

Abstract
Rothmund-Thomson syndrome (RTS) is a rare autosomal recessive disorder with clinical features consisting of poikiloderma, skeletal abnormalities, sparse hair, absent or scanty eyelashes and eyebrows and short stature. Patients with RTS due to genetic mutations of RECQL4 genes carry a high risk of developing osteosarcoma during childhood. Because of this, early genetic diagnosis is important. Here, we describe a 14-year-old white boy who developed an erythematous rash on both cheeks before the age of 3 months and was noted to have absent eyelashes and scanty eyebrows. He was found to have compound heterozygous mutations of the RECQL4 gene alleles at the age of 6 months and was diagnosed to have RTS type II. He subsequently developed osteosarcoma at age 10 which was successfully treated, and currently he has been tumour free for over 3 years.
Keywords: dermatology, paediatric oncology, congenital disorders, genetics
Background
Rothmund-Thomson syndrome (RTS) is a rare autosomal recessive disorder affecting people of all ethnic groups. The disease usually manifests early, between 3 and 6 months of age. The affected children exhibit a characteristic facial rash (poikiloderma) associated with short stature, sparse scalp hair, sparse or absent eyelashes and/or eyebrows, juvenile cataracts, skeletal abnormalities, radial ray defects, premature ageing and a predisposition to cancer.1 According to Larizza et al 1 about 300 cases were reported prior to the year 2010.1 Two subtypes have been recognised (RTSI and RTSII).2 RTSI is characterised by poikiloderma, ectodermal dysplasia and juvenile cataracts. No genetic mutations for this type have been identified. Approximately 35%–40% of reported cases of RTS fall into this subtype. The more common subtype (60%–65%) is RTSII, which is characterised by poikiloderma, congenital bone defects and an increased risk of osteosarcoma in childhood. This subtype is due to compound heterozygous or homozygous mutations in the DNA helicase gene, RECQL4.1 The poikiloderma is typically recognisable during early infancy. As the patients grow, affected skin becomes atrophic and shows telangiectasia, pigmentation and lentiginous depigmentation. Because of the high risk of the affected child developing osteosarcoma during childhood,1 2 early diagnosis is important. We have cared for such a child who was diagnosed with RTSII in early childhood, and while under careful observation, he developed osteosarcoma. We wish to report this relatively rare occurrence as a learning point.
Case presentation
Our patient is a 14-year-old white boy. When he was 3 months old, his parents noticed a persistent erythematous rash on both cheeks. The child’s paediatrician thought that it was eczema, but despite the topical treatment for eczema, the rash failed to improve. Furthermore, the child lacked eyelashes and part of his eyebrows. At 6 months of age, the child was referred to a dermatologist who biopsied the lesion, and the child was diagnosed to have poikiloderma. He was then referred to a geneticist. Genetic studies performed at Baylor College of Medicine laboratory demonstrated that the patient had two mutated alleles of the RECQL4 gene, c.[1568G>C;1573delT];r.(?); p.(Ser523Thr;Cys525Alafs*33) (maternal origin) and c.2269C>T;r.(?);p.(Gln757*) (paternal origin) Thus, this patient carried two mutations both affecting the functionally crucial helicase domain of the RECQL4 protein (encoded by DNA spanning from the end of exon 7 to the two-thirds of exon 15). His two older brothers were not tested. They showed no cutaneous abnormalities. Radiographic imaging of patient’s arms and legs showed no skeletal abnormalities. He has had no cataracts. The patient’s subsequent clinical course was uneventful except that he had absent perspiration and once he would get ‘hot’, it was difficult for him to cool down (heat intolerance). He had telangiectatic lesions on the cheeks and extensor surfaces of the forearms and hands as well as very dry, scaly skin, particularly on the hands and arms (figures 1–4). The patient was subsequently seen by a paediatric oncologist once but has not been regularly followed. When he was 10 years old, he developed pain on his left wrist while he was playing with his older brother. The pain persisted, and his parents noticed significant swelling and limited mobility of the left wrist. An MRI of the left wrist and forearm demonstrated a heterogeneous lesion measuring 6.7×2.8×3.3 cm in the metaphysis of the left radius extending into both diaphysis and epiphysis with considerable periosteal reaction and soft tissue oedema. The biopsy showed osteosarcoma. There were no distant metastases. At this point, he weighed 33.5 kg (59th percentile), and he was 136 cm tall (33rd percentile). Thus, he was not remarkably short. He was treated on the Children’s Oncology Group protocol AOST0331, combination chemotherapy consisting of doxorubicin, cis-platinum and high-dose methotrexate (MAP) for induction (doxorubicin 37.5 mg/m2/day, day 1–2, cis-platinum 60 mg/m2/day, day 1 and 2, methotrexate 12 g/m2/day). He exhibited poor histological tumour response to the induction chemotherapy, and thus following complete resection of the tumour, he was treated with alternate MAP and ifosfamide (2.8 g/m2/dose×5 days on weeks 16, 24 and 32, 3 g/m2/dose×3 days on weeks 20 and 36) (plus etoposide (100 mg/m2/day×5 days on weeks 16, 24 and 32)) for a total of 40 weeks of chemotherapy. He tolerated chemotherapy well, though he frequently developed grade 2–3 stomatitis following doxorubicin infusion, but the dose reduction was not necessary. He has been tumour free for over 3 years. Following chemotherapy, his bone density was normal. However, he has moderate bilateral hearing impairment at the 3000–8000 Hz range due to cis-platinum treatment. His hearing was normal before treatment.
Figure 1.
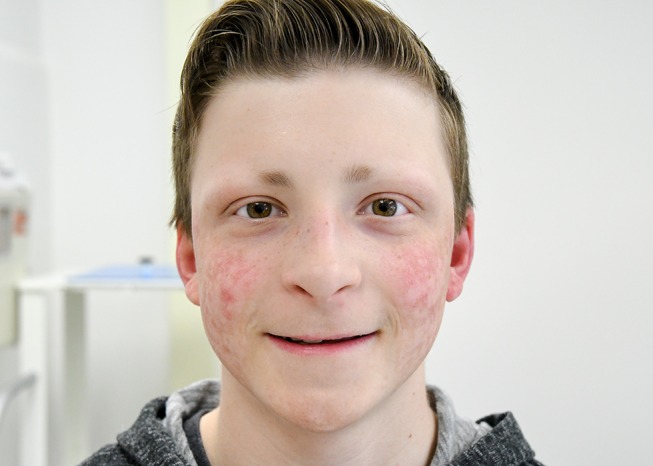
Frontal view of the patient’s face (taken when the patient was 10 years old). Note atrophic erythematous, partially telangiectatic dermal changes of the face bilaterally, absent eyelashes and partial absence of eyebrows.
Figure 2.
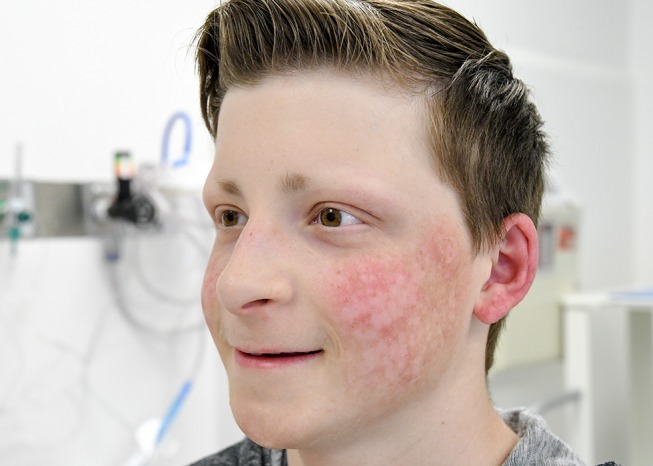
Side view of the patient’s face depicting the same abnormalities.
Figure 3.
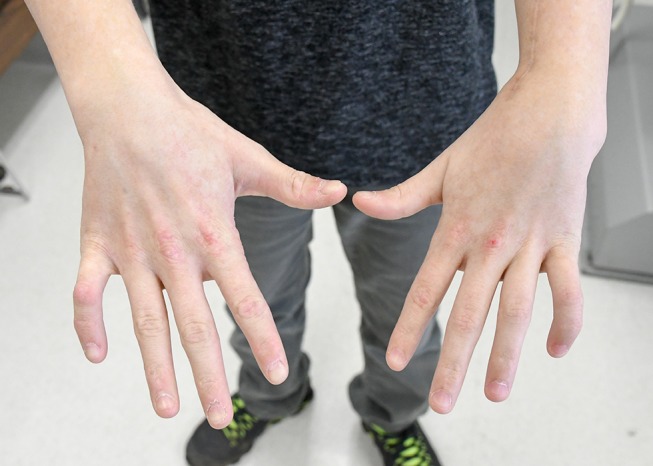
Patient’s hands, dorsal aspect. Note pinkish atrophic skin over the finger joints including metacarpal joints and bilateral clinodactyly of fifth fingers.
Figure 4.
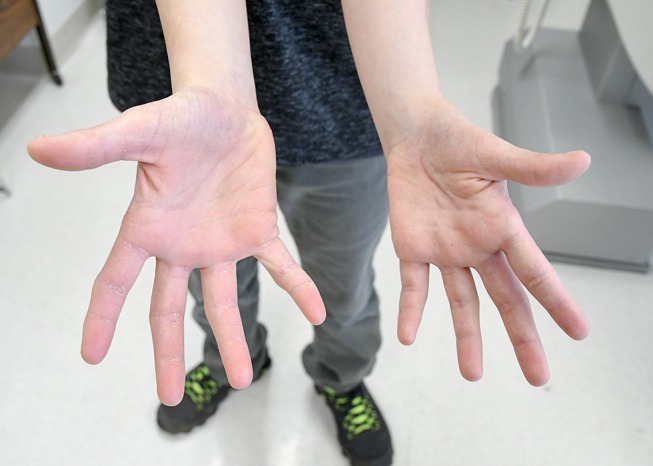
Patient’s palms. Note scaly atrophic skin of all fingers.
Differential diagnosis
Differential diagnosis includes acrogeria, dyskeratosis congenita, Kindler’s syndrome (bullous poikiloderma), xeroderma pigmentosum, Clericuzio-type poikiloderma with neutropaenia and Werner syndrome.1 However, the age and location of skin changes as well as absence of other abnormalities easily excluded other diagnoses. There are, however, many overlapping features with Werner syndrome and RTS. These include poikiloderma, short stature, predisposition to osteosarcoma and cataracts. But genetic testing should lead to correct diagnosis.
RECQL4 scanning that used to be performed for the diagnostic purpose may not be sufficient.1 DNA sequencing of the entire RECQL4 gene is necessary. RECQL4 truncating mutations are believed to be most common. However, Larizza et al stated that it is worth noting that nearly one-third of all the identified RECQL4 mutations are splicing mutations, often not canonical, that is, escaping detection by genomic PCR sequencing by RT PCR.1 3
Outcome and follow-up
The patient has been free of tumour for the past 3 years.
Discussion
Common features of RTSII consist of poikiloderma initially on the face and later on the extensor surface of the arms, legs and buttocks, thin and/or scarce hair, absence of, or thin eyebrows and eyelashes, skeletal abnormalities and small stature. Cataracts are not common in RTS type II but very common in type I.1 Our patient was diagnosed early in life. In Wang et al’s case series of 41 patients,4 the earliest ascertainment of diagnosis was at 9 months of age. Early manifestation of RTS is subtle, and thus diagnosis is often late.4 In the same series,4 13 of 41 subjects (32%) developed osteosarcoma. Patients with RECQL4 mutation are prone to develop malignancy while those without the mutation are not.2 Simon et al 5 suggested that patients with truncating RECQL4 mutation have a high risk, hazard risk of 0.05/year, while patients with non-truncating mutation do not have that risk. Siitonen et al 2 enlisted all reported cases of RECQL4 mutations in 2009 consisting of 41 cases of RTS and 17 cases of RAPADILINO syndrome (shares many clinical features with RTS but lacks dermatological abnormalities). Of the total of 58 cases listed, 10 had mutation at c. 1573 delT (most recurrent mutation), and 8 had c.2269C>T mutation. One patient who developed osteosarcoma at age 9 had homozygous mutations of the latter, while another patient who had the identical compound heterozygous mutations to those of our case developed osteosarcoma at 4 years of age.2 Therefore, whenever a patient is clinically suspected to have RTS, it is advisable to do the genetic testing. The biological characteristics of osteosarcoma developing in RTS are similar to those without RTS, as to the location (femur, tibia), histological type and response to treatment, though peak age seems to be younger than non-syndromic osteosarcoma.6 However, in a case review published by Stinco et al the mean age of development of osteosarcoma was 14.03 years, not too much different from the peak age of non-syndromic paediatric osteosarcoma, though there could be a selection bias.7 Tolerance to chemotherapy among patients with RTS is again variable; some reported extreme toxicity, while others observed no excessive toxicity.5 7 8 The patients with RTS also have a higher risk of developing squamous cell carcinoma, basal cell carcinoma or Bowen’s disease later in life.1 Development of multiple malignancies5 7 and multicentric osteosarcoma9 also have been reported. Our patient did not have congenital cataracts. Although congenital cataracts have been found less often in patients with RECQL4 mutation compared with those without mutation (RTSI), it would still be prudent to have periodic ophthalmological examinations done. A skeletal survey on our patient failed to disclose any abnormality, a rare observation, since Mehollin-Ray et al observed a significant correlation between RECQL4 mutation and the presence of skeletal abnormalities. All 18 subjects with the mutation had skeletal abnormalities, whereas only 3 of 10 without mutation had skeletal abnormalities.10 RECQL4 gene is a DNA helicase gene which participates in DNA repair, recombination and replication processes of cellular DNA.1 Mutation of the same family of genes results in genomic instability and increased risk of malignancy. Bloom’s syndrome (RECQ2, BLM) and Werner syndrome (RECQ3, WRN) are other disorders due to RecQ gene family mutation, constituting cancer predisposition syndromes.
No formal cancer surveillance protocol has been developed for patients with RTS II. Because of the rarity of the disorder, it would be extremely difficult to establish risk (cost)–benefit ratio of a given surveillance protocol. Our patient was unofficially advised to undergo bone screening by MRI every 6 months at a paediatric cancer centre at the time of diagnosis, but the advice was not followed. Recently cancer surveillance recommendations for the RTS type II was published,11 consisting of obtaining a skeletal survey before age 5 years, counselling about risk of osteosarcoma and anticipatory guidance of signs and symptoms of osteosarcoma. The same article states that the benefit of routine screening for osteosarcoma has not yet been determined.
Learning points.
RECQL4 gene mutation in patients with Rothmund-Thomson syndrome (RTS) carries a high risk of developing malignancies. Therefore, it is important to make a molecular diagnosis whenever the clinical picture suggests RTS.
Careful long-term follow-up may be needed after treatment of first malignancy, since there is an increased risk of second and subsequent malignant neoplasia.
The goal is to educate the caregivers about alarming signs and symptoms and when to seek medical advice since it is crucial to detect the malignancy at early stages.
Cisplatin is ototoxic and predictably produces hearing impairment, and thus it is important to do a baseline hearing test before starting therapy as well as to follow with periodic hearing tests after end of therapy.
Footnotes
Contributors: AS wrote the initial manuscript draft. NO read and approved the final product. SI supervised, edited and approved the manuscript from beginning to end.
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis 2010;5:2 10.1186/1750-1172-5-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siitonen HA, Sotkasiira J, Biervliet M, et al. . The mutation spectrum in RECQL4 diseases. Eur J Hum Genet 2009;17:151–8. 10.1038/ejhg.2008.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Colombo EA, Fontana L, Roversi G, et al. . Novel physiological RECQL4 alternative transcript disclosed by molecular characterisation of Rothmund-Thomson syndrome sibs with mild phenotype. Eur J Hum Genet 2014;22:1298–304. 10.1038/ejhg.2014.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang LL, Gannavarapu A, Kozinetz CA, et al. . Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst 2003;95:669–74. 10.1093/jnci/95.9.669 [DOI] [PubMed] [Google Scholar]
- 5. Simon T, Kohlhase J, Wilhelm C, et al. . Multiple malignant diseases in a patient with Rothmund-Thomson syndrome with RECQL4 mutations: case report and literature review. Am J Med Genet A 2010;152A:1575–9. 10.1002/ajmg.a.33427 [DOI] [PubMed] [Google Scholar]
- 6. Hicks MJ, Roth JR, Kozinetz CA, et al. . Clinicopathologic features of osteosarcoma in patients with rothmund-thomson syndrome. J Clin Oncol 2007;25:370–5. 10.1200/JCO.2006.08.4558 [DOI] [PubMed] [Google Scholar]
- 7. Stinco G, Governatori G, Mattighello P, et al. . Multiple cutaneous neoplasms in a patient with Rothmund-Thomson syndrome: case report and published work review. J Dermatol 2008;35:154–61. 10.1111/j.1346-8138.2008.00436.x [DOI] [PubMed] [Google Scholar]
- 8. Borg MF, Olver IN, Hill MP. Rothmund-Thomson syndrome and tolerance of chemoradiotherapy. Australas Radiol 1998;42:216–8. 10.1111/j.1440-1673.1998.tb00496.x [DOI] [PubMed] [Google Scholar]
- 9. Spurney C, Gorlick R, Meyers PA, et al. . Multicentric osteosarcoma, Rothmund-Thomson syndrome, and secondary nasopharyngeal non-Hodgkin’s lymphoma: a case report and review of the literature. J Pediatr Hematol Oncol 1998;20:494–7. [DOI] [PubMed] [Google Scholar]
- 10. Mehollin-Ray AR, Kozinetz CA, Schlesinger AE, et al. . Radiographic abnormalities in Rothmund-Thomson syndrome and genotype-phenotype correlation with RECQL4 mutation status. AJR Am J Roentgenol 2008;191:W62–W66. 10.2214/AJR.07.3619 [DOI] [PubMed] [Google Scholar]
- 11. Walsh MF, Chang VY, Kohlmann WK, et al. . Recommendations for childhood cancer screening and surveillance in dna repair disorders. Clin Cancer Res 2017;23:e23–e31. 10.1158/1078-0432.CCR-17-0465 [DOI] [PMC free article] [PubMed] [Google Scholar]