phenyl-substituted vinyl triflates were used, the OTf groups were formally replaced by carboxylates with mostly retention of the E,Z-geometry. When vinyl triflates derived from acyclic aliphatic ketones were subjected to the reaction conditions, β-ketoacids were obtained instead of the expected α,β-unsaturated carboxylic acids.830 Carboxylation of E- and Z-styrenyl bromides, on the other hand, both afforded the Z-product predominantly.829 Putatively, it was proposed that vinyl triflates undergo a direct two-electron reduction more readily than the bromides, leading to the formatting of vinyl anions; meanwhile, vinyl bromides are presumably reduced to vinyl radicals first—these radical species are more prone to stereo-chemical inversion. Stereoretentive electrochemical carboxylation of β-bromostyrene may be achieved using NiBr2·bpy as the mediator.831 The nickel-catalyzed electrochemical carboxylation could also be applied to convert enol triflates into cyclic α-alkoxyl-α,β-unsaturated carboxylic acids.
The electrochemical carboxylation of pentafluoroarenes has also been investigated (Figure 47C, right).832 These reactions proceed with para selectivity. Although the exact reason for this selectivity is not clear, it was proposed that the para-position experiences the strongest inductive effect from the fluoride substituents, leading to the formation of the most stabilized carbanion.
Senboku et al. choreographed a series of cyclization–carboxylation cascades using an aryl halide as the radical precursor and methyl-4-tert-butylbenzoate as the mediator (Figure 47D).834,835 Cathodic reduction of the aryl ester putatively provides a radical anion which could facilitate the cleavage of C–X bonds in aryl halides; the ensuing radical species can undergo cyclization whereupon the resulting intermediate can participate in carboxylation processes. In a recent example depicted in Figure 47D, the cyclization of an aryl radical onto an alkyne was followed by the fixation of two molecules of carbon dioxide.836 Five- and six-membered heterocycles could be constructed efficiently; subsequent dicarboxylation afforded the corresponding dicarboxylic acids in moderate to good yields.
3.9. Reduction of miscellaneous functional groups
Aside from the examples discussed above, cathodic reductions of other functional groups have been reported within the chosen time frame of this review. For example, advances have been made in the electrochemical reduction of N–N bonds. The reduction of hydrazides into the corresponding amides requires metal catalyzed hydrogenolysis or dissolving metal conditions. In 1978, Horner and co-workers reported the first example of using electrochemistry to convert alkyl and acyl hydrazides into the analogous amides.837 Due to the high reduction potential (absolute value) required, a mercury pool cathode was used to avoid competing hydrogen evolution at the cathode surface. In an effort to render this protocol more environmentally friendly, Breinbauer developed a new process that employs a tin cathode instead (Figure 48A, left).838
Figure 48.
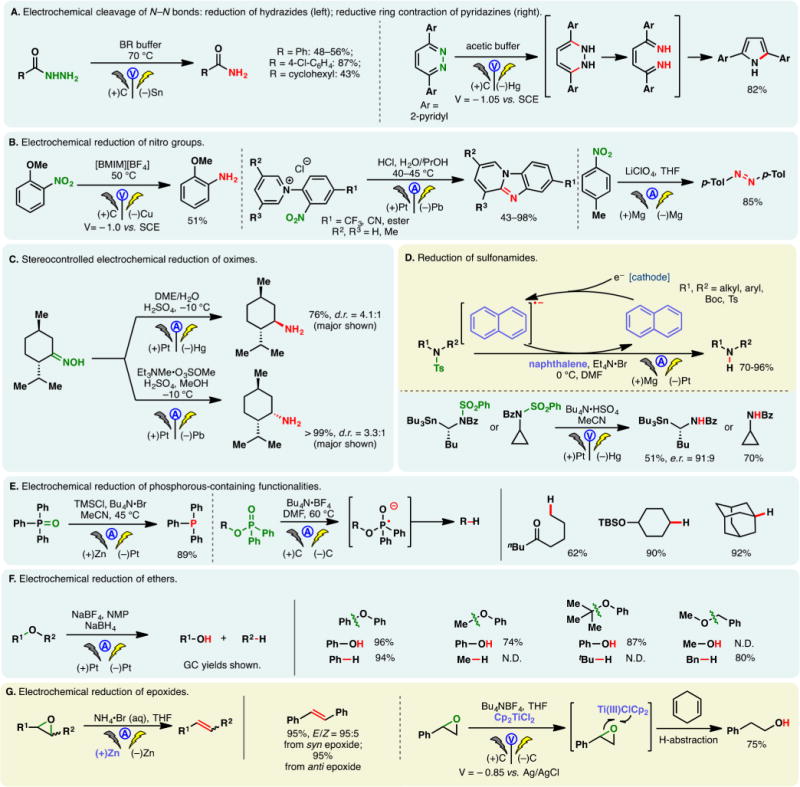
Reduction of miscellaneous functional groups.
Lund explored the electrochemical reduction of pyridazinium salts in 1967, affording pyrroles as products.839 More recently, Pradère and Dubreuil studied the cathodic reduction of pyridazines (Figure 48A, right). Pyrdazines bearing two pyridyl substituents840 and those substituted with two ester groups841–843 at the 2- and 5-positions were subjected to potentiostatic electrolysis using a mercury pool cathode. Pyrroles were afforded in moderate yields.
The reduction of nitroarenes to anilines can also be accomplished electrochemically (Figure 48B); there has been recent interest to conduct the reaction in ionic liquids.844,845 The stepwise cathodic reduction of nitro groups was studied by Haber as early as 1898.846 In an example by Syroeshkin and co-workers, a nitro-arene was reduced in the presence of a pyridinium moiety, allowing the synthesis of pyrido[1,2-a]benzimidazoles.847 Mechanistic studies suggest that the reaction involves the formation of a hydroxylamine intermediate followed by heterocyclization and rearomatization. Kim and co-workers reported a method to reduce nitrobenzenes to azobenzenes with magnesium electrodes.848 Peters and co-workers have reported an electrochemical variant of the Leimgruber-Batcho reaction to synthesize indoles from o-nitrostyrenes through cathodic reduction of the nitro group.849
In 1967, Fry and co-workers studied the electrochemical reduction of camphor and norcamphor oximes with a mercury pool cathode.850 The reductions proceeded in high stereoselectivity; intriguingly, the products showed opposite stereochemistry compared to those formed with dissolving metal reductions (but the stereoselectivity was similar to that of LAH reduction). It was postulated that the substrate could covalently interact with the electrode surface through the formation of organomercury species. Recently, Waldvogel and co-workers probed the cathodic reduction of menthone oxime (Figure 48C).851,852 With a mercury pool cathode, (−)-menthylamine was afforded selectively. However, the stereoselectivity may be shifted to favor (+)-neomenthylamine through the use of a lead cathode in conjunction with a quaternary ammonium salt additive (triethylmethylammonium methylsulfate, MTES, was found to be the optimal choice) at low temperatures. This ammonium salt is crucial as it prevents the corrosion of the toxic cathode material through the formation of a compact salt layer to block the access of protons to the cathode surface. The drain of electric current by H2 evolution is thus prevented. This method was generalized to enable the reduction of menthone oximides with an additional substituent at C8.852
Arenesulfonyl groups such as toluenesulfonyl are useful protecting groups for primary and secondary amines. While electrochemical conditions for their removal have been known since the 1960s, classical examples of cathodic tosyl amide reduction enlist the mercury pool electrode.853,854 Senboku and co-workers developed a mild method for the tosyl deprotection of N,N-disubstituted sulfonamides using naphthalene as the mediator (Figure 47D).855 A naphthalenide is presumably generated in situ. This mediatory system obviated the need for a mercury cathode while tolerating sensitive functionalities such as aziridine. Silvestri reported the detosylation of tetratosylcyclen using carbon cathodes under direct electrolysis.856 Grognec, Quintard, and co-workers described electrochemical conditions for the deprotection of N-phenylsulfonyl N-substituted amines.857 However, when this protocol was extended to cyclopropylamine derivatives, ring fragmentation products were observed. In a follow-up study, they discovered that by introducing a benzoyl group on the sulfonamide nitrogen—the reduction potential of the sulfonamide may be lowered (absolute value) considerably, allowing deprotection to occur without β-fragmentation (Figure 48D, bottom).858 Notably, an α-stannane group was also found to survive the reaction conditions; minimal epimerizations were observed when this method was applied to chiral amines. Cathodic conditions were also utilized by Médebielle and co-workers to remove a sulfone group in a nucleoside derivative through the cleavage of the C(sp3)–S bond.859
Electrochemical methods have found applications in the reduction of pentavalent phosphorus to trivalent species. Cathodic reduction of phosphine oxide has been explored in the 1960s.860 However, as the P=O bond is strong compared to the P–C bonds, phosphine could not be obtained in practical yields; instead, some P–C bond cleavage products were observed. Tanaka devised a workaround by activating the phosphine oxide with TMSCl—interaction of TMSCl with the oxygen atom presumably weakens the P–O bond, allowing efficient reduction of triphenyl phosphine oxide to the corresponding phosphine in good yields.861 The same group also investigated the reduction of triphenylphosphine dichloride and methoxytriphenylphosphonium triflate (Figure 48E, left).862,863
Markó and co-workers studied the cathodic reduction of diphenylphosphinate esters (Figure 48E, right).622 It was discovered that the intermediary radical anion could undergo fragmentation to homolytically cleave the C–O bond. Deoxygenated products can thus be afforded. Primary, secondary, and tertiary alcohols could all be converted into the corresponding diphenylphosphinates where galvanostatic electrolysis with graphite electrodes allowed deoxygenations to occur. Compared to deoxygenation reactions employing toluates (section 3.2),621,864 the use of phosphinates allowed a lower temperature and higher current density. Notably, functional groups such as ketones were left unscathed.
Huang and co-workers developed a method to cleave C–O bonds in aryl and benzyl ethers electrochemically (Figure 48F).865 In the 1980s, Kariv-Miller has used a mercury cathode to effect the reductive cleavage of aryl ether linkages through a Birch-type process;866 together with several other subsequent studies,867–870 these early examples required oxygen-free conditions.865 To this end, Huang et al. discovered that NaBH4 could promote the C–O cleavage in an undivided cell under galvanostatic electrolysis where minimal precautions are necessary. Subjecting diaryl ethers to the reaction conditions yielded a phenol and an arene while reaction of aryl alkyl ethers afforded a phenol and alkane. For benzyl ethers, the benzylic C–O bond was selectively cleaved. Although the exact mechanism is unclear, it was postulated that the NaBH4 could serve two purposes: first, it could help prevent the anodic oxidation of phenol products in the undivided cell; second, it could function as a hydrogen atom donor to quench some of the radical species formed in the reaction. In the reduction of diphenyl ether, deuterium labeling experiments suggested that NaBH4 contributed about 20% of the hydrogen atoms transferred to the intermediary phenyl radical. This technology was applied to break the β-O-4, α-O-4, and 4-O-5 lignin model compounds.
Huang and co-workers also developed a cathodic protocol for the deoxygenation of epoxides, furnishing the corresponding olefins (Figure 48G, left).871 The use of zinc electrodes is critical—this reaction is putatively mediated by Zn(0) generated in situ through the sequential oxidation and reduction of the electrode material. Such zinc deposits were found to adopt a hierarchically organized nanostructure. Lower yields were obtained using commercial zinc powder. Electrochemical reduction of epoxides can also be achieved with a titanocene mediator—in this case, cathodically generated titanium(III) intermediate can trigger a Nugent–RajanBabu reaction (Figure 48G, right).872
3.10. Cathodic generation of oxidants
Molecular oxygen exhibits a low reduction potential (absolute value)—thus, cathodic reduction of O2 offers a synthetically useful means to generate reactive peroxide or superoxide species (Figure 49A).873,874 For example, Chan and co-workers demonstrated that the cathodic reduction of O2 in a mixture of water and an ionic liquid gives rise to hydrogen peroxide via a superoxide intermediate.875 The H2O2 thus generated can then effect Wharton-type epoxidation of enones in situ. Through the inclusion of a Mn2+ catalyst in the reaction system, this method was adapted by the same group as a more general means for olefin epoxidation (Figure 48A).876 Water-soluble alkenes were epoxidized in good yields and current efficiencies; epoxidation of more lipophilic olefins may be achieved through the addition of tBuOH to the solvent mixture. Murray and co-workers were the first to describe such an electrocatalytic approach for epoxidation with electrogenerated H2O2 and a manganese meso-tetraphenylporphyrin catalyst in 1990.877
Figure 49.
Cathodic generation of oxidants.
One electron cathodic reduction of O2 to superoxide has also been exploited in a preparative setting. Casadei and co-workers reported a method for alcohol oxidation using electrogenerated superoxide in combination with CO2 (Figure 49B).878 Primary or secondary benzylic as well as primary allylic alcohols were oxidized into the corresponding aldehydes or ketones in an excell process wherein a solution containing electrogenerated superoxide was added to the substrate. Barba used cathodically generated superoxide to achieve the hydroxylation of aromatic systems.879 The scope of this process is limited to electroreducible arenes with a relatively low (absolute value) reduction potential such as naphthoquinone. Under electrolytic conditions, these arenes are reduced to the corresponding radical anions while superoxide is being generated. Radical recombination then allows the formation of hydroxylated products (Figure 49B, right column). Yoshida and co-workers transformed 4-cyanocinnolines into 4(1H)-cinnolones through this method880 while Fry and co-workers have utilized cathodically generated superoxide ion to induce the desilylation of α-dimethylsilyl esters.881
Takeya and co-workers investigated the electrochemical allylic hydroxylation of cholesteryl acetate under aerobic conditions (Figure 49C).882 Iron picolinate complexes which have been extensively examined in Barton-Gif883 reactions were found to be the optimal catalysts. The allylic hydroxylation product was obtained in moderate yields for cholesteryl acetate and 3-O-acetyl-oleanolate. A complex mechanism was proposed involving both cathodic and anodic processes.
In the 1980s, Minisci reported that Ti(III) could react with hydroxylamine to generate free NH2 radicals.884 Recent efforts by Lisitsyn and co-workers revealed that the NH2 radical could be generated cathodically from hydroxylamine with a Ti(IV) mediator (Figure 49D).885–890 Based on this mode of aminyl radical formation, Lisitsyn achieved amination of olefins, electron-rich arenes, and the α-position of carbonyls. These reactions generally take place under constant current electrolysis in divided cells using a mixture of aqueous H2SO4, acetonitrile, and AcOH as the solvent.
4. PAIRED ELECTROLYSIS
As alluded to in the Introduction, each electrochemical process can be construed as a combination of two half-reactions, oxidation and reduction. Typically, only one of these reactions involves substrates of interest while the other entails the redox transformation of solvent, electrolyte, or other sacrificial species. Paired electrolysis refers to choreographed electrochemical processes where two desirable half reactions are performed simultaneously. The expenditure of electrical power to oxidize/reduce the sacrificial species is averted, thereby maximizing the energy efficiency. This is especially critical in industrial settings.
4.1. Parallel paired electrolysis
Parallel paired electrolysis involves two simultaneous and noninterfering half reactions.891 The most important process in this category is probably the Chlor-alkali process wherein chlorine and sodium hydroxide are formed at the anode and cathode, respectively.892 Millions of tons of NaOH and chlorine have been produced in this fashion.
However, the utility of parallel electrolysis remains under-appreciated in organic synthesis and fine chemical production. This may be ascribed to the misconception that half reactions of “matching potentials” need to be selected. Under galvanostatic conditions, the working potentials of both anode and cathode automatically adjust to the potentials of substrates in solution. Thus, any oxidations and reductions may be paired in a parallel fashion. Kubiak, Moeller, and co-workers recently reported an example where the anodic synthesis of benzimidazole derivatives is combined with the cathodic production of carbon monoxide in a divided cell (Figure 50A, top).893 The anodic process utilized a cerium mediator to facilitate the oxidative condensation between syringaldehyde derived from lignin sawdust and 2-amino aniline. In the cathodic chamber, a rhenium mediator was employed to reduce CO2 to CO. These two seemingly unrelated reactions could be conducted simultaneously, allowing the conservation of electrical energy. Zha, Wang, and co-workers detailed a paired system where halide mediated alcohol oxidation and tin mediated ketone allylation in a divided cell concurrently (Figure 50A bottom).894,895 The same group also reported a protocol to carry out this homoallylic alchol synthesis using an undivided cell, thereby achieving a convergent paired electrolysis (vide infra, section 4.3).896 Fuchigami and Atobe has developed a parallel paired electrolysis system using a microflow reactor—reduction of benzyl halides was combined with the oxidation of secondary benzyl alcohols.897
Figure 50.
Parallel paired electrolysis.
A common starting material can undergo reduction and oxidation, leading to two products in a divergent fashion (termed divergent paired electrolysis by Frontana-Uribe and co-workers).891 For example, De Vos and co-workers devised a system to simultaneously synthesize diacids and diol derivatives from the electrolysis of dienes in the presence of CO2, TFA, and trimethylamine (Figure 50B, top) in an undivided cell.898 The diene can undergo two consecutive anodic oxidations to yield acyl-protected diols. At the cathode, the same diene can undergo reductive carboxylation with CO2, furnishing diacid products. In an earlier example by Atobe, anodic olefin dibromination with electrogenerated Br+ was combined with vanadium catalyzed cathodic epoxidation through the reduction of O2 (Figure 50B, bottom).899 Another example of divergent paired electrolysis is the synthesis of L-cysteine and L-cysteic acid from L-cystine.900
When the same product is produced in both anodic and cathodic reactions, this paired electrolytic process is denoted a linear paired electrolysis.891 The synthesis of D-arabinose from sodium gluconate provides an example—in the catholyte, this oxidative decarboxylation was mediated by iron salts and HO• free radicals while the anodic process is a direct decarboxylation.901
4.2. Sequential anodic oxidation and cathodic reduction
A substrate molecule can undergo anodic and cathodic transformations sequentially in an undivided cell. For example, Waldvogel recently reported an electrochemical method to transform aryl aldoximes to aryl nitriles in a single pot (Figure 51A), accomplishing a formal loss of water.902 Oxidation of the aldoxime occurs at the anode to afford a nitrile oxide which could then undergo cathodic reduction with the lead electrode, furnishing the aryl nitrile product. This reaction utilizes simple galvanostatic conditions with inexpensive electrode materials.
Figure 51.
Sequential anodic oxidation and cathodic reduction.
Nishiyama’s synthesis of biaryl ethers through oxidative C–O dimerization of ortho-dihalo-phenols followed by cathodic reduction was discussed earlier.903 Recently, an improved protocol was reported through linear paired electrolysis, allowing the transformation of phenols to biaryl ethers in a single step (Figure 51B). The use of a microreactor was deemed advantageous, as the short distance between the electrodes in the microreactor enables fast molecular diffusion from anode to cathode.
4.3. Convergent paired electrolysis
Convergent paired electrolysis arises when anodically and cathodically generated intermediates react with each other to afford the product. Yuan and co-workers detailed a method to synthesize amides from methyl ketones and formamides in this manner (Figure 52A).904 The methyl ketone reacted with anodically generated iodine to afford a α,α,α-triiodomethylketone while the accompanying cathodic process reduces the formamide to the corresponding amine. An ensuing iodoform reaction between these two intermediates then furnished the amide product in a convergent fashion.
Figure 52.
Convergent paired electrolysis.
Anodic oxidation of formamide has also been exploited in convergent electrolytic systems. For example, Hilt and co-workers reported an electrochemical carbonyl allylation in DMF (Figure 52B).905 The cathodic Barbier reaction was combined with the Shono oxidation of DMF at the anode. The allyl alcohol produced in the cathodic process was swiftly converted into the corresponding N,O-acetal, thwarting undesired alcohol oxidation. The reaction can thus be conducted in a quasi-divided cell using a platinum-wire anode and a glassy-carbon cathode; the use of a sacrificial anode is not necessary. Shono oxidation of DMF was enlisted in a recent example of benzyl halide carboxylation as well, allowing the in situ protection of cathodically generated carboxylates (Figure 52C, top).906 As with the previous example, this paired electrolytic system makes use of a quasi-divided cell where the anode surface area is much smaller than the cathode. As a result, during the electrolysis, the anode would exhibit higher current densities, allowing the oxidation of solvent molecules (DMF) instead of the substrate. The product is protected from oxidation. Cathodic carboxylation has also been coupled with bromide oxidation at the anode—N-bromo-amino acids were afforded as a result (Figure 52C, bottom).907
The reduction of alkyl esters, a process commonly requiring high reduction potentials (absolute value), can be accomplished with the aid of a magnesium cathode (Figure 52D). Capitalizing on the beneficial effects of Mg electrodes and Mg2+ ions, Ishifune, Kashimura, and co-workers devised electrochemical ester reductions in THF.908,909 The reduction of alkyl esters at the cathode was accompanied by the oxidation of THF to the oxocarbenium ion. As a result, alcohol produced in the cathodic reaction can be readily protected.
In another example of convergent paired electrolysis, oxidation of phenol to quinone was coupled with the reductive formation of quinodimethane, allowing convergent coupling of these intermediates in a Diels–Alder reaction (Figure 52E).910 Anodic oxidation of alcohols and tin-mediated cathodic reduction of nitrate have also been combined in an undivided cell, providing oximes directly from alcohols (Figure 52F).911
Convergent paired electrolysis could also be harnessed to synthesize cyanoacetic acid from acetonitrile and carbon dioxide (Figure 52G, simplified mechanism shown).912 In this case, MeCN is oxidized to the carbon-centered radical in the anodic chamber of a divided cell while CO2 is reduced to the corresponding radical anion in the catholyte. This radical anion was found to diffuse across the partially permeable membrane to react with the acetonitrile radical.
Many examples of anodic oxidations described in this review were aided by electrogenerated bases (EGBs) at the cathode. Strictly speaking, these reactions may all be classified under paired electrolysis. Two additional examples are presented herein to illustrate this (Figure 52H–I). In Figure 51H, an anodically generated thionium was coupled with a nitromethane anion to afford a nitroalkylation product.913 The nucleophile arises through the deprotonation of MeNO2 at the cathode. The example presented in Figure 51H entails the conversion of carbon monoxide to diphenyl carbonate using a palladium–NHC complex as the mediator.914 While this can be largely perceived as an oxidation event, a cathode process allowed the deprotonation of phenols, thereby facilitating nucleophilic attack.
5. CONCLUSION
This extensive review, covering synthetic organic electrochemical methodologies published since 2000, will hopefully serve as a useful starting point for practitioners looking to enter this area. Electrochemistry has many innate advantages, including the ease of scalability, the use of inexpensive and/or recyclable electrodes, and reaction tunability.45 It is a direct method for interacting with organic molecules that in many cases permits one to “dial in” selectivity by choosing the right potential. That said, it is an unassailable truth that for most synthetic organic chemists, electrochemistry represents a technique employed as a “last resort” rather than a go-to method of choice. Looking forward, we see the following challenges preventing the mass adoption of electrochemistry in modern synthesis laboratories: (1) a lack of standardized equipment, (2) a steep learning curve for the underlying physical chemistry principles, and (3) an only limited number of truly broad or compelling transformations that necessitate its adoption. For the first challenge, while compiling this review, we note that each group has its own favorite instrumentation. Thus, reproducibility can be an issue as the underlying engineering aspect is not standardized across the community. Indeed, there is no standard practice concerning the use of certain power sources, electrolytes, or electrodes. The anecdotal nature of this area is undeniably a barrier to the widespread application of electrochemistry in synthesis. Second, many publications in this area are not geared toward practicing synthetic organic chemists—the need to understand mathematical formulas and physical principles can present yet another hurdle. Finally, we have noted that many (but not all) of the reactions described thus far can be accomplished using simple chemical oxidants. In these cases, while the use of electrochemical methods is still advantageous in terms of sustainability and atom economy, the ability to procure the desired products in the most time-efficient manner is nonetheless the overriding concern in most preparative applications. Thus, the community should focus more on examples that are enabled by electrochemistry or provide decisive and powerful advantages over their purely chemical counterparts. In addition to the inherent advantages listed above, one can imagine electrochemically facilitated access to high energy species (organic and organometallic), useful new mediators, and improved variants of organic chemistry’s most used reactions through continuous redox control. If the community embraces these challenges, we anticipate synthetic organic electrochemistry could head into a golden era of vibrant utility.
Acknowledgments
Financial support for this work was provided by the NIH (GM-118176), the Hewitt Foundation (postdoctoral fellowship to Y.K.), and IKA.
Biographies
Ming Yan was born in China in 1989, but was educated in China, Singapore, Germany, and the US. He received his undergraduate education from Lafayette College (2009–2012), which included a research internship at TSRI under the tutelage of Professor Phil S. Baran. He then returned to the Baran laboratory for his doctoral studies, where he is investigating the total synthesis of alkaloids.
Yu Kawamata was born in Japan in 1988, and he completed his undergraduate education at Kyoto University. He obtained his master degree and his Ph.D. at the same university under the supervision of Professor Keiji Maruoka, and he undertook a short-term internship at TSRI working on natural product synthesis with Professor Phil S. Baran. Upon completion of his doctoral studies, he returned to the Baran laboratory as a research associate and currently is pursuing his postdoctoral studies on organic electrochemistry.
Phil S. Baran was born in New Jersey in 1977 and received his undergraduate education from New York University in 1997. After earning his Ph.D. at TSRI in 2001, he pursued postdoctoral studies at Harvard University until 2003, at which point he returned to TSRI to begin his independent career. He was promoted to the rank of Professor in 2008 and is currently the Darlene Shiley Professor of chemistry. The mission of his laboratory is to educate students at the intersection of fundamental organic chemistry and translational science.
Footnotes
ORCID
Phil S. Baran: 0000-0001-9193-9053
Notes
The authors declare no competing financial interest.
References
- 1.Lund H. A Century of Organic Electrochemistry. J Electrochem Soc. 2002;149:S21–S33. [Google Scholar]
- 2.Volta AGA. Nat Philos Chem Arts. 1800;4:179–187. [Google Scholar]
- 3.Faraday M. Ann Phys Leipzig. 1834;47:438. [Google Scholar]
- 4.Kolbe H. J Prakt Chem. 1847;41:138. [Google Scholar]
- 5.Schoenbein ChF. Liebigs Ann Chem. 1845;54:164. [Google Scholar]
- 6.Tafel J, Hahl H. Vollständige Reduktion Des Benzylacetessigesters. Ber Dtsch Chem Ges. 1907;40:3312–3318. [Google Scholar]
- 7.Paidar M, Fateev V, Bouzek K. Membrane Electrolysis—History, Current Status and Perspective. Electrochim Acta. 2016;209:737–756. [Google Scholar]
- 8.Hickling A. Studies in Electrode Polarisation. Part IV—the Automatic Control of the Potential of a Working Electrode. Trans Faraday Soc. 1942;38:27–33. [Google Scholar]
- 9.Lingane JJ, Swain CG, Fields M. Polarographically Controlled Syntheses, with Particular Reference to Organic Chemistry. J Am Chem Soc. 1943;65:1348–1353. [Google Scholar]
- 10.Heyrovsky J. Chem Listy. 1922;16:256–264. doi: 10.1002/tcr.201200103. [DOI] [PubMed] [Google Scholar]
- 11.Heyrovsky J. Philos Mag. 1923;45:303–315. [Google Scholar]
- 12.Randles JEB. A Cathode Ray Polarograph. Part II.—the Current-Voltage Curves. Trans Faraday Soc. 1948;44:327–338. [Google Scholar]
- 13.Simons JH. Production of Fluorocarbons I. the Generalized Procedure and Its Use with Nitrogen Compounds. J Electrochem Soc. 1949;95:47–52. [Google Scholar]
- 14.Baizer MM. CHEMTECH. 1980;161 [Google Scholar]
- 15.Corey EJ, Sauers RR. The Synthesis of Pentacyclosqualene (8,8′-Cycloönocerene) and the α- and β-Onoceradienes. J Am Chem Soc. 1959;81:1739–1743. [Google Scholar]
- 16.Sperry JB, Wright DL. The Application of Cathodic Reductions and Anodic Oxidations in the Synthesis of Complex Molecules. Chem Soc Rev. 2006;35:605–617. doi: 10.1039/b512308a. [DOI] [PubMed] [Google Scholar]
- 17.Le Blanc MZ. Elektrochem Angew Phys Chem. 1900;7:287. [Google Scholar]
- 18.Seo ET, Nelson RF, Fritsch JM, Marcoux LS, Leedy DW, Adams RN. Anodic Oxidation Pathways of Aromatic Amines. Electrochemical and Electron Paramagnetic Resonance Studies. J Am Chem Soc. 1966;88:3498–3503. [Google Scholar]
- 19.Semmelhack MF, Chou CS, Cortes DA. Nitroxyl-Mediated Electrooxidation of Alcohols to Aldehydes and Ketones. J Am Chem Soc. 1983;105:4492–4494. [Google Scholar]
- 20.Steckhan E. Indirect Electroorganic Syntheses—a Modern Chapter of Organic Electrochemistry [New Synthetic Methods (59)] Angew Chem, Int Ed Engl. 1986;25:683–701. [Google Scholar]
- 21.Steckhan E. Organic Syntheses with Electrochemically Regenerable Redox Systems. Top Curr Chem. 1987;142:1–69. [Google Scholar]
- 22.Iversen PE, Lund H. Electrolytic Generation of Strong Bases I. Wittig Reaction. Tetrahedron Lett. 1969;10:3523–3524. [Google Scholar]
- 23.Uneyama K, Isimura A, Fujii K, Torii S. Electrogenerated Acid as a Powerful Catalyst for Transformation of Epoxides to Ketones and Acetonides. Tetrahedron Lett. 1983;24:2857–2860. [Google Scholar]
- 24.Watkins BF, Behling JR, Kariv E, Miller LL. Chiral Electrode. J Am Chem Soc. 1975;97:3549–3550. [Google Scholar]
- 25.Shono T, Hamaguchi H, Matsumura Y. Electroorganic Chemistry. XX. Anodic Oxidation of Carbamates. J Am Chem Soc. 1975;97:4264–4268. [Google Scholar]
- 26.Shono T. Electroorganic Chemistry in Organic Synthesis. Tetrahedron. 1984;40:811–850. [Google Scholar]
- 27.Yoshida J, Murata T, Isoe S. Electrochemical Oxidation of Organosilicon Compounds I. Oxidative Cleavage of Carbon-Silicon Bond in Allylsilanes and Benzylsilanes. Tetrahedron Lett. 1986;27:3373–3376. [Google Scholar]
- 28.Little RD, Schwaebe MK. Reductive Cyclizations at the Cathode. Top Curr Chem. 1997;185:1–48. [Google Scholar]
- 29.Little RD, Fox DP, Van Hijfte L. Electroreductive Cyclization. Ketones and Aldehydes Tethered to α,β-Unsaturated Esters (Nitriles). Fundamental Investigations. J Org Chem. 1988;53:2287–2294. [Google Scholar]
- 30.Sowell CG, Wolin RL, Little RD. Electroreductive Cyclization Reactions. Stereoselection, Creation of Quaternary Centers in Bicyclic Frameworks, and a Formal Total Synthesis of Quadrone. Tetrahedron Lett. 1990;31:485–488. [Google Scholar]
- 31.Schäfer H-J. Recent Contributions of Kolbe Electrolysis to Organic Synthesis. Top Curr Chem. 1990;152:91–151. [Google Scholar]
- 32.Becking L, Scäfer HJ. Pyrrolidines by Intramolecular Addition of Kolbe Radicals Generated From β-Allylaminoalkanoates. Tetrahedron Lett. 1988;29:2797–2800. [Google Scholar]
- 33.Moeller KD. Synthetic Applications of Anodic Electrochemistry. Tetrahedron. 2000;56:9527. [Google Scholar]
- 34.Paddon CA, Atobe M, Fuchigami T, He P, Watts P, Haswell SJ, Pritchard GJ, Bull SD, Marken F. Towards Paired and Coupled Electrode Reactions for Clean Organic Microreactor Electrosyntheses. J Appl Electrochem. 2006;36:617–634. [Google Scholar]
- 35.Steckhan E, Arns T, Heineman WR, Hoormann D, Jörissen J, Kröner L, Lewall B, Pütter H. Environmental Protection and Economization of Resources by Electroorganic and Electroenzymatic Syntheses. Chemosphere. 2001;43:63–73. doi: 10.1016/s0045-6535(00)00325-8. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida J-I, Suga S, Suzuki S, Kinomura N, Yamamoto A, Fujiwara K. Direct Oxidative Carbon—Carbon Bond Formation Using the “Cation Pool” Method. 1. Generation of Iminium Cation Pools and Their Reaction with Carbon Nucleophiles. J Am Chem Soc. 1999;121:9546–9549. [Google Scholar]
- 37.Yoshida J-I, Suga S. Basic Concepts of “Cation Pool” and ‘Cation Flow’ Methods and Their Applications in Conventional and Combinatorial Organic Synthesis. Chem - Eur J. 2002;8:2650–2658. doi: 10.1002/1521-3765(20020617)8:12<2650::aid-chem2650>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 38.Weinberg NL, Weinberg HR. Electrochemical Oxidation of Organic Compounds. Chem Rev. 1968;68:449–523. [Google Scholar]
- 39.Schäfer HJ. Anodic and Cathodic CC-Bond Formation. Angew Chem, Int Ed Engl. 1981;20:911–934. [Google Scholar]
- 40.Francke R, Little RD. Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem Soc Rev. 2014;43:2492–2521. doi: 10.1039/c3cs60464k. [DOI] [PubMed] [Google Scholar]
- 41.Yoshida J-I, Kataoka K, Horcajada R, Nagaki A. Modern Strategies in Electroorganic Synthesis. Chem Rev. 2008;108:2265–2299. doi: 10.1021/cr0680843. [DOI] [PubMed] [Google Scholar]
- 42.Utley J. Trends in Organic Electrosynthesis. Chem Soc Rev. 1997;26:157–167. [Google Scholar]
- 43.Frontana-Uribe BA, Little RD, Ibanez JG, Palma A, Vasquez-Medrano R. Organic Electrosynthesis: a Promising Green Methodology in Organic Chemistry. Green Chem. 2010;12:2099–2119. [Google Scholar]
- 44.Schäfer HJ. Contributions of Organic Electrosynthesis to Green Chemistry. C R Chim. 2011;14:745–765. [Google Scholar]
- 45.Horn EJ, Rosen BR, Baran PS. Synthetic Organic Electrochemistry: an Enabling and Innately Sustainable Method. ACS Cent Sci. 2016;2:302–308. doi: 10.1021/acscentsci.6b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kise N. Electrogenerated Bases and Nucleophiles. Springer New York; New York, NY: 2014. Electrogenerated Acid; pp. 702–706. [Google Scholar]
- 47.Utley J, Nielsen M, Wyatt P. Organic Electrochemistry. 5th. CRC Press; 2015. Electrogenerated Bases and Nucleophiles; pp. 1625–1656. Revised and Expanded. [Google Scholar]
- 48.Kohlmann C, Lütz S. Organic Electrochemistry. 5th. CRC Press; 2015. Electroenzymatic Synthesis; pp. 1511–1541. Revised and Expanded. [Google Scholar]
- 49.Lessard J. Organic Electrochemistry. 5th. CRC Press; 2015. Electrocatalytic Hydrogenation; pp. 1657–1672. Revised and Expanded. [Google Scholar]
- 50.Roth HG, Romero NA, Nicewicz DA. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett. 2016;27:714–723. [Google Scholar]
- 51.Luca OR, Gustafson JL, Maddox SM, Fenwick AQ, Smith DC. Catalysis by Electrons and Holes: Formal Potential Scales and Preparative Organic Electrochemistry. Org Chem Front. 2015;2:823–848. [Google Scholar]
- 52.Schäfer HJ. Recent Synthetic Applications of the Kolbe Electrolysis. Chem Phys Lipids. 1979;24:321–333. [Google Scholar]
- 53.Lateef S, Reddy Krishna Mohan S, Reddy Jayarama Reddy S. Electroorganic Synthesis of Benzathine. Tetrahedron Lett. 2007;48:77–80. [Google Scholar]
- 54.Sugiya M, Nohira H. Synthesis of Optically Pure Bisphosphine Oxides and Related Compounds. Bull Chem Soc Jpn. 2000;73:705–712. [Google Scholar]
- 55.Brakha L, Becker JY. Anodic Oxidation of α-Silylacetic Acid Ph2(Me)SiCH2COOH: Decarboxylation Versus Desilylation. Electrochim Acta. 2012;59:135–139. [Google Scholar]
- 56.Brakha L, Becker JY. Anodic Oxidation of α-Silylacetic Acids: Effects of Anode Material, Current Density and Concentration on Competing Decarboxylation and Desilylation Processes. Electrochim Acta. 2012;77:143–149. [Google Scholar]
- 57.Shtelman AV, Becker JY. Electrochemical Synthesis of 1,2-Disilylethanes From α-Silylacetic Acids. J Org Chem. 2011;76:4710–4714. doi: 10.1021/jo200254z. [DOI] [PubMed] [Google Scholar]
- 58.Schäfer HJ. Electrochemical Conversion of Fatty Acids. Eur J Lipid Sci Technol. 2012;114:2–9. [Google Scholar]
- 59.Behr S, Hegemann K, Schimanski H, Fröhlich R, Haufe G. Synthesis of γ-Lactones From Cycloocta-1,5-Diene–Starting Materials for Natural-Product Synthesis. Eur J Org Chem. 2004;2004:3884–3892. [Google Scholar]
- 60.Brecht Forster A, Fitremann J, Renaud P. Synthesis of (±)-Nephromopsinic, (–)-Phaseolinic, and (–)-Dihydropertusaric Acids. Helv Chim Acta. 2002;85:3965–3974. [Google Scholar]
- 61.Ng’ang’a Wanyoik G, Onomura O, Maki T, Matsumura Y. Highly Enhanced Enantioselectivity in the Memory of Chirality via Acyliminium Ions. Org Lett. 2002;4:1875–1877. doi: 10.1021/ol025865r. [DOI] [PubMed] [Google Scholar]
- 62.Matsumura Y, Shirakawa Y, Satoh Y, Umino M, Tanaka T, Maki T, Onomura O. First Example of Memory of Chirality in Carbenium Ion Chemistry. Org Lett. 2000;2:1689–1691. doi: 10.1021/ol005795t. [DOI] [PubMed] [Google Scholar]
- 63.Onomura O, Ng’ang’a Wanyoike G, Matsumura Y, Kuriyama M. Memory of Chirality in the Electrochemical Oxidation of Thiazolidine-4-Carboxylic Acid Derivatives. Heterocycles. 2010;80:1177–1185. [Google Scholar]
- 64.Kurihara H, Fuchigami T, Tajima T. Kolbe Carbon-Carbon Coupling Electrosynthesis Using Solid-Supported Bases. J Org Chem. 2008;73:6888–6890. doi: 10.1021/jo801016f. [DOI] [PubMed] [Google Scholar]
- 65.Kurihara H, Tajima T, Fuchigami T. Mixed-Kolbe Electrolysis Using Solid-Supported Bases. Electrochemistry. 2006;74:615–617. [Google Scholar]
- 66.Tajima T, Kurihara H, Fuchigami T. Development of an Electrolytic System for Non-Kolbe Electrolysis Based on the Acid—Base Reaction Between Carboxylic Acids as a Substrate and Solid-Supported Bases. J Am Chem Soc. 2007;129:6680–6681. doi: 10.1021/ja070283w. [DOI] [PubMed] [Google Scholar]
- 67.Galicia M, González-Fuentes MA, Valencia DP, González FJ. The Effect of Substituents on the Anodic Oxidation of Aliphatic Carboxylates and the Passage Towards a Pseudo-Kolbe Reaction. J Electroanal Chem. 2012;672:28–33. [Google Scholar]
- 68.Okada Y, Kamimura K, Chiba K. Cycloalkane-Based Thermomorphic Systems for Organic Electrochemistry: an Application to Kolbe-Coupling. Tetrahedron. 2012;68:5857–5862. [Google Scholar]
- 69.Wadhawan JD, Marken F, Compton RG, Bull SD, Davies SG. Sono-Emulsion Electrosynthesis: Electrode-Insensitive Kolbe Reactions. Chem Commun. 2001:87–88. [Google Scholar]
- 70.Becking L, Schäfer HJ. Pyrrolidines by Intramolecular Addition of Kolbe Radicals Generated From β-Allylaminoalkanoates. Tetrahedron Lett. 1988;29:2797–2800. [Google Scholar]
- 71.Lebreux FDR, Buzzo F, Marko IN. Synthesis of Five- and Six-Membered-Ring Compounds by Environmentally Friendly Radical Cyclizations Using Kolbe Electrolysis. Synlett. 2008;2008:2815–2820. [Google Scholar]
- 72.Arai K, Watts K, Wirth T. Difluoro- and Trifluoromethylation of Electron-Deficient Alkenes in an Electrochemical Microreactor. ChemistryOpen. 2014;3:23–28. doi: 10.1002/open.201300039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uneyama K, Nanbu H. Electrochemical 1, 2-Addition of Trifluoromethyl and Acetamide Groups to Methyl Methacrylate. J Org Chem. 1988;53:4598–4599. [Google Scholar]
- 74.Ma X, Luo X, Dochain S, Mathot C, Markó IE. Electrochemical Oxidative Decarboxylation of Malonic Acid Derivatives: a Method for the Synthesis of Ketals and Ketones. Org Lett. 2015;17:4690–4693. doi: 10.1021/acs.orglett.5b02084. [DOI] [PubMed] [Google Scholar]
- 75.Dai J-J, Huang Y-B, Fang C, Guo Q-X, Fu Y. Electrochemical Synthesis of Adiponitrile From the Renewable Raw Material Glutamic Acid. ChemSusChem. 2012;5:617–620. doi: 10.1002/cssc.201100776. [DOI] [PubMed] [Google Scholar]
- 76.Shono T, Matsumura Y, Inoue K. Electroorganic Chemistry. 87. Indirect Electrooxidation of Amines to Nitriles Using Halogen Ions as Mediators. J Am Chem Soc. 1984;106:6075–6076. [Google Scholar]
- 77.Qian P, Bi M, Su J, Zha Z, Wang Z. Electrosynthesis of (E)-Vinyl Sulfones Directly From Cinnamic Acids and Sodium Sulfinates via Decarboxylative Sulfono Functionalization. J Org Chem. 2016;81:4876–4882. doi: 10.1021/acs.joc.6b00661. [DOI] [PubMed] [Google Scholar]
- 78.Luo Y-C, Pan X-J, Yuan G-Q. An Efficient Electrochemical Synthesis of Vinyl Sulfones From Sodium Sulfinates and Olefins. Tetrahedron. 2015;71:2119–2123. [Google Scholar]
- 79.Jiang Y-Y, Liang S, Zeng C-C, Hu L-M, Sun B-G. Electrochemically Initiated Formation of Sulfonyl Radicals: Synthesis of Oxindoles via Difunctionalization of Acrylamides Mediated by Bromide Ion. Green Chem. 2016;18:6311–6319. [Google Scholar]
- 80.Wen J, Shi W, Zhang F, Liu D, Tang S, Wang H, Lin X-M, Lei A. Electrooxidative Tandem Cyclization of Activated Alkynes with Sulfinic Acids to Access Sulfonated Indenones. Org Lett. 2017;19:3131–3134. doi: 10.1021/acs.orglett.7b01256. [DOI] [PubMed] [Google Scholar]
- 81.Zhang C, Chen Y, Yuan G. Electrosynthesis of Arylsulfonamides From Amines and Sodium Sulfinates Using H2O-NaI as the Electrolyte Solution at Room Temperature. Chin J Chem. 2016;34:1277–1282. [Google Scholar]
- 82.O’Brien AG, Maruyama A, Inokuma Y, Fujita M, Baran PS, Blackmond DG. Radical C-H Functionalization of Heteroarenes Under Electrochemical Control. Angew Chem, Int Ed. 2014;53:11868–11871. doi: 10.1002/anie.201407948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tommasino JB, Brondex A, Médebielle M, Thomalla M, Langlois BR, Billard T. Trifluoromethylation Reactions with Potassium Trifluoromethanesulfinate Under Electrochemical Oxidation. Synlett. 2002:1697–1699. [Google Scholar]
- 84.Rosen BR, Werner EW, O’Brien AG, Baran PS. Total Synthesis of Dixiamycin B by Electrochemical Oxidation. J Am Chem Soc. 2014;136:5571–5574. doi: 10.1021/ja5013323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gieshoff T, Schollmeyer D, Waldvogel SR. Access to Pyrazolidin-3,5-Diones Through Anodic N–N Bond Formation. Angew Chem, Int Ed. 2016;55:9437–9440. doi: 10.1002/anie.201603899. [DOI] [PubMed] [Google Scholar]
- 86.Gieshoff T, Kehl A, Schollmeyer D, Moeller KD, Waldvogel SRR. Insights Into the Mechanism of the Anodic N-N Bond Formation by Dehydrogenative Coupling. J Am Chem Soc. 2017;139:12317. doi: 10.1021/jacs.7b07488. [DOI] [PubMed] [Google Scholar]
- 87.Xu H-C, Campbell JM, Moeller KD. Cyclization Reactions of Anode-Generated Amidyl Radicals. J Org Chem. 2014;79:379–391. doi: 10.1021/jo402623r. [DOI] [PubMed] [Google Scholar]
- 88.Xiong P, Xu H-H, Xu H-C. Metal- and Reagent-Free Intramolecular Oxidative Amination of Tri- and Tetrasubstituted Alkenes. J Am Chem Soc. 2017;139:2956–2959. doi: 10.1021/jacs.7b01016. [DOI] [PubMed] [Google Scholar]
- 89.Zhao H-B, Hou Z-W, Liu Z-J, Zhou Z-F, Song J, Xu H-C. Amidinyl Radical Formation Through Anodic N–H Bond Cleavage and Its Application in Aromatic C–H Bond Functionalization. Angew Chem, Int Ed. 2017;56:587–590. doi: 10.1002/anie.201610715. [DOI] [PubMed] [Google Scholar]
- 90.Gieshoff T, Kehl A, Schollmeyer D, Moeller KD, Waldvogel SR. Electrochemical Synthesis of Benzoxazoles From Anilides - a New Approach to Employ Amidyl Radical Intermediates. Chem Commun. 2017;53:2974–2977. doi: 10.1039/c7cc00927e. [DOI] [PubMed] [Google Scholar]
- 91.Hou Z-W, Mao Z-Y, Zhao H-B, Melcamu YY, Lu X, Song J, Xu H-C. Electrochemical C–H/N–H Functionalization for the Synthesis of Highly Functionalized (Aza)Indoles. Angew Chem, Int Ed. 2016;55:9168–9172. doi: 10.1002/anie.201602616. [DOI] [PubMed] [Google Scholar]
- 92.Zhu L, Xiong P, Mao Z-Y, Wang Y-H, Yan X, Lu X, Xu H-C. Electrocatalytic Generation of Amidyl Radicals for Olefin Hydroamidation: Use of Solvent Effects to Enable Anilide Oxidation. Angew Chem, Int Ed. 2016;55:2226–2229. doi: 10.1002/anie.201510418. [DOI] [PubMed] [Google Scholar]
- 93.Xu F, Zhu L, Zhu S, Yan X, Xu H-C. Electrochemical Intramolecular Aminooxygenation of Unactivated Alkenes. Chem - Eur J. 2014;20:12740–12744. doi: 10.1002/chem.201404078. [DOI] [PubMed] [Google Scholar]
- 94.Siu T, Yudin AK. Practical Olefin Aziridination with a Broad Substrate Scope. J Am Chem Soc. 2002;124:530–531. doi: 10.1021/ja0172215. [DOI] [PubMed] [Google Scholar]
- 95.Siu T, Picard CJ, Yudin AK. Development of Electrochemical Processes for Nitrene Generation and Transfer. J Org Chem. 2005;70:932–937. doi: 10.1021/jo048591p. [DOI] [PubMed] [Google Scholar]
- 96.Siu T, Yudin AK. Electrochemical Imination of Sulfoxides Using N-Aminophthalimide. Org Lett. 2002;4:1839–1842. doi: 10.1021/ol0257530. [DOI] [PubMed] [Google Scholar]
- 97.Chen J, Yan W-Q, Lam CM, Zeng C-C, Hu L-M, Little RD. Electrocatalytic Aziridination of Alkenes Mediated by N-Bu4NI: a Radical Pathway. Org Lett. 2015;17:986–989. doi: 10.1021/acs.orglett.5b00083. [DOI] [PubMed] [Google Scholar]
- 98.Watts K, Gattrell W, Wirth T. A Practical Microreactor for Electrochemistry in Flow. Beilstein J Org Chem. 2011;7:1108–1114. doi: 10.3762/bjoc.7.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nishiyama S, Amano Y. Oxidative Synthesis of Azacyclic Derivatives Through the Nitrenium Ion: Application of a Hypervalent Iodine Species Electrochemically Generated From Iodobenzene. Tetrahedron Lett. 2006;47:6505–6507. [Google Scholar]
- 100.Inoue K, Ishikawa Y, Nishiyama S. Synthesis of Tetrahydropyrroloiminoquinone Alkaloids Based on Electrochemically Generated Hypervalent Iodine Oxidative Cyclization. Org Lett. 2010;12:436–439. doi: 10.1021/ol902566p. [DOI] [PubMed] [Google Scholar]
- 101.Kajiyama D, Inoue K, Ishikawa Y, Nishiyama S. A Synthetic Approach to Carbazoles Using Electrochemically Generated Hypervalent Iodine Oxidant. Tetrahedron. 2010;66:9779–9784. [Google Scholar]
- 102.Amano Y, Inoue K, Nishiyama S. Oxidative Access to Quinolinone Derivatives with Simultaneous Rearrangement of Functional Groups. Synlett. 2008;2008:134–136. [Google Scholar]
- 103.Nishiyama S, Amano Y. Effects of Aromatic Substituents of Electrochemically Generated Hypervalent Iodine Oxidant on Oxidation Reactions. Heterocycles. 2008;75:1997. [Google Scholar]
- 104.Broese T, Francke R. Electrosynthesis Using a Recyclable Mediator–Electrolyte System Based on Ionically Tagged Phenyl Iodide and 1,1,1,3,3,3-Hexafluoroisopropanol. Org Lett. 2016;18:5896–5899. doi: 10.1021/acs.orglett.6b02979. [DOI] [PubMed] [Google Scholar]
- 105.Jones AM, Banks CE. The Shono-Type Electroorganic Oxidation of Unfunctionalised Amides. Carbon–Carbon Bond Formation via Electrogenerated N-Acyliminium Ions. Beilstein J Org Chem. 2014;10:3056–3072. doi: 10.3762/bjoc.10.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Onomura O. Anodic Selective Functionalization of Cyclic Amine Derivatives. Heterocycles. 2012;85:2111–2113. [Google Scholar]
- 107.D’Oca MGM, Pilli RA, Pardini VL, Curi D, Comninos FCM. The Addition of Allyltrimethylsilane to Cyclic N-Acyliminium Ions Derived From(S)-(+)-Mandelic Acid and Cyclohexyl-Based Chiral Auxiliaries. J Braz Chem Soc. 2001;12:507–513. [Google Scholar]
- 108.D’Oca M, Pilli RA, Vencato I. The Addition of 2-Tert-Butyldimethylsilyloxyfuran to Cyclic N-Acyliminium Ions Containing Cyclohexyl-Based Chiral Auxiliaries. Tetrahedron Lett. 2000;41:9709–9712. [Google Scholar]
- 109.Schierle Arndt K, Kolter D, Danielmeier K, Steckhan E. Electrogenerated Chiral 4-Methoxy-2-Oxazolidinones as Diastereoselective Amidoalkylation Reagents for the Synthesis of β-Amino Alcohol Precursors. Eur J Org Chem. 2001;2001:2425–2433. [Google Scholar]
- 110.Zelgert M, Nieger M, Lennartz M, Steckhan E. Two Directional Electroorganic Synthesis—Electrochemical Oxidation and Application of a C2-Symmetric Building Block. Tetrahedron. 2002;58:2641–2646. [Google Scholar]
- 111.Sierecki E, Errasti G, Martens T, Royer J. Diastereoselective α-Allylation of Secondary Amines. Tetrahedron. 2010;66:10002–10007. [Google Scholar]
- 112.Sierecki E, Turcaud S, Martens T, Royer J. Phosphorus-Derived Chiral Auxiliaries for α-Alkylation of Secondary Amines by Anodic Oxidation. Synthesis. 2006;2006:3199–3208. [Google Scholar]
- 113.Turcaud S, Martens T, Sierecki E, Pérard-Viret J, Royer J. Anodic Oxidation of Chiral Sulfinylamines: a New Route to Highly Diastereoselective α-Alkylation of Piperidine. Tetrahedron Lett. 2005;46:5131–5134. [Google Scholar]
- 114.Lee D-S. Highly Diastereoselective Synthesis of Pyrido[2,1-B][1,3]Oxazin-4(6H)-One by Intramolecular Anodic Oxidation. Tetrahedron: Asymmetry. 2009;20:2014–2020. [Google Scholar]
- 115.Matsumura Y, Kanda Y, Shirai K, Onomura O, Maki T. A Convenient Method for Synthesis of Optically Active Methylphenidate from N-Methoxycarbonylpiperidine by Utilizing Electrochemical Oxidation and Evans Aldol-type Reaction. Tetrahedron. 2000;56:7411–7422. [Google Scholar]
- 116.Libendi SS, Demizu Y, Matsumura Y, Onomura O. High Regioselectivity in Electrochemical α-Methoxylation of N-Protected Cyclic Amines. Tetrahedron. 2008;64:3935–3942. [Google Scholar]
- 117.Onomura O, Ishida Y, Maki T, Minato D, Demizu Y, Matsumura Y. Electrochemical Oxidation of L-Prolinol Derivative Protected with 1-Alkoxy-2,2,2-Trifluoroethyl Group. Electrochemistry. 2006;74:645–648. [Google Scholar]
- 118.Gallardo I, Vilà N. One-Pot Electrosynthesis of Substituted Imidazolinium and Tetrahydropyrimidinium Salts From Secondary Alkyldiamines: an Electrochemical Route Toward Ionic Liquids. J Org Chem. 2010;75:680–689. doi: 10.1021/jo9017606. [DOI] [PubMed] [Google Scholar]
- 119.Gallardo I, Vilà N. Electrosynthesis of Hindered Alkyl Diamines: Evidence for an Electrocatalytic Anodic Mechanism. J Org Chem. 2008;73:6647–6656. doi: 10.1021/jo8006607. [DOI] [PubMed] [Google Scholar]
- 120.Okimoto M, Yoshida T, Hoshi M, Hattori K, Komata M, Numata K, Tomozawa K. Electrooxidative Cyclization of Hydroquinolyl Alcohols, Hydroquinolylamines, and Dimethyl Aminomalonates. Aust J Chem. 2007;60:236–237. [Google Scholar]
- 121.Okimoto M, Ohashi K, Yamamori H, Nishikawa S, Hoshi M, Yoshida T. Electrooxidative Cyclization of Hydroxyamino Compounds Possessing a Benzyl Group. Synthesis. 2012;44:1315–1322. [Google Scholar]
- 122.Baslé O, Borduas N, Dubois P, Chapuzet JM, Chan T-H, Lessard J, Li C-J. Aerobic and Electrochemical Oxidative Cross-Dehydrogenative-Coupling (CDC) Reaction in an Imidazolium-Based Ionic Liquid. Chem - Eur J. 2010;16:8162–8166. doi: 10.1002/chem.201000240. [DOI] [PubMed] [Google Scholar]
- 123.Kashiwagi Y, Anzai J. Selective Electrocatalytic Oxidation of N-Alkyl-N-Methylanilines to N-Alkylformanilides Using Nitroxyl Radical. Chem Pharm Bull. 2001;49:324–326. doi: 10.1248/cpb.49.324. [DOI] [PubMed] [Google Scholar]
- 124.Baba D, Fuchigami T. Anodic Methoxylation and Acetoxylation of Imines and Imidates. Tetrahedron Lett. 2003;44:3133–3136. [Google Scholar]
- 125.Kirira PG, Kuriyama M, Onomura O. Electrochemical Deallylation of α-Allyl Cyclic Amines and Synthesis of Optically Active Quaternary Cyclic Amino Acids. Chem - Eur J. 2010;16:3970–3982. doi: 10.1002/chem.200903512. [DOI] [PubMed] [Google Scholar]
- 126.Mazurkiewicz R, Adamek J, Październiok-Holewa A, Zielińska K, Simka W, Gajos A, Szymura K. α-Amidoalkylating Agents From N-Acyl-α-Amino Acids: 1-(N-Acylamino)-Alkyltriphenylphosphonium Salts. J Org Chem. 2012;77:1952–1960. doi: 10.1021/jo202534u. [DOI] [PubMed] [Google Scholar]
- 127.Adamek J, Mazurkiewicz R, Październiok-Holewa A, Grymel M, Kuznik A, Zielińska K. 1-(N-Acylamino)Alkyl Sulfones From N-Acyl-α-Amino Acids or N-Alkylamides. J Org Chem. 2014;79:2765–2770. doi: 10.1021/jo500174a. [DOI] [PubMed] [Google Scholar]
- 128.Adamek J, Mazurkiewicz R, Pazdzierniok-Holewa A, Kuźnik A, Grymel M, Zielińska K, Simka W. N-[1-(Benzotriazol-1-yl)Alkyl]Amides From N-Acyl-α-Amino Acids or N-Alkylamides. Tetrahedron. 2014;70:5725–5729. [Google Scholar]
- 129.Tajima T, Nakajima A. Direct Oxidative Cyanation Based on the Concept of Site Isolation. J Am Chem Soc. 2008;130:10496–10497. doi: 10.1021/ja804048a. [DOI] [PubMed] [Google Scholar]
- 130.Asami R, Fuchigami T, Atobe M. Development of an Anodic Substitution Reaction System Using Acoustic Emulsification. Chem Commun. 2008:244–246. doi: 10.1039/b713859h. [DOI] [PubMed] [Google Scholar]
- 131.Kathiresan M, Velayutham D. Ionic Liquids as an Electrolyte for the Electro Synthesis of Organic Compounds. Chem Commun. 2015;51:17499–17516. doi: 10.1039/c5cc06961k. [DOI] [PubMed] [Google Scholar]
- 132.Tajima T, Kurihara H, Shimizu S, Tateno H. Anodic Alkoxylation of Lactams Followed by Reactions with Carbon Nucleophiles in a One-Pot Manner Using HFIP as a Solvent. Electrochemistry. 2013;81:353–355. [Google Scholar]
- 133.Tajima T, Fuchigami T. Development of an Electrolytic System Using Solid-Supported Bases for in Situ Generation of a Supporting Electrolyte From Methanol as a Solvent. J Am Chem Soc. 2005;127:2848–2849. doi: 10.1021/ja0423062. [DOI] [PubMed] [Google Scholar]
- 134.Tajima T, Fuchigami T. Development of a Novel Environmentally Friendly Electrolytic System by Using Recyclable Solid-Supported Bases for in Situ Generation of a Supporting Electrolyte From Methanol as a Solvent: Application for Anodic Methoxylation of Organic Compounds. Chem - Eur J. 2005;11:6192–6196. doi: 10.1002/chem.200500340. [DOI] [PubMed] [Google Scholar]
- 135.Tajima T, Kurihara H. Deprotonation in Anodic Methoxylation of Fluoroethyl Phenyl Sulfides Using Site-Isolated Heterogeneous Bases. Chem Commun. 2008;99:5167–5169. doi: 10.1039/b809998g. [DOI] [PubMed] [Google Scholar]
- 136.Horii D, Fuchigami T, Atobe M. A New Approach to Anodic Substitution Reaction Using Parallel Laminar Flow in a Micro-Flow Reactor. J Am Chem Soc. 2007;129:11692–11693. doi: 10.1021/ja075180s. [DOI] [PubMed] [Google Scholar]
- 137.Horii D, Amemiya F, Fuchigami T, Atobe M. A Novel Electrosynthetic System for Anodic Substitution Reactions by Using Parallel Laminar Flow in a Microflow Reactor. Chem - Eur J. 2008;14:10382–10387. doi: 10.1002/chem.200801511. [DOI] [PubMed] [Google Scholar]
- 138.Libendi SS, Demizu Y, Onomura O. Direct Electrochemical α-Cyanation of N-Protected Cyclic Amines. Org Biomol Chem. 2009;7:351–356. doi: 10.1039/b816598j. [DOI] [PubMed] [Google Scholar]
- 139.Gong M, Huang J-M. Electrochemical Oxidative C–H/N–H Coupling Between γ-Lactams and Anilines. Chem - Eur J. 2016;22:14293–14296. doi: 10.1002/chem.201602454. [DOI] [PubMed] [Google Scholar]
- 140.Fu N, Li L, Yang Q, Luo S. Catalytic Asymmetric Electrochemical Oxidative Coupling of Tertiary Amines with Simple Ketones. Org Lett. 2017;19:2122–2125. doi: 10.1021/acs.orglett.7b00746. [DOI] [PubMed] [Google Scholar]
- 141.Zhang L, Su J-H, Wang S, Wan C, Zha Z, Du J, Wang Z. Direct Electrochemical Imidation of Aliphatic Amines via Anodic Oxidation. Chem Commun. 2011;47:5488–5490. doi: 10.1039/c1cc10871a. [DOI] [PubMed] [Google Scholar]
- 142.Sun H, Martin C, Kesselring D, Keller R, Moeller KD. Building Functionalized Peptidomimetics: Use of Electroauxiliaries for Introducing N-Acyliminium Ions Into Peptides. J Am Chem Soc. 2006;128:13761–13771. doi: 10.1021/ja064737l. [DOI] [PubMed] [Google Scholar]
- 143.Sun H, Moeller KD. Building Functionalized Peptidomimetics: New Electroauxiliaries and the Use of a Chemical Oxidant for Introducing N-Acyliminium Ions Into Peptides. Org Lett. 2003;5:3189–3192. doi: 10.1021/ol034872s. [DOI] [PubMed] [Google Scholar]
- 144.Kamada T, Oku A. Intramolecular Acceleration Effect of a Tosylamide Group on the Electrochemical Oxidation of N-α-Silylalkyl Amides. J Chem Soc, Perkin Trans. 1(2002):1105–1110. [Google Scholar]
- 145.Suga S, Watanabe M, Yoshida J-I. Electroauxiliary-Assisted Sequential Introduction of Two Carbon Nucleophiles on the Same α-Carbon of Nitrogen: Application to the Synthesis of Spiro Compounds. J Am Chem Soc. 2002;124:14824–14825. doi: 10.1021/ja028663z. [DOI] [PubMed] [Google Scholar]
- 146.Suga S, Watanabe M, Song C-H, Yoshida J-I. Electroauxiliary-Assisted Sequential Introduction of Organic Groups on the α-Carbons of Nitrogen. Electrochemistry. 2006;74:672–679. [Google Scholar]
- 147.Kim S, Hayashi K, Kitano Y, Tada M, Chiba K. Anodic Modification of Proline Derivatives Using a Lithium Perchlorate/Nitromethane Electrolyte Solution. Org Lett. 2002;4:3735–3737. doi: 10.1021/ol026713z. [DOI] [PubMed] [Google Scholar]
- 148.Shoji T, Kim S, Yamamoto K, Kawai T, Okada Y, Chiba K. Anodic Substitution Reaction of Proline Derivatives Using the 2,4,6-Trimethoxyphenyl Leaving Group. Org Lett. 2014;16:6404–6407. doi: 10.1021/ol503198p. [DOI] [PubMed] [Google Scholar]
- 149.Chiba K, Kim S. Anodic Carbon-Carbon Bond Formation in Lithium Perchlorate/Nitromethane Electrolyte Solution. Electrochemistry. 2009;77:21–29. [Google Scholar]
- 150.Suga S, Okajima M, Fujiwara K, Yoshida J-I. Cation Flow” Method: a New Approach to Conventional and Combinatorial Organic Syntheses Using Electrochemical Microflow Systems. J Am Chem Soc. 2001;123:7941–7942. doi: 10.1021/ja015823i. [DOI] [PubMed] [Google Scholar]
- 151.Suga S, Okajima M, Yoshida J. Reaction of an Electrogenerated “Iminium Cation Pool”with Organometallic Reagents. Direct Oxidative α-Alkylation and-Arylation of Amine Derivatives. Tetrahedron Lett. 2001;42:2173–2176. [Google Scholar]
- 152.Suga S, Okajima M, Fujiwara K, Yoshida J-I. Electrochemical Combinatorial Organic Syntheses Using Microflow Systems. QSAR Comb Sci. 2005;24:728–741. [Google Scholar]
- 153.Shankaraiah N, Pilli RA, Santos LS. Enantioselective Total Syntheses of Ropivacaine and Its Analogues. Tetrahedron Lett. 2008;49:5098–5100. [Google Scholar]
- 154.Shoji T, Kim S, Chiba K. Synthesis of Azanucleosides by Anodic Oxidation in a Lithium Perchlorate-Nitroalkane Medium and Diversification at the 4′-Nitrogen Position. Angew Chem, Int Ed. 2017;56:4011–4014. doi: 10.1002/anie.201700547. [DOI] [PubMed] [Google Scholar]
- 155.Kim S, Shoji T, Kitano Y, Chiba K. Electrochemical Synthesis of Azanucleoside Derivatives Using a Lithium Perchlorate–Nitromethane System. Chem Commun. 2013;49:6525–6527. doi: 10.1039/c3cc43273d. [DOI] [PubMed] [Google Scholar]
- 156.Shoji T, Haraya S, Kim S, Chiba K. Development of Anodic Modification Reaction of N-Acryloyl-Proline Derivatives Using Lithium Perchlorate-Nitromethane System. Electrochim Acta. 2016;200:290–295. [Google Scholar]
- 157.Nagaki A, Togai M, Suga S, Aoki N, Mae K, Yoshida J-I. Control of Extremely Fast Competitive Consecutive Reactions Using Micromixing. Selective Friedel-Crafts Aminoalkylation. J Am Chem Soc. 2005;127:11666–11675. doi: 10.1021/ja0527424. [DOI] [PubMed] [Google Scholar]
- 158.Ashikari Y, Kiuchi Y, Takeuchi T, Ueoka K, Suga S, Yoshida J-I. Addition of N-Acyliminium Ion Pools to Alkenes Having a Nucleophilic Moiety: Integration of Intermolecular and Intramolecular Reactions. Chem Lett. 2014;43:210–212. [Google Scholar]
- 159.Suga S, Nagaki A, Tsutsui Y, Yoshida J-I. N-Acyliminium Ion Pool” as a Heterodiene in [4 + 2] Cycloaddition Reaction. Org Lett. 2003;5:945–947. doi: 10.1021/ol0341243. [DOI] [PubMed] [Google Scholar]
- 160.Suga S, Nishida T, Yamada D, Nagaki A, Yoshida J-I. Three-Component Coupling Based on the “Cation Pool” Method. J Am Chem Soc. 2004;126:14338–14339. doi: 10.1021/ja0455704. [DOI] [PubMed] [Google Scholar]
- 161.Suga S, Yamada D, Yoshida J-I. Cationic Three-Component Coupling Involving an Optically Active Enamine Derivative. From Time Integration to Space Integration of Reactions. Chem Lett. 2010;39:404–406. [Google Scholar]
- 162.Nagaki A, Kawamura K, Suga S, Ando T, Sawamoto M, Yoshida J-I. Cation Pool-Initiated Controlled/Living Polymerization Using Microsystems. J Am Chem Soc. 2004;126:14702–14703. doi: 10.1021/ja044879k. [DOI] [PubMed] [Google Scholar]
- 163.Suga S, Kageyama Y, Babu G, Itami K, Yoshida J-I. Cationic Carbohydroxylation of Alkenes and Alkynes Using the Cation Pool Method. Org Lett. 2004;6:2709–2711. doi: 10.1021/ol049049q. [DOI] [PubMed] [Google Scholar]
- 164.Maruyama T, Mizuno Y, Shimizu I, Suga S, Yoshida J-I. Reactions of a N-Acyliminium Ion Pool with Benzylsilanes. Implication of a Radical/Cation/Radical Cation Chain Mechanism Involving Oxidative C–Si Bond Cleavage. J Am Chem Soc. 2007;129:1902–1903. doi: 10.1021/ja068589a. [DOI] [PubMed] [Google Scholar]
- 165.Suga S, Shimizu I, Ashikari Y, Mizuno Y, Maruyama T, Yoshida J-I. Electro-Initiated Coupling Reactions of N-Acyliminium Ion Pools with Arylthiomethylsilanes and Aryloxymethylsilanes. Chem Lett. 2008;37:1008–1009. [Google Scholar]
- 166.Maruyama T, Suga S, Yoshida J-I. Distannane Mediated Reaction of N-Acyliminium Ion Pools with Alkyl Halides. a Chain Mechanism Involving Radical Addition Followed by Electron Transfer. Tetrahedron. 2006;62:6519–6525. [Google Scholar]
- 167.Maruyama T, Suga S, Yoshida J-I. Radical Addition to “Cation Pool.” Reverse Process of Radical Cation Fragmentation. J Am Chem Soc. 2005;127:7324–7325. doi: 10.1021/ja0511218. [DOI] [PubMed] [Google Scholar]
- 168.Suga S, Suzuki S, Yoshida J-I. Reduction of a “Cation Pool”: a New Approach to Radical Mediated C–C Bond Formation. J Am Chem Soc. 2002;124:30–31. doi: 10.1021/ja0171759. [DOI] [PubMed] [Google Scholar]
- 169.Kabeshov MA, Musio B, Murray PRD, Browne DL, Ley SV. Expedient Preparation of Nazlinine and a Small Library of Indole Alkaloids Using Flow Electrochemistry as an Enabling Technology. Org Lett. 2014;16:4618–4621. doi: 10.1021/ol502201d. [DOI] [PubMed] [Google Scholar]
- 170.Shankaraiah N, da Silva WA, Andrade CKZ, Santos LS. Enantioselective Total Synthesis of (S)-(–)-Quinolactacin B. Tetrahedron Lett. 2008;49:4289–4291. [Google Scholar]
- 171.Mirabal-Gallardo Y, Piérola J, Shankaraiah N, Santos LS. Enantioselective Total Synthesis of (S)-(+)-Lennoxamine Through Asymmetric Hydrogenation Mediated by L-Proline-Tetrazole Ruthenium Catalyst. Tetrahedron Lett. 2012;53:3672–3675. [Google Scholar]
- 172.Louafi F, Moreau J, Shahane S, Golhen S, Roisnel T, Sinbandhit S, Hurvois J-P. Electrochemical Synthesis and Chemistry of Chiral 1-Cyanotetrahydroisoquinolines. an Approach to the Asymmetric Syntheses of the Alkaloid (–)-Crispine a and Its Natural (+)-Antipode. J Org Chem. 2011;76:9720–9732. doi: 10.1021/jo2017982. [DOI] [PubMed] [Google Scholar]
- 173.Louafi F, Hurvois J-P, Chibani A, Roisnel T. Synthesis of Tetrahydroisoquinoline Alkaloids via Anodic Cyanation as the Key Step. J Org Chem. 2010;75:5721–5724. doi: 10.1021/jo100714y. [DOI] [PubMed] [Google Scholar]
- 174.Girard N, Hurvois J-P, Moinet C, Toupet L. Total Synthesis of (±)-Pumiliotoxin C: an Electrochemical Approach. Eur J Org Chem. 2005;2005:2269–2280. [Google Scholar]
- 175.Kam TS, Lim TM, Tan GH. Electrochemical Oxidation of Aspidofractinine-Type Alkaloids: Formation of Kopsine, Kopsidine, Kopsinitarine and Bisindole Derivatives. J Chem Soc, Perkin Trans. 1(2001):1594–1604. [Google Scholar]
- 176.Paci A, Martens T, Royer J. Anodic Oxidation of Ifosfamide and Cyclophosphamide: a Biomimetic Metabolism Model of the Oxazaphosphorinane Anticancer Drugs. Bioorg Med Chem Lett. 2001;11:1347–1349. doi: 10.1016/s0960-894x(01)00218-9. [DOI] [PubMed] [Google Scholar]
- 177.Tereshchenko AD, Myronchuk JS, Leitchenko LD, Knysh IV, Tokmakova GO, Litsis OO, Tolmachev A, Liubchak K, Mykhailiuk P. Synthesis of 3-Oxadiazolyl/Triazolyl Morpholines: Novel Scaffolds for Drug Discovery. Tetrahedron. 2017;73:750–757. [Google Scholar]
- 178.Furukubo S, Moriyama N, Onomura O, Matsumura Y. Stereoselective Synthesis of Azasugars by Electrochemical Oxidation. Tetrahedron Lett. 2004;45:8177–8181. [Google Scholar]
- 179.Demizu Y, Shiigi H, Mori H, Matsumoto K, Onomura O. Convenient Synthesis of an Enantiomerically Pure Bicyclic Proline and Its N-Oxyl Derivatives. Tetrahedron: Asymmetry. 2008;19:2659–2665. [Google Scholar]
- 180.Bodmann K, Bug T, Steinbeisser S, Kreuder R, Reiser O. Electrochemical Oxidation of 2-Substituted Piperidines as a Key Step Towards the Synthesis of Hydroxylated γ-Amino Acids. Tetrahedron Lett. 2006;47:2061–2064. [Google Scholar]
- 181.Wang X, Li J, Saporito RA, Toyooka N. Enantiodivergent Synthesis of the Quinolizidine Poison Frog Alkaloid 195C. Tetrahedron. 2013;69:10311–10315. [Google Scholar]
- 182.Reuter C, Huy P, Neudörfl J-M, Kühne R, Schmalz H-G. Exercises in Pyrrolidine Chemistry: Gram Scale Synthesis of a Pro-Pro Dipeptide Mimetic with a Polyproline Type II Helix Conformation. Chem - Eur J. 2011;17:12037–12044. doi: 10.1002/chem.201101704. [DOI] [PubMed] [Google Scholar]
- 183.Beal LM, Bin Liu, Chu W, Moeller KD. Anodic Amide Oxidation/Olefin Metathesis Strategies: Developing a Unified Approach to the Synthesis of Bicyclic Lactam Peptidomimetics. Tetrahedron. 2000;56:10113–10125. [Google Scholar]
- 184.Lennartz M, Steckhan E. Synthesis of Bicyclic Lactams via Ring Closing Olefin Metathesis and Intramolecular Heck Reaction. Synlett. 2000:319–322. [Google Scholar]
- 185.Frankowski KJ, Liu R, Milligan GL, Moeller KD, Aubé J. Practical Electrochemical Anodic Oxidation of Polycyclic Lactams for Late Stage Functionalization. Angew Chem, Int Ed. 2015;54:10555–10558. doi: 10.1002/anie.201504775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Faust MR, HOfner G, Pabel J, Wanner KT. Azetidine Derivatives as Novel γ-Aminobutyric Acid Uptake Inhibitors: Synthesis, Biological Evaluation, and Structure-Activity Relationship. Eur J Med Chem. 2010;45:2453–2466. doi: 10.1016/j.ejmech.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 187.Madelaine C, Six Y, Buriez O. Electrochemical Aerobic Oxidation of Aminocyclopropanes to Endoperoxides. Angew Chem, Int Ed. 2007;46:8046–8049. doi: 10.1002/anie.200702903. [DOI] [PubMed] [Google Scholar]
- 188.Kubota J, Shimizu Y, Mitsudo K, Tanaka H. Water-Soluble N-Oxyl Compounds-Mediated Electrooxidation of Alcohols in Water: a Prominent Access to a Totally Closed System. Tetrahedron Lett. 2005;46:8975–8979. [Google Scholar]
- 189.Demizu Y, Shiigi H, Oda T, Matsumura Y, Onomura O. Efficient Oxidation of Alcohols Electrochemically Mediated by Azabicyclo-N-Oxyls. Tetrahedron Lett. 2008;49:48–52. [Google Scholar]
- 190.Rafiee M, Miles KC, Stahl SS. Electrocatalytic Alcohol Oxidation with TEMPO and Bicyclic Nitroxyl Derivatives: Driving Force Trumps Steric Effects. J Am Chem Soc. 2015;137:14751–14757. doi: 10.1021/jacs.5b09672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hickey DP, Schiedler DA, Matanovic I, Doan PV, Atanassov P, Minteer SD, Sigman MS. Predicting Electrocatalytic Properties: Modeling Structure–Activity Relationships of Nitroxyl Radicals. J Am Chem Soc. 2015;137:16179–16186. doi: 10.1021/jacs.5b11252. [DOI] [PubMed] [Google Scholar]
- 192.Palma A, Cárdenas J, Frontana-Uribe BA. Comparative Study of the N-Isobutyl-(2E,6Z)-Dodecadienamide Chemical and Electrochemical Syntheses. Green Chem. 2009;11:283–293. [Google Scholar]
- 193.Kuroboshi M, Yoshihisa H, Cortona MN, Kawakami Y. Electro-Oxidative Kinetic Resolution of Sec-Alcohols by Using an Optically Active N-Oxyl Mediator. Tetrahedron Lett. 2000;41:8131–8135. [Google Scholar]
- 194.Tanaka H, Kawakami Y, Goto K, Kuroboshi M. An Aqueous Silica Gel Disperse Electrolysis System. N-Oxyl-Mediated Electrooxidation of Alcohols. Tetrahedron Lett. 2001;42:445–448. [Google Scholar]
- 195.Tanaka H, Kubota J, Itogawa SJ, Ido T, Kuroboshi M, Shimamura K, Uchida T. Electrooxidation of Alcoholsin a Disperse System with N-Oxyl-Immobilized PolyethyleneParticles as Disperse Phase and Aqueous NaHCO3/NaBras Disperse Media: a Totally Closed Electrolysis System. Synlett. 2003:951–954. [Google Scholar]
- 196.Yoshida T, Kuroboshi M, Oshitani J, Gotoh K, Tanaka H. Electroorganic Synthesis in Oil-in-Water Nanoemulsion: TEMPO-Mediated Electrooxidation of Amphiphilic Alcohols in Water. Synlett. 2007;2007:2691–2694. [Google Scholar]
- 197.Hill-Cousins JT, Kuleshova J, Green RA, Birkin PR, Pletcher D, Underwood TJ, Leach SG, Brown RCD. TEMPO-Mediated Electrooxidation of Primary and Secondary Alcohols in a Microfluidic Electrolytic Cell. ChemSusChem. 2012;5:326–331. doi: 10.1002/cssc.201100601. [DOI] [PubMed] [Google Scholar]
- 198.Kubota J, Ido T, Kuroboshi M, Tanaka H, Uchida T, Shimamura K. Electrooxidation of Alcohols in an N-Oxyl-Immobilized Rigid Network Polymer Particles/Water Disperse System. Tetrahedron. 2006;62:4769–4773. [Google Scholar]
- 199.Tanaka H, Kubota J, Miyahara S, Kuroboshi M. Electrooxidation of Alcohols in an N-Oxyl-Immobilized Poly(Ethylene-Co-Acrylic Acid)/Water Disperse System. Bull Chem Soc Jpn. 2005;78:1677–1684. [Google Scholar]
- 200.Geneste F, Moinet C, Ababou-Girard S, Solal F. Covalent Attachment of TEMPO Onto a Graphite Felt Electrode and Application in Electrocatalysis. New J Chem. 2005;29:1520–1526. [Google Scholar]
- 201.Belgsir EM, Schäfer HJ. Selective Oxidation of Carbohydrates on Nafion®–TEMPO-Modified Graphite Felt Electrodes. Electrochem Commun. 2001;3:32–35. [Google Scholar]
- 202.Palmisano G, Ciriminna R, Pagliaro M. Waste-Free Electrochemical Oxidation of Alcohols in Water. Adv Synth Catal. 2006;348:2033–2037. [Google Scholar]
- 203.Kashiwagi Y, Kurashima F, Chiba S, Anzai JI, Osa T, Bobbitt JM. Asymmetric Electrochemical Lactonization of Diols on a Chiral 1-Azaspiro[5.5]Undecane N-Oxyl Radical Mediator-Modified Graphite Felt Electrode. Chem Commun. 2003:114–115. doi: 10.1039/b209871g. [DOI] [PubMed] [Google Scholar]
- 204.Das A, Stahl SS. Noncovalent Immobilization of Molecular Electrocatalysts for Chemical Synthesis: Efficient Electrochemical Alcohol Oxidation with a Pyrene-TEMPO Conjugate. Angew Chem, Int Ed. 2017;56:8892–8897. doi: 10.1002/anie.201704921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hickey DP, Milton RD, Chen D, Sigman MS, Minteer SD. TEMPO-Modified Linear Poly(Ethylenimine) for Immobilization-Enhanced Electrocatalytic Oxidation of Alcohols. ACS Catal. 2015;5:5519–5524. [Google Scholar]
- 206.Schämann M, Schäfer HJ. TEMPO-Mediated Anodic Oxidation of Methyl Glycosides and 1-Methyl and 1-Azido Disaccharides. Eur J Org Chem. 2003;2003:351–358. [Google Scholar]
- 207.Badalyan A, Stahl SS. Cooperative Electrocatalytic Alcohol Oxidation with Electron-Proton-Transfer Mediators. Nature. 2016;535:406–410. doi: 10.1038/nature18008. [DOI] [PubMed] [Google Scholar]
- 208.Christopher C, Lawrence S, Kulandainathan MA, Kulangiappar K, Raja ME, Xavier N, Raja S. Electrochemical Selective Oxidation of Aromatic Alcohols with Sodium Nitrate Mediator in Biphasic Medium at Ambient Temperature. Tetrahedron Lett. 2012;53:2802–2804. [Google Scholar]
- 209.Christopher C, Lawrence S, Bosco AJ, Xavier N, Raja S. Selective Oxidation of Benzyl Alcohol by Two Phase Electrolysis Using Nitrate as Mediator. Catal Sci Technol. 2012;2:824–824. [Google Scholar]
- 210.Khan FN, Jayakumar R, Pillai CN. Electrocatalytic Oxidative Cleavage by Electrogenerated Periodate. J Mol Catal A: Chem. 2003;195:139–145. [Google Scholar]
- 211.Blanco M, de Lima DP, Maia G. A Novel Compound From the Electrosynthesis of Bioactive (+)-10β, 14-Dihydroxy-Allo-Aromadendrane. J Electroanal Chem. 2001;512:49–55. [Google Scholar]
- 212.Maki T, Iikawa S, Mogami G, Harasawa H, Matsumura Y, Onomura O. Efficient Oxidation of 1,2-Diols Into α-Hydroxyketones Catalyzed by Organotin Compounds. Chem - Eur J. 2009;15:5364–5370. doi: 10.1002/chem.200900159. [DOI] [PubMed] [Google Scholar]
- 213.Minato D, Arimoto H, Nagasue Y, Demizu Y, Onomura O. Asymmetric Electrochemical Oxidation of 1,2-Diols, Aminoalcohols, and Aminoaldehydes in the Presence of Chiral Copper Catalyst. Tetrahedron. 2008;64:6675–6683. [Google Scholar]
- 214.Medici A, Pedrini P, De Battisti A, Fantin G. Anodic Electrochemical Oxidation of Cholic Acid. Steroids. 2001;66:63–69. doi: 10.1016/s0039-128x(00)00185-9. [DOI] [PubMed] [Google Scholar]
- 215.Carr JP, Hampson NA. Lead Dioxide Electrode. Chem Rev. 1972;72:679–703. [Google Scholar]
- 216.Lybaert J, Trashin S, Maes BUW, De Wael K, Abbaspour Tehrani K. Cooperative Electrocatalytic and Chemoselective Alcohol Oxidation by Shvo’s Catalyst. Adv Synth Catal. 2017;359:919–925. [Google Scholar]
- 217.Zhu Y, Zhang J, Chen Z, Zhang A, Ma C. Synthesis of Nitrocarbazole Compounds and Their Electrocatalytic Oxidation of Alcohol. Chin J Catal. 2016;37:533–538. [Google Scholar]
- 218.Hosokawa Y-Y, Hakamata H, Murakami T, Aoyagi S, Kuroda M, Mimaki Y, Ito A, Morosawa S, Kusu F. Electrochemical Oxidation of Cholesterol in Acetonitrile Leads to the Formation of Cholesta-4,6-Dien-3-One. Electrochim Acta. 2009;54:6412–6416. [Google Scholar]
- 219.Hosokawa Y-Y, Hakamata H, Murakami T, Kusu F. Electrosynthesis of Cholesta-4,6-Dien-3-One From Cholesterol on a Laboratory Synthetic Scale. Tetrahedron Lett. 2010;51:129–132. [Google Scholar]
- 220.Markó IE. Electrochemical Oxidative Cyclisation of δ-Hydroxy-Tetrahydropyrans to Spiroketals. Tetrahedron Lett. 2000;41:4383–4387. [Google Scholar]
- 221.Shirai K, Hamamoto T, Maki T, Onomura O. Effects of Trifluoroethanol as a Co-Solvent on the Electrochemical Oxidation of Hardly Oxidizable Organic Compounds. J Electroanal Chem. 2001;507:191–197. [Google Scholar]
- 222.Suga S, Suzuki S, Yamamoto A, Yoshida J-I. Electrooxidative Generation and Accumulation of Alkoxycarbenium Ions and Their Reactions with Carbon Nucleophiles. J Am Chem Soc. 2000;122:10244–10245. [Google Scholar]
- 223.Suzuki S, Matsumoto K, Kawamura K, Suga S, Yoshida J-I. Generation of Alkoxycarbenium Ion Pools From Thioacetals and Applications to Glycosylation Chemistry. Org Lett. 2004;6:3755–3758. doi: 10.1021/ol048524h. [DOI] [PubMed] [Google Scholar]
- 224.Okajima M, Suga S, Itami K, Yoshida J-I. Cation Pool” Method Based on C-C Bond Dissociation. Effective Generation of Monocations and Dications. J Am Chem Soc. 2005;127:6930–6931. doi: 10.1021/ja050414y. [DOI] [PubMed] [Google Scholar]
- 225.Yamago S, Kokubo K, Hara O, Masuda S, Yoshida J-I. Electrochemistry of Chalcogenoglycosides. Rational Design of Iterative Glycosylation Based on Reactivity Control of Glycosyl Donors and Acceptors by Oxidation Potentials. J Org Chem. 2002;67:8584–8592. doi: 10.1021/jo0261350. [DOI] [PubMed] [Google Scholar]
- 226.France RR, Rees NV, Wadhawan JD, Fairbanks AJ, Compton RG. Selective Activation of Glycosyl Donors Utilising Electrochemical Techniques: a Study of the Thermodynamic Oxidation Potentials of a Range of Chalcoglycosides. Org Biomol Chem. 2004;2:2188–2202. doi: 10.1039/b316720h. [DOI] [PubMed] [Google Scholar]
- 227.Tanaka N, Ohnishi F, Uchihata D, Torii S, Nokami J. Electrochemical O-Glycosylation Using Thioglycosides as Glycosyl Donors in the Presence of a Catalytic Amount of Sodium Trifluoromethanesulfonate as a Supporting Electrolyte. Tetrahedron Lett. 2007;48:7383–7387. [Google Scholar]
- 228.Nokami T, Shibuya A, Tsuyama H, Suga S, Bowers AA, Crich D, Yoshida J-I. Electrochemical Generation of Glycosyl Triflate Pools. J Am Chem Soc. 2007;129:10922–10928. doi: 10.1021/ja072440x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Nokami T, Shibuya A, Manabe S, Ito Y, Yoshida J-I. α- and β-Glycosyl Sulfonium Ions: Generation and Reactivity. Chem - Eur J. 2009;15:2252–2255. doi: 10.1002/chem.200802293. [DOI] [PubMed] [Google Scholar]
- 230.Nokami T, Nozaki Y, Saigusa Y, Shibuya A, Manabe S, Ito Y, Yoshida J-I. Glycosyl Sulfonium Ions as Storable Intermediates for Glycosylations. Org Lett. 2011;13:1544–1547. doi: 10.1021/ol200242u. [DOI] [PubMed] [Google Scholar]
- 231.Mitsudo K, Kawaguchi T, Miyahara S, Matsuda W, Kuroboshi M, Tanaka H. Electrooxidative Glycosylation Through C–S Bond Cleavage of 1-Arylthio-2,3-Dideoxyglycosides. Synthesis of 2′,3′-Dideoxynucleosides. Org Lett. 2005;7:4649–4652. doi: 10.1021/ol051776d. [DOI] [PubMed] [Google Scholar]
- 232.Nokami T, Hayashi R, Saigusa Y, Shimizu A, Liu C-Y, Mong K-KT, Yoshida J-I. Automated Solution-Phase Synthesis of Oligosaccharides via Iterative Electrochemical Assembly of Thioglycosides. Org Lett. 2013;15:4520–4523. doi: 10.1021/ol402034g. [DOI] [PubMed] [Google Scholar]
- 233.Nokami T, Isoda Y, Sasaki N, Takaiso A, Hayase S, Itoh T, Hayashi R, Shimizu A, Yoshida J-I. Automated Electrochemical Assembly of the Protected Potential TMG-Chitotriomycin Precursor Based on Rational Optimization of the Carbohydrate Building Block. Org Lett. 2015;17:1525–1528. doi: 10.1021/acs.orglett.5b00406. [DOI] [PubMed] [Google Scholar]
- 234.Nokami T, Tsuyama H, Shibuya A, Nakatsutsumi T. Oligosaccharide Synthesis Based on a One-Pot Electrochemical Glycosylation–Fmoc Deprotection Sequence. Chem Lett. 2008;37:942–943. [Google Scholar]
- 235.Drouin L, Compton RG, Fairbanks AJ. Electrochemical Glycosylation in the Presence of a Catalytic Chemical Mediator. J Phys Org Chem. 2008;21:516–522. [Google Scholar]
- 236.France RR, Compton RG, Davis BG, Fairbanks AJ, Rees NV, Wadhawan JD. Selective Electrochemical Glycosylation by Reactivity Tuning. Org Biomol Chem. 2004;2:2195–2202. doi: 10.1039/b316728c. [DOI] [PubMed] [Google Scholar]
- 237.Yoshida J-I, Watanabe M, Toshioka H, Imagawa M, Suga S. Selective Electrochemical Oxidation of Heteroatom Compounds Having Both Silicon and Tin on the Same Carbon as Electroauxiliaries. J Electroanal Chem. 2001;507:55–65. [Google Scholar]
- 238.Suga S, Suzuki S, Yoshida J-I. Intramolecular Participation in Alkoxycarbenium Ion Pools. Org Lett. 2005;7:4717–4720. doi: 10.1021/ol051915r. [DOI] [PubMed] [Google Scholar]
- 239.Chiba K, Uchiyama R, Kim S, Kitano Y, Tada M. Benzylic Intermolecular Carbon–Carbon Bond Formation by Selective Anodic Oxidation of Dithioacetals. Org Lett. 2001;3:1245–1248. doi: 10.1021/ol015734a. [DOI] [PubMed] [Google Scholar]
- 240.Honjo E, Kutsumura N, Ishikawa Y, Nishiyama S. Synthesis of a Spiroacetal Moiety of Antitumor Antibiotic Ossamycin by Anodic Oxidation. Tetrahedron. 2008;64:9495–9506. [Google Scholar]
- 241.Barnhurst LA, Wan Y, Kutateladze AG. Efficient Electrochemical Deprotection of Carboxylic and Amino Acids From Their 2-(Hydroxymethyl)-1,3-Dithiane (Dim) Esters. Org Lett. 2000;2:799–801. doi: 10.1021/ol005537w. [DOI] [PubMed] [Google Scholar]
- 242.Tanaka H, Tokumaru Y, Fukui K-I, Kuroboshi M, Torii S, Jutand A, Amatore C. Chemo- and Product-Selective Electrooxidation of 3-(Arylthiomethyl)-Δ3-Cephems. Synthesis. 2009;2009:3449–3459. [Google Scholar]
- 243.Batanero B, Picazo O, Barba F. Facile and Efficient Transformation of Xanthates Into Thiocarbonates by Anodic Oxidation. J Org Chem. 2001;66:320–322. doi: 10.1021/jo001206l. [DOI] [PubMed] [Google Scholar]
- 244.Kaimakliotis C, Fry AJ. Anodic Oxidation of Methyl α-Dimethylsilyldihydrocinnamate. a Novel Silicon γ -Aryl Effect. J Org Chem. 2003;68:9893–9898. doi: 10.1021/jo030264e. [DOI] [PubMed] [Google Scholar]
- 245.Watanabe M, Suga S, Yoshida J. Intramolecular Assistance of Electron Transfer From Heteroatom Compounds. Electrochemical Oxidation of 2-(2-Pyridyl) Ethyl-Substituted Ethers, Sulfides, and Selenides. Bull Chem Soc Jpn. 2000;73:243–247. [Google Scholar]
- 246.Finney EE, Ogawa KA, Boydston AJ. Organocatalyzed Anodic Oxidation of Aldehydes. J Am Chem Soc. 2012;134:12374–12377. doi: 10.1021/ja304716r. [DOI] [PubMed] [Google Scholar]
- 247.Green RA, Pletcher D, Leach SG, Brown RCD. N-Heterocyclic Carbene-Mediated Oxidative Electrosynthesis of Esters in a Microflow Cell. Org Lett. 2015;17:3290–3293. doi: 10.1021/acs.orglett.5b01459. [DOI] [PubMed] [Google Scholar]
- 248.Ogawa KA, Boydston AJ. Organocatalyzed Anodic Oxidation of Aldehydes to Thioesters. Org Lett. 2014;16:1928–1931. doi: 10.1021/ol500459x. [DOI] [PubMed] [Google Scholar]
- 249.Green RA, Pletcher D, Leach SG, Brown RCD. N-Heterocyclic Carbene-Mediated Microfluidic N-Heterocyclic Carbene-Mediated Oxidative Electrosynthesis. Org Lett. 2016;18:1198–1201. doi: 10.1021/acs.orglett.6b00339. [DOI] [PubMed] [Google Scholar]
- 250.Qu Q, Gao X, Gao J, Yuan G. A Highly Efficient Electrochemical Route for the Conversion of Aldehydes to Nitriles. Sci China: Chem. 2015;58:747–750. [Google Scholar]
- 251.Liang S, Zeng C-C, Tian H-Y, Sun B-G, Luo X-G, Ren F-Z. Electrochemically Oxidative α-C–H Functionalization of Ketones: a Cascade Synthesis of α-Amino Ketones Mediated by NH4I. J Org Chem. 2016;81:11565–11573. doi: 10.1021/acs.joc.6b01595. [DOI] [PubMed] [Google Scholar]
- 252.Gao W-J, Lam CM, Sun B-G, Little RD, Zeng C-C. Selective Electrochemical C O Bond Cleavage of β-O-4 Lignin Model Compounds Mediated by Iodide Ion. Tetrahedron. 2017;73:2447–2454. [Google Scholar]
- 253.Elinson MN, Feducovich SK, Dorofeev AS. Indirect Electrochemical Oxidation of Aryl Alkyl Ketones Mediated by NaI–NaOH System: Facile and Effective Way to α-Hydroxyketals. Tetrahedron. 2000;56:9999–10003. [Google Scholar]
- 254.Elinson MN, Feducovich SK, Dmitriev DE. Stereoselective Electrochemical Transformation of 4-Substituted Cyclohexanones Into Cis-5-Substituted-2, 2-Dimethoxycyclohexanols. Tetrahedron Lett. 2001;42:5557–5559. [Google Scholar]
- 255.Elinson MN, Dorofeev AS, Feducovich SK, Nasybullin RF, Litvin EF, Kopyshev MV, Nikishin GI. Indirect Electrochemical Oxidation of Piperidin-4-Ones Mediated by Sodium Halide-Base System. Tetrahedron. 2006;62:8021–8028. [Google Scholar]
- 256.Kang L-S, Luo M-H, Lam CM, Hu L-M, Little RD, Zeng C-C. Electrochemical C–H Functionalization and Subsequent C–S and C–N Bond Formation: Paired Electrosynthesis of 3-Amino-2-Thiocyanato-α,β-Unsaturated Carbonyl Derivatives Mediated by Bromide Ions. Green Chem. 2016;18:3767–3774. [Google Scholar]
- 257.Pan X-J, Gao J, Yuan G-Q. An Efficient Electrochemical Synthesis of β-Keto Sulfones From Sulfinates and 1,3-Dicarbonyl Compounds. Tetrahedron. 2015;71:5525–5530. [Google Scholar]
- 258.Tsuchida K, Kochi T, Kakiuchi F. Copper-Catalyzed Electrochemical Chlorination of 1,3-Dicarbonyl Compounds Using Hydrochloric Acid. Asian J Org Chem. 2013;2:935–937. [Google Scholar]
- 259.Yao C, Wang Y, Li T, Yu C, Li L, Wang C. A Pseudo Multi-Component Electrochemical Synthesis of Spiro Dihydrofuran Derivatives. Tetrahedron. 2013;69:10593–10597. [Google Scholar]
- 260.Elinson MN, Dorofeev AS, Feducovich SK, Belyakov PA, Nikishin GI. Stereoselective Electrocatalytic Oxidative Coupling of Phenylacetonitriles: Facile and Convenient Way to Trans-α,β-Dicyanostilbenes. Eur J Org Chem. 2007;2007:3023–3027. [Google Scholar]
- 261.Gao H, Zha Z, Zhang Z, Ma H, Wang Z. A Simple and Efficient Approach to Realize Difunctionalization of Arylketones with Malonate Esters via Electrochemical Oxidation. Chem Commun. 2014;50:5034–5036. doi: 10.1039/c4cc01277a. [DOI] [PubMed] [Google Scholar]
- 262.Okimoto M, Yamamori H, Ohashi K, Nishikawa S, Hoshi M, Yoshida T. Anodic Cyclization of 1,7-Diarylheptane-1,7-Diones to the Corresponding 1,2-Diaroylcyclopentanes. Synlett. 2012;23:2544–2548. [Google Scholar]
- 263.Minato D, Mizuta S, Kuriyama M, Matsumura Y, Onomura O. Diastereoselective Construction of Azetidin-2-Ones by Electrochemical Intramolecular C-C Bond Forming Reaction. Tetrahedron. 2009;65:9742–9748. [Google Scholar]
- 264.Okimoto M, Yamamori H, Ohashi K, Hoshi M, Yoshida T. Anodic Cyclization of Dimethyl 2-(3-Oxo-3-Arylpropyl) Malonates Into the Corresponding Dimethyl 2-Aroylcyclopropane-1,1-Dicarboxylates. Synlett. 2013;24:1568–1572. [Google Scholar]
- 265.Elinson MN, Feducovich SK, Lizunova TL, Nikishin GI. Electrochemical Transformation of Malononitrile and Carbonyl Compounds Into Functionally Substituted Cyclopropanes: Electrocatalytic Variant of the Wideqvist Reaction. Tetrahedron. 2000;56:3063–3069. [Google Scholar]
- 266.Elinson MN, Feducovich SK, Starikova ZA, Vereshchagin AN, Nikishin GI. Stereoselective Electrocatalytic Transformation of Arylidenemalononitriles and Malononitrile Into (1R,5S,6R)*-6-Aryl-2-Amino-4,4-Dialkoxy-1,5-Dicyano-3-Azabicyclo[3.1.0]Hex-2-Enes. Tetrahedron. 2004;60:11743–11749. [Google Scholar]
- 267.Elinson MN, Feducovich SK, Starikova ZA, Vereshchagin AN, Belyakov PA, Nikishin GI. Stereoselective Electrocatalytic Transformation of Malonate and Alkylidenecyanoacetates Into (E)-3-Substituted 2-Cyanocyclopropane-1,1,2-Tricarboxylates. Tetrahedron. 2006;62:3989–3996. [Google Scholar]
- 268.Vereshchagin AN, Elinson MN, Zaimovskaya TA, Nikishin GI. Electrocatalytic Cascade Multicomponent Assembling: Stereoselective One-Pot Synthesis of the Substituted 3-Azabicyclo[3.1.0]Hexane-1-Carboxylate System From Aldehyde, Malononitrile, Malonate and Methanol. Tetrahedron. 2008;64:9766–9770. [Google Scholar]
- 269.Vereshchagin AN, Elinson MN, Dorofeeva EO, Demchuk DV, Bushmarinov IS, Goloveshkin AS, Nikishin GI. Chemical and Electrocatalytic Cascade Cyclization of Guareschi Imides: “One-Pot” Simple and Efficient Way to the 2,4-Dioxo-3-Azabicyclo[3.1.0]Hexane Scaffold. Tetrahedron. 2013;69:5234–5241. [Google Scholar]
- 270.Elinson MN, Feducovich SK, Starikova ZA, Vereshchagin AN, Gorbunov SV, Nikishin GI. Stereoselective Electrocatalytic Transformation of Arylidene- or Alkylidenemalononitriles and Malonate Into Alkyl (1R,5R,6R)* 6-Substituted 5-Cyano-4,4-Dialkoxy-2-Oxo-3-Azabicyclo[3.1.0]Hexane-1-Carboxylates. Tetrahedron Lett. 2005;46:6389–6393. [Google Scholar]
- 271.Vereshchagin AN, Elinson MN, Dorofeeva EO, Nasybullin RF, Bushmarinov IS, Goloveshkin AS, Egorov MP. Electrocatalytic Cyclization of 3-(5-Hydroxy-3-Methylpyrazol-4-yl)-3-Arylpropionitriles: “One-Pot” Simple Fast and Efficient Way to Substituted Spirocyclopropylpyrazolones. Electrochim Acta. 2015;165:116–121. [Google Scholar]
- 272.Elinson MN, Dorofeeva EO, Vereshchagin AN, Nasybullin RF, Egorov MP. Electrocatalytic Stereoselective Transformation of Aldehydes and Two Molecules of Pyrazolin-5-One Into (R*,R*)-Bis(Spiro-2,4-Dihydro-3H-Pyrazol-3-One)-Cyclopropanes. Catal Sci Technol. 2015;5:2384–2387. [Google Scholar]
- 273.Zhang Z, Su J, Zha Z, Wang Z. Electrochemical Synthesis of the Aryl α-Ketoesters From Acetophenones Mediated by KI. Chem - Eur J. 2013;19:17711–17714. doi: 10.1002/chem.201302307. [DOI] [PubMed] [Google Scholar]
- 274.Zhang Z, Su J, Zha Z, Wang Z. A Novel Approach for the One-Pot Preparation of α-Ketoamides by Anodic Oxidation. Chem Commun. 2013;49:8982–8984. doi: 10.1039/c3cc43685c. [DOI] [PubMed] [Google Scholar]
- 275.Xu K, Zhang Z, Qian P, Zha Z, Wang Z. Electrosynthesis of Enaminones Directly From Methyl Ketones and Amines with Nitromethane as a Carbon Source. Chem Commun. 2015;51:11108–11111. doi: 10.1039/c5cc02730f. [DOI] [PubMed] [Google Scholar]
- 276.Qian P, Su J-H, Wang Y, Bi M, Zha Z, Wang Z. Electrocatalytic C-H/N-H Coupling of 2′-Aminoacetophenones for the Synthesis of Isatins. J Org Chem. 2017;82:6434–6440. doi: 10.1021/acs.joc.7b00635. [DOI] [PubMed] [Google Scholar]
- 277.Wu Z-J, Xu H-C. Synthesis of C3-Fluorinated Oxindoles Through Reagent-Free Cross-Dehydrogenative Coupling. Angew Chem, Int Ed. 2017;56:4734–4738. doi: 10.1002/anie.201701329. [DOI] [PubMed] [Google Scholar]
- 278.Green RA, Jolley KE, Al-Hadedi AAM, Pletcher D, Harrowven DC, De Frutos O, Mateos C, Klauber DJ, Rincón JA, Brown RCD. Electrochemical Deprotection of Para-Methoxybenzyl Ethers in a Flow Electrolysis Cell. Org Lett. 2017;19:2050–2053. doi: 10.1021/acs.orglett.7b00641. [DOI] [PubMed] [Google Scholar]
- 279.Meng L, Su J, Zha Z, Zhang L, Zhang Z, Wang Z. Direct Electrosynthesis of Ketones From Benzylic Methylenes by Electrooxidative C–H Activation. Chem - Eur J. 2013;19:5542–5545. doi: 10.1002/chem.201204207. [DOI] [PubMed] [Google Scholar]
- 280.Li C, Zeng C-C, Hu L-M, Yang F-L, Yoo SJ, Little RD. Electrochemically Induced C–H Functionalization Using Bromide Ion/2,2,6,6-Tetramethylpiperidinyl-N-Oxyl Dual Redox Catalysts in a Two-Phase Electrolytic System. Electrochim Acta. 2013;114:560–566. [Google Scholar]
- 281.Utley JHP, Rozenberg GG. Electroorganic Reactions. Part 57. DDQ Mediated Anodic Oxidation of 2-Methyl- and 2-Benzylnaphthalenes. J Appl Electrochem. 2003;33:525–532. [Google Scholar]
- 282.Hruszkewycz DP, Miles KC, Thiel OR, Stahl SS. Co/NHPI-Mediated Aerobic Oxygenation of Benzylic C–H Bonds in Pharmaceutically Relevant Molecules. Chem Sci. 2017;8:1282–1287. doi: 10.1039/c6sc03831j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Zeng C-C, Zhang N-T, Lam CM, Little RD. Novel Triarylimidazole Redox Catalysts: Synthesis, Electrochemical Properties, and Applicability to Electrooxidative C–H Activation. Org Lett. 2012;14:1314–1317. doi: 10.1021/ol300195c. [DOI] [PubMed] [Google Scholar]
- 284.Lu N-N, Yoo SJ, Li L-J, Zeng C-C, Little RD. A Comparative Study of Organic Electron Transfer Redox Mediators: Electron Transfer Kinetics for Triarylimidazole and Triarylamine Mediators in the Oxidation of 4-Methoxybenzyl Alcohol. Electrochim Acta. 2014;142:254–260. [Google Scholar]
- 285.Lu N-N, Zhang N-T, Zeng C-C, Hu L-M, Yoo SJ, Little RD. Electrochemically Induced Ring-Opening/Friedel–Crafts Arylation of Chalcone Epoxides Catalyzed by a Triarylimidazole Redox Mediator. J Org Chem. 2015;80:781–789. doi: 10.1021/jo5022184. [DOI] [PubMed] [Google Scholar]
- 286.Yoo SJ, Li L-J, Zeng C-C, Little RD. Polymeric Ionic Liquid and Carbon Black Composite as a Reusable Supporting Electrolyte: Modification of the Electrode Surface. Angew Chem, Int Ed. 2015;54:3744–3747. doi: 10.1002/anie.201410207. [DOI] [PubMed] [Google Scholar]
- 287.Francke R, Little RD. Optimizing Electron Transfer Mediators Based on Arylimidazoles by Ring Fusion: Synthesis, Electrochemistry, and Computational Analysis of 2-Aryl-1-Methylphenanthro[9,10-D]Imidazoles. J Am Chem Soc. 2014;136:427–435. doi: 10.1021/ja410865z. [DOI] [PubMed] [Google Scholar]
- 288.Okajima M, Soga K, Nokami T, Suga S, Yoshida J-I. Oxidative Generation of Diarylcarbenium Ion Pools. Org Lett. 2006;8:5005–5007. doi: 10.1021/ol061647c. [DOI] [PubMed] [Google Scholar]
- 289.Nokami T, Watanabe T, Terao K, Soga K, Ohata K, Yoshida J-I. Multiple Alkylation of Thiophene Derivatives with Simple and Extended Diarylcarbenium Ion Pools. Electrochemistry. 2013;81:399–401. [Google Scholar]
- 290.Nokami T, Ohata K, Inoue M, Tsuyama H, Shibuya A, Soga K, Okajima M, Suga S, Yoshida J-I. Iterative Molecular Assembly Based on the Cation-Pool Method. Convergent Synthesis of Dendritic Molecules. J Am Chem Soc. 2008;130:10864–10865. doi: 10.1021/ja803487q. [DOI] [PubMed] [Google Scholar]
- 291.Nokami T, Watanabe T, Musya N, Morofuji T, Tahara K, Tobe Y, Yoshida J-I. Direct Dendronization of Polystyrenes Using Dendritic Diarylcarbenium Ion Pools. Chem Commun. 2011;47:5575–5577. doi: 10.1039/c1cc10923e. [DOI] [PubMed] [Google Scholar]
- 292.Nokami T, Watanabe T, Musya N, Suehiro T, Morofuji T, Yoshida J-I. Electrochemical Synthesis of Dendritic Diarylcarbenium Ion Pools. Tetrahedron. 2011;67:4664–4671. [Google Scholar]
- 293.Nokami T, Musya N, Morofuji T, Takeda K, Takumi M, Shimizu A, Yoshida J-I. Redox Active Dendronized Polystyrenes Equipped with Peripheral Triarylamines. Beilstein J Org Chem. 2014;10:3097–3103. doi: 10.3762/bjoc.10.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Ashikari Y, Nokami T, Yoshida J-I. Integrated Electrochemical–Chemical Oxidation Mediated by Alkoxysulfonium Ions. J Am Chem Soc. 2011;133:11840–11843. doi: 10.1021/ja202880n. [DOI] [PubMed] [Google Scholar]
- 295.Hayashi R, Shimizu A, Yoshida J-I. The Stabilized Cation Pool Method: Metal- and Oxidant-Free Benzylic C–H/Aromatic C–H Cross-Coupling. J Am Chem Soc. 2016;138:8400–8403. doi: 10.1021/jacs.6b05273. [DOI] [PubMed] [Google Scholar]
- 296.Hayashi R, Shimizu A, Song Y, Ashikari Y, Nokami T, Yoshida J-I. Metal-Free Benzylic C–H Amination via Electrochemically Generated Benzylaminosulfonium Ions. Chem - Eur J. 2017;23:61–64. doi: 10.1002/chem.201604484. [DOI] [PubMed] [Google Scholar]
- 297.Tajima T, Kishi Y, Nakajima A. Anodic Acyloxylation Based on the Acid–Base Reactions Between Acetic Acid or Trifluoroacetic Acid and Solid-Supported Bases. Electrochim Acta. 2009;54:5959–5963. [Google Scholar]
- 298.De Lamo Marin S, Martens T, Mioskowski C, Royer J. Efficient N-p-Methoxyphenyl Amine Deprotection Through Anodic Oxidation. J Org Chem. 2005;70:10592–10595. doi: 10.1021/jo051867o. [DOI] [PubMed] [Google Scholar]
- 299.Vaxelaire C, Souquet F, Lannou M-I, Ardisson J, Royer J. Anodic Oxidation: an Attractive Alternative to CAN-Mediated Cleavage of Para-Methoxyphenyl Ethers. Eur J Org Chem. 2009;2009:3138–3140. [Google Scholar]
- 300.Ding H, DeRoy PL, Perreault C, Larivée A, Siddiqui A, Caldwell CG, Harran S, Harran PG. Electrolytic Macrocyclizations: Scalable Synthesis of a Diazonamide-Based Drug Development Candidate. Angew Chem, Int Ed. 2015;54:4818–4822. doi: 10.1002/anie.201411663. [DOI] [PubMed] [Google Scholar]
- 301.Wang P, Tang S, Huang P, Lei A. Electrocatalytic Oxidant-Free Dehydrogenative C–H/S–H Cross-Coupling. Angew Chem, Int Ed. 2017;56:3009–3013. doi: 10.1002/anie.201700012. [DOI] [PubMed] [Google Scholar]
- 302.Wang P, Tang S, Lei A. Electrochemical Intramolecular Dehydrogenative C–S Bond Formation for the Synthesis of Benzothiazoles. Green Chem. 2017;19:2092–2095. [Google Scholar]
- 303.Qian X-Y, Li S-Q, Song J, Xu H-C. TEMPO-Catalyzed Electrochemical C–H Thiolation: Synthesis of Benzothiazoles and Thiazolopyridines From Thioamides. ACS Catal. 2017;7:2730–2734. [Google Scholar]
- 304.Sumi T, Saitoh T, Natsui K, Yamamoto T, Atobe M, Einaga Y, Nishiyama S. Anodic Oxidation on a Boron-Doped Diamond Electrode Mediated by Methoxy Radicals. Angew Chem, Int Ed. 2012;51:5443–5446. doi: 10.1002/anie.201200878. [DOI] [PubMed] [Google Scholar]
- 305.Dolson MG, Swenton JS. Product and Mechanistic Studies of the Anodic Oxidation of Methoxylated Naphthalenes. the EECrCp Mechanism. J Am Chem Soc. 1981;103:2361–2371. [Google Scholar]
- 306.Yajima S, Saitoh T, Kawa K, Nakamura K, Nagase H, Einaga Y, Nishiyama S. Asymmetric Induction in Cyclohexadienones Carrying α- D -Glucopyranosyl Moiety. Tetrahedron. 2016;72:8428–8435. [Google Scholar]
- 307.Carreño MC, Ribagorda M. Anodic Oxidation of N-Protected 4-Methoxy Anilines: Improved Synthesis of Quinone Imine Acetals. J Org Chem. 2000;65:1231–1234. doi: 10.1021/jo991398o. [DOI] [PubMed] [Google Scholar]
- 308.Li L-J, Jiang Y-Y, Lam CM, Zeng C-C, Hu L-M, Little RD. Aromatic C–H Bond Functionalization Induced by Electrochemically in Situ Generated Tris(p-Bromophenyl)Aminium Radical Cation: Cationic Chain Reactions of Electron-Rich Aromatics with Enamides. J Org Chem. 2015;80:11021–11030. doi: 10.1021/acs.joc.5b02222. [DOI] [PubMed] [Google Scholar]
- 309.Gao W-J, Li W-C, Zeng C-C, Tian H-Y, Hu L-M, Little RD. Electrochemically Initiated Oxidative Amination of Benzoxazoles Using Tetraalkylammonium Halides as Redox Catalysts. J Org Chem. 2014;79:9613–9618. doi: 10.1021/jo501736w. [DOI] [PubMed] [Google Scholar]
- 310.Nieto-Mendoza E, Guevara-Salazar JA, Ram rez-Apan MAT, Frontana-Uribe BA, Cogordan JA, Crdenas J. Electro-Oxidation of Hispanolone and Anti-Inflammatory Properties of the Obtained Derivatives. J Org Chem. 2005;70:4538–4541. doi: 10.1021/jo0503308. [DOI] [PubMed] [Google Scholar]
- 311.Albert M, De Souza D, Feiertag P, Hönig H. A New Concept for the Preparation of β-L-And β-D-2′, 3′-Dideoxynucleoside Analogues. Org Lett. 2002;4:3251–3254. doi: 10.1021/ol026460+. [DOI] [PubMed] [Google Scholar]
- 312.Laurencé C, Rivard M, Lachaise I, Bensemhoun J, Martens T. Preparative Access to Transformation Products (TPs) of Furosemide: a Versatile Application of Anodic Oxidation. Tetrahedron. 2011;67:9518–9521. [Google Scholar]
- 313.Nad S, Breinbauer R. Electroorganic Synthesis on the Solid Phase Using Polymer Beads as Supports. Angew Chem, Int Ed. 2004;43:2297–2299. doi: 10.1002/anie.200352674. [DOI] [PubMed] [Google Scholar]
- 314.Nad S, Roller S, Haag R, Breinbauer R. Electrolysis as an Efficient Key Step in the Homogeneous Polymer-Supported Synthesis of N-Substituted Pyrroles. Org Lett. 2006;8:403–406. doi: 10.1021/ol052096d. [DOI] [PubMed] [Google Scholar]
- 315.Hatta H, Zhou L, Mori M, Teshima S-I, Nishimoto S-I. N(1)–C(5′)-Linked Dimer Hydrates of 5-Substituted Uracils Produced by Anodic Oxidation in Aqueous Solution. J Org Chem. 2001;66:2232–2239. doi: 10.1021/jo0011282. [DOI] [PubMed] [Google Scholar]
- 316.Morofuji T, Shimizu A, Yoshida J-I. Metal- and Chemical-Oxidant-Free C-H/C-H Cross-Coupling of Aromatic Compounds: the Use of Radical-Cation Pools. Angew Chem, Int Ed. 2012;51:7259–7262. doi: 10.1002/anie.201202788. [DOI] [PubMed] [Google Scholar]
- 317.Arai T, Tateno H, Nakabayashi K, Kashiwagi T, Atobe M. An Anodic Aromatic C,C Cross-Coupling Reaction Using Parallel Laminar Flow Mode in a Flow Microreactor. Chem Commun. 2015;51:4891–4894. doi: 10.1039/c5cc01253h. [DOI] [PubMed] [Google Scholar]
- 318.Morofuji T, Shimizu A, Yoshida J-I. Electrochemical C–H Amination: Synthesis of Aromatic Primary Amines via N-Arylpyridinium Ions. J Am Chem Soc. 2013;135:5000–5003. doi: 10.1021/ja402083e. [DOI] [PubMed] [Google Scholar]
- 319.Herold S, Möhle S, Zirbes M, Richter F, Nefzger H, Waldvogel SR. Electrochemical Amination of Less-Activated Alkylated Arenes Using Boron-Doped Diamond Anodes. Eur J Org Chem. 2016;2016:1274–1278. [Google Scholar]
- 320.Möhle S, Herold S, Richter F, Nefzger H, Waldvogel SR. Twofold Electrochemical Amination of Naphthalene and Related Arenes. ChemElectroChem. 2017;4:2196. [Google Scholar]
- 321.Morofuji T, Shimizu A, Yoshida J-I. Electrochemical Intramolecular C–H Amination: Synthesis of Benzoxazoles and Benzothiazoles. Chem - Eur J. 2015;21:3211–3214. doi: 10.1002/chem.201406398. [DOI] [PubMed] [Google Scholar]
- 322.Wesenberg L, Waldvogel SR, Herold S, Shimizu A, Yoshida J-I. New Approach to 1,4-Benzoxazin-3-Ones by Electrochemical C,H-Amination. Chem - Eur J. 2017;23:12096. doi: 10.1002/chem.201701979. [DOI] [PubMed] [Google Scholar]
- 323.Morofuji T, Shimizu A, Yoshida J-I. Direct C–N Coupling of Imidazoles with Aromatic and Benzylic Compounds via Electrooxidative C–H Functionalization. J Am Chem Soc. 2014;136:4496–4499. doi: 10.1021/ja501093m. [DOI] [PubMed] [Google Scholar]
- 324.Morofuji T, Shimizu A, Yoshida J-I. Heterocyclization Approach for Electrooxidative Coupling of Functional Primary Alkylamines with Aromatics. J Am Chem Soc. 2015;137:9816–9819. doi: 10.1021/jacs.5b06526. [DOI] [PubMed] [Google Scholar]
- 325.Yamamura S, Nishiyama S. Anodic Oxidation of Phenols Towards the Synthesis of Bioactive Natural Products. Synlett. 2002;2002:533–543. [Google Scholar]
- 326.Quideau S, Pouységu L. Chemical and Electrochemical Oxidative Activation of Arenol Derivatives for Carbon-Carbon Bond Formation. Curr Org Chem. 2004;8:113–148. [Google Scholar]
- 327.Barjau J, Schnakenburg G, Waldvogel SR. Diversity-Oriented Synthesis of Polycyclic Scaffolds by Modification of an Anodic Product Derived From 2,4-Dimethylphenol. Angew Chem, Int Ed. 2011;50:1415–1419. doi: 10.1002/anie.201006637. [DOI] [PubMed] [Google Scholar]
- 328.Mirion M, Andernach L, Stobe C, Barjau J, Schollmeyer D, Opatz T, Lützen A, Waldvogel SR. Synthesis and Isolation of Enantiomerically Enriched Cyclopenta[B]Benzofurans Based on Products From Anodic Oxidation of 2,4-Dimethylphenol. Eur J Org Chem. 2015;2015:4876–4882. [Google Scholar]
- 329.Barjau J, Schnakenburg G, Waldvogel S. Short Domino Sequence to Dioxa[4.3. 3]Propellanes Synthesis. 2011;2011:2054–2061. [Google Scholar]
- 330.Barjau J, Königs P, Kataeva O, Waldvogel S. Reinvestigation of Highly Diastereoselective Pentacyclic Spirolactone Formation by Direct Anodic Oxidation of 2,4-Dimethylphenol. Synlett. 2008;2008:2309–2312. [Google Scholar]
- 331.Waldvogel SR, Mirk D. Handbook of C–H Transformations. Wiley-VCH; Weinheim: 2005. pp. 251–262. [Google Scholar]
- 332.Malkowsky IM, Rommel CE, Fröhlich R, Griesbach U, Pütter H, Waldvogel SR. Novel Template-Directed Anodic Phenol-Coupling Reaction. Chem - Eur J. 2006;12:7482–7488. doi: 10.1002/chem.200600375. [DOI] [PubMed] [Google Scholar]
- 333.Kirste A, Nieger M, Malkowsky IM, Stecker F, Fischer A, Waldvogel SR. Ortho-Selective Phenol-Coupling Reaction by Anodic Treatment on Boron-Doped Diamond Electrode Using Fluorinated Alcohols. Chem - Eur J. 2009;15:2273–2277. doi: 10.1002/chem.200802556. [DOI] [PubMed] [Google Scholar]
- 334.Malkowsky IM, Griesbach U, Pütter H, Waldvogel SR. Unexpected Highly Chemoselective Anodic Ortho-Coupling Reaction of 2,4-Dimethylphenol on Boron-Doped Diamond Electrodes. Eur J Org Chem. 2006;2006:4569–4572. [Google Scholar]
- 335.Kirste A, Schnakenburg G, Waldvogel SR. Anodic Coupling of Guaiacol Derivatives on Boron-Doped Diamond Electrodes. Org Lett. 2011;13:3126–3129. doi: 10.1021/ol201030g. [DOI] [PubMed] [Google Scholar]
- 336.Elsler B, Schollmeyer D, Dyballa KM, Franke R, Waldvogel SR. Metal- and Reagent-Free Highly Selective Anodic Cross-Coupling Reaction of Phenols. Angew Chem, Int Ed. 2014;53:5210–5213. doi: 10.1002/anie.201400627. [DOI] [PubMed] [Google Scholar]
- 337.Riehl B, Dyballa K, Franke R, Waldvogel S. Electro-Organic Synthesis as a Sustainable Alternative for Dehydrogenative Cross-Coupling of Phenols and Naphthols. Synthesis. 2016;49:252–259. [Google Scholar]
- 338.Wiebe A, Schollmeyer D, Dyballa KM, Franke R, Waldvogel SR. Selective Synthesis of Partially Protected Non-symmetric Biphenols by Reagent- and Metal-Free Anodic Cross-Coupling Reaction. Angew Chem, Int Ed. 2016;55:11801–11805. doi: 10.1002/anie.201604321. [DOI] [PubMed] [Google Scholar]
- 339.Kirste A, Schnakenburg G, Stecker F, Fischer A, Waldvogel SR. Anodic Phenol-Arene Cross-Coupling Reaction on Boron-Doped Diamond Electrodes. Angew Chem, Int Ed. 2010;49:971–975. doi: 10.1002/anie.200904763. [DOI] [PubMed] [Google Scholar]
- 340.Kirste A, Elsler B, Schnakenburg G, Waldvogel SR. Efficient Anodic and Direct Phenol-Arene C,C Cross-Coupling: the Benign Role of Water or Methanol. J Am Chem Soc. 2012;134:3571–3576. doi: 10.1021/ja211005g. [DOI] [PubMed] [Google Scholar]
- 341.Elsler B, Wiebe A, Schollmeyer D, Dyballa KM, Franke R, Waldvogel SR. Source of Selectivity in Oxidative Cross-Coupling of Aryls by Solvent Effect of 1,1,1,3,3,3-Hexafluoropropan-2-Ol. Chem - Eur J. 2015;21:12321–12325. doi: 10.1002/chem.201501604. [DOI] [PubMed] [Google Scholar]
- 342.Schulz L, Enders M, Elsler B, Schollmeyer D, Dyballa KM, Franke R, Waldvogel SR. Reagent- and Metal-Free Anodic C-C Cross-Coupling of Aniline Derivatives. Angew Chem, Int Ed. 2017;56:4877–4881. doi: 10.1002/anie.201612613. [DOI] [PubMed] [Google Scholar]
- 343.Lips S, Wiebe A, Elsler B, Schollmeyer D, Dyballa KM, Franke R, Waldvogel SR. Synthesis of Meta-Terphenyl-2,2“-”Diols by Anodic C-C Cross-Coupling Reactions. Angew Chem, Int Ed. 2016;55:10872–10876. doi: 10.1002/anie.201605865. [DOI] [PubMed] [Google Scholar]
- 344.Kirste A, Hayashi S, Schnakenburg G, Malkowsky IM, Stecker F, Fischer A, Fuchigami T, Waldvogel SR. Highly Selective Electrosynthesis of Biphenols on Graphite Electrodes in Fluorinated Media. Chem - Eur J. 2011;17:14164–14169. doi: 10.1002/chem.201102182. [DOI] [PubMed] [Google Scholar]
- 345.Mori K, Takahashi M, Yamamura S, Nishiyama S. Anodic Oxidation of Monohalogenated Phenols. Tetrahedron. 2001;57:5527–5532. [Google Scholar]
- 346.Kawabata Y, Naito Y, Saitoh T, Kawa K, Fuchigami T, Nishiyama S. Synthesis of (+)-O-Methylthalibrine by Employing a Stereocontrolled Bischler-Napieralski Reaction and an Electrochemically Generated Diaryl Ether. Eur J Org Chem. 2014;2014:99–104. [Google Scholar]
- 347.Naito Y, Tanabe T, Kawabata Y, Ishikawa Y, Nishiyama S. Electrochemical Construction of the Diaryl Ethers: a Synthetic Approach to O-Methylthalibrine. Tetrahedron Lett. 2010;51:4776–4778. [Google Scholar]
- 348.Tanabe T, Doi F, Ogamino T, Nishiyama S. A Total Synthesis of Verbenachalcone, a Bioactive Diaryl Ether From Verbena Littoralis. Tetrahedron Lett. 2004;45:3477–3480. [Google Scholar]
- 349.Uno K, Tanabe T, Nishiyama S. Electrochemical Multi-Step Approach Towards Isodityrosine: a Component of Biologically Important Cyclic Peptides. ECS Trans. 2009:91–95. doi: 10.1149/1.3312767. [DOI] [Google Scholar]
- 350.Mori K, Yamamura S, Nishiyama S. Synthesis of Spirodienone Derivatives and Their Conversion Into Dihydrobenzopyrans. Tetrahedron. 2001;57:5533–5542. [Google Scholar]
- 351.Deffieux D, Fabre I, Titz A, Léger J-M, Quideau S. Electrochemical Synthesis of Dimerizing and Nondimerizing Orthoquinone Monoketals. J Org Chem. 2004;69:8731–8738. doi: 10.1021/jo048677i. [DOI] [PubMed] [Google Scholar]
- 352.Ogamino T, Obata R, Nishiyama S. Asymmetric Synthesis of Aerothionin, a Marine Dimeric Spiroisoxazoline Natural Product, Employing Optically Active Spiroisoxazoline Derivative. Tetrahedron Lett. 2006;47:727–731. [Google Scholar]
- 353.Ogamino T, Nishiyama S. A New Ring-Opening Access to Aeroplysinin-1, a Secondary Metabolite of Verongia Aerophoba. Tetrahedron. 2003;59:9419–9423. [Google Scholar]
- 354.Doi F, Ogamino T, Sugai T, Nishiyama S. Synthesis of Bioactive Sesquiterpene Heliannuol E Involving a Ring-Expansion Reaction of Spirodienones. Synlett. 2003:411–413. [Google Scholar]
- 355.Doi F, Ogamino T, Sugai T, Nishiyama S. Enantioselective Synthesis of Heliannuol E; Structural Consideration of Natural Molecule. Tetrahedron Lett. 2003;44:4877–4880. [Google Scholar]
- 356.El-Seedi HR, Yamamura S, Nishiyama S. Reactivity of Naphthol Towards Nucleophiles in Anodic Oxidation. Tetrahedron. 2002;58:7485–7489. [Google Scholar]
- 357.Kim S, Hirose K, Uematsu J, Mikami Y, Chiba K. Electrochemically Active Cross-Linking Reaction for Fluorescent Labeling of Aliphatic Alkenes. Chem - Eur J. 2012;18:6284–6288. doi: 10.1002/chem.201103630. [DOI] [PubMed] [Google Scholar]
- 358.Kim S, Noda S, Hayashi K, Chiba K. An Oxidative Carbon–Carbon Bond Formation System in Cycloalkane-Based Thermomorphic Multiphase Solution. Org Lett. 2008;10:1827–1829. doi: 10.1021/ol8004408. [DOI] [PubMed] [Google Scholar]
- 359.Khodaei MM, Alizadeh A, Pakravan N. Polyfunctional Tetrazolic Thioethers Through Electrooxidative/Michael-Type Sequential Reactions of 1,2- and 1,4-Dihydroxybenzenes with 1-Phenyl-5-Mercaptotetrazole. J Org Chem. 2008;73:2527–2532. doi: 10.1021/jo702327m. [DOI] [PubMed] [Google Scholar]
- 360.Zeng C-C, Liu F-J, Ping D-W, Hu L-M, Cai Y-L, Zhong R-G. Electrochemical Synthesis of 1,3,4-Thiadiazol-2-ylthio-Substituted Catechols in Aqueous Medium. Tetrahedron. 2009;65:4505–4512. [Google Scholar]
- 361.Zeng C-C, Ping D-W, Zhang S-C, Zhong R-G, Becker JY. Electrochemical Synthesis of Polyhydroxylated Aromatic Derivatives Substituted with Mono- and Dipyrimidinyl Thioethers in Aqueous Medium. J Electroanal Chem. 2008;622:90–96. [Google Scholar]
- 362.Zeng C-C, Liu F-J, Ping D-W, Cai Y-L, Zhong R-G, Becker JY. Electrochemical Oxidation of Catechols in the Presence of 4-Amino-3-Methyl-5-Mercapto-1,2,4-Triazole Bearing Two Nucleophilic Groups. J Electroanal Chem. 2009;625:131–137. [Google Scholar]
- 363.Nematollahi D, Tammari E. Electroorganic Synthesis of Catecholthioethers. J Org Chem. 2005;70:7769–7772. doi: 10.1021/jo0508301. [DOI] [PubMed] [Google Scholar]
- 364.Zeng C-C, Liu C-F, Zeng J, Zhong R-G. Electrochemical Synthesis of 6-Arylsulfonyl Caffeic Acid Derivatives in Aqueous Medium. J Electroanal Chem. 2007;608:85–90. [Google Scholar]
- 365.Shahrokhian S, Hamzehloei A. Electrochemical Oxidation of Catechol in the Presence of 2-Thiouracil: Application to Electro-Organic Synthesis. Electrochem Commun. 2003;5:706–710. [Google Scholar]
- 366.Shabani-Nooshabadi M, Moradian M, Dadkhah-Tehrani S. A Practical One-Pot Electrochemical Synthesis of Pyrimido[4,5-B]Indole Derivatives. Bull Chem Soc Jpn. 2017;90:68–73. [Google Scholar]
- 367.Nematollahi D, Hosseiny Davarani SS, Mirahmadpour P. A Green Approach for the Electroorganic Synthesis of New Dihydroxyphenyl-Indolin-2-One Derivatives. ACS Sustainable Chem Eng. 2014;2:579–583. [Google Scholar]
- 368.Salehzadeh H, Nematollahi D, Hesari H. An Efficient Electrochemical Method for the Atom Economical Synthesis of Some Benzoxazole Derivatives. Green Chem. 2013;15:2441–2446. [Google Scholar]
- 369.Zeng C-C, Liu F-J, Ping D-W, Hu L-M, Cai Y-L, Zhong R-G. One-Pot Electrochemical Synthesis of Fused Indole Derivatives Containing Active Hydroxyl Groups in Aqueous Medium. J Org Chem. 2009;74:6386–6389. doi: 10.1021/jo901091s. [DOI] [PubMed] [Google Scholar]
- 370.Bai Y-X, Ping D-W, Little RD, Tian H-Y, Hu L-M, Zeng C-C. Electrochemical Oxidation of Catechols in the Presence of Ketene N,O-Acetals: Indole Formation Versus α-Arylation. Tetrahedron. 2011;67:9334–9341. [Google Scholar]
- 371.Zeng C-C, Ping D-W, Hu L-M, Song X-Q, Zhong R-G. Anodic Oxidation of Catechols in the Presence of α-Oxoketene N,N-Acetals with a Tetrahydropyrimidine Ring: Selective α-Arylation Reaction. Org Biomol Chem. 2010;8:2465–2468. doi: 10.1039/c001847c. [DOI] [PubMed] [Google Scholar]
- 372.Zeng C-C, Ping D-W, Xu Y-S, Hu L-M, Zhong R-G. A Facile Synthesis of α-Aryl α-Oxoheterocyclic Ketene N,N-Acetals Bearing an Electron-Rich Catechol Subunit-an Electrochemical Oxidative Approach. Eur J Org Chem. 2009;2009:5832–5840. [Google Scholar]
- 373.Zhang N-T, Gao X-G, Zeng C-C, Hu L-M, Tian H-Y, She Y-B. Electrochemical Oxidation of Catechols in the Presence of Enaminone: Exclusive α-Arylation. RSC Adv. 2012;2:298–306. [Google Scholar]
- 374.Nematollahi D, Habibi D, Rahmati M, Rafiee M. A Facile Electrochemical Method for Synthesis of New Benzofuran Derivatives. J Org Chem. 2004;69:2637–2640. doi: 10.1021/jo035304t. [DOI] [PubMed] [Google Scholar]
- 375.Hosseiny Davarani SS, Nematollahi D, Shamsipur M, Najafi NM, Masoumi L, Ramyar S. Electrochemical Oxidation of 2,3-Dimethylhydroquinone in the Presence of 1,3-Dicarbonyl Compounds. J Org Chem. 2006;71:2139–2142. doi: 10.1021/jo0523767. [DOI] [PubMed] [Google Scholar]
- 376.Xiao H-L, Yang C-W, Zhang N-T, Zeng C-C, Hu L-M, Tian H-Y, Little RD. Electrochemical Oxidation of Aminophenols in the Presence of Benzenesulfinate. Tetrahedron. 2013;69:658–663. [Google Scholar]
- 377.Salahifar E, Nematollahi D, Bayat M, Mahyari A, Amiri Rudbari H. Regioselective Green Electrochemical Approach to the Synthesis of Nitroacetaminophen Derivatives. Org Lett. 2015;17:4666–4669. doi: 10.1021/acs.orglett.5b01837. [DOI] [PubMed] [Google Scholar]
- 378.Amani A, Nematollahi D. Electrochemical Synthesis Based on the Oxidation of 1-(4-(4-Hydroxyphenyl)Piperazin-1-yl)Ethanone in the Presence of Nucleophiles. J Org Chem. 2012;77:11302–11306. doi: 10.1021/jo302418p. [DOI] [PubMed] [Google Scholar]
- 379.Blattes E, Fleury M-B, Largeron M. Simultaneously Electrogenerated Diene and Dienophile: a Unique Access to Novel Polyfunctionalized 1,4-Benzoxazine Derivatives as Neuroprotective Agents. Electrochim Acta. 2005;50:4902–4910. [Google Scholar]
- 380.Xu D, Chiaroni A, Largeron M. A One-Pot Regiospecific Synthesis of Highly Functionalized 1,4-Benzodioxin Derivatives From an Electrochemically Induced Diels–Alder Reaction. Org Lett. 2005;7:5273–5276. doi: 10.1021/ol052146e. [DOI] [PubMed] [Google Scholar]
- 381.Jensen KL, Franke PT, Nielsen LT, Daasbjerg K, Jørgensen KA. Anodic Oxidation and Organocatalysis: Direct Regio- and Stereoselective Access to Meta-Substituted Anilines by α-Arylation of Aldehydes. Angew Chem, Int Ed. 2010;49:129–133. doi: 10.1002/anie.200904754. [DOI] [PubMed] [Google Scholar]
- 382.Maleki A, Nematollahi D. Electrochemical Synthesis and Mechanestic Study of Quinone Imines Exploiting the Dual Character of N,N-Dialkyl-p-Phenylenediamines. Org Lett. 2011;13:1928–1931. doi: 10.1021/ol103146k. [DOI] [PubMed] [Google Scholar]
- 383.Salehzadeh H, Rafiee M, Nematollahi D. Electro-Organic Synthesis of New Esculetin Derivatives Based on 1, 6-Conjugate Addition. Curr Org Chem. 2013;17:848–852. [Google Scholar]
- 384.Nematollahi D, Ghasemi F, Sharafi-Kolkeshvandi M, Varmaghani F. Insight Into the Electrochemical Oxidation of N,N-Dialkyl-p-Phenylenediamines in the Presence of Malononitrile and Methyl Cyanoacetate. a Convergent Paired Electrochemical Method for the Synthesis of Cyanide and Dicyanide Derivatives of Phenylcarbonimidoyl. J Electroanal Chem. 2016;775:299–305. [Google Scholar]
- 385.Kaihani S, Salehzadeh H, Nematollah D. Electrochemical Synthesis of N1,N4-Diphenyl-2-(Phenylsulfonyl)Benzene-1,4-Diamine Derivatives: Introducing an Example of ECDispCMich Mechanism. Electrochim Acta. 2015;157:166–174. [Google Scholar]
- 386.Nematollahi D, Esmaili R. A Green Approach for the Electrochemical Synthesis of 4-Morpholino-2-(Arylsulfonyl)-Benzenamines. Tetrahedron Lett. 2010;51:4862–4865. [Google Scholar]
- 387.Sharafi-Kolkeshvandi M, Nematollahi D, Nikpour F, Salahifar E. Electrochemical Synthesis of 1-N-Phenyl-4-(Sulfonyl)-Benzene-1,2-Diamine Derivatives: a Mild and Regioselective Protocol. New J Chem. 2016;40:5442–5447. [Google Scholar]
- 388.Sharafi-Kolkeshvandi M, Nematollahi D, Nikpour F. A Regioselective and Convergent Paired Electrochemical Synthesis of N,N′-Diphenyl-3-Sulfonyl-[1,1′-Biphenyl]-4,4′-Diamines. Synthesis. 2017;49:1555–1560. [Google Scholar]
- 389.Jamshidi M, Nematollahi D, Bayat M, Salahifar E. Unsymmetrical Diaryl Sulfones Through Electrochemical Oxidation of Fast Violet B in the Presence of Aryl Sulfinic Acids. J Electrochem Soc. 2016;163:G211–G218. [Google Scholar]
- 390.Moutiers G, Pinson J, Terrier F, Goumont R. Electrochemical Oxidation of Sigma-Complex-Type Intermediates in Aromatic Nucleophilic Substitutions. Chem - Eur J. 2001;7:1712–1719. doi: 10.1002/1521-3765(20010417)7:8<1712::aid-chem17120>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 391.Gallardo I, Guirado G. Thermodynamic Study of σH Complexes in Nucleophilic Aromatic Substitution Reactions: Relative Stabilities of Electrochemically Generated Radicals. Eur J Org Chem. 2008;2008:2463–2472. [Google Scholar]
- 392.Charushin VN, Chupakhin ON. Topics in Heterocyclic Chemistry. Vol. 37. Springer International Publishing; Cham: 2013. Metal-Free C–H Functionalization of Aromatic Compounds Through the Action of Nucleophilic Reagents; pp. 1–50. [Google Scholar]
- 393.Gallardo I, Guirado G, Marquet J. Nucleophilic Aromatic Substitution of Hydrogen: a Novel Electrochemical Approach to the Cyanation of Nitroarenes. Chem - Eur J. 2001;7:1759–1765. doi: 10.1002/1521-3765(20010417)7:8<1759::aid-chem17590>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 394.Gallardo I, Guirado G, Marquet J. Nucleophilic Aromatic Substitution for Heteroatoms: an Oxidative Electrochemical Approach. J Org Chem. 2002;67:2548–2555. doi: 10.1021/jo010847t. [DOI] [PubMed] [Google Scholar]
- 395.Gallardo I, Guirado G, Marquet J. Electrochemical Synthesis of Alkyl Nitroaromatic Compounds. J Org Chem. 2003;68:631–633. doi: 10.1021/jo025898k. [DOI] [PubMed] [Google Scholar]
- 396.Gallardo I, Guirado G, Marquet J. Alkylation of Nitroaromatics with Tetraalkylborate Ion via Electrochemical Oxidation. J Org Chem. 2003;68:7334–7341. doi: 10.1021/jo030158c. [DOI] [PubMed] [Google Scholar]
- 397.Cruz H, Gallardo I, Guirado G. Electrochemical Synthesis of Organophosphorus Compounds Through Nucleophilic Aromatic Substitution: Mechanistic Investigations and Synthetic Scope. Eur J Org Chem. 2011;2011:7378–7389. [Google Scholar]
- 398.Gallardo I, Guirado G, Marquet J. Electrochemical Synthesis of Nitroanilines. Eur J Org Chem. 2002;2002:251–259. [Google Scholar]
- 399.Gallardo I, Guirado G, Marquet J. Electrochemical Synthesis of Nitroaromatic Ketones. Eur J Org Chem. 2002;2002:261–267. doi: 10.1021/jo025898k. [DOI] [PubMed] [Google Scholar]
- 400.Cruz H, Gallardo I, Guirado G. Electrochemically Promoted Nucleophilic Aromatic Substitution in Room Temperature Ionic Liquids—an Environmentally Benign Way to Functionalize Nitroaromatic Compounds. Green Chem. 2011;13:2531–2542. [Google Scholar]
- 401.Shchepochkin AV, Chupakhin ON, Charushin VN, Steglenko DV, Minkin VI, Rusinov GL, Matern AI. C–H Functionalization of Azines. Anodic Dehydroaromatization of 9-(Hetero)Aryl-9,10-Dihydroacridines. RSC Adv. 2016;6:77834–77840. [Google Scholar]
- 402.Lyalin BV, Petrosyan VA. Electrochemical Halogenation of Organic Compounds. Russ J Electrochem. 2013;49:497–529. [Google Scholar]
- 403.Lyalin BV, Petrosyan VA, Ugrak BI. Electrosynthesis of 4-Chloropyrazolecarboxylic Acids. Russ Chem Bull. 2009;58:291–296. [Google Scholar]
- 404.Lyalin BV, Petrosyan VA, Ugrak BI. Electrosynthesis of 4-Iodopyrazole and Its Derivatives. Russ Chem Bull. 2010;59:1549–1555. [Google Scholar]
- 405.Lyalin BV, Petrosyan VA, Ugrak BI. Electrosynthesis of 4-Chloro Derivatives of Pyrazole and Alkylpyrazoles. Russ J Electrochem. 2008;44:1320–1326. [Google Scholar]
- 406.Stevanović D, Damljanović I, Vukićević M, Manojlović N, Radulović NS, Vukićević RD. Electrochemical Chlorination of Physcion–an Approach to Naturally Occurring Chlorinated Secondary Metabolites of Lichens. Helv Chim Acta. 2011;94:1406–1415. [Google Scholar]
- 407.Raju T, Kulangiappar K, Anbu Kulandainathan M, Uma U, Malini R, Muthukumaran A. Site Directed Nuclear Bromination of Aromatic Compounds by an Electrochemical Method. Tetrahedron Lett. 2006;47:4581–4584. [Google Scholar]
- 408.Sawamura T, Takahashi K, Inagi S, Fuchigami T. Sodium Salts Dissolution in an Aprotic Solvent by Coordination of Poly-(Ethylene Glycol) for Effective Anodic Reactions of Organic Compounds. Electrochemistry. 2013;81:365–367. [Google Scholar]
- 409.Fotouhi L, Nikoofar K. Electrochemical Thiocyanation of Nitrogen-Containing Aromatic and Heteroaromatic Compounds. Tetrahedron Lett. 2013;54:2903–2905. [Google Scholar]
- 410.Kokorekin VA, Sigacheva VL, Petrosyan VA. New Data on Heteroarene Thiocyanation by Anodic Oxidation of NH4SCN. the Processes of Electroinduced Nucleophilic Aromatic Substitution of Hydrogen. Tetrahedron Lett. 2014;55:4306–4309. [Google Scholar]
- 411.Gitkis A, Becker JY. Anodic Thiocyanation of Mono- and Disubstituted Aromatic Compounds. Electrochim Acta. 2010;55:5854–5859. [Google Scholar]
- 412.Tan Z, Liu Y, Helmy R, Rivera NR, Hesk D, Tyagarajan S, Yang L, Su J. Electrochemical Bromination of Late Stage Intermediates and Drug Molecules. Tetrahedron Lett. 2017;58:3014–3018. [Google Scholar]
- 413.Kataoka K, Hagiwara Y, Midorikawa K, Suga S, Yoshida J-I. Practical Electrochemical Iodination of Aromatic Compounds. Org Process Res Dev. 2008;12:1130–1136. [Google Scholar]
- 414.Midorikawa K, Suga S, Yoshida JI. Selective Monoiodination of Aromatic Compounds with Electrochemically Generated I+ Using Micromixing. Chem Commun. 2006:3794–3796. doi: 10.1039/b607284d. [DOI] [PubMed] [Google Scholar]
- 415.Raju T, Kulangiappar K, Kulandainathan MA, Muthukumaran A. A Simple and Regioselective α-Bromination of Alkyl Aromatic Compounds by Two-Phase Electrolysis. Tetrahedron Lett. 2005;46:7047–7050. [Google Scholar]
- 416.Kulangiappar K, Karthik G, Kulandainathan MA. Electrochemical Method for the Preparation of Dibromomethyl, Bis-(Bromomethyl), and Bis(Dibromomethyl) Arenes. Synth Commun. 2009;39:2304–2309. [Google Scholar]
- 417.Fuchigami T, Inagi S. Selective Electrochemical Fluorination of Organic Molecules and Macromolecules in Ionic Liquids. Chem Commun. 2011;47:10211–10223. doi: 10.1039/c1cc12414e. [DOI] [PubMed] [Google Scholar]
- 418.Fuchigami T, Tajima T. Highly Selective Electrochemical Fluorination of Organic Compounds in Ionic Liquids. J Fluorine Chem. 2005;126:181–187. [Google Scholar]
- 419.Noel M, Suryanarayanan V. Current Approaches to the Electrochemical Synthesis of Organo-Fluorine Compounds. J Appl Electrochem. 2004;34:357–369. [Google Scholar]
- 420.Ishii H, Yamada N, Fuchigami T. Electrolytic Partial Fluorination of Organic Compound. Part: 53 Highly Regioselective Anodic Mono- and Difluorination of 4-Arylthio-1,3-Dioxolan-2-Ones. a Marked Solvent Effect on Fluorinated Product Selectivity. Tetrahedron. 2001;57:9067–9072. [Google Scholar]
- 421.Ishii H, Yamada N, Fuchigami T. Highly Selective Anodic Monofluorination of 4-Arylthio-1,3-Dioxolan-2-Ones: a Marked Solvent Effect on Product Selectivity. Chem Commun. 2000:1617–1618. [Google Scholar]
- 422.Hasegawa M, Ishii H, Fuchigami T. Selective Anodic Fluorination of Phthalides in Ionic Liquids. Green Chem. 2003;5:512–515. [Google Scholar]
- 423.Hasegawa M, Fuchigami T. Electroorganic Reactions in Ionic Liquids. Electrochim Acta. 2004;49:3367–3372. [Google Scholar]
- 424.Suzuki K, Fuchigami T. Electrolytic Partial Fluorination of Organic Compounds.71. Highly Diastereoselective Anodic Fluorination of Sulfides Having Oxygen-Containing Heterocyclic Groups. J Org Chem. 2004;69:1276–1282. doi: 10.1021/jo035291j. [DOI] [PubMed] [Google Scholar]
- 425.Sawamura T, Takahashi K, Inagi S, Fuchigami T. Electrochemical Fluorination Using Alkali-Metal Fluorides. Angew Chem, Int Ed. 2012;51:4413–4416. doi: 10.1002/anie.201200438. [DOI] [PubMed] [Google Scholar]
- 426.Sunaga T, Atobe M, Inagi S, Fuchigami T. Highly Efficient and Selective Electrochemical Fluorination of Organosulfur Compounds in Et3N·3HF Ionic Liquid Under Ultrasonication. Chem Commun. 2009:956–958. doi: 10.1039/b817860g. [DOI] [PubMed] [Google Scholar]
- 427.Tajima T, Nakajima A, Doi Y, Fuchigami T. Anodic Fluorination Based on Cation Exchange Between Alkali-Metal Fluorides and Solid-Supported Acids. Angew Chem, Int Ed. 2007;46:3550–3552. doi: 10.1002/anie.200700037. [DOI] [PubMed] [Google Scholar]
- 428.Sawamura T, Kuribayashi S, Inagi S, Fuchigami T. Use of Task-Specific Ionic Liquid for Selective Electrocatalytic Fluorination. Org Lett. 2010;12:644–646. doi: 10.1021/ol9028836. [DOI] [PubMed] [Google Scholar]
- 429.Sawamura T, Kuribayashi S, Inagi S, Fuchigami T. Recyclable Polymer-Supported Iodobenzene-Mediated Electrocatalytic Fluorination in Ionic Liquid. Adv Synth Catal. 2010;352:2757–2760. [Google Scholar]
- 430.Fuchigami T, Tetsu M, Tajima T, Ishii H. Indirect Anodic Monofluorodesulfurization of β-Phenylsulfenyl β-Lactams Using a Triarylamine Mediator. Synlett. 2001;2001:1269–1271. [Google Scholar]
- 431.Tanigawa M, Kuriyama Y, Inagi S, Fuchigami T. Electrochemical Properties and Reactions of Sulfur-Containing Organoboron Compounds. Electrochim Acta. 2016;199:314–318. [Google Scholar]
- 432.Suzuki J, Shida N, Inagi S, Fuchigami T. Electrochemical Properties and Reactions of Organoboronic Acids in the Presence of Fluoride Ions. Electroanalysis. 2016;28:2797–2801. [Google Scholar]
- 433.Cao Y, Suzuki K, Tajima T, Fuchigami T. Electrolytic Partial Fluorination of Organic Compounds. Part 78: Regioselective Anodic Fluorination of 2-Oxazolidinones. Tetrahedron. 2005;61:6854–6859. [Google Scholar]
- 434.Fukuhara T, Akiyama Y, Yoneda N, Tada T, Hara S. Effective Synthesis of Difluorocyclohexadienones by Electrochemical Oxidation of Phenols. Tetrahedron Lett. 2002;43:6583–6585. [Google Scholar]
- 435.Yin B, Wang L, Inagi S, Fuchigami T. Electrosynthesis of Fluorinated Indole Derivatives. Tetrahedron. 2010;66:6820–6825. [Google Scholar]
- 436.Tajima T, Ishii H, Fuchigami T. Anodic Benzylic Fluorination of Toluene, Ethylbenzene, and Cumene Derivatives. Electrochem Commun. 2002;4:589–592. [Google Scholar]
- 437.Aoyama M, Fukuhara T, Hara S. Selective Fluorination of Adamantanes by an Electrochemical Method. J Org Chem. 2008;73:4186–4189. doi: 10.1021/jo8004759. [DOI] [PubMed] [Google Scholar]
- 438.Monoi M, Hara S. Electrochemical Synthesis of Methyl-3,5,7-Trifluoroadamantane-1-Carboxylate Under Recycling Use of Ionic Liquid Media. J Fluorine Chem. 2012;140:28–30. [Google Scholar]
- 439.Hasegawa M, Ishii H, Fuchigami T. Electroorganic Synthesis Under Solvent-Free Conditions. Highly Regioselective Anodic Monofluorination of Cyclic Ethers, Lactones, and a Cyclic Carbonate. Tetrahedron Lett. 2002;43:1503–1505. [Google Scholar]
- 440.Dinoiu V, Fukuhara T, Hara S, Yoneda N. Electrochemical Partial Fluorination of Conjugated Diene Esters. J Fluorine Chem. 2000;103:75–80. [Google Scholar]
- 441.Tajima T, Nakajima A, Fuchigami T. Electrolytic Partial Fluorination of Organic Compounds. 83. Anodic Fluorination of N-Substituted Pyrroles and Its Synthetic Applications to Gem-Difluorinated Heterocyclic Compounds. J Org Chem. 2006;71:1436–1441. doi: 10.1021/jo0520745. [DOI] [PubMed] [Google Scholar]
- 442.Dawood KM, Fuchigami T. Electrochemical Partial Fluorination of Organic Compounds. 74. Efficient Anodic Synthesis of 2-Fluoro- and 2,3-Difluoro-2,3-Dihydrobenzofuran Derivatives. J Org Chem. 2004;69:5302–5306. doi: 10.1021/jo035871g. [DOI] [PubMed] [Google Scholar]
- 443.Yin B, Inagi S, Fuchigami T. Electrosynthesis of Fluorinated Benzo[B]Thiophene Derivatives. Synlett. 2011;2011:1313–1317. [Google Scholar]
- 444.Baba D, Ishii H, Higashiya S, Fujisawa K, Fuchigami T. Electroytic Partial Fluorination of Organic Compounds. 52. Regio- and Diastereoselective Anodic Fluorination of Thiazolidines. J Org Chem. 2001;66:7020–7024. doi: 10.1021/jo010472b. [DOI] [PubMed] [Google Scholar]
- 445.Frey DA, Krishna Reddy SH, Moeller KD. Intramolecular Anodic Olefin Coupling Reactions: the Use of Allylsilane Coupling Partners with Allylic Alkoxy Groups. J Org Chem. 1999;64:2805–2813. doi: 10.1021/jo982280v. [DOI] [PubMed] [Google Scholar]
- 446.Moeller K. Intramolecular Anodic Olefin Coupling Reactions: Using Radical Cation Intermediates to Trigger New Umpolung Reactions. Synlett. 2009;2009:1208–1218. [Google Scholar]
- 447.Tang F, Chen C, Moeller K. Electrochemistry and Umpolung Reactions: New Tools for Solving Synthetic Challenges of Structure and Location. Synthesis. 2007;2007:3411–3420. [Google Scholar]
- 448.Wu H, Moeller KD. Anodic Coupling Reactions: a Sequential Cyclization Route to the Arteannuin Ring Skeleton. Org Lett. 2007;9:4599–4602. doi: 10.1021/ol702118n. [DOI] [PubMed] [Google Scholar]
- 449.Sun Y, Liu B, Kao J, d’Avigno DA, Moeller KD. Anodic Cyclization Reactions: Reversing the Polarity of Ketene Dithioacetal Groups. Org Lett. 2001;3:1729–1732. doi: 10.1021/ol015925d. [DOI] [PubMed] [Google Scholar]
- 450.Huang Y-T, Moeller KD. Anodic Coupling Reactions: the Use of N,O-Ketene Acetal Coupling Partners. Org Lett. 2004;6:4199–4202. doi: 10.1021/ol048450+. [DOI] [PubMed] [Google Scholar]
- 451.Huang Y-T, Moeller KD. Anodic Cyclization Reactions: Probing the Chemistry of N,O-Ketene Acetal Derived Radical Cations. Tetrahedron. 2006;62:6536–6550. [Google Scholar]
- 452.Redden A, Perkins RJ, Moeller KD. Oxidative Cyclization Reactions: Controlling the Course of a Radical Cation-Derived Reaction with the Use of a Second Nucleophile. Angew Chem, Int Ed. 2013;52:12865–12868. doi: 10.1002/anie.201308739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Sun Y, Moeller KD. Anodic Olefin Coupling Reactions Involving Ketene Dithioacetals: Evidence for a “Radical-Type”Cyclization. Tetrahedron Lett. 2002;43:7159–7161. [Google Scholar]
- 454.Tang F, Moeller KD. Anodic Oxidations and Polarity: Exploring the Chemistry of Olefinic Radical Cations. Tetrahedron. 2009;65:10863–10875. [Google Scholar]
- 455.Campbell JM, Smith JA, Gonzalez L, Moeller KD. Competition Studies and the Relative Reactivity of Enol Ether and Allylsilane Coupling Partners Toward Ketene Dithioacetal Derived Radical Cations. Tetrahedron Lett. 2015;56:3595–3599. [Google Scholar]
- 456.Tang F, Moeller KD. Intramolecular Anodic Olefin Coupling Reactions: the Effect of Polarization on Carbon–Carbon Bond Formation. J Am Chem Soc. 2007;129:12414–12415. doi: 10.1021/ja076172e. [DOI] [PubMed] [Google Scholar]
- 457.Sperry JB, Wright DL. Synthesis of the Hamigeran Skeleton Through an Electro-Oxidative Coupling Reaction. Tetrahedron Lett. 2005;46:411–414. [Google Scholar]
- 458.Sperry JB, Ghiviriga I, Wright DL. Electrochemical Annulation of Five-Membered Rings Through Dearomatization of Furans and Thiophenes. Chem Commun. 2006:194–196. doi: 10.1039/b513532j. [DOI] [PubMed] [Google Scholar]
- 459.Sperry JB, Wright DL. The Gem-Dialkyl Effect in Electron Transfer Reactions: Rapid Synthesis of Seven-Membered Rings Through an Electrochemical Annulation. J Am Chem Soc. 2005;127:8034–8035. doi: 10.1021/ja051826+. [DOI] [PubMed] [Google Scholar]
- 460.Sperry JB, Wright DL. Annulated Heterocycles Through a Radical-Cation Cyclization: Synthetic and Mechanistic Studies. Tetrahedron. 2006;62:6551–6557. [Google Scholar]
- 461.Sperry JB, Whitehead CR, Ghiviriga I, Walczak RM, Wright DL. Electrooxidative Coupling of Furans and Silyl Enol Ethers: Application to the Synthesis of Annulated Furans. J Org Chem. 2004;69:3726–3734. doi: 10.1021/jo049889i. [DOI] [PubMed] [Google Scholar]
- 462.Whitehead CR, Sessions EH, Ghiviriga I, Wright DL. Two-Step Electrochemical Annulation for the Assembly of Polycyclic Systems. Org Lett. 2002;4:3763–3765. doi: 10.1021/ol026771k. [DOI] [PubMed] [Google Scholar]
- 463.Redden A, Moeller KD. Anodic Coupling Reactions: Exploring the Generality of Curtin–Hammett Controlled Reactions. Org Lett. 2011;13:1678–1681. doi: 10.1021/ol200182f. [DOI] [PubMed] [Google Scholar]
- 464.Mihelcic J, Moeller KD. Anodic Cyclization Reactions: the Total Synthesis of Alliacol A. J Am Chem Soc. 2003;125:36–37. doi: 10.1021/ja029064v. [DOI] [PubMed] [Google Scholar]
- 465.Miller AK, Hughes CC, Kennedy-Smith JJ, Gradl SN, Trauner D. Total Synthesis of (–)-Heptemerone B and (–)-Guanacastepene E. J Am Chem Soc. 2006;128:17057–17062. doi: 10.1021/ja0660507. [DOI] [PubMed] [Google Scholar]
- 466.Hughes CC, Miller AK, Trauner D. An Electrochemical Approach to the Guanacastepenes. Org Lett. 2005;7:3425–3428. doi: 10.1021/ol047387l. [DOI] [PubMed] [Google Scholar]
- 467.Sutterer A, Moeller KD. Reversing the Polarity of Enol Ethers: an Anodic Route to Tetrahydrofuran and Tetrahydropyran Rings. J Am Chem Soc. 2000;122:5636–5637. [Google Scholar]
- 468.Anderson LA, Redden A, Moeller KD. Connecting the Dots: Using Sunlight to Drive Electrochemical Oxidations. Green Chem. 2011;13:1652–1653. [Google Scholar]
- 469.Nguyen BH, Perkins RJ, Smith JA, Moeller KD. Photovoltaic-Driven Organic Electrosynthesis and Efforts Toward More Sustainable Oxidation Reactions. Beilstein J Org Chem. 2015;11:280–287. doi: 10.3762/bjoc.11.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Liu B, Duan S, Sutterer AC, Moeller KD. Oxidative Cyclization Based on Reversing the Polarity of Enol Ethers and Ketene Dithioacetals. Construction of a Tetrahydrofuran Ring and Application to the Synthesis of (+)-Nemorensic Acid. J Am Chem Soc. 2002;124:10101–10111. doi: 10.1021/ja026739l. [DOI] [PubMed] [Google Scholar]
- 471.Xu H-C, Brandt JD, Moeller KD. Anodic Cyclization Reactions and the Synthesis of (–)-Crobarbatic Acid. Tetrahedron Lett. 2008;49:3868–3871. [Google Scholar]
- 472.Duan S, Moeller KD. Anodic Cyclization Reactions: Capitalizing on an Intramolecular Electron Transfer to Trigger the Synthesis of a Key Tetrahydropyran Building Block. J Am Chem Soc. 2002;124:9368–9369. doi: 10.1021/ja027227+. [DOI] [PubMed] [Google Scholar]
- 473.Xu G, Moeller KD. Anodic Coupling Reactions and the Synthesis of C-Glycosides. Org Lett. 2010;12:2590–2593. doi: 10.1021/ol100800u. [DOI] [PubMed] [Google Scholar]
- 474.Brandt JD, Moeller KD. Oxidative Cyclization Reactions: Amide Trapping Groups and the Synthesis of Furanones. Org Lett. 2005;7:3553–3556. doi: 10.1021/ol051296m. [DOI] [PubMed] [Google Scholar]
- 475.Perkins RJ, Xu H-C, Campbell JM, Moeller KD. Anodic Coupling of Carboxylic Acids to Electron-Rich Double Bonds: a Surprising Non-Kolbe Pathway to Lactones. Beilstein J Org Chem. 2013;9:1630–1636. doi: 10.3762/bjoc.9.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Xu H-C, Moeller KD. Intramolecular Anodic Olefin Coupling Reactions: the Use of a Nitrogen Trapping Group. J Am Chem Soc. 2008;130:13542–13543. doi: 10.1021/ja806259z. [DOI] [PubMed] [Google Scholar]
- 477.Xu H-C, Moeller KD. Intramolecular Anodic Olefin Coupling Reactions and the Synthesis of Cyclic Amines. J Am Chem Soc. 2010;132:2839–2844. doi: 10.1021/ja910586v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Xu H-C, Moeller KD. Intramolecular Anodic Olefin Coupling Reactions: Using Competition Studies to Probe the Mechanism of Oxidative Cyclization Reactions. Org Lett. 2010;12:1720–1723. doi: 10.1021/ol100317t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Campbell JM, Xu H-C, Moeller KD. Investigating the Reactivity of Radical Cations: Experimental and Computational Insights Into the Reactions of Radical Cations with Alcohol and p-Toluene Sulfonamide Nucleophiles. J Am Chem Soc. 2012;134:18338–18344. doi: 10.1021/ja307046j. [DOI] [PubMed] [Google Scholar]
- 480.Xu H-C, Moeller KD. Intramolecular Anodic Olefin Coupling Reactions: Use of the Reaction Rate to Control Substrate/Product Selectivity. Angew Chem, Int Ed. 2010;49:8004–8007. doi: 10.1002/anie.201003924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Smith JA, Moeller KD. Oxidative Cyclizations, the Synthesis of Aryl-Substituted C-Glycosides, and the Role of the Second Electron Transfer Step. Org Lett. 2013;15:5818–5821. doi: 10.1021/ol402826z. [DOI] [PubMed] [Google Scholar]
- 482.Chiba K, Miura T, Kim S, Kitano Y, Tada M. Electrocatalytic Intermolecular Olefin Cross-Coupling by Anodically Induced Formal [2 + 2] Cycloaddition Between Enol Ethers and Alkenes. J Am Chem Soc. 2001;123:11314–11315. doi: 10.1021/ja016885b. [DOI] [PubMed] [Google Scholar]
- 483.Okada Y, Akaba R, Chiba K. Electrocatalytic Formal [2 + 2] Cycloaddition Reactions Between Anodically Activated Aliphatic Enol Ethers and Unactivated Olefins Possessing an Alkoxyphenyl Group. Org Lett. 2009;11:1033–1035. doi: 10.1021/ol802984n. [DOI] [PubMed] [Google Scholar]
- 484.Okada Y, Nishimoto A, Akaba R, Chiba K. Electron-Transfer-Induced Intermolecular [2 + 2] Cycloaddition Reactions Based on the Aromatic “Redox Tag” Strategy. J Org Chem. 2011;76:3470–3476. doi: 10.1021/jo200490q. [DOI] [PubMed] [Google Scholar]
- 485.Okada Y, Yamaguchi Y, Ozaki A, Chiba K. Aromatic “Redox Tag-”Assisted Diels–Alder Reactions by Electrocatalysis. Chem Sci. 2016;7:6387–6393. doi: 10.1039/c6sc02117d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Miura T, Kim S, Kitano Y, Tada M, Chiba K. Electrochemical Enol Ether/Olefin Cross-Metathesis in a Lithium Perchlorate/Nitromethane Electrolyte Solution. Angew Chem, Int Ed. 2006;45:1461–1463. doi: 10.1002/anie.200503656. [DOI] [PubMed] [Google Scholar]
- 487.Okada Y, Chiba K. Electron Transfer-Induced Four-Membered Cyclic Intermediate Formation: Olefin Cross-Coupling vs. Olefin Cross-Metathesis. Electrochim Acta. 2011;56:1037–1042. [Google Scholar]
- 488.Moore JC, Davies ES, Walsh DA, Sharma P, Moses JE. Formal Synthesis of Kingianin a Based Upon a Novel Electrochemically-Induced Radical Cation Diels–Alder Reaction. Chem Commun. 2014;50:12523–12525. doi: 10.1039/c4cc05906a. [DOI] [PubMed] [Google Scholar]
- 489.Stewart MP, Lam K, Chong D, Geiger WE. Electron-Transfer Catalyzed Cycloaddition Reactions of Unactivated Cyclic Olefins in Weakly Coordinating Anion Electrolyte. J Electroanal Chem. 2015;743:68–77. [Google Scholar]
- 490.Chong D, Stewart M, Geiger WE. Cycloaddition Reactions of Unactivated Olefins Catalyzed by an Organorhenium Electron-Transfer Mediator. J Am Chem Soc. 2009;131:7968–7969. doi: 10.1021/ja9032296. [DOI] [PubMed] [Google Scholar]
- 491.Bui N-N, Ho X-H, Mho S-I, Jang H-Y. Organocatalyzed α-Oxyamination of Aldehydes Using Anodic Oxidation. Eur J Org Chem. 2009;2009:5309–5312. [Google Scholar]
- 492.Ho XH, Mho SI, Kang H, Jang HY. Electro-Organocatalysis: Enantioselective α-Alkylation of Aldehydes. Eur J Org Chem. 2010:4436–4441. [Google Scholar]
- 493.Mori N, Furuta A, Watanabe H. Electochemical Asymmetric Dimerization of Cinnamic Acid Derivatives and Application to the Enantioselective Syntheses of Furofuran Lignans. Tetrahedron. 2016;72:8393–8399. [Google Scholar]
- 494.Hong F-J, Low Y-Y, Chong K-W, Thomas NF, Kam T-S. Biomimetic Oxidative Dimerization of Anodically Generated Stilbene Radical Cations: Effect of Aromatic Substitution on Product Distribution and Reaction Pathways. J Org Chem. 2014;79:4528–4543. doi: 10.1021/jo500559r. [DOI] [PubMed] [Google Scholar]
- 495.Chong K-W, Hong F-J, Thomas NF, Low Y-Y, Kam T-S. Electrochemically Mediated Oxidative Transformations of Substituted 4-Methoxystilbenes: Effect of Ortho-Substituted Nucleophilic Groups. J Org Chem. 2017;82:6172–6191. doi: 10.1021/acs.joc.7b00753. [DOI] [PubMed] [Google Scholar]
- 496.Wu X, Davis AP, Fry AJ. Electrocatalytic Oxidative Cleavage of Electron-Deficient Substituted Stilbenes in Acetonitrile–Water Employing a New High Oxidation Potential Electrocatalyst. an Electrochemical Equivalent of Ozonolysis. Org Lett. 2007;9:5633–5636. doi: 10.1021/ol7026416. [DOI] [PubMed] [Google Scholar]
- 497.Ashikari Y, Nokami T, Yoshida J-I. Oxidative Hydroxylation Mediated by Alkoxysulfonium Ions. Org Lett. 2012;14:938–941. doi: 10.1021/ol203467v. [DOI] [PubMed] [Google Scholar]
- 498.Ashikari Y, Nokami T, Yoshida J-I. Integration of Electrooxidative Cyclization and Chemical Oxidation via Alkoxysulfonium Ions. Synthesis of Exocyclic Ketones From Alkenes with Cyclization. Org Biomol Chem. 2013;11:3322–3331. doi: 10.1039/c3ob40315g. [DOI] [PubMed] [Google Scholar]
- 499.Baumer U-St, Schäfer HJ. Cleavage of Olefinic Double Bonds by Mediated Anodic Oxidation. Electrochim Acta. 2003;48:489–495. [Google Scholar]
- 500.Bäumer US, Schäfer HJ. Cleavage of Alkenes by Anodic Oxidation. J Appl Electrochem. 2005;35:1283–1292. [Google Scholar]
- 501.Lambert PC, Fry AJ. Anodic Oxidation of a Tetrasubstituted Cyclooctatetraene. Multiple Carbon–Carbon Bond Cleavage and Aromatization. Tetrahedron Lett. 2011;52:5281–5284. [Google Scholar]
- 502.Bours J, Morton M, Fry AJ. Electrochemical Oxidation of Cyclooctatetraene in the Presence of Allyltrimethylsilane. Anodic Trialkylation with Bicyclization. Tetrahedron Lett. 2012;53:1015–1017. [Google Scholar]
- 503.Ashikari Y, Shimizu A, Nokami T, Yoshida J-I. Halogen and Chalcogen Cation Pools Stabilized by DMSO. Versatile Reagents for Alkene Difunctionalization. J Am Chem Soc. 2013;135:16070–16073. doi: 10.1021/ja4092648. [DOI] [PubMed] [Google Scholar]
- 504.Shimizu A, Hayashi R, Ashikari Y, Nokami T, Yoshida J-I. Switching the Reaction Pathways of Electrochemically Generated β-Haloalkoxysulfonium Ions–Synthesis of Halohydrins and Epoxides. Beilstein J Org Chem. 2015;11:242–248. doi: 10.3762/bjoc.11.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Fujie S, Matsumoto K, Suga S, Nokami T, Yoshida J-I. Addition of ArSSAr to Carbon-Carbon Multiple Bonds Using Electrochemistry. Tetrahedron. 2010;66:2823–2829. [Google Scholar]
- 506.Do QT, Elothmani D, Simonet J, Guillanton GL. The Electrochemical Oxidation of Dimethyl Disulfide–Anodic Methylsulfanylation of Phenols and Aromatic Ethers. Electrochim Acta. 2005;50:4792–4799. [Google Scholar]
- 507.Matsumoto K, Sanada T, Shimazaki H, Shimada K, Hagiwara S, Fujie S, Ashikari Y, Suga S, Kashimura S, Yoshida J-I. The Addition of ArSSAr to Alkenes: the Implications of a Cationic Chain Mechanism Initiated by Electrogenerated ArS(ArSSAr) Asian J Org Chem. 2013;2:325–329. [Google Scholar]
- 508.Matsumoto K, Shimazaki H, Sanada T, Shimada K, Hagiwara S, Suga S, Kashimura S, Yoshida J-I. Electrogenerated Acid (EGA)-Catalyzed Addition of Diaryl Disulfides to Carbon–Carbon Multiple Bonds. Chem Lett. 2013;42:843–845. [Google Scholar]
- 509.Fujie S, Matsumoto K, Suga S, Yoshida J-I. Thiofluorination of Carbon–Carbon Multiple Bonds Using Electrochemically Generated ArS(ArSSAr) BF4. Chem Lett. 2009;38:1186–1187. [Google Scholar]
- 510.Matsumoto K, Suga S, Yoshida J-I. Organic Reactions Mediated by Electrochemically Generated ArS. Org Biomol Chem. 2011;9:2586–2596. doi: 10.1039/c0ob01070g. [DOI] [PubMed] [Google Scholar]
- 511.Matsumoto K, Kozuki Y, Ashikari Y, Suga S, Kashimura S, Yoshida J-I. Electrophilic Substitution Reactions Using an Electrogenerated ArS(ArSSAr)+ Cation Pool as an ArS+ Equivalent. Tetrahedron Lett. 2012;53:1916–1919. [Google Scholar]
- 512.Matsumoto K, Ueoka K, Suzuki S, Suga S, Yoshida J-I. Direct and Indirect Electrochemical Generation of Alkoxycarbenium Ion Pools From Thioacetals. Tetrahedron. 2009;65:10901–10907. [Google Scholar]
- 513.Matsumoto K, Ueoka K, Fujie S, Suga S, Yoshida J-I. Synthesis of Thiochromans Based on Indirect Cation Pool Method. Heterocycles. 2008;76:1103–1119. [Google Scholar]
- 514.Suga S, Matsumoto K, Ueoka K, Yoshida J-I. Indirect Cation Pool Method. Rapid Generation of Alkoxycarbenium Ion Pools From Thioacetals. J Am Chem Soc. 2006;128:7710–7711. doi: 10.1021/ja0625778. [DOI] [PubMed] [Google Scholar]
- 515.Saito K, Saigusa Y, Nokami T, Yoshida J-I. Electrochemically Generated ArS(ArSSAr) + B(C6F5)4–As an Activator of Thioglycosides for Glycosylation. Chem Lett. 2011;40:678–679. [Google Scholar]
- 516.Saito K, Ueoka K, Matsumoto K, Suga S, Nokami T, Yoshida J-I. Indirect Cation-Flow Method: Flash Generation of Alkoxycarbenium Ions and Studies on the Stability of Glycosyl Cations. Angew Chem, Int Ed. 2011;50:5153–5156. doi: 10.1002/anie.201100854. [DOI] [PubMed] [Google Scholar]
- 517.Matsumoto K, Fujie S, Suga S, Nokami T, Yoshida JI. Addition of ArSSAr to Dienes via Intramolecular C–C Bond Formation Initiated by a Catalytic Amount of ArS. Chem Commun. 2009:5448–5450. doi: 10.1039/b910821a. [DOI] [PubMed] [Google Scholar]
- 518.Matsumoto K, Fujie S, Ueoka K, Suga S, Yoshida J-I. An Electroinitiated Cation Chain Reaction: Intramolecular Carbon–Carbon Bond Formation Between Thioacetal and Olefin Groups. Angew Chem, Int Ed. 2008;47:2506–2508. doi: 10.1002/anie.200705748. [DOI] [PubMed] [Google Scholar]
- 519.Röse P, Emge S, Yoshida J-I, Hilt G. Electrochemical Selenium-and Iodonium-Initiated Cyclisation of Hydroxy-Functionalised 1,4-Dienes. Beilstein J Org Chem. 2015;11:174–183. doi: 10.3762/bjoc.11.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Liang S, Zeng C-C, Luo X-G, Ren F-Z, Tian H-Y, Sun B-G, Little RD. Electrochemically Catalyzed Amino-Oxygenation of Styrenes: N-Bu4NI Induced C–N Followed by a C–O Bond Formation Cascade for the Synthesis of Indolines. Green Chem. 2016;18:2222–2230. [Google Scholar]
- 521.Yan W-Q, Lin M-Y, Little RD, Zeng C-C. Electrochemical Regioselective Azidoiodination of Alkenes. Tetrahedron. 2017;73:764–770. [Google Scholar]
- 522.Tanaka H, Kuroboshi M, Takeda H, Kanda H, Torii S. Electrochemical Asymmetric Epoxidation of Olefins by Using an Optically Active Mn-Salen Complex. J Electroanal Chem. 2001;507:75–81. [Google Scholar]
- 523.Nishiguchi I, Kanbe O, Itoh K, Maekawa H. Facile Iodination of Terminal Acetylenes by Anodic Oxidation in the Presence of NaI. Synlett. 2000:89–91. [Google Scholar]
- 524.Fu N, Sauer GS, Saha A, Loo A, Lin S. Metal-Catalyzed Electrochemical Diazidation of Alkenes. Science. 2017;357:575–579. doi: 10.1126/science.aan6206. [DOI] [PubMed] [Google Scholar]
- 525.Li W-C, Zeng C-C, Hu L-M, Tian H-Y, Little RD. Efficient Indirect Electrochemical Synthesis of 2-Substituted Benzoxazoles Using Sodium Iodide as Mediator. Adv Synth Catal. 2013;355:2884–2890. [Google Scholar]
- 526.Shih Y, Ke C, Pan C, Huang Y. Transition-Metal Catalyst Free C=N Coupling with Phenol/Phenoxide: a Green Synthesis of a Benzoxazole Scaffold by an Anodic Oxidation Reaction. RSC Adv. 2013;3:7330–7337. [Google Scholar]
- 527.Kang L-S, Xiao H-L, Zeng C-C, Hu L-M, Little RD. Electrochemical Synthesis of Benzoxazoles Mediated by 2,3-Dichloro-5,6-Dicyano-p-Hydroquinone (DDH) as a Redox Catalyst. J Electroanal Chem. 2016;767:13–17. [Google Scholar]
- 528.Luca OR, Wang T, Konezny SJ, Batista VS, Crabtree RH. DDQ as an Electrocatalyst for Amine Dehydrogenation, a Model System for Virtual Hydrogen Storage. New J Chem. 2011;35:998–999. [Google Scholar]
- 529.Largeron M, Fleury M-B. A Biomimetic Electrocatalytic System for the Atom-Economical Chemoselective Synthesis of Secondary Amines. Org Lett. 2009;11:883–886. doi: 10.1021/ol802885b. [DOI] [PubMed] [Google Scholar]
- 530.Largeron M, Neudorffer A, Fleury M-B. Oxidation of Unactivated Primary Aliphatic Amines Catalyzed by an Electrogenerated 3,4-Azaquinone Species: a Small-Molecule Mimic of Amine Oxidases. Angew Chem, Int Ed. 2003;42:1026–1029. doi: 10.1002/anie.200390263. [DOI] [PubMed] [Google Scholar]
- 531.Largeron M, Chiaroni A, Fleury M-B. Environmentally Friendly Chemoselective Oxidation of Primary Aliphatic Amines by Using a Biomimetic Electrocatalytic System. Chem - Eur J. 2008;14:996–1003. doi: 10.1002/chem.200700876. [DOI] [PubMed] [Google Scholar]
- 532.Largeron M. Regiospecific Inverse-Electron-Demand Diels - Alder Reaction of Simultaneously Electrogenerated Diene and Dienophile: an Expeditious Route to Polyfunctionalized 1, 4-Benzoxazine Derivatives. Angew Chem, Int Ed. 2002;41:824–827. doi: 10.1002/1521-3773(20020301)41:5<824::aid-anie824>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 533.Xu D, Chiaroni A, Fleury M-B, Largeron M. Electrochemically Induced Cascade Reaction for the Assembly of Libraries of Biologically Relevant 1,4-Benzoxazine Derivatives. J Org Chem. 2006;71:6374–6381. doi: 10.1021/jo060452f. [DOI] [PubMed] [Google Scholar]
- 534.Singh S, Sharma LK, Saraswat A, Siddiqui IR, Kehri HK, Singh RKP. Electrosynthesis and Screening of Novel 1,3,4-Oxadiazoles as Potent and Selective Antifungal Agents. RSC Adv. 2013;3:4237–4239. [Google Scholar]
- 535.Xiao H-L, Zeng C-C, Tian H-Y, Hu L-M, Little RD. Electrochemical Synthesis of 3,5-Disubstituted Isoxazoles. J Electroanal Chem. 2014;727:120–124. [Google Scholar]
- 536.Wang H-B, Huang J-M. Decarboxylative Coupling of α-Keto Acids with Ortho-Phenylenediamines Promoted by an Electrochemical Method in Aqueous Media. Adv Synth Catal. 2016;358:1975–1981. [Google Scholar]
- 537.Kowalski J, Płoszyńska J, Sobkowiak A, Morzycki JW, Wilczewska AZ. Direct Electrochemical Acetoxylation of Cholesterol at the Allylic Position. J Electroanal Chem. 2005;585:275–280. [Google Scholar]
- 538.Horn EJ, Rosen BR, Chen Y, Tang J, Chen K, Eastgate MD, Baran PS. Scalable and Sustainable Electrochemical Allylic C–H Oxidation. Nature. 2016;533:77–81. doi: 10.1038/nature17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Kawamata Y, Yan M, Liu Z, Bao D-H, Chen J, Starr JT, Baran PS. Scalable, Electrochemical Oxidation of Unactivated C–H Bonds. J Am Chem Soc. 2017;139:7448–7451. doi: 10.1021/jacs.7b03539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Kitada S, Takahashi M, Yamaguchi Y, Okada Y, Chiba K. Soluble-Support-Assisted Electrochemical Reactions: Application to Anodic Disulfide Bond Formation. Org Lett. 2012;14:5960–5963. doi: 10.1021/ol302863r. [DOI] [PubMed] [Google Scholar]
- 541.Takahashi M, Okada Y, Kitano Y, Chiba K. Phase-Transfer-Mediated Electrochemical Reaction: Anodic Disulfide Bond Formation Under Biphasic Condition. Tetrahedron Lett. 2014;55:3622–3624. [Google Scholar]
- 542.Tomkiel AM, Brzezinski K, Łotowskia Z, Siergiejczyk L, Wałejkoa P, Witkowski SA, Kowalski J, Płoszyńskab J, Sobkowiak A, Morzycki JW. Electrochemical Synthesis of Glycoconjugates of 3β-Hydroxy-Δ 5-Steroids by Using Non-Activated Sugars and Steroidal Thioethers. Tetrahedron. 2013;69:8904–8913. [Google Scholar]
- 543.Tomkiel AM, Kowalski J, Płoszyńska J, Siergiejczyk L, Łotowski Z, Sobkowiak A, Morzycki JW. Electrochemical Synthesis of Glycoconjugates From Activated Sterol Derivatives. Steroids. 2014;82:60–67. doi: 10.1016/j.steroids.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 544.Maeda H, Ohmori H. Electrochemistry of Phosphorus and Sulfur Compounds: a Unique Tool for Organic Synthesis. Acc Chem Res. 1999;32:72–80. [Google Scholar]
- 545.Nikitin EV, Zagumennov VA. Initiation of Homolytic Addition to Alkenes by Means of Organophosphorus Radical-Cations. Electrochim Acta. 2000;45:3983–3992. [Google Scholar]
- 546.Sheremetev AB, Lyalin BV, Kozeev AM, Palysaeva NV, Struchkova MI, Suponitsky KY. A Practical Anodic Oxidation of Aminofurazans to Azofurazans: an Environmentally Friendly Route. RSC Adv. 2015;5:37617–37625. [Google Scholar]
- 547.Abaci S, Tamer U, Pekmez K, Yildiz A. Electrosynthesis of 4,4′-Dinitroazobenzene on PbO2 Electrodes. J Appl Electrochem. 2002;32:193–196. [Google Scholar]
- 548.Ye J-Q, Zhang Z-L, Zha Z-G, Wang Z-Y. A Green and Efficient Access to Aryl Nitriles via an Electrochemical Anodic Oxidation. Chin Chem Lett. 2014;25:1112–1114. [Google Scholar]
- 549.Shusterman-Honger Y, Becker JY. Electrochemical Oxidation of Organic Compounds Containing CN Double Bonds. J Electroanal Chem. 2015;740:105–113. [Google Scholar]
- 550.Nokami T, Soma R, Yamamoto Y, Kamei T, Itami K, Yoshida J-I. Generation of Pyridyl Coordinated Organosilicon Cation Pool by Oxidative Si-Si Bond Dissociation. Beilstein J Org Chem. 2007;3:7. doi: 10.1186/1860-5397-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Qi H-L, Chen D-S, Ye J-S, Huang J-M. Electrochemical Technique and Copper-Promoted Transformations: Selective Hydroxylation and Amination of Arylboronic Acids. J Org Chem. 2013;78:7482–7487. doi: 10.1021/jo400981f. [DOI] [PubMed] [Google Scholar]
- 552.Park YS, Little RD. Redox Electron-Transfer Reactions: Electrochemically Mediated Rearrangement, Mechanism, and a Total Synthesis of Daucene. J Org Chem. 2008;73:6807–6815. doi: 10.1021/jo801199s. [DOI] [PubMed] [Google Scholar]
- 553.Park YS, Wang SC, Tantillo DJ, Little RD. A Highly Selective Rearrangement of a Housane-Derived Cation Radical: an Electrochemically Mediated Transformation. J Org Chem. 2007;72:4351–4357. doi: 10.1021/jo070190x. [DOI] [PubMed] [Google Scholar]
- 554.Jutand A. Contribution of Electrochemistry to Organometallic Catalysis. Chem Rev. 2008;108:2300–2347. doi: 10.1021/cr068072h. [DOI] [PubMed] [Google Scholar]
- 555.Fultz ML, Durst RA. Mediator Compounds for the Electrochemical Study of Biological Redox Systems: A Compilation. Anal Chim Acta. 1982;140:1–18. [Google Scholar]
- 556.Tsuji J, Minato M. Oxidation of Olefins to Ketones in Combination with Electrooxidation. Tetrahedron Lett. 1987;28:3683–3686. [Google Scholar]
- 557.Mitsudo K, Kaide T, Nakamoto E, Yoshida K, Tanaka H. Electrochemical Generation of Cationic Pd Catalysts and Application to Pd/TEMPO Double-Mediatory Electrooxidative Wacker-Type Reactions. J Am Chem Soc. 2007;129:2246–2247. doi: 10.1021/ja069043r. [DOI] [PubMed] [Google Scholar]
- 558.Mitsudo K, Fukunaga S, Fujita T, Mandai H, Suga S, Tanaka H. Recyclable Palladium Catalyst in PEG/CH3CN Biphasic System for Electro-Oxidative Wacker-Type Reaction. Electrochemistry. 2013;81:347–349. [Google Scholar]
- 559.Mitsudo K, Ishii T, Tanaka H. Pd/TEMPO Double-Mediatory Electrooxidative Wacker-Type Cyclizations. Electrochemistry. 2008;76:859–861. [Google Scholar]
- 560.Mitsudo K, Shiraga T, Kagen D, Shi D, Becker JY, Tanaka H. Pd/TEMPO-Catalyzed Electrooxidative Synthesis of Biaryls From Arylboronic Acids or Arylboronic Esters. Tetrahedron. 2009;65:8384–8388. [Google Scholar]
- 561.Mitsudo K, Shiraga T, Tanaka H. Electrooxidative Homo-Coupling of Arylboronic Acids Catalyzed by Electrogenerated Cationic Palladium Catalysts. Tetrahedron Lett. 2008;49:6593–6595. [Google Scholar]
- 562.Amatore C, Cammoun C, Jutand A. Pd(OAc)2/p-Benzoquinone-Catalyzed Anaerobic Electrooxidative Homocoupling of Arylboronic Acids, Arylboronates and Aryltrifluoroborates in DMF and/or Water. Eur J Org Chem. 2008;2008:4567–4570. [Google Scholar]
- 563.Mitsudo K, Shiraga T, Mizukawa J-I, Suga S, Tanaka H. Electrochemical Generation of Silver Acetylides From Terminal Alkynes with a Ag Anode and Integration Into Sequential Pd-Catalysed Coupling with Arylboronic Acids. Chem Commun. 2010;46:9256–9258. doi: 10.1039/c0cc02633f. [DOI] [PubMed] [Google Scholar]
- 564.Amatore C, Cammoun C, Jutand A. Electrochemical Recycling of Benzoquinone in the Pd/Benzoquinone-Catalyzed Heck-Type Reactions From Arenes. Adv Synth Catal. 2007;349:292–296. [Google Scholar]
- 565.Amatore C, Cammoun C, Jutand A. Palladium/Benzoquinone-Catalyzed Electrochemical Oxidation of Alcohols Under Anaerobic Conditions. Synlett. 2007;2007:2173–2178. [Google Scholar]
- 566.Peterson KP, Larock RC. Palladium-Catalyzed Oxidation of Primary and Secondary Allylic and Benzylic Alcohols. J Org Chem. 1998;63:3185–3189. [Google Scholar]
- 567.Kakiuchi F, Kochi T, Mutsutani H, Kobayashi N, Urano S, Sato M, Nishiyama S, Tanabe T. Palladium-Catalyzed Aromatic C–H Halogenation with Hydrogen Halides by Means of Electrochemical Oxidation. J Am Chem Soc. 2009;131:11310–11311. doi: 10.1021/ja9049228. [DOI] [PubMed] [Google Scholar]
- 568.Grayaznova TV, Dudkina YB, Islamov DR, Kataeva ON, Sinyashin OG, Vicic DA, Budnikova YH. Pyridine-Directed Palladium-Catalyzed Electrochemical Phosphonation of C(Sp2)-H Bond. J Organomet Chem. 2015;785:68–71. [Google Scholar]
- 569.Dudkina YB, Mikhaylov DY, Gryaznova TV, Tufatullin AI, Kataeva ON, Vicic DA, Budnikova YH. Electrochemical Ortho Functionalization of 2-Phenylpyridine with Perfluorocarboxylic Acids Catalyzed by Palladium in Higher Oxidation States. Organometallics. 2013;32:4785–4792. [Google Scholar]
- 570.Dudkina YB, Gryaznova TV, Sinyashin OG, Budnikova YH. Ligand-Directed Electrochemical Functionalization of C(Sp2)—H Bonds in the Presence of the Palladium and Nickel Compounds. Russ Chem Bull. 2015;64:1713–1725. [Google Scholar]
- 571.Dudkina YB, Mikhaylov DY, Gryaznova TV, Sinyashin OG, Vicic DA, Budnikova YH. MII/MIII-Catalyzed Ortho-Fluoroalkylation of 2-Phenylpyridine. Eur J Org Chem. 2012;2012:2114–2117. [Google Scholar]
- 572.Aiso H, Kochi T, Mutsutani H, Tanabe T, Nishiyama S, Kakiuchi F. Catalytic Electrochemical C–H Iodination and One-Pot Arylation by ON/OFF Switching of Electric Current. J Org Chem. 2012;77:7718–7724. doi: 10.1021/jo3012286. [DOI] [PubMed] [Google Scholar]
- 573.Saito F, Aiso H, Kochi T, Kakiuchi F. Palladium-Catalyzed Regioselective Homocoupling of Arenes Using Anodic Oxidation: Formal Electrolysis of Aromatic Carbon–Hydrogen Bonds. Organometallics. 2014;33:6704–6707. [Google Scholar]
- 574.Chen X, Hao XS, Goodhue CE, Yu J-Q. Cu(II)-Catalyzed Functionalizations of Aryl C–H Bonds Using O2 As an Oxidant. J Am Chem Soc. 2006;128:6790–6791. doi: 10.1021/ja061715q. [DOI] [PubMed] [Google Scholar]
- 575.Hull KL, Lanni EL, Sanford MS. Highly Regioselective Catalytic Oxidative Coupling Reactions: Synthetic and Mechanistic Investigations. J Am Chem Soc. 2006;128:14047–14049. doi: 10.1021/ja065718e. [DOI] [PubMed] [Google Scholar]
- 576.Mitsudo K, Kamimoto N, Murakami H, Mandai H, Wakamiya A, Murata Y, Suga S. Site-Selective Sequential Coupling Reactions Controlled by “Electrochemical Reaction Site Switching”: a Straightforward Approach to 1,4-Bis(Diaryl)Buta-1,3-Diynes. Org Biomol Chem. 2012;10:9562–9568. doi: 10.1039/c2ob26567b. [DOI] [PubMed] [Google Scholar]
- 577.Yang Q-L, Li Y-Q, Ma C, Fang P, Zhang X-J, Mei T-S, Palladium-Catalyzed C. Sp3)—H Oxygenation via Electrochemical Oxidation. J Am Chem Soc. 2017;139:3293–3298. doi: 10.1021/jacs.7b01232. [DOI] [PubMed] [Google Scholar]
- 578.Li Y-Q, Yang Q-L, Fang P, Mei T-S, Zhang D, Palladium-Catalyzed C. Sp2)–H Acetoxylation via Electrochemical Oxidation. Org Lett. 2017;19:2905–2908. doi: 10.1021/acs.orglett.7b01138. [DOI] [PubMed] [Google Scholar]
- 579.Eberson L, Schäfer H. Organic Electrochemistry. 1. Vol. 21. Springer, Berlin, Heidelberg; Berlin/Heidelberg: 1971. Organic Electrochemistry; pp. 1–182. (Fortschritte der Chemischen Forschung). [Google Scholar]
- 580.Kronenwetter H, Husek J, Etz B, Jones A, Manchanayakage R. Electrochemical Pinacol Coupling of Aromatic Carbonyl Compounds in a [BMIM][BF4]–H2O Mixture. Green Chem. 2014;16:1489–1495. [Google Scholar]
- 581.André F, Hapiot P, Lagrost C. Dimerization of Ion Radicals in Ionic Liquids. an Example of Favourable “Coulombic” Solvation. Phys Chem Chem Phys. 2010;12:7506–7512. doi: 10.1039/b920552g. [DOI] [PubMed] [Google Scholar]
- 582.Lagrost C, Hapiot P, Vaultier M. The Influence of Room-Temperature Ionic Liquids on the Stereoselectivity and Kinetics of the Electrochemical Pinacol Coupling of Acetophenone. Green Chem. 2005;7:468–474. [Google Scholar]
- 583.Heimann J, Schäfer HJ, Fr hlich R, Wibbeling B. Cathodic Cyclisation of N-(Oxoalkyl)Pyridinium Salts–Formation of Tricyclic Indolizidine and Quinolizidine Derivatives in Aqueous Medium. Eur J Org Chem. 2003;2003:2919–2932. [Google Scholar]
- 584.Kise N, Agui S, Morimoto S, Ueda N. Electroreductive Acylation of Aromatic Ketones with Acylimidazoles. J Org Chem. 2005;70:9407–9410. doi: 10.1021/jo051498w. [DOI] [PubMed] [Google Scholar]
- 585.Kise N, Arimoto K, Ueda N. Electroreductive Intramolecular Coupling of Aromatic δ-and ε-Keto Esters. Tetrahedron Lett. 2003;44:6281–6284. [Google Scholar]
- 586.Kise N, Shiozawa Y, Ueda N. Trans-Stereoselective Intramolecular Crossed Pinacol Coupling of Aromatic 1,4-, 1,5-, and 1,6-Diketones by Electroreduction. Tetrahedron Lett. 2004;45:7599–7603. [Google Scholar]
- 587.Kise N, Shiozawa Y, Ueda N. Electroreductive Crossed Pinacol Coupling of Aromatic Ketones with Aliphatic Ketones and Aldehydes. Tetrahedron. 2007;63:5415–5426. [Google Scholar]
- 588.Kise N, Hamada Y, Sakurai T. Electroreductive Coupling of Aromatic Ketones, Aldehydes, and Aldimines with α, β-Unsaturated Esters: Synthesis of 5-Aryl Substituted γ-Butyrolactones and Lactams. Tetrahedron. 2017;73:1143–1156. [Google Scholar]
- 589.Kise N, Hamada Y, Sakurai T. Electroreductive Coupling of Optically Active α,β-Unsaturated Carbonyl Compounds with Diaryl Ketones: Asymmetric Synthesis of 4,5,5-Trisubstituted γ-Butyrolactones. Org Lett. 2014;16:3348–3351. doi: 10.1021/ol5013789. [DOI] [PubMed] [Google Scholar]
- 590.Kise N, Miyamoto H, Hamada Y, Sakurai T. Electroreductive Coupling of 1,3-Dimethyluracils with Aromatic Ketones: Synthesis of 6-Substituted 1,3-Dimethyluracils. Tetrahedron Lett. 2015;56:4599–4602. [Google Scholar]
- 591.Kise N, Hamada Y, Sakurai T. Electroreductive Intermolecular Coupling of Uracils with Aromatic Ketones: Synthesis of 6-Substituted and Cis-5,6-Disubstituted 5,6-Dihydro-1,3-Dimethyluracils and Their Transformation to 6-Substituted 1,3-Dimethyluracils, Trans-5,6-Disubstituted 5,6-Dihydro-1,3-Dimethyluracils, and 4,5,5-Trisubstituted 3-Methyloxazolizin-2-Ones. J Org Chem. 2016;81:5101–5119. doi: 10.1021/acs.joc.6b00670. [DOI] [PubMed] [Google Scholar]
- 592.Kise N, Ozaki H, Moriyama N, Kitagishi Y, Ueda N. Electroreductive Intramolecular Coupling of Chiral α-Imino Esters: Stereoselective Synthesis of Mixed Ketals Of cis-2,4-Disubstituted Azetidine-3-Ones. J Am Chem Soc. 2003;125:11591–11596. doi: 10.1021/ja036975b. [DOI] [PubMed] [Google Scholar]
- 593.Kise N, Ohya K, Arimoto K, Yamashita Y, Hirano Y, Ono T, Ueda N. Electroreductive Intramolecular Coupling of Aromatic β- and γ-Imino Esters: a New Synthetic Method for N-Alkoxycarbonyl-2-Aryl-3-Ones and Cis-2-Aryl-3-Ols of Pyrrolidines and Piperidines. J Org Chem. 2004;69:7710–7719. doi: 10.1021/jo0488917. [DOI] [PubMed] [Google Scholar]
- 594.Takenaga Y, Umesaki Y, Sakurai T, Kise N. Electroreductive Five-and Six-Membered Cyclization of Aromatic β-and γ-Imino Esters Derived From (S)-Aspartic Acid and (S)-Glutamic Acid. Tetrahedron. 2012;68:2579–2589. [Google Scholar]
- 595.Kise N, Hirano Y, Tanaka Y. Electroreductive Intramolecular Coupling of Aromatic Imino Esters: Is Four-Membered Cyclization Much More Favorable Than Six-Membered Cyclization? Org Lett. 2006;8:1323–1325. doi: 10.1021/ol053144x. [DOI] [PubMed] [Google Scholar]
- 596.Kise N, Morimoto S. Electroreductive Acylation of Aromatic Imines with Acylimidazoles. Tetrahedron. 2008;64:1765–1771. [Google Scholar]
- 597.Kise N, Sueyoshi A, Takeuchi S-I, Sakurai T. Electroreductive Intermolecular Coupling of 3-Methoxycarbonylindoles with Ketones. Org Lett. 2013;15:2746–2749. doi: 10.1021/ol4010799. [DOI] [PubMed] [Google Scholar]
- 598.Kise N, Mano T, Sakurai T. Electroreductive Intramolecular Coupling of 1-Indolealkanones. Org Lett. 2008;10:4617–4620. doi: 10.1021/ol801876k. [DOI] [PubMed] [Google Scholar]
- 599.Kise N, Isemoto S, Sakurai T. Electroreductive Intermolecular Coupling of Phthalimides with Aldehydes: Application to the Synthesis of Alkylideneisoindolin-1-Ones. Tetrahedron. 2012;68:8805–8816. [Google Scholar]
- 600.Kise N, Sakurai T. Electroreductive Intramolecular Coupling of N-(Oxoalkyl)Phthalimides: Complementary Method to Samarium-(II) Iodide Reduction. Tetrahedron Lett. 2010;51:70–74. [Google Scholar]
- 601.Kise N, Fukazawa K, Sakurai T. Electroreductive Intramolecular Coupling of Aliphatic Cyclic Imides with Ketones and O-Methyloximes. Tetrahedron Lett. 2010;51:5767–5770. [Google Scholar]
- 602.Kise N, Isemoto S, Sakurai T. Electroreductive Coupling of Phthalimides with α,β-Unsaturated Esters: Unusual Rearrangement of Resulting Silyl Ketene Acetals. Org Lett. 2009;11:4902–4905. doi: 10.1021/ol902016a. [DOI] [PubMed] [Google Scholar]
- 603.Kise N, Isemoto S, Sakurai T. Electroreductive Intramolecular Coupling of Phthalimides with Aromatic Aldehydes: Application to the Synthesis of Lennoxamine. J Org Chem. 2011;76:9856–9860. doi: 10.1021/jo2018735. [DOI] [PubMed] [Google Scholar]
- 604.Parrish JD, Little RD. Electrochemical Generation of Low-Valent Lanthanides. Tetrahedron Lett. 2001;42:7767–7770. [Google Scholar]
- 605.Regio- and Stereoselective Intramolecular Cyclization of Some Unconjugated Ketones at Mercury Pool Cathode. Synlett. 2000:1199–1201. [Google Scholar]
- 606.Yadav AK, Manju M, Kumar M, Yadav T, Jain R. A New Diastereoselective Intramolecular Electroreductive Coupling of Unsaturated β-Ketoesters and β-Ketoamides in Ionic Liquids at a Tin Cathode. Tetrahedron Lett. 2008;49:5724–5726. [Google Scholar]
- 607.Miyazaki T, Maekawa H, Hisatomi T, Nishiguchi I. Highly Stereo- and Regio-Selective Intramolecular Cyclization with Diastereoselective Asymmetric Induction by Electroreduction of Optically Active N-Alkenyl-2-Acylpyrrolidines. Electrochemistry. 2011;79:447–449. [Google Scholar]
- 608.Andreu R, Pletcher D. Ytterbium(II) as a Mediator in Organic Electrosynthesis—Possibilities and Limitations. Electrochim Acta. 2003;48:1065–1071. [Google Scholar]
- 609.Sahloul K, Sun L, Requet A, Chahine Y, Mellah M. A Samarium “Soluble” Anode: a New Source of SmI2 Reagent for Electrosynthetic Application. Chem - Eur J. 2012;18:11205–11209. doi: 10.1002/chem.201201390. [DOI] [PubMed] [Google Scholar]
- 610.Sun L, Mellah M. Efficient Electrosynthesis of SmCl2, SmBr2, And Sm(OTf)2From a “Sacrificial” Samarium Anode: Effect of nBu4NPF6 On the Reactivity. Organometallics. 2014;33:4625–4628. [Google Scholar]
- 611.Yadav AK, Manju M, Chhinpa PR. Enantioselective Cathodic Reduction of Some Prochiral Ketones in the Presence of (–)-N,N′-Dimethylquininium Tetrafluoroborate at Mercury Cathode. Tetrahedron: Asymmetry. 2003;14:1079–1081. [Google Scholar]
- 612.Yadav AK, Singh A. Enantioselective Cathodic Reduction of Some Prochiral Ketones in the Presence of (1R,2S)-(–)-N,N-Dimethylephedrinium Tetrafluoroborate at a Mercury Pool Cathode. Bull Chem Soc Jpn. 2002;75:587–588. [Google Scholar]
- 613.Batanero B, Barba F. Facile Conversion of O-Quinones Into 1,3-Dioxoles. Org Lett. 2005;7:2567–2569. doi: 10.1021/ol050614e. [DOI] [PubMed] [Google Scholar]
- 614.Batanero B, Barba F. Electron Transfer in the Cathodic Reduction of α-Dicarbonyl Compounds. Tetrahedron. 2008;64:1834–1838. [Google Scholar]
- 615.Batanero B, Saez R, Barba F. Electroreduction of Quinones Under Aprotic Conditions. Electrochim Acta. 2009;54:4872–4879. [Google Scholar]
- 616.Batanero B, Barba F, Ranz F, Barba I, Elinson MN. Synthesis of New Derivatives of a Representative O-Quinone Scaffold by Reduction at the Electrode. Tetrahedron. 2012;68:5979–5983. [Google Scholar]
- 617.Batanero B, Ramirez-Moreno M, Barba F. Electroinduced Carbene Formation in the Cathodic Reduction of 1,2-Dicarbonyl Compounds via Electron-Transfer to the Solvent. Electrochim Acta. 2015;167:207–212. [Google Scholar]
- 618.Batanero B, Barba F, Martin A. One-Pot Formation of 1,3,4-Oxadiazol-2(3H)-Ones and Dibenzo[C,E]Azepines by Concomitant Cathodic Reduction of Diazonium Salts and Phenanthrenequinones. J Org Chem. 2013;78:9477–9481. doi: 10.1021/jo401264w. [DOI] [PubMed] [Google Scholar]
- 619.Shono T, Masuda H, Murase H, Shimomura M, Kashimura S. Electroorganic Chemistry 0.134. Facile Electroreduction of Methyl-Esters and N,N-Dimethylamides of Aliphatic Carboxylic-Acids to Primary Alcohols. J Org Chem. 1992;57:1061–1063. [Google Scholar]
- 620.Lam K, Markó IE. Using Toluates as Simple and Versatile Radical Precursors. Org Lett. 2008;10:2773–2776. doi: 10.1021/ol800944p. [DOI] [PubMed] [Google Scholar]
- 621.Lam K, Markó IE. Organic Electrosynthesis Using Toluates as Simple and Versatile Radical Precursors. Chem Commun. 2009:95–97. doi: 10.1039/b813545b. [DOI] [PubMed] [Google Scholar]
- 622.Lam K, Markó IE. Novel Electrochemical Deoxygenation Reaction Using Diphenylphosphinates. Org Lett. 2011;13:406–409. doi: 10.1021/ol102714s. [DOI] [PubMed] [Google Scholar]
- 623.Lam K, Markó IE. Electrochemical Deoxygenation of Primary Alcohols. Synlett. 2012;23:1235–1239. [Google Scholar]
- 624.Lam K, Markó IE. Chemoselective Chemical and Electrochemical Deprotections of Aromatic Esters. Org Lett. 2009;11:2752–2755. doi: 10.1021/ol900828x. [DOI] [PubMed] [Google Scholar]
- 625.Ozaki S, Yoshinaga H, Matsui E, Adachi M. Synthesis of Cyclic Ketones by Electrochemical Reduction of S-(2-Methoxycarbonyl)Phenyl Thiolesters. J Org Chem. 2001;66:2503–2505. doi: 10.1021/jo001578u. [DOI] [PubMed] [Google Scholar]
- 626.Costero AM, Rodríguez-Muñiz GM, Gaviña P, Gil S, Domenech A. Biphenylthioureas as Organocatalysts for Electrochemical Reductions. Tetrahedron Lett. 2007;48:6992–6995. [Google Scholar]
- 627.Popp FD, Schultz HP. Electrolytic Reduction of Organic Compounds. Chem Rev. 1962;62:19–40. [Google Scholar]
- 628.Edinger C, Waldvogel SR. Electrochemical Deoxygenation of Aromatic Amides and Sulfoxides. Eur J Org Chem. 2014;2014:5144–5148. [Google Scholar]
- 629.Edinger C, Grimaudo V, Broekmann P, Waldvogel SR. Stabilizing Lead Cathodes with Diammonium Salt Additives in the Deoxygenation of Aromatic Amides. ChemElectroChem. 2014;1:1018–1022. [Google Scholar]
- 630.Fechete I, Jouikov V. Double Decarbonylation of Phthalimide Revisited: a Facile Cathodic Synthesis of Isoindoline. Electrochim Acta. 2008;53:7107–7110. [Google Scholar]
- 631.Utley J, Little R, Nielsen M. Organic Electrochemistry. 5th. CRC Press; 2015. Reductive Coupling; pp. 621–704. (Revised and Expanded). [Google Scholar]
- 632.Brosa C, Rodr guez-Santamarta C, Pilard JF, Simonet J. The Cathodic Reduction of Activated Olefins. Experimental Conditions Allowing the Specific Hydrogenation of the Enone Derived From Ergosterol. Phys Chem Chem Phys. 2001;3:2655–2661. [Google Scholar]
- 633.Miura Y, Tateno H, Tajima T. An Electrolytic System Based on the Acid-Base Reaction Between Solid-Supported Acids and Water. Electrochemistry. 2013;81:371–373. [Google Scholar]
- 634.Tomida S, Tsuda R, Furukawa S, Saito M, Tajima T. Electroreductive Hydrogenation of Activated Olefins Using the Concept of Site Isolation. Electrochem Commun. 2016;73:46–49. [Google Scholar]
- 635.Vilar M, Oliveira JL, Navarro M. Investigation of the Hydrogenation Reactivity of Some Organic Substrates Using an Electrocatalytic Method. Appl Catal A. 2010;372:1–7. [Google Scholar]
- 636.da Silva AP, Maia ACS, Navarro M. Homogeneous Electromediated Reduction of the 2-Cyclohexen-1-One by Transition Metals. Tetrahedron Lett. 2005;46:3233–3235. [Google Scholar]
- 637.da Silva AP, Mota SDC, Bieber LW, Navarro M. Homogeneous Electro-Mediated Reduction of Unsaturated Compounds Using Ni and Fe as Mediators in DMF. Tetrahedron. 2006;62:5435–5440. [Google Scholar]
- 638.Fussing I, Güllü M, Hammerich O, Hussain A, Nielsen MF, Utley JHP. Stereoselectivity and Mechanism in the Electrohydrodimerisation of Esters of Cinnamic Acid. J Chem Soc Perkin Trans 2. 1996;38:649–658. [Google Scholar]
- 639.Kratschmer S, Schäfer HJ, Fröhlich R. Cathodically Initiated Cyclodimerization of Trimethyl Aconitate. J Electroanal Chem. 2001;507:2–10. [Google Scholar]
- 640.Kojima T, Obata R, Saito T, Einaga Y, Nishiyama S. Cathodic Reductive Coupling of Methyl Cinnamate on Boron-Doped Diamond Electrodes and Synthesis of New Neolignan-Type Products. Beilstein J Org Chem. 2015;11:200–203. doi: 10.3762/bjoc.11.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Chen A-H, Cheng C-Y, Chen C-W. Synthesis of 2,2′-Biflavanones From Flavone via Electrolytic Reductive Coupling. J Chin Chem Soc. 2002;49:1105–1109. [Google Scholar]
- 642.Jones A, Kronenwetter H, Manchanayakage R. Electrochemical Reductive Coupling of 2-Cyclohexen-1-One in a Mixture of Ionic Liquid and Water. Electrochem Commun. 2012;25:8–10. [Google Scholar]
- 643.Utley JHP, Smith CZ, Motevalli M. Electro-Organic Reactions. Part 51. Reactivity and Stereochemical Pathways for Cathodically Generated Radical-Anions of α,β-Unsaturated Ketones in Aqueous Media. J Chem Soc, Perkin Trans 2. 2000:1053–1057. [Google Scholar]
- 644.Handy ST, Omune D. The Intramolecular Reductive Cyclization of Cyclic Enones. Tetrahedron. 2007;63:1366–1371. [Google Scholar]
- 645.Kise N, Kitagishi Y, Ueda N. Diastereoselective Dl-Hydrocoupling of Benzalacetones by Electroreduction. J Org Chem. 2004;69:959–963. doi: 10.1021/jo035280q. [DOI] [PubMed] [Google Scholar]
- 646.Weßling M, Schäfer HJ. Cathodic Hydrodimerization of Nitroolefins. Beilstein J Org Chem. 2015;11:1163–1174. doi: 10.3762/bjoc.11.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Handy ST, Omune D. A Chelation Effect on the Pathway Between Intramolecular Hydrodimerization and Pinacol Coupling. Org Lett. 2005;7:1553–1555. doi: 10.1021/ol0502026. [DOI] [PubMed] [Google Scholar]
- 648.Kise N, Iitaka S, Iwasaki K, Ueda N. Stereoselective Hydrocoupling of Cinnamic Acid Esters by Electroreduction: Application to Asymmetric Synthesis of Hydrodimers. J Org Chem. 2002;67:8305–8315. doi: 10.1021/jo026183k. [DOI] [PubMed] [Google Scholar]
- 649.Roh Y, Jang H-Y, Lynch V, Bauld NL, Krische MJ. Anion Radical Chain Cycloaddition of Tethered Enones: Intramolecular Cyclobutanation and Diels–Alder Cycloaddition. Org Lett. 2002;4:611–613. doi: 10.1021/ol0172065. [DOI] [PubMed] [Google Scholar]
- 650.Felton GAN, Bauld NL. Efficient Electrocatalytic Intramolecular Anion Radical Cyclobutanation Reactions. Tetrahedron. 2004;60:10999–11010. [Google Scholar]
- 651.Yang J, Cauble DF, Berro AJ, Bauld NL, Krische MJ. Anion Radical [2 + 2] Cycloaddition as a Mechanistic Probe: Stoichiometry- and Concentration-Dependent Partitioning of Electron-Transfer and Alkylation Pathways in the Reaction of the Gilman Reagent Me2CuLi·LiI with Bis(Enones) J Org Chem. 2004;69:7979–7984. doi: 10.1021/jo048499t. [DOI] [PubMed] [Google Scholar]
- 652.Yang J, Felton GAN, Bauld NL, Krische MJ. Chemically Induced Anion Radical Cycloadditions: Intramolecular Cyclobutanation of Bis(Enones) via Homogeneous Electron Transfer. J Am Chem Soc. 2004;126:1634–1635. doi: 10.1021/ja030543j. [DOI] [PubMed] [Google Scholar]
- 653.Bergamini JF, Delaunay J, Hapiot P, Hillebrand M, Lagrost C, Simonet J, Volanschi E. Catalytic Cathodic Cyclodimerization of Vinylarylsulfones. J Electroanal Chem. 2004;569:175–184. [Google Scholar]
- 654.Felton GAN. Electrocatalytic Reactions: Anion Radical Cyclobutanation Reactions and Electrogenerated Base Reactions. Tetrahedron Lett. 2008;49:884–887. [Google Scholar]
- 655.Miranda JA, Wade CJ, Little RD. Indirect Electroreductive Cyclization and Electrohydrocyclization Using Catalytic Reduced Nickel(II) Salen. J Org Chem. 2005;70:8017–8026. doi: 10.1021/jo051148+. [DOI] [PubMed] [Google Scholar]
- 656.Ohno T, Sakai M, Ishino Y, Shibata T, Maekawa H, Nishiguchi I. Mg-Promoted Regio- and Stereoselective C-Acylation of Aromatic α,β-Unsaturated Carbonyl Compounds. Org Lett. 2001;3:3439–3442. doi: 10.1021/ol016376e. [DOI] [PubMed] [Google Scholar]
- 657.Nishiguchi I, Yamamoto Y, Sakai M, Ohno T, Ishino Y, Maekawa H. Regioselective Mg-Promoted C-Acylation of Stilbene and Acenaphthylene Derivatives. Synlett. 2002;2002:759–762. [Google Scholar]
- 658.Yamamoto Y, Maekawa H, Goda S, Nishiguchi I. Novel One-Pot Vicinal Double C-Acylation of Styrenes and Methacrylates by Electroreduction. Org Lett. 2003;5:2755–2758. doi: 10.1021/ol035020v. [DOI] [PubMed] [Google Scholar]
- 659.Nishiguchi I, Sunderrao KP, Yamamoto U, Yamamoto Y, Uchida T, Maekawa H. One-Pot Vicinal and Geminal Double Carboalkoxylation with N-Carboalkoxy-Imidazole by Electroreduction of Aromatic Vinyl and Imine Derivatives. Electrochemistry. 2006;74:680–684. [Google Scholar]
- 660.Kendrekar PS, Yamamoto Y, Dhiman A, Maekawa H, Nishiguchi M. Stereoselective Synthesis of Spiro-Lactones Possessing Cyclopropane Rings Through Electroreductive Cross Coupling of Styrene Derivatives with Diphenyl Succinate or Glutarate. Electrochemistry. 2007;75:813–818. [Google Scholar]
- 661.Franco D, Duñach E. New and Mild Allyl Carbamate Deprotection Method Catalyzed by Electrogenerated Nickel Complexes. Tetrahedron Lett. 2000;41:7333–7336. [Google Scholar]
- 662.Franco D, Riahi A, Hénin F, Muzart J, Duñach E. Electrochemical Reduction of a Racemic Allyl β-Keto Ester Catalyzed by Nickel Complexes: Asymmetric Induction. Eur J Org Chem. 2002;2002:2257–2259. [Google Scholar]
- 663.Solis-Oba A, Hudlicky T, Koroniak L, Frey D. Selective Electrochemical Reduction of Cinnamyl Ethers in the Presence of Other Allylic CO Bonds. Tetrahedron Lett. 2001;42:1241–1243. [Google Scholar]
- 664.Hansen J, Freeman S, Hudlicky T. Selective Electrochemical Deprotection of Cinnamyl Ethers, Esters, and Carbamates. Tetrahedron Lett. 2003;44:1575–1578. [Google Scholar]
- 665.Cankař P, Dubas D, Banfield SC, Chahma M, Hudlicky T. Selectivity in the Electrochemical Deprotection of Cinnamyl Groups From Oxygen and Nitrogen Functionalities: Carbonates Versus Carbamates. Tetrahedron Lett. 2005;46:6851–6854. [Google Scholar]
- 666.Chai D, Genders D, Weinberg N, Zappi G, Bernasconi E, Lee J, Roletto J, Sogli L, Walker D, Martin CR, et al. Ceftibuten: Development of a Commercial Process Based on Cephalosporin C. Part IV. Pilot-Plant Scale Electrochemical Reduction of 3-Acetoxymethyl-7(R)-Glutaroylaminoceph-3-Em-4-Carboxylic Acid 1(S)-Oxide. Org Process Res Dev. 2002;6:178–183. [Google Scholar]
- 667.Benkeser RA, Kaiser EM. An Electrochemical Method of Reducing Aromatic Compounds Selectively to Dihydro or Tetrahydro Products. J Am Chem Soc. 1963;85:2858–2859. [Google Scholar]
- 668.Chaussard J, Combellas C, Thiebault A. Electrochemical Reductions in Liquid-Ammonia - Electrolytic Birch Reactions and Chemical-Bond Fissions. Tetrahedron Lett. 1987;28:1173–1174. [Google Scholar]
- 669.Sternberg HW, Markby RE, Wender I. Reduction of the Benzene Ring and of the Olefinic Double Bond by Electrolytically Generated Electrons. J Am Chem Soc. 1969;91:4191–4194. [Google Scholar]
- 670.Coleman JP, Wagenknecht JH. Reduction of Benzene and Related Compounds in Aqueous Solution and Undivided Cells. J Electrochem Soc. 1981;128:322–326. [Google Scholar]
- 671.Swenson KE, Zemach D, Nanjundiah C, Kariv-Millar E. Birch Reductions of Methoxyaromatics in Aqueous-Solution. J Org Chem. 1983;48:1777–1779. [Google Scholar]
- 672.Kariv-Miller E, Swenson KE. Cathodic Birch Reduction of Methoxy Aromatics and Steroids in Aqueous Solution. J Org Chem. 1983;48:4210–4214. [Google Scholar]
- 673.Ishifune M, Yamashita H, Kera Y, Yamashita N, Hirata K, Murase H, Kashimura S. Electroreduction of Aromatics Using Magnesium Electrodes in Aprotic Solvents Containing Alcoholic Proton Donors. Electrochim Acta. 2003;48:2405–2409. [Google Scholar]
- 674.Kita Y, Maekawa H, Yamasaki Y, Nishiguchi I. Highly Selective and Facile Synthesis of Dihydro-and Tetrahydropyridine Dicarboxylic Acid Derivatives Using Electroreduction as a Key Step. Tetrahedron. 2001;57:2095–2102. [Google Scholar]
- 675.Pasciak EM, Rittichier JT, Chen C-H, Mubarak MS, VanNieuwenhze MS, Peters DG. Electroreductive Dimerization of Coumarin and Coumarin Analogues at Carbon Cathodes. J Org Chem. 2015;80:274–280. doi: 10.1021/jo502272g. [DOI] [PubMed] [Google Scholar]
- 676.Gütz C, Selt M, Bänziger M, Bucher C, Römelt C, Hecken N, Gallou F, Galvão TR, Waldvogel SR. A Novel Cathode Material for Cathodic Dehalogenation of 1,1-Dibromo Cyclopropane Derivatives. Chem - Eur J. 2015;21:13878–13882. doi: 10.1002/chem.201502064. [DOI] [PubMed] [Google Scholar]
- 677.Gütz C, Bänziger M, Bucher C, Galvão TR, Waldvogel SR. Development and Scale-Up of the Electrochemical Dehalogenation for the Synthesis of a Key Intermediate for NS5A Inhibitors. Org Process Res Dev. 2015;19:1428–1433. [Google Scholar]
- 678.Shen Y, Inagi S, Atobe M, Fuchigami T. Electrocatalytic Debromination of Open-Chain and Cyclic Dibromides in Ionic Liquids with Cobalt (II) Salen Complex as Mediator. Res Chem Intermed. 2013;39:89–99. [Google Scholar]
- 679.Parrish JD, Little RD. Electrochemical Formation of Glycals in THF. Tetrahedron Lett. 2001;42:7371–7374. [Google Scholar]
- 680.Mikhaylov D, Gryaznova T, Dudkina Y, Khrizanphorov M, Latypov S, Kataeva O, Vicic DA, Sinyashin OG, Budnikova Y. Electrochemical Nickel-Induced Fluoroalkylation: Synthetic, Structural and Mechanistic Study. Dalton Trans. 2012;41:165–172. doi: 10.1039/c1dt11299f. [DOI] [PubMed] [Google Scholar]
- 681.Utley J, Oguntoye E, Smith CZ, Wyatt PB. Electro-Organic Reactions. Part 52: Diels–Alder Reactions in Aqueous Solution via Electrogenerated Quinodimethanes. Tetrahedron Lett. 2000;41:7249–7254. [Google Scholar]
- 682.Utley JHP, Ramesh S, Salvatella X, Szunerits S, Motevalli M, Nielsen MF. Electro-Organic Reactions. Part 54. Quinodimethane Chemistry. Part 2. Electrogeneration and Reactivity of O-Quinodimethanes. J Chem Soc, Perkin Trans. 2(2001):153–163. [Google Scholar]
- 683.Habibi D, Pakravan N, Nematollahi D. Green and Efficient One-Pot Diels-Alder Electro-Organic Cyclization Reaction of 1,2-Bis(Bromomethyl)Benzene with Naphthoquinone Derivatives. J Electroanal Chem. 2015;759:190–193. [Google Scholar]
- 684.Utley JHP, Gruber J. Electrochemical Synthesis of Poly(p-Xylylenes) (PPXs) and Poly(p-Phenylenevinylenes) (PPVs) and the Study of Xylylene (Quinodimethane) Intermediates; an Underrated Approach. J Mater Chem. 2002;12:1613–1624. [Google Scholar]
- 685.Hosoi K, Inagi S, Kubo T, Fuchigami T. O-Carborane as an Electron-Transfer Mediator in Electrocatalytic Reduction. Chem Commun. 2011;47:8632–8633. doi: 10.1039/c1cc12912k. [DOI] [PubMed] [Google Scholar]
- 686.Magdesieva TV, Graczyk M, Vallat A, Nikitin OM, Demyanov PI, Butin KP, Vorotyntsev MA. Electrochemically Reduced Titanocene Dichloride as a Catalyst of Reductive Dehalogenation of Organic Halides. Electrochim Acta. 2006;52:1265–1280. [Google Scholar]
- 687.He P, Watts P, Marken F, Haswell SJ. Self-Supported and Clean One-Step Cathodic Coupling of Activated Olefins with Benzyl Bromide Derivatives in a Micro Flow Reactor. Angew Chem, Int Ed. 2006;45:4146–4149. doi: 10.1002/anie.200600951. [DOI] [PubMed] [Google Scholar]
- 688.Hilt G, Smolko KI. Electrochemical Regeneration of Low-Valent Indium(I) Species as Catalysts for C–C Bond Formations. Angew Chem, Int Ed. 2001;40:3399–3402. doi: 10.1002/1521-3773(20010917)40:18<3399::aid-anie3399>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 689.Zha Z, Hui A, Zhou Y, Miao Q, Wang Z, Zhang H. A Recyclable Electrochemical Allylation in Water. Org Lett. 2005;7:1903–1905. doi: 10.1021/ol050483h. [DOI] [PubMed] [Google Scholar]
- 690.Huang JM, Dong Y. Zn-Mediated Electrochemical Allylation of Aldehydes in Aqueous Ammonia. Chem Commun. 2009:3943–3943. doi: 10.1039/b905553c. [DOI] [PubMed] [Google Scholar]
- 691.Huang J-M, Ren H-R. Electrochemical Allylation of Carbonyl Compounds in Aqueous Electrolyte Catalyzed by Zinc. Chem Commun. 2010;46:2286–3. doi: 10.1039/b922897g. [DOI] [PubMed] [Google Scholar]
- 692.Sinha AK, Mondal B, Kundu M, Chakraborty B, Roy UK. Recyclable Electrochemical Allylation in Aqueous ZnCl2 Medium: Synthesis and Reactivity of a Wire-Shaped Nano Zinc Architecture. Org Chem Front. 2014;1:1270–1275. [Google Scholar]
- 693.Sun L, Sahloul K, Mellah M. Use of Electrochemistry to Provide Efficient SmI2 Catalytic System for Coupling Reactions. ACS Catal. 2013;3:2568–2573. [Google Scholar]
- 694.Durandetti M, Meignein C, Périchon J. Iron-Catalyzed Electrochemical Allylation of Carbonyl Compounds by Allylic Acetates. J Org Chem. 2003;68:3121–3124. doi: 10.1021/jo026782r. [DOI] [PubMed] [Google Scholar]
- 695.de Souza RFM, Areias MCC, Bieber LW, Navarro M. Inversion of Regioselectivity in the Electrochemical Prenylation of Benzaldehyde on a Graphite Powder Cathode. RSC Adv. 2013;3:6526–6530. [Google Scholar]
- 696.Amemiya F, Fuse K, Fuchigami T, Atobe M. Chemoselective Reaction System Using a Two Inlet Micro-Flow Reactor: Application to Carbonyl Allylation. Chem Commun. 2010;46:2730–2732. doi: 10.1039/b926943f. [DOI] [PubMed] [Google Scholar]
- 697.Amemiya F, Matsumoto H, Fuse K, Kashiwagi T, Kuroda C, Fuchigami T, Atobe M. Product Selectivity Control Induced by Using Liquid–Liquid Parallel Laminar Flow in a Microreactor. Org Biomol Chem. 2011;9:4256–4265. doi: 10.1039/c1ob05174a. [DOI] [PubMed] [Google Scholar]
- 698.Hilt G, Smolko KI, Waloch C. Indium-Catalyzed Allylation of Imines with Electrochemically Assisted Catalyst Regeneration. Tetrahedron Lett. 2002;43:1437–1439. [Google Scholar]
- 699.Huang J-M, Wang X-X, Dong Y. Electrochemical Allylation Reactions of Simple Imines in Aqueous Solution Mediated by Nanoscale Zinc Architectures. Angew Chem, Int Ed. 2011;50:924–927. doi: 10.1002/anie.201004852. [DOI] [PubMed] [Google Scholar]
- 700.Godeau J, Pintaric C, Olivero S, Duñach E. Electrochemical Preparation of Pinacol Allylboronic Esters. Electrochim Acta. 2009;54:5116–5119. [Google Scholar]
- 701.Laza C, Pintaric C, Olivero S, Duñach E. Electrochemical Reduction of Polyhalogenated Aryl Derivatives in the Presence of Pinacolborane: Electrosynthesis of Functionalised Arylboronic Esters. Electrochim Acta. 2005;50:4897–4901. [Google Scholar]
- 702.Laza C, Duñach E. New Electrosynthesis of Arylboronic Esters From Aromatic Halides and Pinacolborane. Adv Synth Catal. 2003;345:580–583. [Google Scholar]
- 703.Laza C, Duñach E, Serein-Spirau F, Moreau JJE, Vellutini L. Novel Synthesis of Arylboronic Acids by Electroreduction of Aromatic Halides in the Presence of Trialkyl Borates. New J Chem. 2002;26:373–375. [Google Scholar]
- 704.Guirado A, Martiz B, Andreu R, Sánchez JIL, Bautista D, Gálvez J. On the Diastereoselective Electrogeneration of (2SR,3SR)-1,5-Diaryl-2-(2,2-Dichlorovinyl)-3-(Trichloromethyl)Pentane-1,5-Diones From 2,2,2-Trichloroethylideneacetophenones. a Combined Experimental and Density Functional Computational Study. Electrochim Acta. 2013;102:409–415. [Google Scholar]
- 705.Schwarz M, Speiser B. Combinatorial Micro-Electrochemistry. Part 5. Electrosynthesis Screening of the Electroreductive Coupling of α,β-Unsaturated Esters and Allyl Bromides in a Room Temperature Ionic Liquid. Electrochim Acta. 2009;54:3735–3744. [Google Scholar]
- 706.Durandetti M, Meignein C, Périchon J. Iron-Mediated Electrochemical Reaction of α-Chloroesters with Carbonyl Compounds. Org Lett. 2003;5:317–320. doi: 10.1021/ol0273046. [DOI] [PubMed] [Google Scholar]
- 707.Areias MCC, Bieber LW, Navarro M, Diniz FB. Metal-Free Electrochemical Reformatsky Reaction in Water: Further Evidence for a Radical Mechanism. J Electroanal Chem. 2003;558:125–130. [Google Scholar]
- 708.de Souza CA, de Souza RFM, Navarro M, Malvestiti I, da Silva APF, Bieber LW, Areias MCC. Electrochemical Reformatsky Reaction of α-Haloketones and Benzaldehyde on a Graphite Powder Cathode Free of Organic Solvents. Electrochim Acta. 2013;89:631–634. [Google Scholar]
- 709.Areias MCC, Navarro M, Bieber LW, Diniz FB, Léonel E, Cachet-Vivier C, Nédélec J-Y. A Novel Electrosynthesis Cell with a Compressed Graphite Powder Cathode and Minimal Organic Solvent Content: Application to the Reformatsky Reaction. Electrochim Acta. 2008;53:6477–6483. [Google Scholar]
- 710.Batanero B, Elinson MN, Barba F. Electrochemical Dimerization of Phenacyl Bromides N-Acylhydrazones—a New Way to 1-N-Acylamino-2,5-Diaryl-Pyrroles. Tetrahedron. 2004;60:10787–10792. [Google Scholar]
- 711.Pasciak EM, Peters DG. Reduction of Substituted Phenyl 2-Chloroacetates at Silver Cathodes: Electrosynthesis of Coumarins. J Electrochem Soc. 2014;161:G98–G102. [Google Scholar]
- 712.Quintanilla G, Pérez I, Záková L, Uth C, Barba F. Electrosynthesis of Halogenated δ-Lactones. Eur J Org Chem. 2011;2011:4681–4686. [Google Scholar]
- 713.Guirado A, Andreu R, Gálvez J, Jones PG. Electrochemical Generation of 4-Amino-2-Aryl-2-Oxazolines. Tetrahedron. 2002;58:9853–9858. [Google Scholar]
- 714.Batanero B, Barba F. Electrosynthesis of 3-Chloro-1,4-Disubstituted-2(1 H)-Quinolinones and 3,3-Dichloro-4-Hydroxy-1,4-Disubstituted-3,4-Dihydro-2(1 H)-Quinolinones, as Well as a New Convenient Process to Dioxindoles. J Org Chem. 2003;68:3706–3709. doi: 10.1021/jo0267403. [DOI] [PubMed] [Google Scholar]
- 715.Dolly, Batanero B, Barba F. Cathodic Reduction of Hydroxycarbonyl Compound Trichloroacetyl Esters. Tetrahedron. 2003;59:9161–9165. [Google Scholar]
- 716.Nishiguchi I. Selective Synthesis of α-Chloroepoxides by Electroreductive Cross-Coupling of Methyl Trichloroacetate with Aliphatic Aldehydes Using Reactive Metal Anodes. J Electroanal Chem. 2001;507:185–190. [Google Scholar]
- 717.Adouama C, Keyrouz R, Pilet G, Monnereau C, Gueyrard D, Noël T, Médebielle M. Access to Cyclic Gem-Difluoroacyl Scaffolds via Electrochemical and Visible Light Photocatalytic Radical Tandem Cyclization of Heteroaryl Chlorodifluoromethyl Ketones. Chem Commun. 2017;53:5653–5656. doi: 10.1039/c7cc02979a. [DOI] [PubMed] [Google Scholar]
- 718.Shimakoshi H, Luo Z, Inaba T, Hisaeda Y. Electrolysis of Trichloromethylated Organic Compounds Under Aerobic Conditions Catalyzed by the B12 Model Complex for Ester and Amide Formation. Dalton Trans. 2016;45:10173–10180. doi: 10.1039/c6dt00556j. [DOI] [PubMed] [Google Scholar]
- 719.Giedyk M, Shimakoshi H, Goliszewska K, Gryko D, Hisaeda Y. Electrochemistry and Catalytic Properties of Amphiphilic Vitamin B12 Derivatives in Nonaqueous Media. Dalton Trans. 2016;45:8340–8346. doi: 10.1039/c6dt00355a. [DOI] [PubMed] [Google Scholar]
- 720.Sengmany S, Léonel E, Paugam JP, Nédélec JY. Cyclopropane Formation by Copper-Catalysed Indirect Electroreductive Coupling of Activated Olefins and Activated α,α,α-Trichloro or Gem-Dichloro Compounds. Synthesis. 2002;2002:533–537. [Google Scholar]
- 721.Oudeyer S, Léonel E, Paugam JP, Sulpice-Gaillet C, Nédélec J-Y. Formation of Polysubstituted Chlorocyclopropanes From Electrophilic Olefins and Activated Trichloromethyl Compounds. Tetrahedron. 2006;62:1583–1589. [Google Scholar]
- 722.Oudeyer S, Léonel E, Paugam JP, Nédélec JY. Copper-Catalyzed Electrosynthesis of 1-Acyl-2, 2-Diphenylcyclopropanes and Their Behaviour in Acidic Medium. Tetrahedron. 2003;59:1073–1081. [Google Scholar]
- 723.Ribeiro RRT, de Mattos IL, Sengmany S, Barhdadi R, Léonel E, Cachet-Vivier C, Navarro M. Iron Role in the Electrochemical Cyclopropanation Reaction of Activated Olefins and Halogenated Compounds. Electrochim Acta. 2011;56:7352–7360. [Google Scholar]
- 724.Oudeyer S, Léonel E, Paugam JP, Nédélec JY. Epoxide Formation by Indirect Electroreductive Coupling Between Aldehydes or Ketones and Activated Gem-Dichloro Compounds. Synthesis. 2004:389–400. [Google Scholar]
- 725.Njue CK, Nuthakki B, Vaze A, Bobbitt JM. Vitamin B 12-Mediated Electrochemical Cyclopropanation of Styrene. Electrochem Commun. 2001;3:733–736. [Google Scholar]
- 726.Duquenne C, Goumain S, Jubault P, Feasson C, Quirion J-C. Electrosynthesis of α-Arylated β-Substituted Cyclopropylphosphonates. Synthesis of a Phosphonic Analogue of Minalcipran. Org Lett. 2000;2:453–455. doi: 10.1021/ol991301k. [DOI] [PubMed] [Google Scholar]
- 727.Condon S, Zou C, Nédélec J-Y. Electrochemical Alkyl Transfer Reaction From Trialkylboranes to Polyhalo Compounds. J Organomet Chem. 2006;691:3245–3250. [Google Scholar]
- 728.Sengmany S, Léonel E, Paugam JP, Nédélec JY. Cyclopropane Formation by Nickel-Catalysed Electroreductive Coupling of Activated Olefins and Unactivated Gem-Dibromo Compounds. Tetrahedron. 2002;58:271–277. [Google Scholar]
- 729.Kweon D, Jang Y, Kim H. Organic Electrochemical Synthesis Utilizing Mg Electrodes (1)-Facile Reductive Coupling Reactions of Aromatic Halides. Bull Korean Chem Soc. 2003;34:1049–1050. [Google Scholar]
- 730.Mellah M, Gmouh S, Vaultier M, Jouikov V. Electrocatalytic Dimerisation of PhBr and PhCH2Br in [BMIM]+NTf2–Ionic Liquid. Electrochem Commun. 2003;5:591–593. [Google Scholar]
- 731.Xu Y, Ding X, Ma H, Chu Y, Ma C. Selective Hydrodechlorination of 3,5,6-Trichloropicolinic Acid at an Activated Silver Cathode: Synthesis of 3,5-Dichloropicolinic Acid. Electrochim Acta. 2015;151:284–288. [Google Scholar]
- 732.Feroci M, Orsini M, Palombi L, Sotgiu G, Inesi A. Electrochemically Induced Hydrogenolysis of 1,1-Dibromoalkenes to Vinyl Bromides. Electrochim Acta. 2004;49:635–640. [Google Scholar]
- 733.Nascimento W, Oliveira J, Freitas J, Navarro M, Menezes P. Electrochemical Synthesis of Potassium Aryltrifluoroborates. Synthesis. 2014;46:2579–2584. [Google Scholar]
- 734.Mitsudo K, Okada T, Shimohara S, Mandai H, Suga S. Electro-Reductive Halogen-Deuterium Exchange and Methylation of Aryl Halides in Acetonitrile. Electrochemistry. 2013;81:362–364. [Google Scholar]
- 735.Mitsudo K, Nakagawa Y, Mizukawa J-I, Tanaka H, Akaba R, Okada T, Suga S. Electro-Reductive Cyclization of Aryl Halides Promoted by Fluorene Derivatives. Electrochim Acta. 2012;82:444–449. [Google Scholar]
- 736.Kurono N, Honda E, Komatsu F, Orito K, Tokuda M. Regioselective Synthesis of Substituted 1-Indanols, 2,3-Dihydrobenzofurans and 2,3-Dihydroindoles by Electrochemical Radical Cyclization Using an Arene Mediator. Tetrahedron. 2004;60:1791–1801. [Google Scholar]
- 737.Kurono N, Honda E, Komatsu F, Orito K, Tokuda M. Regioselective Radical Cyclization by Electrochemical Reduction Using an Arene Mediator. Environmentally Benign Method. Chem Lett. 2003;32:720–721. [Google Scholar]
- 738.Olivero S, Perriot R, Duñach E, Baru A, Bell E, Mohan R. An Environmentally Friendly Synthesis of Functionalized Indanes Using Electrochemical Cyclization of Ortho-Halo-Substituted Homoallyl Ethers and Esters. Synlett. 2006;2006:2021–2026. [Google Scholar]
- 739.de Mendonça Cavalcanti JC, Goulart M, Léonel E. Synthesis of 6-, 7-, and 8-Membered Lactones via the Nickel-Catalysed Electrochemical Arylation of Electron-Deficient Olefins. Tetrahedron Lett. 2002;43:6343–6345. [Google Scholar]
- 740.Duñach E, Esteves AP, Medeiros MJ, dos Santos Neves CS, Olivero S. Radical-Type Reactions in Protic and Aprotic Media: Comparisons in Nickel-Catalysed Electrochemical Reductive Cyclisations. C R Chim. 2009;12:889–894. [Google Scholar]
- 741.Pelletier JRM, Olivero S, Duñach E. Nickel–Catalyzed Electrochemical Synthesis of Dihydro-Benzo[B]Thiophene Derivatives. Synth Commun. 2004;34:3343–3348. [Google Scholar]
- 742.Esteves AP, Goken DM, Klein LJ, Lemos MA, Medeiros MJ, Peters DG. Electroreductive Intramolecular Cyclization of a Bromo Propargyloxy Ester Catalyzed by Nickel(I) Tetramethylcyclam Electrogenerated at Carbon Cathodes in Dimethylformamide. J Org Chem. 2003;68:1024–1029. doi: 10.1021/jo026102k. [DOI] [PubMed] [Google Scholar]
- 743.Toyota M, Ilangovan A, Kashiwagi Y, Ihara M. One-Pot Assembly of Tricyclo[6.2.1.01,6]Undecan-4-One and Related Polycyclic Compounds by Tandem Electroreductive Cyclization. Org Lett. 2004;6:3629–3632. doi: 10.1021/ol0484660. [DOI] [PubMed] [Google Scholar]
- 744.Ischay MA, Mubarak MS, Peters DG. Catalytic Reduction and Intramolecular Cyclization of Haloalkynes in the Presence of Nickel(I) Salen Electrogenerated at Carbon Cathodes in Dimethylformamide. J Org Chem. 2006;71:623–628. doi: 10.1021/jo052007a. [DOI] [PubMed] [Google Scholar]
- 745.Takasu K, Ohsato H, Kuroyanagi J-I, Ihara M. Novel Intramolecular [4 + 1] and [4 + 2] Annulation Reactions Employing Cascade Radical Cyclizations. J Org Chem. 2002;67:6001–6007. doi: 10.1021/jo0258397. [DOI] [PubMed] [Google Scholar]
- 746.Sun G, Ren S, Zhu X, Huang M, Wan Y. Direct Arylation of Pyrroles via Indirect Electroreductive C–H Functionalization Using Perylene Bisimide as an Electron-Transfer Mediator. Org Lett. 2016;18:544–547. doi: 10.1021/acs.orglett.5b03581. [DOI] [PubMed] [Google Scholar]
- 747.LeStrat F, Murphy JA, Hughes M. Direct Electroreductive Preparation of Indolines and Indoles From Diazonium Salts. Org Lett. 2002;4:2735–2738. doi: 10.1021/ol0262643. [DOI] [PubMed] [Google Scholar]
- 748.Barba F, Ranz F, Batanero B. Electrochemical Transformation of Diazonium Salts Into Diaryl Disulfides. Tetrahedron Lett. 2009;50:6798–6799. [Google Scholar]
- 749.Nédélec J-Y, Perichon J, Troupel M. Organic Electroreductive Coupling Reactions Using Transition Metal Complexes as Catalysts. Top Curr Chem. 1997;185:141–173. [Google Scholar]
- 750.Duñach E, Franco D, Olivero S. Carbon–Carbon Bond Formation with Electrogenerated Nickel and Palladium Complexes. Eur J Org Chem. 2003;2003:1605–1622. [Google Scholar]
- 751.Condon S, Dupré D, Falgayrac G, Nédélec J-Y. Nickel-Catalyzed Electrochemical Arylation of Activated Olefins. Eur J Org Chem. 2002;2002:105–111. [Google Scholar]
- 752.Barhdadi R, Courtinard C, Nédélec JY, Troupel M. Room-Temperature Ionic Liquids as New Solvents for Organic Electrosynthesis. the First Examples of Direct or Nickel-Catalysed Electroreductive Coupling Involving Organic Halides. Chem Commun. 2003:1434–1435. doi: 10.1039/b302944a. [DOI] [PubMed] [Google Scholar]
- 753.Condon S, Dupré D, Lachaise I, Nédélec J-Y. Nickel Catalyzed Electrochemical Heteroarylation of Activated Olefins. Synthesis. 2002;2002:1752–1758. [Google Scholar]
- 754.Condon S, Dupré D, Nédélec JY. Nickel-Catalyzed Arylation of Acrolein Diethyl Acetal: a Substitute to the 1,4-Addition of Arylhalides to Acrolein. Org Lett. 2003;5:4701–4703. doi: 10.1021/ol035877s. [DOI] [PubMed] [Google Scholar]
- 755.Métay E, Léonel E, Sulpice-Gaillet C, Nédélec JY. Synthesis of Precursors for Medium-Ring Aromatic Lactones. Synthesis. 2005:1682–1688. [Google Scholar]
- 756.Dolhem E, Barhdadi R, Folest JC, Nédélec JY. Nickel Catalysed Electrosynthesis of Ketones From Organic Halides and Iron Pentacarbonyl. Part 2: Unsymmetrical Ketones. Tetrahedron. 2001;57:525–529. [Google Scholar]
- 757.Durandetti M, Nédélec J-Y, Périchon J. An Electrochemical Coupling of Organic Halide with Aldehydes, Catalytic in Chromium and Nickel Salts. the Nozaki–Hiyama–Kishi Reaction. Org Lett. 2001;3:2073–2076. doi: 10.1021/ol016033g. [DOI] [PubMed] [Google Scholar]
- 758.Cannes C, Condon S, Durandetti M, Nédélec JY. Nickel-Catalyzed Electrochemical Couplings of Vinyl Halides: Synthetic and Stereochemical Aspects. J Org Chem. 2000;65:4575–4583. doi: 10.1021/jo000182f. [DOI] [PubMed] [Google Scholar]
- 759.Oliveira JL, Le Gall E, Sengmany S, Léonel E, Dubot P, Cénédèse P, Navarro M. A Graphite Powder Cavity Cell as an Efficient Tool of Sustainable Chemistry: Electrocatalytic Homocoupling of 2-Halopyridines. Electrochim Acta. 2015;173:465–475. [Google Scholar]
- 760.Oliveira JL, Silva MJ, Florêncio T, Urgin K, Sengmany S, Léonel E, Nédélec J-Y, Navarro M. Electrochemical Coupling of Mono and Dihalopyridines Catalyzed by Nickel Complex in Undivided Cell. Tetrahedron. 2012;68:2383–2390. [Google Scholar]
- 761.Sengmany S, Vasseur S, Lajnef A, Le Gall E, Léonel E. Beneficial Effects of Electrochemistry in Cross-Coupling Reactions: Electroreductive Synthesis of 4-Aryl- or 4-Heteroaryl-6-Pyrrolylpyrimidines. Eur J Org Chem. 2016;2016:4865–4871. [Google Scholar]
- 762.Gosmini C, Nédélec JY, Périchon J. Electrochemical Cross-Coupling Between Functionalized Aryl Halides and 2-Chloropyrimidine or 2-Chloropyrazine Catalyzed by Nickel 2, 2′-Bipyridine Complex. Tetrahedron Lett. 2000;41:201–203. [Google Scholar]
- 763.Sengmany S, Léonel E, Polissaint F, Nédélec J-Y, Pipelier M, Thobie-Gautier C, Dubreuil D. Preparation of Functionalized Aryl- and Heteroarylpyridazines by Nickel-Catalyzed Electrochemical Cross-Coupling Reactions. J Org Chem. 2007;72:5631–5636. doi: 10.1021/jo070429+. [DOI] [PubMed] [Google Scholar]
- 764.Sengmany S, Vitu-Thiebaud A, Le Gall E, Condon S, Léonel E, Thobie-Gautier C, Pipelier M, Lebreton J, Dubreuil D. An Electrochemical Nickel-Catalyzed Arylation of 3-Amino-6-Chloropyridazines. J Org Chem. 2013;78:370–379. doi: 10.1021/jo3022428. [DOI] [PubMed] [Google Scholar]
- 765.Perkins RJ, Pedro DJ, Hansen EC. Electrochemical Nickel Catalysis for sp2-sp3 Cross-Electrophile Coupling Reactions of Unactivated Alkyl Halides. Org Lett. 2017;19:3755–3758. doi: 10.1021/acs.orglett.7b01598. [DOI] [PubMed] [Google Scholar]
- 766.Moreau C, Serein-Spirau F, Bordeau M, Biran C. Electrochemical Synthesis of Functional Aryl- and Heteroarylchlorosilanes. Application to the Preparation of Donor–Acceptor or Donor–Donor Organosilicon Molecules. Organometallics. 2001;20:1910–1917. [Google Scholar]
- 767.Gosmini C, Rollin Y, Nédélec J-Y, Périchon J. New Efficient Preparation of Arylzinc Compounds From Aryl Halides Using Cobalt Catalysis and Sacrificial Anode Process. J Org Chem. 2000;65:6024–6026. doi: 10.1021/jo000459b. [DOI] [PubMed] [Google Scholar]
- 768.Le Gall E, Gosmini C, Nédélec JY, Périchon J. Synthesis of Functionalized 4-Phenyl-Pyridines via Electrochemically Prepared Organozinc Reagents. Tetrahedron. 2001;57:1923–1927. [Google Scholar]
- 769.Fillon H, Gosmini C, Périchon J. A Convenient Method for the Preparation of Aromatic Ketones From Acyl Chlorides and Arylzinc Bromides Using a Cobalt Catalysis. Tetrahedron. 2003;59:8199–8202. [Google Scholar]
- 770.Gomes P, Gosmini C, Périchon J. Cobalt-Catalyzed Direct Electrochemical Cross-Coupling Between Aryl or Heteroaryl Halides and Allylic Acetates or Carbonates. J Org Chem. 2003;68:1142–1145. doi: 10.1021/jo026421b. [DOI] [PubMed] [Google Scholar]
- 771.Gomes P, Gosmini C, Périchon J. Cobalt-Catalyzed Electrochemical Vinylation of Aryl Halides Using Vinylic Acetates. Tetrahedron. 2003;59:2999–3002. [Google Scholar]
- 772.Gomes P, Gosmini C, Nédélec JY, Périchon J. Cobalt Bromide as Catalyst in Electrochemical Addition of Aryl Halides Onto Activated Olefins. Tetrahedron Lett. 2000;41:3385–3388. [Google Scholar]
- 773.Polleux L, Labbé E, Buriez O, Perichon J. CoI- And Co0-Bipyridine Complexes Obtained by Reduction of CoBr2Bpy: Electrochemical Behaviour and Investigation of Their Reactions with Aromatic Halides and Vinylic Acetates. Chem - Eur J. 2005;11:4678–4686. doi: 10.1002/chem.200400971. [DOI] [PubMed] [Google Scholar]
- 774.Gomes P, Fillon H, Gosmini C, Labbé E, Périchon J. Synthesis of Unsymmetrical Biaryls by Electroreductive Cobalt-Catalyzed Cross-Coupling of Aryl Halides. Tetrahedron. 2002;58:8417–8424. [Google Scholar]
- 775.Le Gall E, Gosmini C, Nédélec JY, Périchon J. Cobalt-Catalyzed Electrochemical Cross-Coupling of Functionalized Phenyl Halides with 4-Chloroquinoline Derivatives. Tetrahedron Lett. 2001;42:267–269. [Google Scholar]
- 776.Torii S, Tanaka H, Morisaki K. Pd (0)-Catalyzed Electro-Reductive Coupling of Aryl Halides. Tetrahedron Lett. 1985;26:1655–1658. [Google Scholar]
- 777.Amatore C, Carré E, Jutand A, Tanaka H, Ren Q, Torii S. Oxidative Addition of Aryl Halides to Transient Anionic σ-Aryl–Palladium(0) Intermediates—Application to Palladium-Catalyzed Reductive Coupling of Aryl Halides. Chem - Eur J. 1996;2:957–966. [Google Scholar]
- 778.Pachón LD, Elsevier CJ, Rothenberg G. Electroreductive Palladium-Catalysed Ullmann Reactions in Ionic Liquids: Scope and Mechanism. Adv Synth Catal. 2006;348:1705–1710. [Google Scholar]
- 779.Kuroboshi M, Shiba T, Tanaka H. Viologen as Catalytic Organic Reductant: Electro-Reductive Dimerization of Aryl Bromides in a Pd/Viologen Double Mediatory System. Tetrahedron Lett. 2013;54:3666–3668. [Google Scholar]
- 780.Kuroboshi M, Kobayashi R, Nakagawa T, Tanaka H. Electroreductive Generation of Recyclable Organic Reductant From N,N′-Dioctyl-4,4′-Bipyridinium and Pd-Catalyzed Reductive Coupling of Aryl Halides. Synlett. 2009;2009:85–88. [Google Scholar]
- 781.Tanaka H, Kuroboshi M, Kataoka R, Suzuki R. Electro-Reductive Homo-Coupling Reaction of Aryl Bromides in PdCl2(PPh3)2/Pyridinium Salt Double Mediatory Systems. Electrochemistry. 2013;81:356–358. [Google Scholar]
- 782.Kuroboshi M, Kondo T, Tanaka H. Diquat Derivatives, a Precursor of Organic Reductant. Heterocycles. 2015;90:723–729. [Google Scholar]
- 783.Lai Y-L, Huang J-M. Palladium-Catalyzed Electrochemical Allylic Alkylation Between Alkyl and Allylic Halides in Aqueous Solution. Org Lett. 2017;19:2022–2025. doi: 10.1021/acs.orglett.7b00473. [DOI] [PubMed] [Google Scholar]
- 784.Moeller KD. Electrochemically Generated Organometallic Reagents and Site-Selective Synthesis on a Microelectrode Array. Organometallics. 2014;33:4607–4616. [Google Scholar]
- 785.Tian J, Moeller KD. Electrochemically Assisted Heck Reactions. Org Lett. 2005;7:5381–5383. doi: 10.1021/ol0519487. [DOI] [PubMed] [Google Scholar]
- 786.Matthessen R, Fransaer J, Binnemans K, De Vos DE. Electrocarboxylation: Towards Sustainable and Efficient Synthesis of Valuable Carboxylic Acids. Beilstein J Org Chem. 2014;10:2484–2500. doi: 10.3762/bjoc.10.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 787.Senboku H, Komatsu H, Fujimura Y, Tokuda M. Efficient Electrochemical Dicarboxylation of Phenyl-Substituted Alkenes: Synthesis of 1-Phenylalkane-1, 2-Dicarboxylic Acids. Synlett. 2001;2001:418–420. [Google Scholar]
- 788.Ballivet-Tkatchenko DB, Folest JC, Tanji J. Electrocatalytic Reduction of CO2 For the Selective Carboxylation of Olefins. Appl Organomet Chem. 2000;14:847–849. [Google Scholar]
- 789.Yuan G-Q, Jiang H-F, Lin C, Liao S-J. Efficient Electrochemical Synthesis of 2-Arylsuccinic Acids From CO2 And Aryl-Substituted Alkenes with Nickel as the Cathode. Electrochim Acta. 2008;53:2170–2176. [Google Scholar]
- 790.Wang H, Lin M-Y, Fang HJ, Chen TT, Lu J-X. Electrochemical Dicarboxylation of Styrene: Synthesis of 2-Phenylsuccinic Acid. Chin J Chem. 2007;25:913–916. [Google Scholar]
- 791.Li C-H, Yuan G-Q, Ji X-C, Wang X-J, Ye J-S, Jiang H-F. Highly Regioselective Electrochemical Synthesis of Dioic Acids From Dienes and Carbon Dioxide. Electrochim Acta. 2011;56:1529–1534. [Google Scholar]
- 792.Matthessen R, Fransaer J, Binnemans K, Vos DED. Electrochemical Dicarboxylation of Conjugated Fatty Acids as an Efficient Valorization of Carbon Dioxide. RSC Adv. 2013;3:4634–4639. [Google Scholar]
- 793.Zhang K, Xiao Y, Lan Y, Zhu M, Wang H, Lu J. Electrochemical Reduction of Aliphatic Conjugated Dienes in the Presence of Carbon Dioxide. Electrochem Commun. 2010;12:1698–1702. [Google Scholar]
- 794.Bringmann J, Dinjus E. Electrochemical Synthesis of Carboxylic Acids From Alkenes Using Various Nickel-Organic Mediators: CO2 As C1-Synthon. Appl Organomet Chem. 2001;15:135–140. [Google Scholar]
- 795.Wang H, Du YF, LIN MY, Zhang K. Electrochemical Reduction and Carboxylation of Ethyl Cinnamate in MeCN. Chin J Chem. 2008;26:1745–1748. [Google Scholar]
- 796.Wang H, Zhang G, Liu Y, Luo Y, Lu J. Electrocarboxylation of Activated Olefins in Ionic Liquid BMIMBF4. Electrochem Commun. 2007;9:2235–2239. [Google Scholar]
- 797.Wang H, Zhang K, Liu Y-Z, Lin M-Y, Lu J-X. Electrochemical Carboxylation of Cinnamate Esters in MeCN. Tetrahedron. 2008;64:314–318. [Google Scholar]
- 798.Orsini M, Feroci M, Sotgiu G, Inesi A. Stereoselective Electrochemical Carboxylation: 2-Phenylsuccinates From Chiral Cinnamic Acid Derivatives. Org Biomol Chem. 2005;3:1202–1207. doi: 10.1039/b500570a. [DOI] [PubMed] [Google Scholar]
- 799.Chowdhury MA, Senboku H, Tokuda M. Electrochemical Carboxylation of Bicyclo[N.1.0]Alkylidene Derivatives. Tetrahedron. 2004;60:475–481. [Google Scholar]
- 800.Köster F, Dinjus E, Duñach E. Electrochemical Selective Incorporation of CO2 Into Terminal Alkynes and Diynes. Eur J Org Chem. 2001;2001:2507–2511. [Google Scholar]
- 801.Derien S, Duñach E, Périchon J. From Stoichiometry to Catalysis: Electroreductive Coupling of Alkynes and Carbon Dioxide with Nickel-Bipyridine Complexes. Magnesium Ions as the Key for Catalysis. J Am Chem Soc. 1991;113:8447–8454. [Google Scholar]
- 802.Yuan G-Q, Jiang H-F, Lin C. Efficient Electrochemical Dicarboxylations of Arylacetylenes with Carbon Dioxide Using Nickel as the Cathode. Tetrahedron. 2008;64:5866–5872. [Google Scholar]
- 803.Li C, Yuan G, Jiang H. Electrocarboxylation of Alkynes with Carbon Dioxide in the Presence of Metal Salt Catalysts. Chin J Chem. 2010;28:1685–1689. [Google Scholar]
- 804.Senboku H, Tokuda M. Electrochemical Organic Synthesis in Supercritical Carbon Dioxide. Fine Chemicals. 2002;31:50–60. [Google Scholar]
- 805.Yuan G, Li L, Jiang H, Qi C, Xie F. Electrocarboxylation of Carbon Dioxide with Polycyclic Aromatic Hydrocarbons Using Ni as the Cathode. Chin J Chem. 2010;28:1983–1988. [Google Scholar]
- 806.Senboku H, Yamauchi Y, Kobayashi N, Fukui A, Hara S. Electrochemical Carboxylation of Flavones: Facile Synthesis of Flavanone-2-Carboxylic Acids. Electrochemistry. 2011;79:862–864. [Google Scholar]
- 807.Senboku H, Yamauchi Y, Kobayashi N, Fukui A, Hara S. Some Mechanistic Studies on Electrochemical Carboxylation of Flavones to Yield Flavanone-2-Carboxylic Acids. Electrochim Acta. 2012;82:450–456. [Google Scholar]
- 808.Yuan G, Li Z, Jiang H. Electrosyntheses of α-Hydroxycarboxylic Acids From Carbon Dioxide and Aromatic Ketones Using Nickel as the Cathode. Chin J Chem. 2009;27:1464–1470. [Google Scholar]
- 809.Wang H, Xu X-M, Lan Y-C, Wang H-M, Lu J-X. Electrocarboxylation of Haloacetophenones at Silver Electrode. Tetrahedron. 2014;70:1140–1143. [Google Scholar]
- 810.Feng Q, Huang K, Liu S, Yu J, Liu F. Electrocatalytic Carboxylation of Aromatic Ketones with Carbon Dioxide in Ionic Liquid 1-Butyl-3-Methylimidazoliumtetrafluoborate to α-Hydroxy-Carboxylic Acid Methyl Ester. Electrochim Acta. 2011;56:5137–5141. [Google Scholar]
- 811.Chen B-L, Tu Z-Y, Zhu H-W, Sun W-W, Wang H, Lu J-X. CO2 As a C1-Organic Building Block: Enantioselective Electrocarboxylation of Aromatic Ketones with CO2 Catalyzed by Cinchona Alkaloids Under Mild Conditions. Electrochim Acta. 2014;116:475–483. [Google Scholar]
- 812.Senboku H, Yamauchi Y, Fukuhara T, Hara S. Electrochemical Carboxylation of Aliphatic Ketones: Synthesis of β-Keto Carboxylic Acids. Electrochemistry. 2006;74:612–614. [Google Scholar]
- 813.Isse AA, Gennaro A. Electrocatalytic Carboxylation of Benzyl Chlorides at Silver Cathodes in Acetonitrile. Chem Commun. 2002:2798–2799. doi: 10.1039/b206746c. [DOI] [PubMed] [Google Scholar]
- 814.Scialdone O, Galia A, Errante G, Isse AA, Gennaro A, Filardo G. Electrocarboxylation of Benzyl Chlorides at Silver Cathode at the Preparative Scale Level. Electrochim Acta. 2008;53:2514–2528. [Google Scholar]
- 815.Niu D-F, Xiao L-P, Zhang A-J, Zhang G-R, Tan Q-Y, Lu J-X. Electrocatalytic Carboxylation of Aliphatic Halides at Silver Cathode in Acetonitrile. Tetrahedron. 2008;64:10517–10520. [Google Scholar]
- 816.Niu D, Zhang J, Zhang K, Xue T. Electrocatalytic Carboxylation of Benzyl Chloride at Silver Cathode in Ionic Liquid BMImBF4. Chin J Chem. 2009;27:1041–1044. [Google Scholar]
- 817.Raju RR, Lateef SK, Mohan S, Reddy S. Synthesis of Aryl-2-Propionic Acids by Electrocarboxylation of Methyl Aryl Bromides. Arkivoc. 2006:76–80. [Google Scholar]
- 818.Yamauchi Y, Hara S, Senboku H. Synthesis of 2-Aryl-3,3,3-Trifluoropropanoic Acids Using Electrochemical Carboxylation of (1-Bromo-2,2,2-Trifluoroethyl)Arenes and Its Application to the Synthesis of β,β,β-Trifluorinated Non-Steroidal Anti-Inflammatory Drugs. Tetrahedron. 2010;66:473–479. [Google Scholar]
- 819.Yang H-P, Yue Y-N, Sun Q-L, Feng Q, Wang H, Lu J-X. Entrapment of a Chiral Cobalt Complex Within Silver: a Novel Heterogeneous Catalyst for Asymmetric Carboxylation of Benzyl Bromides with CO2. Chem Commun. 2015;51:12216–12219. doi: 10.1039/c5cc04554a. [DOI] [PubMed] [Google Scholar]
- 820.Tateno H, Nakabayashi K, Kashiwagi T, Senboku H, Atobe M. Electrochemical Fixation of CO2 To Organohalides in Room-Temperature Ionic Liquids Under Supercritical CO2. Electrochim Acta. 2015;161:212–218. [Google Scholar]
- 821.Anandhakumar S, Sripriya R, Chandrasekaran M, Govindu S, Noel M. Electrocarboxylation and Related Radical Coupling Processes of Aryl and Benzyl Halides in Microemulsion. J Appl Electrochem. 2009;39:463–465. [Google Scholar]
- 822.Senboku H, Yoneda K, Hara S. Electrochemical Direct Carboxylation of Benzyl Alcohols Having an Electron-Withdrawing Group on the Phenyl Ring: One-Step Formation of Phenylacetic Acids From Benzyl Alcohols Under Mild Conditions. Tetrahedron Lett. 2015;56:6772–6776. [Google Scholar]
- 823.Ohkoshi M, Michinishi J-Y, Hara S, Senboku H. Electrochemical Carboxylation of Benzylic Carbonates: Alternative Method for Efficient Synthesis of Arylacetic Acids. Tetrahedron. 2010;66:7732–7737. [Google Scholar]
- 824.Yamauchi Y, Fukuhara T, Hara S, Senboku H. Electrochemical Carboxylation of α,α-Difluorotoluene Derivatives and Its Application to the Synthesis of α-Fluorinated Nonsteroidal Anti-Inflammatory Drugs. Synlett. 2008;2008:438–442. [Google Scholar]
- 825.Feroci M, Inesi A, Orsini M, Palombi L. Electrochemical Carboxylation of N-(2-Bromopropionyl)-4 R-Phenyloxazolidin-2-One: an Efficient Route to Unsymmetrical Methylmalonic Ester Derivatives. Org Lett. 2002;4:2617–2620. doi: 10.1021/ol025939z. [DOI] [PubMed] [Google Scholar]
- 826.Feroci M, Orsini M, Palombi L, Sotgiu G, Colapietro M, Inesi A. Diastereoselective Electrochemical Carboxylation of Chiral α -Bromocarboxylic Acid Derivatives: an Easy Access to Unsymmetrical Alkylmalonic Ester Derivatives. J Org Chem. 2004;69:487–494. doi: 10.1021/jo0343836. [DOI] [PubMed] [Google Scholar]
- 827.Stepanov AA, Volodin YY, Grachev MK. Reactions of Direct Electrocarboxylation of 1-Phenylethylbromide and 1-(4-Isobutylphenyl) Ethyl Chloride in the Presence of β-Cyclodextrin. Russ J Electrochem. 2002;38:1346–1352. [Google Scholar]
- 828.Chen B-L, Zhu H-W, Xiao Y, Sun Q-L, Wang H, Lu J-X. Asymmetric Electrocarboxylation of 1-Phenylethyl Chloride Catalyzed by Electrogenerated Chiral [CoI(salen)]–Complex. Electrochem Commun. 2014;42:55–59. [Google Scholar]
- 829.Senboku H, Kanaya H, Fujimura Y, Tokuda M. Stereochemical Study on Electrochemical Carboxylation of Vinyl Triflates. J Electroanal Chem. 2001;507:82–88. [Google Scholar]
- 830.Senboku H, Fujimura Y, Kamekawa H, Tokuda M. Divergent Electrochemical Carboxylation of Vinyl Triflates: New Electrochemical Synthesis of Phenyl-Substituted α, β-Unsaturated Carboxylic Acids and Aliphatic β-Keto Carboxylic Acids. Electrochim Acta. 2000;45:2995–3003. [Google Scholar]
- 831.Kuang C, Yang Q, Senboku H, Tokuda M. Stereospecific Electrochemical Carboxylation of β-Bromostyrene by Use of Nickel(II) Catalyst. Chem Lett. 2005;34:528–529. [Google Scholar]
- 832.Senboku H, Yoneda K, Hara S. Regioselective Electrochemical Carboxylation of Polyfluoroarenes. Electrochemistry. 2013;81:380–382. [Google Scholar]
- 833.Wu W-B, Li M-L, Huang J-M. Electrochemical Hydrodefluorination of Fluoroaromatic Compounds. Tetrahedron Lett. 2015;56(12):1520–1523. [Google Scholar]
- 834.Senboku H, Michinishi J-Y, Hara S. Facile Synthesis of 2,3-Dihydrobenzofuran-3-ylacetic Acids by Novel Electrochemical Sequen-tial Aryl Radical Cyclization-Carboxylation of 2-Allyloxybromobenzenes Using Methyl 4-Tert-Butylbenzoate as an Electron-Transfer Mediator. Synlett. 2011;2011:1567–1572. [Google Scholar]
- 835.Katayama A, Senboku H. Sequential Vinyl Radical Cyclization/Fixation of Carbon Dioxide Through Electrochemical Reduction of Vinyl Bromide in the Presence of an Electron-Transfer Mediator. ChemElectroChem. 2016;3:2052–2057. [Google Scholar]
- 836.Katayama A, Senboku H, Hara S. Aryl Radical Cyclization with Alkyne Followed by Tandem Carboxylation in Methyl 4-Tert-Butylbenzoate-Mediated Electrochemical Reduction of 2-(2-Propynyloxy)Bromobenzenes in the Presence of Carbon Dioxide. Tetrahedron. 2016;72:4626–4636. [Google Scholar]
- 837.Horner L, Jordan M. Studien Zum Vorgang Der Wasserstoffübertragung, 52. Zur Kenntnis Der Elektroreduktiven Spaltung Der N - N-Bindung. Eur J Org Chem. 1978;1978:1505–1517. [Google Scholar]
- 838.Mentel M, Beier M, Breinbauer R. An Environmentally Benign Electrochemical Process for the Reduction of Carboxylic Acid Hydrazides to Amides. Synthesis. 2009;2009:1463–1468. [Google Scholar]
- 839.Lund H, Lunde P. Quaternization Reactions. II. Pyridazines. Acta Chem Scand. 1967;21:1067–1080. [Google Scholar]
- 840.Bakkali H, Marie C, Ly A, Thobie-Gautier C, Graton J, Pipelier M, Sengmany S, Léonel E, Nédélec J-Y, Evain M, et al. Functionalized 2,5-Dipyridinylpyrroles by Electrochemical Reduction of 3,6-Dipyridinylpyridazine Precursors. Eur J Org Chem. 2008;2008:2156–2166. [Google Scholar]
- 841.Manh GT, Hazard R, Pradère JP, Tallec A, Raoult E, Dubreuil D. Activated Pyrroles From Pyridazines: Nitrogen Extrusion by Electroreduction. Tetrahedron Lett. 2000;41:647–650. [Google Scholar]
- 842.Joshi U, Josse S, Pipelier M, Chevallier F, Pradère J-P, Hazard R, Legoupy S, Huet F, Dubreuil D. Novel Pyrrole C-Nucleosides by Nitrogen Extrusion From Pyridazine C-Nucleosides. Tetrahedron Lett. 2004;45:1031–1033. [Google Scholar]
- 843.Manh GT, Hazard R, Tallec A, Pradère JP, Dubreuil D. Electrochemical Reduction of Substituted Pyridazines: a New Access to Activated Pyrroles. Electrochim Acta. 2002;47:2833–2841. [Google Scholar]
- 844.Liu Y-Z, Lin M-Y, Xiao L-P, Zhang K, Lu J-X. Electrochemical Reduction of 2-Nitroanisole in Ionic Liquid BMIm-BF4: Synthesis of 2-Anisidine. Chin J Chem. 2008;26:1168–1172. [Google Scholar]
- 845.Silvester DS, Wain AJ, Aldous L, Hardacre C, Compton RG. Electrochemical Reduction of Nitrobenzene and 4-Nitrophenol in the Room Temperature Ionic Liquid [C4Dmim][N(Tf)2] J Electroanal Chem. 2006;596:131–140. [Google Scholar]
- 846.Haber FZ. Elektrochem Angew Phys Chem. 1898;5:235. [Google Scholar]
- 847.Sokolov AA, Syroeshkin MA, Solkan VN, Shebunina TV, Begunov RS, Mikhal’chenko LV, Leonova MY, Gultyai VP. Efficient Electrochemical Synthesis of Pyrido[1,2-a]Benzimidazoles. Russ Chem Bull. 2014;63:372–380. [Google Scholar]
- 848.Won S, Kim W, Kim H. Electro Organic Synthesis Utilizing Mg Electrodes (II) - Novel Synthesis of Symmetric Azobenzenes From Nitrobenzenes. Bull Korean Chem Soc. 2006;27:195–196. [Google Scholar]
- 849.Du P, Brosmer JL, Peters DG. Electrosynthesis of Substituted 1 H-Indoles From O-Nitrostyrenes. Org Lett. 2011;13:4072–4075. doi: 10.1021/ol2006049. [DOI] [PubMed] [Google Scholar]
- 850.Fry AJ, Newberg JH. Stereoselective Electrochemical Reductions of Camphor Oxime and Norcamphor Oxime. J Am Chem Soc. 1967;89:6374–6374. [Google Scholar]
- 851.Kulisch J, Nieger M, Stecker F, Fischer A, Waldvogel SR. Efficient and Stereodivergent Electrochemical Synthesis of Optically Pure Menthylamines. Angew Chem, Int Ed. 2011;50:5564–5567. doi: 10.1002/anie.201101330. [DOI] [PubMed] [Google Scholar]
- 852.Edinger C, Kulisch J, Waldvogel SR. Stereoselective Cathodic Synthesis of 8-Substituted (1R,3R,4S)-Menthylamines. Beilstein J Org Chem. 2015;11:294–301. doi: 10.3762/bjoc.11.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 853.Horner L, Neumann H. Studien Zum Vorgang Der Wasserstoffübertragung, XII: Hydrierende Spaltung Von Sulfonen Mit Tetramethylammonium Als Elektronenüberträger. Chem Ber. 1965;98:1715–1721. [Google Scholar]
- 854.Roemmele RC, Rapoport H. Removal of N-Arylsulfonyl Groups From Hydroxy α-Amino Acids. J Org Chem. 1988;53:2367–2371. [Google Scholar]
- 855.Senboku H, Nakahara K, Fukuhara T, Hara S. Hg Cathode-Free Electrochemical Detosylation of N,N-Disubstituted p-Toluenesulfonamides: Mild, Efficient, and Selective Removal of N-Tosyl Group. Tetrahedron Lett. 2010;51:435–438. [Google Scholar]
- 856.Scialdone O, Belfiore C, Filardo G, Galia A, Sabatino MA, Silvestri G. Direct Electrochemical Detosylation of Tetratosylcyclen to Cyclen with Carbon Cathodes. Electrochim Acta. 2005;51:598–604. [Google Scholar]
- 857.Coeffard V, Thobie-Gautier C, Beaudet I, Le Grognec E, Quintard J-P. Mild Electrochemical Deprotection of N-Phenylsulfonyl N-Substituted Amines Derived From (R)-Phenylglycinol. Eur J Org Chem. 2008;2008:383–391. [Google Scholar]
- 858.Viaud P, Coeffard V, Thobie-Gautier C, Beaudet I, Galland N, Quintard J-P, Le Grognec E. Electrochemical Cleavage of Sulfonamides: an Efficient and Tunable Strategy to Prevent β-Fragmentation and Epimerization. Org Lett. 2012;14:942–945. doi: 10.1021/ol300003f. [DOI] [PubMed] [Google Scholar]
- 859.Rayala R, Giuglio-Tonolo A, Broggi J, Terme T, Vanelle P, Theard P, Médebielle M, Wnuk SF. Studies Toward the Oxidative and Reductive Activation of C–S Bonds in 2′-S-Aryl-2′-Thiouridine Derivatives. Tetrahedron. 2016;72:1969–1977. doi: 10.1016/j.tet.2016.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 860.Santhanam KSV, Bard AJ. Electrochemistry of Organophosphorus Compounds. II. Electroreduction of Triphenylphosphine and Triphenylphosphine Oxide. J Am Chem Soc. 1968;90:1118–1122. [Google Scholar]
- 861.Kawakubo H, Kuroboshi M, Yano T, Kobayashi K, Kamenoue S, Akagi T, Tanaka H. Electroreduction of Triphenylphosphine Oxide to Triphenylphosphine in the Presence of Chlorotrimethylsilane. Synthesis. 2011;2011:4091–4098. [Google Scholar]
- 862.Kuroboshi M, Yano T, Kamenoue S, Kawakubo H, Tanaka H. Electroreduction of Tetra-Coordinate Phosphonium Derivatives; One-Pot Transformation of Triphenylphosphine Oxide Into Triphenylphosphine. Tetrahedron. 2011;67:5825–5831. [Google Scholar]
- 863.Yano T, Kuroboshi M, Tanaka H. Electroreduction of Triphenylphosphine Dichloride and the Efficient One-Pot Reductive Conversion of Phosphine Oxide to Triphenylphosphine. Tetrahedron Lett. 2010;51:698–701. [Google Scholar]
- 864.Lam K, Markó IE. Toluates: Unexpectedly Versatile Reagents. Tetrahedron. 2009;65:10930–10940. [Google Scholar]
- 865.Wu W-B, Huang J-M. Electrochemical Cleavage of Aryl Ethers Promoted by Sodium Borohydride. J Org Chem. 2014;79:10189–10195. doi: 10.1021/jo5018537. [DOI] [PubMed] [Google Scholar]
- 866.Pacut RI, Kariv-Miller E. Birch-Type Reductions in Aqueous Media. Benzo[B]Thiophene and Diphenyl Ether. J Org Chem. 1986;51:3468–3470. [Google Scholar]
- 867.Janissek PR, Pardini VL, Viertler H. Anodic Cleavage of Carbon–Oxygen Bonds in Diaryl Ethers. J Chem Soc, Chem Commun. 1987;0:576–577. [Google Scholar]
- 868.Dabo P, Cyr A, Lessard J, Brossard L, Ménard H. Electrocatalytic Hydrogenation of 4-Phenoxyphenol on Active Powders Highly Dispersed in a Reticulated Vitreous Carbon Electrode. Can J Chem. 1999;77:1225–1229. [Google Scholar]
- 869.Thornton TA, Ross GA, Patil D, Mukaida K, Warwick JO, Woolsey NF, Bartak DE. Carbon-Oxygen Bond-Cleavage Reactions by Electron Transfer. 4. Electrochemical and Alkali-Metal Reductions of Phenoxynaphthalenes. J Am Chem Soc. 1989;111:2434–2440. [Google Scholar]
- 870.Thornton TA, Woolsey NF, Bartak DE. Carbon-Oxygen Bond-Cleavage Reactions by Electron Transfer. 3. Electrochemical Formation and Decomposition of the Diphenyl Ether Radical Anion. J Am Chem Soc. 1986;108:6497–6502. [Google Scholar]
- 871.Huang J-M, Lin Z-Q, Chen D-S. Electrochemically Supported Deoxygenation of Epoxides Into Alkenes in Aqueous Solution. Org Lett. 2012;14:22–25. doi: 10.1021/ol2026944. [DOI] [PubMed] [Google Scholar]
- 872.Nikitin OM, Magdesieva TV. Electrochemically Induced Titanocene-Mediated Reductive Opening of Epoxides. Mendeleev Commun. 2011;21:194–195. [Google Scholar]
- 873.Peover ME, White BS. Electrolytic Reduction of Oxygen in Aprotic Solvents: the Superoxide Ion. Electrochim Acta. 1966;11:1061–1067. [Google Scholar]
- 874.Dietz R, Forno AEJ, Larcombe BE, Peover ME. Nucleophilic Reactions of Electrogenerated Superoxide Ion. J Chem Soc B. 1970:816–820. [Google Scholar]
- 875.Tang MC-Y, Wong KY, Chan TH. Electrosynthesis of Hydrogen Peroxide in Room Temperature Ionic Liquids and in Situ Epoxidation of Alkenes. Chem Commun. 2005:1345–1347. doi: 10.1039/b416837b. [DOI] [PubMed] [Google Scholar]
- 876.Ho K-P, Chan T-H, Wong K-Y. An Environmentally Benign Catalytic System for Alkene Epoxidation with Hydrogen Peroxide Electrogenerated in Situ. Green Chem. 2006;8:900–906. [Google Scholar]
- 877.Nishihara H, Pressprich K, Murray RW, Collman JP. Electrochemical Olefin Epoxidation with Manganese Meso-Tetraphenylporphyrin Catalyst and Hydrogen Peroxide Generation at Polymer-Coated Electrodes. Inorg Chem. 1990;29:1000–1006. [Google Scholar]
- 878.Casadei MA. Reactivity of the Electrogenerated O2·–/CO2 System Towards Alcohols. Eur J Org Chem. 2001;2001:1689–1693. [Google Scholar]
- 879.Barba F, Batanero B, Barba I. Electrogenerated Superoxide Anion: Hydroxylation of Electroreducible Substrates in Aprotic Solvent. J Electroanal Chem. 2017;793:66–69. [Google Scholar]
- 880.Matsubara Y, Matsuda T, Kato A, Yamaguchi Y, Yoshida Z-I. Electrochemical Transformation of 4-Cyanocinnolines Into 4 (1H)-Cinnolones. Tetrahedron Lett. 2000;41:7901–7904. [Google Scholar]
- 881.Kaimakliotis C, Fry AJ. Novel Desilylation of α-Dimethylsilyl Esters by Electrochemically Generated Superoxide Ion. Tetrahedron Lett. 2003;44:5859–5861. [Google Scholar]
- 882.Okamoto I, Funaki W, Nakaya K, Kotani E, Takeya T. A New Electrochemical System for Stereoselective Allylic Hydroxylation of Cholesteryl Acetate with Dioxygen Induced by Iron Picolinate Complexes. Chem Pharm Bull. 2004;52:756–759. doi: 10.1248/cpb.52.756. [DOI] [PubMed] [Google Scholar]
- 883.Barton D, Doller D. The Selective Functionalization of Saturated Hydrocarbons: Gif Chemistry. Acc Chem Res. 1992;25:504–512. [Google Scholar]
- 884.Minisci F, Citterio A, Vismara E, Giordano C. Polar Effects in Free-Radical Reactions. New Synthetic Developments in the Functionalization of Heteroaromatic Bases by Nucleophilic Radicals. Tetrahedron. 1985;41:4157–4170. [Google Scholar]
- 885.Lisitsyn YA, Kargin YM. Electrochemical Amination of Unsaturated and Aromatic Compounds. Russ J Electrochem. 2000;36:89–99. [Google Scholar]
- 886.Lisitsyn YA, Grigor’eva LV. Electrochemical Amination. Dilute Aqueous Organic Solutions of Sulfuric Acid. Russ J Electrochem. 2009;45:132–138. [Google Scholar]
- 887.Lisitsyn YA, Sukhov AV. Electrochemical Amination. Functionalization of Anisole in Solutions of 4.0–6.0 M H2So4 And Acetic Acid. Russ J Electrochem. 2011;47:1180–1185. [Google Scholar]
- 888.Lisitsyn YA, Sukhov AV. Electrochemical Amination of Anisole in 4–6 M Solutions of H2So4 And Acetonitrile. Russ J Electrochem. 2013;49:91–95. [Google Scholar]
- 889.Lisitsyn YA, Sukhov AV. Indirect Cathode Amination of Anisole in Dilute Sulfuric Acid and Acetonitrile Solutions. Russ J Gen Chem. 2012;82:1315–1316. [Google Scholar]
- 890.Lisitsyn YA, Sukhov AV. Electrochemical Amination. Synthesis of Aniline in Aqueous–Acetonitrile Solutions of Sulfuric Acid. Russ J Electrochem. 2015;51:1092–1095. [Google Scholar]
- 891.Ibanez JG, Frontana-Uribe BA, Vasquez-Medrano R. Paired Electrochemical Processes: Overview, Systematization, Selection Criteria, Design Strategies, and Projection. J Mex Chem Soc. 2016;60:247–260. [Google Scholar]
- 892.Botte GG. Electrochemical Manufacturing in the Chemical Industry. Electrochem Soc Interface. 2014;23:49–55. [Google Scholar]
- 893.Llorente MJ, Nguyen BH, Kubiak CP, Moeller KD. Paired Electrolysis in the Simultaneous Production of Synthetic Intermediates and Substrates. J Am Chem Soc. 2016;138:15110–15113. doi: 10.1021/jacs.6b08667. [DOI] [PubMed] [Google Scholar]
- 894.Zhang L, Zha Z, Wang Z, Fu S. Aqueous Electrosynthesis of Carbonyl Compounds and the Corresponding Homoallylic Alcohols in a Divided Cell. Tetrahedron Lett. 2010;51:1426–1429. [Google Scholar]
- 895.Zhang L, Zha Z, Wang Z. An Efficient Electrochemical Method for the Paired Synthesis of Carbonyl Compounds and Homoallylic Alcohols in a Simple Home-Made Cell. Synlett. 2010;2010:1915–1918. [Google Scholar]
- 896.Zhang L, Zha Z, Zhang Z, Li Y, Wang Z. An Electrochemical Tandem Reaction: One-Pot Synthesis of Homoallylic Alcohols From Alcohols in Aqueous Media. Chem Commun. 2010;46:7196–7198. doi: 10.1039/c0cc01964j. [DOI] [PubMed] [Google Scholar]
- 897.Amemiya F, Horii D, Fuchigami T, Atobe M. Self-Supported Paired Electrosynthesis Using a Microflow Reactor Without Intentionally Added Electrolyte. J Electrochem Soc. 2008;155:E162–E164. [Google Scholar]
- 898.Matthessen R, Fransaer J, Binnemans K, De Vos DE. Paired Electrosynthesis of Diacid and Diol Precursors Using Dienes and CO2 as the Carbon Source. ChemElectroChem. 2015;2:73–76. [Google Scholar]
- 899.Shen Y, Atobe M, Li W, Nonaka T. Paired Electrosynthesis of Epoxides and Dibromides from Olefinic Compounds. Electrochim Acta. 2003;48:1041. [Google Scholar]
- 900.Wang X, Zhao J. Synthesis of L-Cysteine and L-Cysteic Acid by Paired Electrolysis Method. Chem Lett. 2004;33:332–333. [Google Scholar]
- 901.Chou CF, Chou TC. Paired Electrooxidation IV. Decarboxylation of Sodium Gluconate to D-Arabinose. J Appl Electrochem. 2003;33:741–745. [Google Scholar]
- 902.Hartmer MF, Waldvogel SR. Electroorganic Synthesis of Nitriles via a Halogen-Free Domino Oxidation–Reduction Sequence. Chem Commun. 2015;51:16346–16348. doi: 10.1039/c5cc06437f. [DOI] [PubMed] [Google Scholar]
- 903.Kashiwagi T, Fuchigami T, Saito T, Nishiyama S, Atobe M. Sequential Paired Electrosynthesis of a Diaryl Ether Derivative Using an Electrochemical Microreactor. Chem Lett. 2014;43:799–801. [Google Scholar]
- 904.Huang H, Yuan G, Li X, Jiang H. Electrochemical Synthesis of Amides: Direct Transformation of Methyl Ketones with Formamides. Tetrahedron Lett. 2013;54:7156–7159. [Google Scholar]
- 905.Hilt G. Convergent Paired Electrolysis for the Three-Component Synthesis of Protected Homoallylic Alcohols. Angew Chem, Int Ed. 2003;42:1720–1721. doi: 10.1002/anie.200350892. [DOI] [PubMed] [Google Scholar]
- 906.Senboku H, Nagakura K, Fukuhara T, Hara S. Three-Component Coupling Reaction of Benzylic Halides, Carbon Dioxide, and N,N-Dimethylformamide by Using Paired Electrolysis: Sacrificial Anode-Free Efficient Electrochemical Carboxylation of Benzylic Halides. Tetrahedron. 2015;71:3850–3856. [Google Scholar]
- 907.Li C-H, Song X-Z, Tao L-M, Li Q-G, Xie J-Q, Peng M-N, Pan L, Jiang C, Peng Z-Y, Xu M-F. Electrogenerated-Bases Promoted Electrochemical Synthesis of N-Bromoamino Acids From Imines and Carbon Dioxide. Tetrahedron. 2014;70:1855–1860. [Google Scholar]
- 908.Ishifune M, Yamashita H, Matsuda M, Ishida H, Yamashita N, Kera Y, Kashimura S, Masuda H, Murase H. Electroreduction of Aliphatic Esters Using New Paired Electrolysis Systems. Electrochim Acta. 2001;46:3259–3264. [Google Scholar]
- 909.Kashimura S, Yamashita H, Murai Y, Kera Y. Magnesium and Lanthanide Ions Promoted Electrochemical Coupling of Aliphatic Esters and Tetrahydrofuran for the Synthesis of Acetals. Electrochim Acta. 2002;48:7–10. [Google Scholar]
- 910.Habibi D, Pakravan N, Nematollahi D. The Green and Convergent Paired Diels-Alder Electro-Synthetic Reaction of 1,4-Hydroquinone with 1,2-Bis(Bromomethyl)Benzene. Electrochem Commun. 2014;49:65–69. [Google Scholar]
- 911.Zhang L, Chen H, Zha Z, Wang Z. Electrochemical Tandem Synthesis of Oximes From Alcohols Using KNO3 As the Nitrogen Source, Mediated by Tin Microspheres in Aqueous Medium. Chem Commun. 2012;48:6574–3. doi: 10.1039/c2cc32800c. [DOI] [PubMed] [Google Scholar]
- 912.Batanero B, Barba F, Sánchez-Sánchez CM, Aldaz A. Paired Electrosynthesis of Cyanoacetic Acid. J Org Chem. 2004;69:2423–2426. doi: 10.1021/jo0358473. [DOI] [PubMed] [Google Scholar]
- 913.Kim S, Uchiyama R, Kitano Y, Tada M. Benzylic Nitroalkylation by Paired Electrolysis of Benzyl Sulfides in Nitroalkanes. J Electroanal Chem. 2001;507:152–156. [Google Scholar]
- 914.Kanega R, Hayashi T, Yamanaka I. Pd(NHC) Electrocatalysis for Phosgene-Free Synthesis of Diphenyl Carbonate. ACS Catal. 2013;3:389–392. [Google Scholar]