

Abstract
Background and Purpose
Autotaxin (ATX) is a secreted phospholipase which hydrolyses lysophosphatidylcholine to generate lysophosphatidic acid (LPA). The extracellular signalling molecule LPA exerts its biological actions through activation of six GPCRs expressed in various cell types including fibroblasts. Multiple preclinical studies using knockout animals, LPA receptor antagonists or ATX inhibitors have provided evidence for a potential role of the ATX/LPA axis in tissue fibrosis. Despite growing evidence for a correlation between ATX levels and the degree of fibrosis in chronic liver diseases, including viral hepatitis and hepatocellular carcinoma, the role of ATX in non‐alcoholic steatohepatitis (NASH) remains unclear.
Experimental Approach
The relevance of ATX in the pathogenesis of liver fibrosis was investigated by oral administration of Ex_31, a selective ATX inhibitor, in a 10 week model of carbon tetrachloride‐induced liver injury and in a 14 week model of choline‐deficient amino acid‐defined diet‐induced liver injury in rats.
Key Results
Oral administration of Ex_31, a selective ATX inhibitor, at 15 mg·kg−1 twice daily in therapeutic intervention mode resulted in efficient ATX inhibition and more than 95% reduction in plasma LPA levels in both studies. Treatment with Ex_31 had no effect on biomarkers of liver function, inflammation, or fibrosis and did not result in histological improvements in diseased animals.
Conclusions and Implications
Our findings question the role of ATX in the pathogenesis of hepatic fibrosis and the potential of small molecule ATX inhibitors for the treatment of patients with NASH and advanced stages of liver fibrosis.
Abbreviations
- ACTA2
α smooth muscle actin gene
- ADME
absorption, distribution, metabolism, and excretion
- ALT
alanine aminotransferase
- ATX
autotaxin
- CCl4
carbon tetrachloride
- CDAA
choline‐deficient amino acid‐defined
- CSAA
choline‐supplemented amino acid‐defined
- CTGF
connective tissue growth factor
- Emr1
epidermal growth factor‐like module‐containing mucin‐like hormone receptor‐like 1
- Ex_31
Example 31(24)
- FCS
fetal calf serum
- H&E
haematoxylin and eosin
- HSC
hepatic stellate cell
- Itgam
integrin α M
- LPA
lysophosphatidic acid
- LPC
lysophosphatidylcholine
- NAS
non‐alcoholic steatohepatitis activity score
- NASH
non‐alcoholic steatohepatitis
- TRAIL
tumour necrosis factor‐related apoptosis‐inducing ligand
- αSMA
α smooth muscle actin
Introduction
Autotaxin (ATX) is a secreted enzyme of the phospholipase superfamily which hydrolyses lysophospholipids to generate lysophosphatidic acid (LPA) (Perrakis and Moolenaar, 2014). Mice deficient in ATX die during embryonic development, whereas heterozygous mice survive to adulthood and display plasma LPA levels that are approximately half that of wild‐type mice, implying that ATX is the major source of plasma LPA in mice (Tanaka et al., 2006 ; van Meeteren et al., 2006). Recently, it was demonstrated that anti‐ATX DNA aptamers block LPA production by more than 90% in human serum, showing that ATX is a major source of LPA in human plasma (Kato et al., 2016). LPA acts as an extracellular signalling molecule and exerts its biological actions through the activation of GPCRs. To date, six GPCRs have been described that are involved in LPA signalling (LPA1, LPA2, LPA3, LPA4, LPA5 and LPA6 receptors) and each is coupled to various signalling cascades including Gα12/13, Gαq, Gαs and GαI (Kihara et al., 2014). Elevated plasma LPA concentrations and increased LPA receptor expression have been observed in patients suffering from chronic inflammatory diseases, fibrosis and cancer (reviewed in Sevastou et al., 2013; Chu et al., 2015; Leblanc and Peyruchaud, 2015). Associations between LPA receptor signalling and various diseases have stimulated interest within the pharmaceutical industry for the development of LPA antagonists (Llona‐Minguez et al., 2015). However, it has proven challenging to identify potent and selective LPA receptor antagonists with few compounds having been advanced to clinical development (Pasquinelli, 2013; Khanna et al., 2014).
Autotaxin as the major source of plasma LPA has attracted the interest of researchers and the pharmaceutical industry. Several small molecule ATX inhibitors have been developed for use in various inflammatory and fibrotic diseases (reviewed in Barbayianni et al., 2015; Castagna et al., 2016). Of the candidates, GLPG1690 is currently being investigated in a Phase II trial in patients with idiopathic pulmonary fibrosis. Beyond that, there is growing evidence to support the involvement of ATX in the pathogenesis of liver fibrosis related to viral hepatitis and hepatocellular carcinoma (Nakagawa et al., 2011; Kondo et al., 2014; Pleli et al., 2014). However, the role of ATX in non‐alcoholic steatohepatitis (NASH) remains unclear.
The elevated ATX and LPA levels in patients with chronic liver diseases from different aetiologies have prompted efforts to explore the role of the LPA/ATX axis in the pathogenesis of NASH‐related fibrosis. The aim of the present study was to investigate the effect of LPA in primary human hepatic stellate cells (HSCs) in vitro and to characterize the properties of Example 31(24) (Ex_31), a selective small molecule ATX inhibitor, in preclinical models of advanced liver fibrosis.
Methods
Synthesis and purification of Ex_31
The ATX inhibitor Ex_31 was synthesized according to the procedures described in patent WO 2012/005227 and EP 2592081 A1 respectively (Supporting Information Figure S9). The synthesis of Ex_31 started from commercially available 7‐azaindole S1, which upon treatment with dimethylamine and formaldehyde underwent a Mannich reaction to give 7‐azagramine S2. The corresponding nitroethyl‐derivative S3 was next obtained through reaction with nitromethane and dimethylsulfate under basic conditions. Subsequently, the nitro group present in S3 was converted to the amine S4 through hydrogenation using Pearlman's catalyst. In order to construct the tricyclic core present in S5, the aza‐tryptamine S4 was heated with formaldehyde under acidic conditions in a sealed tube, which triggered the desired Pictet–Spengler cyclization to yield S5. Boc‐protection of S5 afforded S6, the latter which was alkylated with 1‐(bromomethyl)‐4‐chloro‐2‐fluorobenzene under basic conditions to give S7. Deprotection of the Boc group was achieved under acidic conditions and yielded the tricyclic amine S8. The latter was coupled with carboxylic acid S9 (synthesized in three steps from commercially available trans‐1,4‐cyclohexanedicarboxylic acid monomethyl ester according to procedures described in patent WO 2012/005227 and EP 2592081 A1 respectively) using HOBt and EDC to yield an amide, which was subsequently subjected to ester hydrolysis to give the final carboxylic acid Ex_31. The final crude compound was purified by silica gel column chromatography (eluent: 1% MeOH : dichloromethane) to provide Ex_31 as a white solid.
Primary human hepatic stellate cell culture
Primary human HSCs (Innoprot, Spain) were maintained in humidified cell culture incubators at 37°C and 5% CO2. Cells were cultured in standard cell culture plastic ware according to the manufacturer's instructions. Cells were grown in SteCM medium (ScienCell, USA) containing growth supplements and FCS (2%). Cells were split when they reached sub‐confluence.
Real‐time PCR analysis of gene expression in primary human hepatic stellate cells
For gene expression studies, HSCs were seeded in 12‐well plates and serum starved for 18 h before treatment with LPA (1‐oleoyl LPA, 10 μM 18:1; Cayman Chemicals, USA) for 1, 2, 4 and 6 h. Cells were lysed in RLT‐buffer, and total RNA was isolated using RNeasy Mini Kit (Qiagen, Germany) according to the manufacturer's instructions. Concentrations of RNA were analysed with a NanoDrop® ND‐1000 UV–Vis spectrophotometer at 260 nm (Thermo Fisher Scientific, USA). Purity of RNA was confirmed using 260/280 nm ratios (pure samples show a range of 1.8–2.1), and samples were stored at −80°C before further processing. For RT‐PCR, RNA (50–2000 ng) was reverse‐transcribed into cDNA with a high capacity cDNA archive kit (Thermo Fisher Scientific) in accordance with the manufacturer's instructions, and cDNAs were stored at −20°C. For the analysis of gene‐specific mRNA expression, cDNA was amplified by real‐time PCR with specific primers and fluorescently labelled probes (Supporting Information Table S8) using a thermal cycler (Eppendorf, Germany). Results were evaluated using SDS software version 2.2 (Thermo Fisher Scientific). Expression levels were normalized by reference to the house keeping gene RNA polymerase 2. For calculation of relative changes in gene expression, values of individual samples were divided by the mean value of untreated samples at time zero.
Measurement of TRAIL‐induced hepatic stellate cell apoptosis
For the measurement of TNF‐related apoptosis‐inducing ligand (TRAIL)‐induced apoptosis, white opaque 384‐well plates were seeded with HSCs at a density of 2000 cells per well in SteCM medium supplemented with FCS (2%) and growth supplements (1%). Six hours later, medium was replaced by SteCM medium without supplements and cells were serum‐starved for 6 h. For the apoptosis assay, cells were stimulated for 24 h with different concentrations of LPA alone or in combination with TRAIL (30 ng·mL−1; R&D Systems, Germany). Caspase 3/7 activity was measured using a luminescence‐based Caspase‐Glo 3/7 assay system (Promega, USA) according to the manufacturer's instructions.
In vitro ADME assays
Plasma protein binding (PPB)
The plasma protein binding was determined by equilibrium dialysis. Teflon dialysis cells with a cut‐off of 5–10 kDa were used. EDTA plasma was spiked with 10 mM Ex_31 and transferred to the donor chamber. The acceptor chamber was filled with PBS buffer (pH 7.4) supplemented with dextran. The chamber was incubated for 3 h under rotation at 37°C. Aliquots from both chambers were collected and analysed by LC–MS/MS for calculation of the PPB.
The following assays were performed as described previously (Luippold et al., 2011):
Cytochrome P450 assay
Cytochrome P450 oxygenase‐specific substrates were incubated with Ex_31 at 37°C with liver microsomes. The assay was performed in 0.1 M Tris buffer supplemented with 5 mM MgCl2 and 1 mM NADPH. Ex_31 was tested in a range of 0–50 μM.
Caco‐2 assay
A 10 μM solution of EX_31 (pH 7.4) was added to a donor chamber. Samples at multiple time points (up to 90 min) were collected from the donor and receiver chamber for analysis of the permeability and efflux ratio.
Animals
All animal care and experimental protocols were approved by the ethics review committee for animal experimentation of Boehringer Ingelheim Pharma GmbH & Co. KG. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). The pharmacokinetic study was carried out at Boehringer Ingelheim (Biberach, Germany) according to licence 14‐009‐G and national animal welfare guidelines. The carbon tetrachloride (CCl4) study was conducted at Boehringer Ingelheim (Biberach, Germany) according to licence 13‐011‐G and national animal welfare guidelines. The choline‐deficient amino acid‐defined (CDAA) study was conducted at Gubra ApS (Hørsholm, Denmark) according to licence 2013‐15‐2934‐00784 and national welfare guidelines. Details on animals, experimental procedures, housing and husbandry as well as animals numbers are disclosed in the following methods sections.
Pharmacokinetic profile of Ex_31 and target engagement in healthy rats
The ATX inhibitor Ex_31 was dissolved in water containing cyclodextrin (20%; 1 mg in 4 mL) for i.v. administration. For p.o. administration, Ex_31 was dissolved in water supplemented with Tween (0.1%) and Natrosol (0.5%; 6 mg in 2.5 mL). These solutions were administered to fasted male Han Wistar rats (180–200 g) at doses of 1 μmol·kg−1 (i.v.) or 10 μmol·kg−1 (p.o.) respectively. Blood samples (150 μL) were taken sublingually at 0, 5, 15 and 30 min and 1, 2, 4, 8 and 24 h. Plasma was isolated by centrifugation and immediately frozen until analysis of LPA levels and compound exposure as described below.
Quantification of Ex_31
For the quantification of the exposure of Ex_31 in liver the tissue was homogenized in a mixture of 0.1% formic acid and 30% acetonitrile/methanol (1:1) in a ratio of 1:4. The homogenate was prepared in a dry iced cooled Precyllis Evolution (Bertin Technologies) instrument.
Five microlitres of this homogenate or 5 μL of EDTA plasma was precipitated with 70 μL 0.1% formic acid and 70% acetonitrile/methanol. Protein precipitate was removed by centrifugation. Subsequently, a 30 μL aliquot of the supernatant was diluted in 150 μL 0.1% formic acid. Calibrants from 0.5 nM to 10 μM and quality controls at 10, 100 and 1000 nM were prepared in EDTA rat plasma. Calibrants, quality controls and samples were supplemented with internal standard and subjected to LC–MS/MS analysis on an API 4000 (ABSciex, Germany). The instrument was equipped with a 1200 LC‐system (Agilent, Germany), a Kinetex C18 column (50 × 2.1 mm, 2,6 μm, 100 Å Phenomenex), Solvent A (0.1% formic acid) and Solvent B (1:1 acentonitrile/methanol). A gradient profile was applied, starting at 10% Solvent B, increasing within 2.6 min to 95% and a decrease to 10% from 3.3 to 4 min. Ex_31 was monitored by recording the MRM trace 484.2/143.0 with a declustering potential of 71 and a collision energy of 57. The lower limit of quantification of Ex_31 was 5 nM.
Carbon tetrachloride‐induced hepatic fibrosis in rats
CCl4‐induced liver fibrosis is one of the most commonly used animal models to mimic liver fibrosis in rodents. This model is often used to investigate the role of signalling pathways or individual proteins in fibrosis development by using knockout animals or pharmacological tools. Male Sprague–Dawley rats, 6–7 weeks of age (JANVIER LABS, France), were housed in pairs in a controlled environment (12 h light/dark cycle). All animals had ad libitum access to normal chow (KLIBA 3438; Provimi Kliba AG, Switzerland) and tap water. Animals received carbon tetrachloride (CCl4; 0.25 mL·kg−1) diluted in olive oil by p.o. administration three times a week for 6 weeks, followed by 4 weeks of p.o. administration of Vehicle or Ex_31 (15 mg·kg−1) twice daily while maintaining the CCl4 regimen. Ten animals received olive oil as a control, whereas 14 animals per treatment group received CCl4. A higher number of CCl4‐treated animals was used due to the increased likelihood of losing animals under CCl4 treatment during the course of the study. In total, two animals had to be killed due to complications directly after one CCl4 injection. The remaining animals were allocated to the vehicle and the EX_31 group respectively, resulting in 13 animals in each in these groups. Animals received the compound (5 mL·kg−1) suspended in Natrosol (0.5%)/Tween 80 (0.01%). Before compound treatment, plasma samples were analysed for collagen IV and LPA levels and animals were stratified based on these parameters (Supporting Information Table S4). Plasma was obtained by sublingual bleeding from isofluorane (2–3% in oxygen) anaesthetized animals. Depth of anaesthesia was assessed by controlling reflexes (stimulated movement reflex, palpebral reflex, toe withdrawal reflex). At the end of the study, the animals were killed by final bleeding under pentobarbital anaesthesia. Livers were weighed, and blood and liver samples were used for further analysis. The CCl4 study was conducted at Boehringer Ingelheim (Biberach, Germany) according to licence 13‐011‐G and national animal welfare guidelines.
Choline‐deficient L‐amino acid‐defined diet‐induced liver injury in rats
As shown in previous studies, rats fed a CDAA diet develop a hepatic phenotype that closely mirrors the human NASH pathology including steatosis, inflammation and fibrosis. Additionally, animals fed a CDAA diet do not suffer from the massive body weight loss that is observed in animals fed a methionine‐ and choline‐deficient diet. Male Wistar rats, 6 weeks of age (JANVIER LABS), were housed in pairs in a controlled environment (12 h light/dark cycle). After acclimatizing to their environment, animals were fed either a CDAA diet containing cholesterol (1%; E15666‐94, ssniff Spezialdiäten GmbH, Germany) or a choline‐supplemented L‐amino acid‐defined (CSAA) diet (E15668‐04, ssniff Spezialdiäten GmbH) ad libitum for 6 weeks. All animals had ad libitum access to tap water. Following diet‐induced liver injury, liver pre‐biopsies were obtained with animals maintained under isoflurane anaesthesia as previously described (Clapper et al., 2013; Kristiansen et al., 2016). Rats were anaesthetized with isofluorane (3–5%), and a midline abdominal incision was made to expose the left lateral lobe. A tissue biopsy of 50–100 mg was taken and fixed in 4% paraformaldehyde overnight for histological assessment. The remaining cut surfaces were electro‐coagulated using an ERBE VIO 100C electrosurgical unit (Erbe Elektromedizin, Germany), the liver returned to the abdominal cavity and both the abdominal wall and the skin were sutured. The animals received carprofen (5 mg·kg−1; Pfizer, USA) and enrofloxacin (5 mg·kg−1; Bayer, Germany) both before surgery and on postoperative days 1 and 2 to control postoperative pain and infection. Animals were single‐housed after the surgical procedure. Liver biopsies were used for histological stratification of animals into groups showing similar collagen 1a1 content (Supporting Information Table S5). In each group, 12 animals received the indicated diet and treatment. From week 7, animals received Vehicle or Ex_31 (15 mg·kg−1, p.o.) suspended in 0.5% Natrosol/0.01% Tween 80 twice daily and were maintained on the CDAA diet. Control animals received Vehicle and were kept on the CSAA diet. After 5 weeks, animals were killed by cardiac puncture under isofluorane anaesthesia. The liver tissue was excised and weighed, and blood and liver samples were taken for further analysis. The CDAA study was conducted at Gubra ApS (Hørsholm, Denmark) according to licence 2013‐15‐2934‐00784 and national welfare guidelines.
Measurement of plasma alanine aminotransferase and collagen IV
Plasma aminotransferase (ALT) activity was measured using 80 μL samples collected into EDTA tubes using a Cobas Integra 400 or Cobas C‐111 (Roche Diagnostics, Germany). Collagen IV was measured from 50 μL of EDTA plasma using the Exocell Immunoassay collagen IV M (Exocell, USA) according to the instruction manual. Plasma samples and collagen IV standards were incubated overnight with anti‐collagen IV antibody in a murine collagen IV pre‐coated 96‐well plate. After a washing step, goat anti‐rabbit IgG HRP conjugate was added to the samples followed by a second incubation for 2 h at 24°C on a shaker set to 450 rpm. Wells were washed again before addition of HRP substrate (100 μL). After 10–12 min, reactions were stopped by the addition of sulphuric acid (100 μl, 2 M), and sample absorbance was determined at 450 nm using a SpectraMax microplate reader (Molecular Devices, USA).
Quantification of plasma and liver lysophosphatidic acid
LPA was extracted as described previously (Scherer et al., 2009). In brief, 35 μL of plasma was transferred to a 96‐deep‐well plate together with disodium hydrogen phosphate buffer (200 μL of 40 mM) containing 30 mM citric acid (pH 4), followed by addition of 1 μM LPA internal standard. To extract the phospholipids, 500 μL 1‐butanol was added to samples, shaken vigorously for 10 min on a monoshake (Thermo Electron, Germany) and centrifuged at 4°C for 10 min. A sample of the upper butanolic phase (400 μL) was transferred to a 96‐deep‐well plate and evaporated by an ultravap (Porvair, UK) with a 60°C heated nitrogen flow (103.425 kPa for 45 min) until dry. The extract was dissolved in ethanol (100 μL), and LPA levels were determined by MS as described previously (Bretschneider et al., 2017). An API 6500 mass spectrometer (AB Sciex, Germany) was equipped with an Agilent 1290 LC system, a CTC autosampler and an Atlantis 50 × 2.1 mm, 3 μm HILIC LC column (Waters, UK) was used to determine levels of LPA. The instrument operated in negative mode with a source temperature of 300°C, cad gas = 50, gas 1 = 60, gas 2 = 60 and voltage of −4500 V. Declustering potential was set to −150 and collision energy to −28. The following MS transitions for the LPAs were recorded: 16:0 LPA: 409.2/152.8; 18:0 LPA: 437.3/152.8; 18:1 LPA: 435.3/152.8; 18:2 LPA: 433.2/152.8; 20:4 LPA: 457.2/152.8; and 17:0 LPA: 423.5/152.8.
The bi mobile phase chromatographic system was equipped with solvent A (0.2% formic acid and 50 mM ammonium formate in water) and solvent B (0.2% formic acid in acetonitrile). Separation of LPA and lysophosphatidylcholine (LPC) was achieved with a gradient starting from 95% solvent B, which decreasing to 75% over 90 s and to 50% solvent B over a further 12 s, with an increase in the flow rate from 500 to 700 μL·min−1. The column was re‐equilibrated at 108 s by set back of the solvent B concentration to 95%, which was maintained for 42 s. LPA was eluted at 117 s and LPC at 126 s with a peak width of 5 s. Quality control samples from 0.5 nM to 10 μM were measured to check the linearity of the LC–MS system and to control the robustness of the system. The lower limit of quantification was 12.5 nM. The absolute LPA levels were calculated and normalized to the mean of the control group to provide comparable datasets.
Histology assessment of livers in hepatic fibrosis models
For histological analysis of the CCl4‐treated animals, the right lobe of the liver was sectioned and fixed in phosphate‐buffered 10% formaldehyde. Each formaldehyde‐fixed sample was embedded in paraffin, cut into 4 μm‐thick sections and stained with haematoxylin and eosin (H&E) and Masson's trichrome according to standard procedures. All slides were scored by the same pathologist using the NASH activity score (NAS) as described previously (Kleiner et al., 2005). A semi‐quantitative analysis of steatosis, lobular inflammation and hepatocellular ballooning was assessed using the H&E‐stained sections. Fibrosis staging was performed on the Masson's trichrome stained liver samples. For quantitative analysis of collagen positive area and degree of steatosis, histological slides were systematically scanned with a Zeiss AxioScan.Z1 microscope (Zeiss, Germany). Images were analysed using a script based on HDevelop ImageAnalysis Toolbox (MVTec, Germany). Image segmentation was performed using texture and colour information from colour space transformation RGB to HSI and from colour deconvolution. In the images, liver sections were segmented and the area covered by liver segmented into mosaic tiles of size 1024*1024 pixels (from 500 to 1200 tiles per slide). For each tissue tile, the total area of tissue and the area with collagen rich tissue were detected and used to calculate a value describing fibrosis. The median value of all tiles was reported.
For the histological assessment of samples from the CDAA study, liver pre‐ and post‐biopsies from the left lateral lobe were fixed in paraformaldehyde (4%) overnight before paraffin embedding and sectioning (3 μm in depth). Sections were stained with H&E, Sirius Red (Sigma‐Aldrich, Germany) and anti‐collagen 1a1 antibody (1:300, Southern Biotech, USA; 2° antibody Bright Vision anti‐goat, ImmunoLogic, Netherlands). The stained sections were used for histological assessment and scoring as described above. In addition, collagen 1a1‐stained slices were analysed with Visiomorph software (Visiopharm) for quantification of collagen‐positive area.
Biochemical quantification of hepatic hydroxyproline content
Hepatic collagen content in samples from the CCl4 study was determined using 50–100 mg liver samples from two different lobes after hydrolysis in hydrochloric acid (6 M) for 16 h at 120°C. Samples were cooled to room temperature and centrifuged at 18 000× g for 10 min. Standards and samples were transferred to 96‐well plates and 50 μL of citrate‐acetate buffer was added. After addition of chloramine T solution (100 μL), plates were incubated for 20 min at room temperature before the addition of Ehrlich's reagent (100 μL; p–dimethylaminobenzaldehyde in ethanol:hydrochloric acid). Assay plates were incubated at 65°C for 15 min and then cooled to room temperature. Sample absorbance was measured at 558 nm using a SpectraMax microplate reader (Molecular Devices).
From the CDAA study, 25 mg liver tissue samples were transferred to FastPrep tubes containing zirconium beads and snap frozen in liquid nitrogen. Hydrochloric acid (20 μL per μg liver tissue, 6 M) was added to each sample tube and homogenized using the FastPrep homogenizer (MP Biomedicals, USA) for 1 min, followed by 3 min pause and further homogenization (1 min). The samples were incubated overnight at 95°C in a Binder oven (Binder, Germany). The following day, the samples were cooled to room temperature, mixed on a vortex and centrifuged (17 900× g for 10 min). Supernatants were transferred to tubes pre‐filled with charcoal (Sigma‐Aldrich), mixed on a vortex and centrifuged (17 900× g for 10 min) and the supernatants transferred to Micronic tubes (In Vitro Technologies, USA). Hydroxyproline content was determined using a Hydroxyproline Assay Kit (QuickZyme, Netherlands) according to the manufacturer's instructions.
Quantification of hepatic α smooth muscle actin content
Liver extracts (50 mg) were prepared by suspending them in 700 μL of MSD lysis buffer (Meso Scale Discovery, USA) supplemented with protease inhibitor cocktails (Thermo Fisher Scientific and Sigma‐Aldrich). Samples were homogenized for 30 s at 6000× g, 4°C, using a FastPrep homogenizer (MP Biomedicals, USA). Homogenates were centrifuged at 10 000× g at 4°C for 10 min. Supernatants were adjusted to a protein concentration of 3 mg·mL−1. An MSD Western‐replacement method (Meso Scale Discovery) using specific anti‐α smooth muscle actin (αSMA) antibody was used to quantify SMA in protein lysates. Samples (25 μL) were added to multi‐array 96‐well plates (high bind, Meso Scale Discovery) and incubated for 2 h at room temperature with gentle shaking. Non‐specific antibody binding was prevented by incubation of 150 μL of 3% blocking buffer (0.6 g of MSD Blocker A in 20 mL bidest) for 1 h at room temperature. For the detection of bound αSMA, an anti‐αSΜΑ antibody (1:5000; Sigma‐Aldrich) and goat anti‐mouse sulfo‐TAG antibody (1:167; Meso Scale Discovery) mixture was prepared in blocking buffer (1.5 mL, 3%) and Tris (4.47 mL, pH 7.4) wash buffer and 25 μL added per well for 1 h at room temperature. Plates were washed three times using 200 μL of Tris pH 7.4 wash buffer between all steps. The final detection reaction was initiated by the addition of 150 μL of MSD read buffer T, after which plates were analysed on a SECTOR S 600 plate reader (Meso Scale Discovery).
Real‐time PCR analysis of gene expression in rat livers
Tissue samples were treated prior to the isolation of nucleic acids with a phenol‐chloroform extraction protocol. First, samples were homogenized in Lysing Matrix D Tubes containing 700 μL RLT‐buffer (Qiagen) for 3 min at 3000× g, using a FastPrep®‐24 homogenizer (MP Biomedicals). Supernatants were transferred to fresh tubes, and 700 μL phenol‐chloroform‐isoamyl alcohol was added. Tubes were mixed and centrifuged at 12 000× g for 5 min before addition of chloroform‐isoamyl alcohol (500 μL). Tubes were incubated for 3 min at room temperature followed by another centrifugation step (12 000× g for 5 min). Upper phases were used for the extraction of RNA. Total RNA was isolated using RNeasy 96 Kit (Qiagen) according to the manufacturer's instructions. RNA concentrations were analysed by NanoDrop ND‐1000 UV–Vis spectrophotometer at 260 nm (Thermo Fisher Scientific). The purity of the RNA was confirmed using 260/280 nm ratios, and samples were stored at −80°C before further processing. For RT‐PCR, RNA samples (50–2000 ng) were reverse‐transcribed into cDNA with a high‐capacity cDNA archive kit (Thermo Fisher Scientific) in accordance with the manufacturer's protocol and the cDNA produced was stored at −20°C. For the analysis of gene‐specific mRNA expression, cDNA was amplified by real‐time PCR with specific primers and fluorescent‐labelled probes (Supporting Information Table S8) using a thermal cycler (Eppendorf). Results were evaluated using SDS software version 2.2 (Thermo Fisher Scientific). Expression levels were normalized against levels of 18 s RNA. For calculation of relative changes in gene expression, values of individual samples were divided by the mean value of samples from control animals.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data were analysed using GraphPad Prism 7.00 software. Results are shown as individual values or as mean ± SEM or SD. If not otherwise stated, ordinary one‐way ANOVA with Tukey's multiple comparison tests were used to evaluate statistical significance between control and treatment group data (*P < 0.05). In all experiments where Tukey's multiple comparison tests were applied, F‐test of the ANOVA demonstrated statistical significance (P < 0.05).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Effect of LPA on gene expression in primary human hepatic stellate cells
In primary human HSCs, 18:1 LPA at a concentration of 10 μM induced a time‐dependent increase in α smooth muscle actin gene 2 (ACTA2) expression reaching a maximum of −4.4‐fold at 6 h (Figure 1A). Levels of mRNA for connective tissue growth factor (CTGF) had increased after 1 h (7.6‐fold; Figure 1B). Expression of CTGF remained elevated in LPA treated cells until 6 h. A transient increase in expression of CCL2 and CXCL1 was observed, reaching maximal levels after 2 h of stimulation (18.8‐fold and 6.6‐fold, respectively; Figure 1C,D). By the 4 and 6 h time points, expression levels of CCL2 and CXCL1 were lower than at 2 h.
Figure 1.
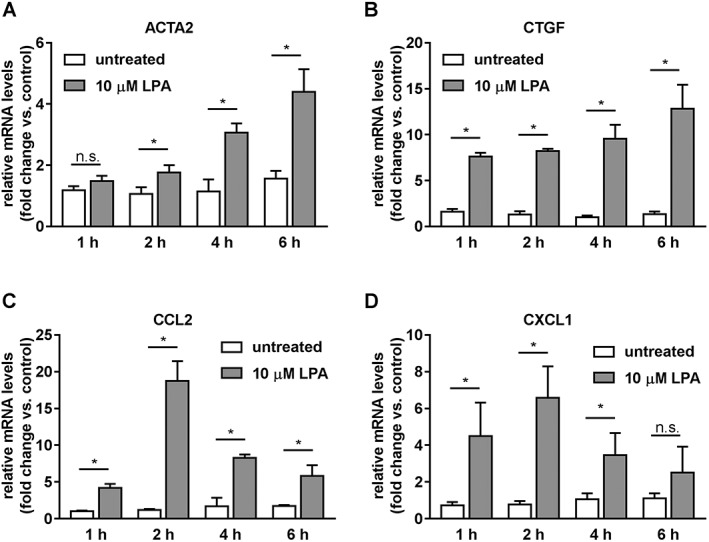
Effect of LPA on expression of fibrosis and inflammation marker genes in primary human HSCs. (A) ACTA2. (B) CTGF. (C) CCL2. (D) CXCL1. Data represent relative mRNA levels normalized versus levels of RNA‐polymerase II and calculated as fold change compared to untreated samples at time zero. Data represent mean ± SD of one representative experiment (n = 6 samples per group, each measured in technical duplicate), *P < 0.05.
Effect of LPA on TRAIL‐induced hepatic stellate cell apoptosis
Treatment of primary human HSCs with 30 ng·mL−1 TRAIL for 24 h induced apoptosis, as determined by a 7.1‐fold increase in caspase3/7 activity (Figure 2). LPA caused a concentration‐dependent suppression of TRAIL‐induced apoptosis with an estimated 50% inhibitory concentration (IC50) value of 3 μM, although at concentrations of up to 30 μM, 18:1 LPA had no effect on basal caspase3/7 activity.
Figure 2.
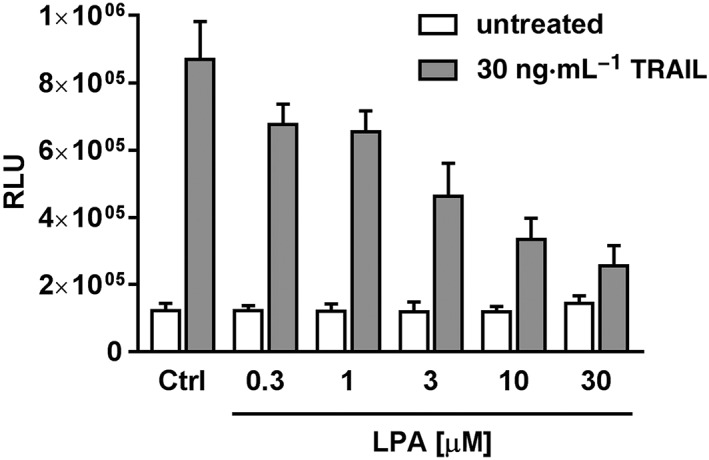
Effect of LPA on TRAIL‐induced apoptosis in primary human HSCs. Data represent relative fluorescence units (RFU) measured with a caspase3/7 activity assay after 24 h incubation. Data represent mean ± SD of one representative experiment (n = 7 samples per group, each measured in technical duplicate).
Characterization of a potent and selective autotaxin inhibitor
The potency of the ATX inhibitor Ex_31 was determined in vitro in a biochemical ATX assay (IC50 27 nM) and a rat whole blood assay (IC50 10 nM) (Table 1, Supporting Information Figure S2, Bretschneider et al., 2017). The selectivity of Ex_31 was evaluated by screening the compound at 10 μM in a diversity target profile (Eurofin Cerep, France, Supporting Information Table S3). The only target which was inhibited by more than 50% was PDE4D2. The IC50 of Ex_31 on PDE4D2 was determined at 4 μM. Clearance of Ex_31 after single i.v. dose (1 μmol·kg−1) was 13 mg·mL−1·kg−1 with a half‐life of 294 min. Dose normalized maximal concentration of 248 nM at 24 min (0.4 h) was achieved after a single administration of Ex_31 (10 μmol·kg−1). The in vivo IC50 of Ex_31 was estimated to be 33 nM (data not shown).
Table 1.
In vitro characterization of Ex_31 and in vivo pharmacokinetic properties in rats after single i.v. or p.o. dose
| Parameter | Ex_31 | |
|---|---|---|
| In vitro | ||
| Plasma protein binding [%] | 97.5 | |
| Cytochrome P450 inhibition IC50 [μM] | – | |
| 3A4/2D6/2C9/2C8/2C19 | >50/>50/>50/47/30 | |
| Caco permeability [10−6 cm·s−1] | 118 | |
| Caco efflux ratio | 1.1 | |
| IC50 potency on rat ATX [nM] | 27 | |
| IC50 potency on ATX in rat whole blood [nM] | 10 | |
| In vivo | i.v. | p.o. |
| Dose [mg·kg−1] | 1 | 10 |
| Dose normalized AUC [nM·h−1] | 857 | 551 |
| Clearance [mL·(min·Kg)−1] | 13 | – |
| % QH [%] | 18 | – |
| Vss [L·Kg−1] | 3.3 | – |
| Dose normalized cmax [nM] | – | 248 |
| Tmax [h] | 0.4 | |
| t 1/2 [h] | 4.9 | 6.2 |
| F [%] | – | 64 |
| IC50 potency on ATX [nM] | – | 33 |
Pharmacokinetic profile of Ex_31 and target engagement in healthy rats
Plasma exposure of Ex_31 reached more than 2 μM (~60‐fold in vivo IC50) 30 min after a single oral dose (10 μmol·kg−1, Figure 3). At 30 to 240 min post‐dosing, a reduction of 90% in maximal plasma LPA compared to baseline was observed. Based on these results, estimates suggested that administration of 15 mg·kg−1 Ex_31 twice daily would result in >95% reduction in plasma LPA over a 24 h period.
Figure 3.
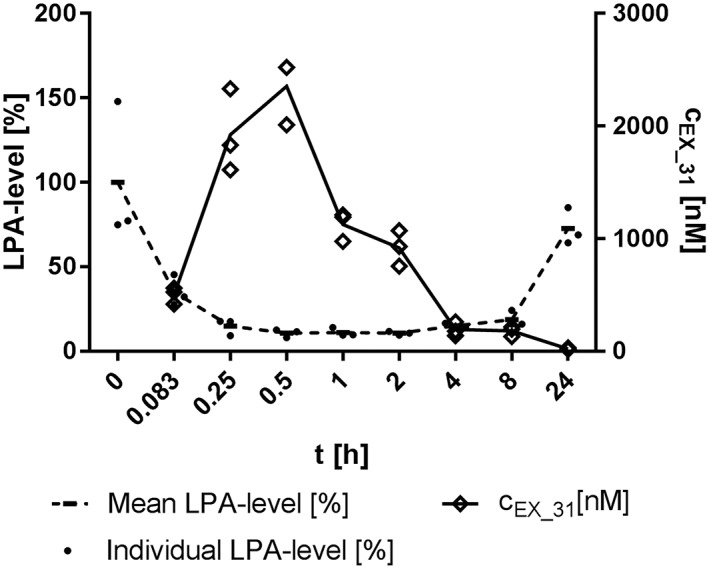
Pharmacokinetic profile of Ex_31 and plasma LPA levels after a 10 μmol·kg−1 single oral dose in rats. Relative levels of the sum of 16:0, 18:0, 18:1, 18:2 and 20:4 LPA normalized versus controls at time 0 are shown. CEX_31: plasma exposures of Ex_31. Data represent values from three individual animals.
Hepatic expression of LPA1 receptor and quantification of plasma and liver LPA levels in models of CCL4‐ and CDAA diet‐induced liver injury
To determine whether pharmacological inhibition of ATX prevents fibrosis development, we tested Ex_31 in a 10 week model of CCl4‐induced liver injury and in a 14 week model of CDAA diet‐induced liver injury. At the end of the CCl4 study, exposures of Ex_31 were 3525 ± 978 nM in plasma and 17 223 ± 2591 nM in livers 15–16 h after last compound administration. In the CDAA study, exposures of Ex_31 were 360 ± 113 nM in plasma and 3332 ± 767 nM in livers 15–16 h after last compound administration. In CCl4‐treated rats, LPAR1 mRNA expression was increased 25.8‐fold compared with control animals (P < 0.05, Figure 4A). Exposure of CCl4‐treated rats to Ex_31 did not result in significant changes in LPAR1 mRNA expression. LPA levels were increased in plasma (+36% compared to control, P < 0.05, Figure 4B) and in liver samples (+62% compared with control, P < 0.05, Figure 4C) of CCl4‐treated rats. Exposure of CCl4‐treated rats to Ex_31 resulted in a decrease in plasma LPA levels of >95%, whereas levels of LPA in liver homogenates were unchanged. In CDAA diet‐fed rats, LPAR1 mRNA expression was increased 12.2‐fold compared with CSAA diet‐fed animals (P < 0.05, Figure 4D). Exposure of CDAA diet‐fed rats to Ex_31 did not result in significant changes in LPAR1 mRNA expression. LPA levels in CDAA diet‐fed rats were increased in plasma (+39% compared with control, P < 0.05, Figure 4E) and in liver samples (+40% compared with control, P < 0.05, Figure 4F). Exposure of CDAA diet‐fed rats to Ex_31 resulted in a decrease in plasma LPA levels by more than 95%, whereas levels of LPA in liver homogenates were unchanged.
Figure 4.
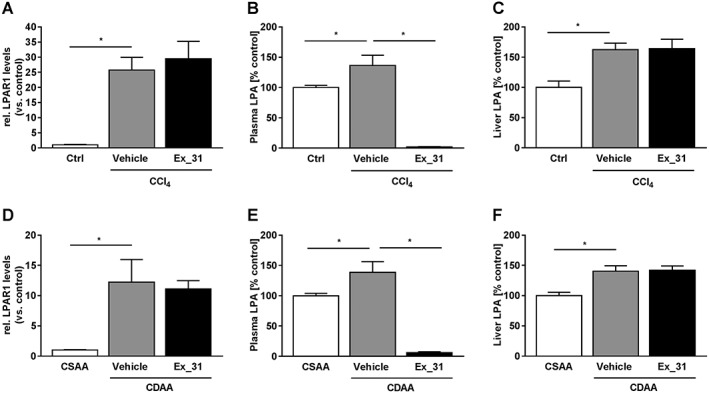
Expression levels of LPA receptors and LPA levels in models of chronic liver injury in rats. (A) Expression of LPA1 receptor (LPAR1) in control, CCl4/Vehicle‐ and CCl4/Ex_31‐treated rats. Levels of LPA in plasma (B) and livers (C) of control, CCl4/Vehicle‐ and CCl4/Ex_31‐treated rats. (D) Expression of LPAR1 in CSAA or CDAA diet‐fed rats treated with Vehicle or Ex_31. Levels of LPA in plasma (E) and livers (F) of CSAA or CDAA diet‐fed rats treated with Vehicle or Ex_31. Data in (A) and (D) represent relative mRNA levels normalized versus levels of 18 s RNA and calculated as fold change compared to controls. Data in B,C and E,F represent relative levels of the sum of 16:0 LPA, 18:0 LPA, 18:1 LPA, 18:2 LPA and 20:4 LPA normalized versus controls. Data represent mean ± SEM, n = 10 (control) and n = 13 (CCl4‐treated), *P < 0.05.
Effects of Ex_31 on body weight, liver weight and plasma alanine aminotransferase in models of CCL4‐ and CDAA diet‐induced of liver injury
Before Ex_31 administration and at study termination, body weights of CCl4‐treated rats and control animals were comparable (Table 2). At the end of the study, CCl4‐treated rats displayed signs of liver injury, including elevated plasma ALT levels (365.5 vs. 62.6 U·L−1; P < 0.05) and increased liver weights (25.8 vs. 18.0 g; P < 0.05) compared to control animals. Exposure of CCl4‐treated rats to Ex_31 did not result in significant changes in body weight, liver weight or plasma ALT levels. In the CDAA study, body weights between CDAA and CSAA diet‐fed rats were not significantly different; however, CDAA diet‐fed animals had elevated plasma ALT levels (56.3 vs. 40.5 U·L−1; P < 0.05) and increased liver weights (24.9 vs. 15.4 g; P < 0.05) compared to CSAA diet‐fed rats. Exposure of CDAA diet‐fed rats to Ex_31 did not result in significant changes in body weight, liver weight or plasma ALT levels.
Table 2.
Body weights, liver weights and plasma parameters in Control, CCl4/Vehicle and CCl4/Ex_31 treated rats and in CSAA or CDAA diet‐fed rats treated with Vehicle or Ex_31
| CCl4 study | CDAA study | |||||
|---|---|---|---|---|---|---|
| No CCl4 | CCl4 | CCl4 | CSAA | CDAA | CDAA | |
| Vehicle | Ex_31 | Vehicle | Ex_31 | |||
| Body weight at start of cpd treatment [g] | 551.6 ± 13.9 | 542.1 ± 8.8 | 534.5 ± 11.4 | 549.3 ± 9.3 | 530.6 ± 11.2 | 509.4 ± 15.2 |
| Body weight at study termination [g] | 608.3 ± 12.8 | 569.3 ± 12.5 | 571.4 ± 13.5 | 591.1 ± 12.4 | 592.0 ± 15.1 | 568.9 ± 21.4 |
| Liver weight [g] | 18.0 ± 0.5 | 25.8 ± 1.9* | 25.3 ± 2.2 | 15.4 ± 0.4 | 24.9 ± 1.4* | 23.8 ± 1.5 |
| ALT [U.L‐1] | 62.6 ± 2.1 | 365.5 ± 21.7* | 309.1 ± 23.2 | 40.5 ± 2.2 | 56.3 ± 2.1* | 63.6 ± 6.3 |
Values represent mean ± SEM, n = 10 (control), n = 13 (CCl4‐treated) and n = 12 (CSAA‐ and CDAA‐fed),
P < 0.05.
Effects of Ex_31 on histology in a rat model of CCL4‐induced liver injury
Rats treated with CCl4 presented signs of liver injury and bridging fibrosis (Figure 5A). In CCl4‐treated rats, NAS ranged from 4 to 6 (5.1 ± 0.14; Figure 5B) and fibrosis scores from 3 to 4 (3.5 ± 0.1, Figure 5C). Quantitative histological assessment based on Masson's trichrome staining revealed collagen‐positive area of 3.7 ± 0.5% in CCl4‐treated rats, as compared with 0.2 ± 0.0% in control rats (Figure 5D). Exposure of CCl4‐treated rats to Ex_31 did not result in significant changes in NAS (5.2 ± 0.2), fibrosis score (3.1 ± 0.3) or area of collagen‐positive staining (3.6 ± 0.9%).
Figure 5.
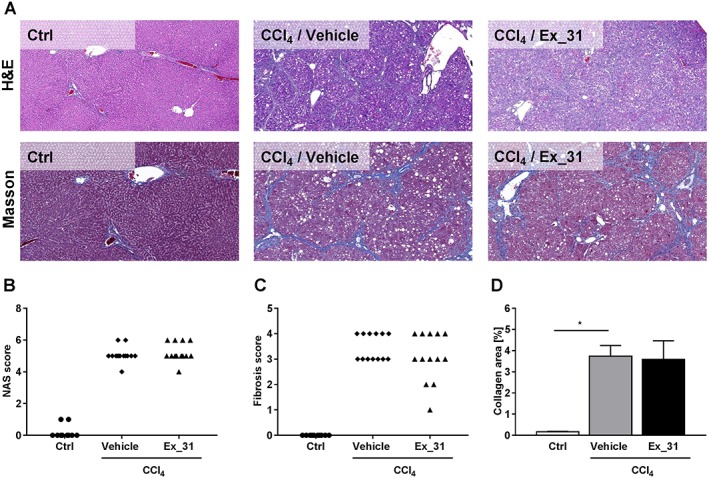
Histology assessment of liver sections from control rats and from rats with CCl4‐induced liver injury. (A) Representative images of H&E stain and Masson's trichrome stain (Masson). (B) NAS. (C) Fibrosis scores. (D) Image‐based quantification of collagen‐positive area in Masson's trichrome‐stained liver sections. Data in (B) and (C) represent scores of individual animals. Data in (D) represent mean ± SEM, n = 10 [control (Ctrl)] and n = 13 (CCl4‐treated), *P < 0.05.
Effects of Ex_31 on histology in a rat model of CDAA diet‐induced liver injury
Rats fed a CDAA diet presented signs of NASH and bridging fibrosis (Figure 6A). In CDAA diet‐fed rats, NAS scores ranged from 5 to 7 (5.8 ± 0.3; Figure 6B) and fibrosis scores from 0 to 4 (2.7 ± 0.4; Figure 6C). Quantitative histological assessment based on collagen I staining revealed collagen‐positive area of 15.2 ± 2.6% in CDAA diet‐fed rats, as compared with 4.7 ± 0.2% in CSAA diet‐fed animals (Figure 6D). Exposure of CDAA diet‐fed rats to Ex_31 resulted in NAS of 3 to 6 (4.8 ± 0.3), fibrosis scores of 1 to 4 (3.5 ± 0.3) and area of collagen‐positive staining of 18.6 ± 2.7%.
Figure 6.
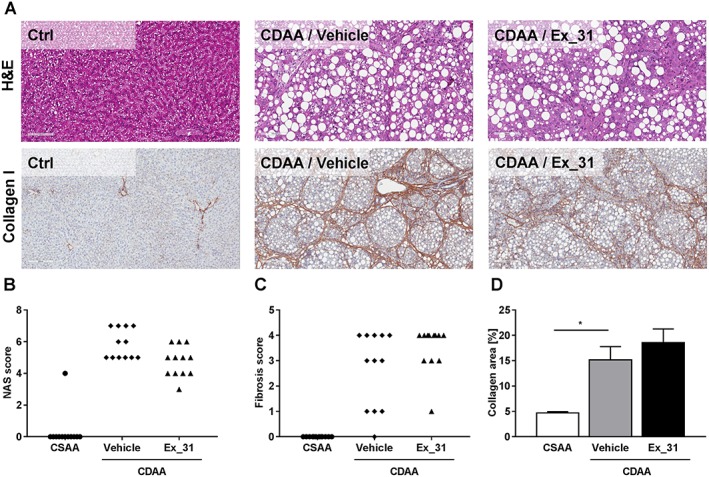
Histology assessment of liver sections from rats fed CSAA or CDAA diet. (A) Representative images of H&E stain and Masson's trichrome stain (Masson). (B) NAS. (C) Kleiner fibrosis scores. (D) Image‐based quantification of collagen‐positive area in Masson's trichrome stained liver sections. Data in (B) and (C) represent scores of individual animals. Data in (D) represent mean ± SEM, n = 12, *P < 0.05.
Effect of Ex_31 on hepatic hydroxyproline and α smooth muscle actin content in models of liver injury
In CCl4‐treated rats, hepatic hydroxyproline content was 3.3‐fold higher (P < 0.05; Figure 7A) and αSMA protein levels were 3.5‐fold higher (P < 0.05; Figure 7B) than in control animals. Exposure of CCl4‐treated rats to Ex_31 did not result in significant changes in hepatic hydroxyproline or αSMA contents. In CDAA diet‐fed rats, hepatic hydroxyproline content was 5.5‐fold higher (P > 0.05; Figure 7C) and αSMA protein levels were 2.9‐fold higher (P < 0.05; Figure 7D) than in CSAA diet‐fed animals. Exposure of CDAA diet‐fed rats to Ex_31 did not result in significant changes in hepatic hydroxyproline or αSMA contents.
Figure 7.
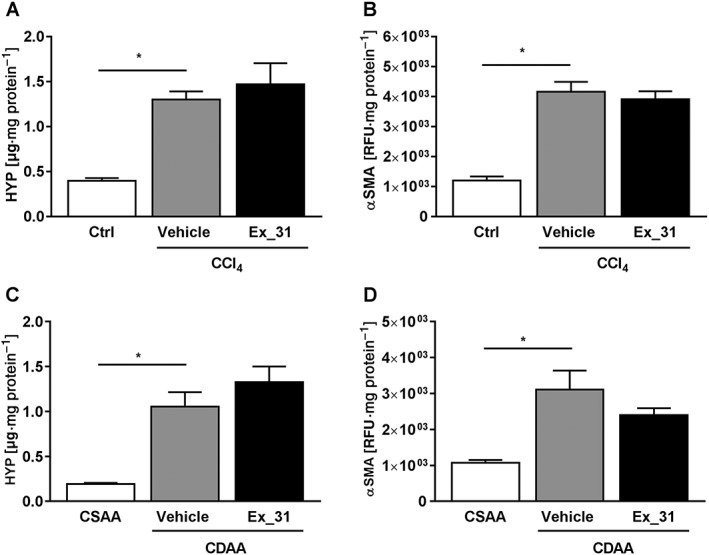
Biochemical quantification of hepatic hydroxyproline (HYP) content and αSMA in models of chronic liver injury in rats. (A) Hepatic HYP content in control, CCl4/Vehicle‐ and CCl4/Ex_31‐treated rats. (B) Hepatic αSMA content in control, CCl4/Vehicle‐ and CCl4/Ex_31‐treated rats. (C) Hepatic HYP content in CSAA or CDAA diet‐fed rats treated with Vehicle or Ex_31. (D) Hepatic αSMA content in CSAA or CDAA diet‐fed rats treated with Vehicle or Ex_31. Values are normalized versus liver protein content. Data represent mean ± SEM, n = 10 (control), n = 13 (CCl4‐treated) and n = 12 (CSAA‐ and CDAA‐fed), *P < 0.05.
Effect of Ex_31 on expression of inflammation and fibrosis marker genes in models of liver injury
In livers of CCl4‐treated rats, mRNA levels of Ccl2, Cxcl1, intergin α M (Itgam), EGF‐like module‐containing mucin‐like hormone receptor‐like 1 (Emr1), TNFα (Tnfa) and TGFβ (Tgfb) were increased 6.5‐, 3.1‐, 25.0‐, 6.0‐, 9.2‐ and 33.0‐fold respectively (P < 0.05 for all genes; Figure 8A). Exposure of CCl4‐treated rats to Ex_31 did not result in significant changes in mRNA expression levels of these genes. In livers of CDAA diet‐fed rats, mRNA levels of Ccl2, Cxcl1, Itgam, Emr1, Tnfa and Tgfb were increased 8.3‐, 4.3‐, 12.8‐, 1.8‐, 5.3‐ and 4.6‐fold, respectively (P < 0.05 for all genes; Figure 8B). Exposure of CDAA diet‐fed rats to Ex_31 did not result in significant changes in mRNA expression levels of these genes.
Figure 8.
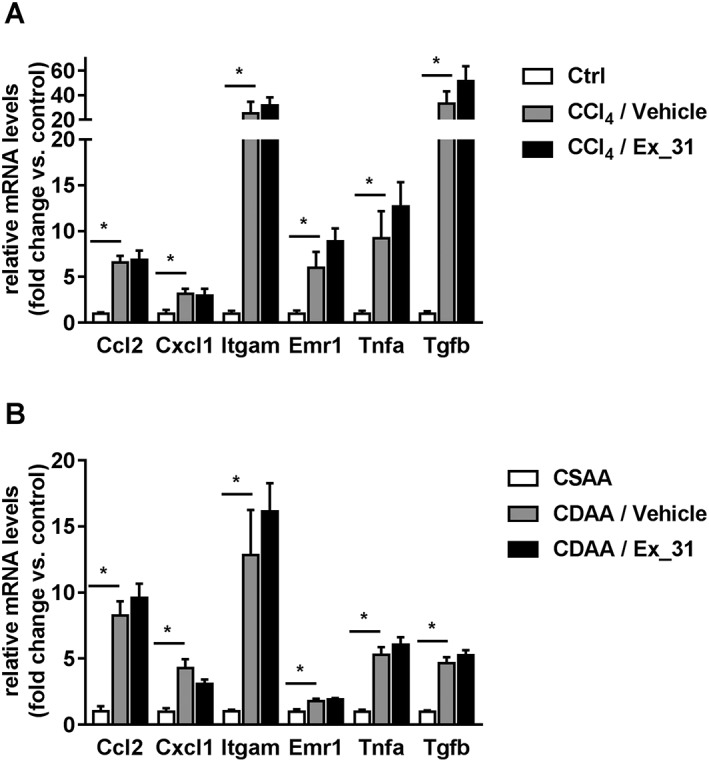
Hepatic expression levels of inflammation and fibrosis marker genes in models of chronic liver injury in rats. (A) Ccl2, Cxcl1, Itgam, Emr1, Tnfa and Tgfb expression in livers of control (Ctrl), CCl4/Vehicle‐ and CCl4/Ex_31‐treated rats. (B) Gene expression in livers of CSAA or CDAA diet‐fed rats treated with Vehicle or Ex_31. Data represent relative mRNA levels normalized versus levels of 18 s RNA and calculated as fold change compared to controls. Data represent mean ± SEM, n = 10 (Ctrl), n = 13 (CCl4‐treated) and n = 12 (CSAA‐ and CDAA‐fed). Unpaired t‐test for each gene, *P < 0.05.
Discussion
The aim of the present work was to explore the role of the LPA/ATX axis in the pathogenesis of NASH‐related fibrosis by characterization of the properties of a selective small molecule ATX inhibitor in preclinical models of advanced liver fibrosis.
NASH is a progressive liver disease characterized by hepatic fat accumulation and lobular hepatitis in the absence of viral hepatitis, biliary disease or a history of alcoholism. Persistent inflammation and oxidative stress can result in hepatocyte damage and HSC activation, ultimately resulting in fibrogenesis (Maher, 2016). Expression of LPA receptors in fibroblasts has previously been demonstrated (Stortelers et al., 2008), and several effects of LPA on HSCs have been described that could contribute to the development of fibrosis (Yanase et al., 2000; Tangkijvanich et al., 2002).
Effects of LPA on cytokine, chemokine and fibrosis marker gene expression have been described in mouse embryonic fibroblasts (Stortelers et al., 2008). We have demonstrated expression of LPA1 receptors and to a lesser extent LPA6 receptors in culture‐activated primary human HSCs from three different donors (Supporting Information Figure S1). To investigate the effect of LPA in primary human HSCs, we have treated cells with 10 μM LPA. While normal serum LPA concentrations are in the range of 1 μM (Baker et al., 2001), these may be higher under pathological conditions. Based on previously published in vitro studies, 10 μM represents a 90% effective concentration (Tangkijvanich et al., 2002). We have observed up‐regulation of fibrosis related genes (ACTA2 and CTGF) and chemokine‐encoding genes (CCL2 and CXCL1) in 18:1 LPA‐treated primary human HSCs. These results suggest that in vivo, elevated LPA levels could stimulate HSC activation and chemokine‐dependent immune cell recruitment, both of which are hallmarks of human NASH. Since the elimination of HSCs by TRAIL‐induced apoptosis has been reported to protect rats from CCl4‐induced liver injury (Xu et al., 2016), we investigated the effect of LPA on TRAIL‐induced HSC apoptosis in vitro. We observed the effective suppression of TRAIL‐induced HSC apoptosis by 18:1 LPA, which may result in increased HSC survival in pathogenic settings and thereby promote the development of fibrosis. These in vitro observations prompted us to evaluate the role of the main LPA producing enzyme ATX in the pathogenesis of liver fibrosis in vivo.
Elevated plasma LPA levels have been observed in different acute and subchronic models of liver injury (Watanabe et al., 2007). To evaluate the potential role of the ATX/LPA axis in the pathogenesis of liver fibrosis, we tested a selective small molecule ATX inhibitor, Ex_31, in a 10 week model of CCl4‐induced liver fibrosis and in a more metabolic driven 14 week model of CDAA diet‐induced liver injury. The rat was used as the species for proof of concept studies based on the observation that ATX inhibition resulted in an almost complete depletion in plasma LPA.
In the present study, we used Ex_31 as a selective ATX inhibitor with oral bioavailability, favourable pharmacokinetic properties and in vivo target inhibition. In the CCl4 study, we observed increased hepatic LPAR1 expression and plasma LPA levels, indicating an up‐regulation of the LPA signalling pathway as previously described in several preclinical models of liver injury (Watanabe et al., 2007). LPA levels were elevated not only in plasma but also in liver tissue of diseased animals. Interestingly, while plasma LPA was reduced by >95% in animals exposed to Ex_31, LPA levels in liver tissue of these animals remained unchanged, indicating an ATX‐independent pathway for intracellular LPA production. Indeed, different enzymes including an acylglycerol kinase that produce intracellular LPA have been described (Pages et al., 2001; Bektas et al., 2005). To further address this point, we have measured ATX activity ex vivo in plasma and homogenates of un‐perfused and perfused rat liver sections. As expected, we observed 18:2 LPA synthesis after incubation of plasma with 10 μM 18:2 LPC (Supporting Information Figure S11). While we also observed 18:2 LPA synthesis in homogenates of un‐perfused livers incubated with 18:2 LPC, we did not detect any LPA synthesis in homogenates of perfused livers. These results indicate that in healthy rats, liver ATX activity is restricted to the plasma and that intracellular LPA must be synthesized through ATX‐independent pathways. These findings may explain our observation that liver LPA levels were not reduced in Ex_31‐treated rats in spite of high liver compound exposure.
In the CCl4‐induced liver injury study, efficient pharmacological inhibition of ATX did not result in the consistent improvement of markers for liver injury, inflammation or fibrosis. The lack of any signs of anti‐inflammatory or anti‐fibrotic efficacy of Ex_31, despite excellent target engagement, prompted us to challenge the relevance of the CCl4 model for the evaluation of a potential role of the ATX/LPA axis in the pathogenesis of hepatic fibrosis. Continuous CCl4 challenges could result in the activation of HSCs, which may mask a potential pathophysiological role for LPA, such as HSC activation or protection of HSCs from TRAIL‐induced apoptosis as shown in our in vitro studies. Indeed, strong induction of HSC proliferation followed by apoptosis has been observed after single dose administration of CCl4 in Sprague–Dawley rats (Lee et al., 2003).We also tested Ex_31 in a second, diet‐induced model of liver injury in rats. Multiple NASH‐related models have previously been described in the literature (Ibrahim et al., 2016). Here, we performed a 14 week study with a modified version of a CDAA diet supplemented with 1% cholesterol. This model was recently used to characterize the efficacy of the PPAR agonist Elafibranor (Noel et al., 2015). In that study, supplementation of a classical CDAA diet with 1% cholesterol markedly increased hepatocyte ballooning and fibrosis development. In line with these observations, the addition of 1% cholesterol to a 15% high‐fat diet has previously been shown to trigger fibrosis development over the course of 30 weeks of exposure (Savard et al., 2013). In the present CDAA study, plasma LPA levels were increased moderately after 14 weeks of CDAA diet feeding. However, levels of the ATX substrate LPC were markedly lower in CDAA diet‐fed rats compared with rats receiving CSAA diet (Supporting Information Figure S6). As a consequence, plasma LPA/LPC ratios were significantly higher in CDAA diet‐fed rats than in CSAA diet‐fed animals, indicating increased plasma ATX activity. As observed in the CCl4 study, LPA levels were not only increased in plasma but also in the livers of CDAA diet‐fed rats. While exposure of CDAA diet‐fed animals to Ex_31 resulted in a decrease in plasma LPA by >95%, no lowering of liver LPA was observed. The CDAA study confirmed the observation made in the CCl4 model that ATX inhibition by Ex_31 had no beneficial effects on a majority of markers of liver injury, inflammation and fibrosis. The finding that liver tissue LPA levels were increased in both models of liver injury, but unchanged in animals exposed to Ex_31, raises the question of whether or not intracellular LPA could contribute to the pathogenesis of liver injury and fibrosis. Indeed, intracellular pro‐inflammatory effects of LPA have previously been reported in bronchial epithelial cells (Kalari et al., 2009). To investigate the potential of intracellular LPA to diffuse outside the cell and bind to LPA receptors, we investigated passive permeability of different LPA species in a parallel artificial membrane permeability assay. All tested LPA species showed a very good permeability at pH 5, 6.5, 7.4 (Supporting Information Table S7). Therefore, intracellular LPA could be released from the cells and subsequently contribute to classical LPA receptor‐dependent signalling.
A different ATX small molecule inhibitor developed by PharmAkea (PAT‐505) was recently tested in two diet‐induced models of liver injury in mice (Bain et al., 2017). In the STAM™ model, administration of PAT‐505 (10 mg·kg−1) once per day resulted in a significant reduction in NAS and fibrotic area. However, it was noted that the observed effects were not dose dependent and thus inconclusive. In a second CDAA high‐fat diet model, PAT‐505 significantly reduced fibrosis development but had no effect on steatosis, inflammation or hepatocyte ballooning. When comparing these results with our studies, it is worth mentioning that the mouse models were characterized by lower degrees of fibrosis development.
The role of ATX in liver injury has also been investigated with hepatocyte‐specific conditional ATX deletion in a 4 week model of CCl4‐induced liver injury in AlbENPP2−/− mice (Kaffe et al., 2017). In this study, mice deficient in hepatic ATX displayed reduced markers of fibrosis compared to control mice following CCl4 treatment. In contrast, ATX deficiency had no significant effect on markers of inflammation. In their study, substantial transient increase in hepatic ATX mRNA expression and protein peaking at 4 weeks of CCl4 treatment was shown. Elevated plasma ATX activity and plasma LPA levels were reduced to baseline in CCl4‐treated (4 weeks) AlbENPP2−/− mice. Interestingly, plasma ATX and LPA levels were unaffected in AlbENPP2−/− control animals not receiving CCl4. These data suggest that the observed increase in plasma ATX and LPA in CCl4‐treated mice is derived from hepatocytes. However, hepatocytes appear not to contribute to basal plasma ATX and LPA levels in healthy animals. In conclusion, the observation that liver ATX mRNA and protein levels returned to baseline at 8 and 12 weeks of CCl4 treatment indicates that hepatocytes may only produce relevant amounts of ATX during the initiation phase of CCl4‐induced liver injury, but not at later stages. This hypothesis is in alignment with our data, since in our study, we treated the animals with ATX inhibitor in a therapeutic intervention mode from week 6 to 10 of CCl4 treatment. This may explain lack of liver LPA lowering by the ATX inhibitor in our study.
Similar effects as described with AlbENPP2−/− mice were observed in CCl4‐treated mice exposed to the ATX inhibitor PF‐8380 (30 mg·kg−1, intraperitoneally, twice daily). Plasma ATX activity and liver LPA levels were reduced by approximately 50% in PF‐8380‐treated mice. These results are in contrast to our studies, where almost complete inhibition of plasma ATX did not result in reduced LPA levels in liver tissue. As discussed above, due to the transient increase in liver ATX expression in CCl4‐treated mice peaking at 4 weeks, the duration of the CCl4 treatment in the two studies may explain the different outcomes. Furthermore, we used a different species, and the relevance of ATX for the generation of LPA in the liver may be different in mice and rats.
In summary, we characterized the properties of a selective ATX inhibitor, Ex_31, in two models of advanced liver fibrosis in rats. In both studies, exposure of animals to Ex_31 did not result in significant changes in inflammation and fibrosis markers in spite of efficient target inhibition resulting in >95% reduction in plasma LPA levels. These results are in contrast to previous publications (Bain et al., 2017; Kaffe et al., 2017), where anti‐inflammatory and anti‐fibrotic effects of ATX inhibition or depletion have been shown in models of liver injury in mice. However, in contrast to the published studies, the species we used was rats, and our models were characterized by advanced stages of liver fibrosis up to cirrhosis. Both differences may account for the observed discrepancies between the studies. Taken together, our findings question the value of ATX as a new target for the treatment of advanced stages of NASH‐related liver fibrosis.
Author contributions
M.B. was responsible for the general conception of the studies, in vitro studies with hepatic stellate cells and wrote the manuscript; T.B. was responsible for the in vitro characterization of Ex_31, pharmacokinetic studies, LPA and LPC analytics; A.B. was responsible for the CCl4 study and interaction with the contract research organization Gubra for the CDAA study; J.F.R. contributed to biochemical readouts and gene expression analysis of the in vivo studies; B.S. did the histology assessments; C.A.K. selected the ATX inhibitor and oversaw its synthesis; M.M. revised the design of the studies and the manuscript.
Conflict of interest
All authors are full employees of Boehringer Ingelheim Pharma GmbH & Co. KG.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Expression of LPA receptors (LPAR) in primary human hepatic stellate cells from 3 different donors. Data represent relative mRNA levels normalized versus levels of RNA‐polymerase II (RNAPol2). Dots represent three individual donors.
Figure S2 IC50 curves of EX_31 from the (A) biochemical and (B) rat whole blood assay. The potency of Ex_31 was determined as described recently (Bretschneider et al., 2017). Briefly, the reaction buffer consisted of 50 mM Tris (pH 8.0), 3 mM KCl, 1 mM CaCl2, 1 mM Mg Cl2, 0.14 mM NaCl and 0.1% bovine serum albumin, which was supplemented with 5 nM recombinant rat ATX. Ex_31 was added to the assay solution in a concentration range from 0.1 nM to 10 μM. To 2.5 μL of this solution, 2.5 μL of a10 μM 18:1 LPC substrate solution was added to initiate the biochemical reaction. After 2 h incubation at room temperature the samples were analysed by LC–MS. The whole blood potency was determined by incubation of 45 μL heparinized rat whole blood with a serial dilution of Ex_31 (0.12 to 100 μM). After 1 h at 37°C the reaction was stopped by addition of 100 μL 40 mM disodium hydrogen phosphate buffer containing 30 mM citric acid (pH 4) and 1 μM 17:0 LPA (internal standard). Subsequently, LPA was extracted by liquid–liquid extraction using 500 μL butanol. The samples were analysed by LC–MS. IC50 values were calculated by applying a sigmoidal Hill equation.
Table S3 Eurofin Cerep profile of Ex_31. Values represent relative activities in Ex_31 treated samples (10 μM) compared to untreated controls.
Table S4 In the CCl4 study, male Sprague Dawley rats were administered CCl4 (0.25 mL kg−1 in olive oil, twice weekly) for a total duration of 10 weeks. After 6 weeks, animals were stratified based on plasma collagen IV (Col. IV) and 18:1 LPA levels. Each row represents one individual animal, which was assigned to the Vehicle (columns 1 and 2) or Ex_31 (columns 3 and 4) group.
Table S5 Male Wistar rats were fed a CDAA diet for a total duration of 14 weeks. After 9 weeks, animals were stratified based on histology assessment of liver biopsies. Values represent quantification of fibrotic area based on collagen I‐stained liver sections. Each row represents one individual animal, which was assigned to the Vehicle (column 1) or Ex_31 (column 2) group.
Figure S6 Normalized plasma lysophosphytidylcholine (LPC) levels and lysophosphatidic acid to LPC ratios in choline‐deficient amino acid‐defined diet‐induced liver injury model. Data represent mean ± SEM, n = 12, **P < 0.01.
Table S7 Permeability of LPA species in cm ·sec−1.
Table S8 List of TaqMan assays and primers/probes for RT‐PCR.
Figure S9 Synthesis scheme for ATX inhibitor Ex_31 (Example 31(24)).
Figure S10 Analytical Data for Ex_31: Rf = 0.4 (10% MeOH:Dichloromethane); m.p.: 176–180°C; 1H–NMR (400 MHz, DMSO‐d6) δ ppm 12.01 (s, 1 H), 8.20 (dd, J = 4.7, 1.3 Hz, 1 H), 7.93–7.90 (m, 1 H), 7.50–7.46 (m, 1 H), 7.20–7.17 (m, 1 H), 7.11 (dd, J = 7.8, 4.8 Hz, 1 H), 6.90–6.83 (m, 1 H), 5.53–5.48 (m, 2 H), 4.66 (s, 2 H), 3.82–3.75 (m, 2 H), 2.79–2.66 (m, 2 H), 2.34–2.06 (m, 3 H), 1.90–1.65 (m, 5 H), 1.34–1.22 (m, 2 H), 1.05–0.86 (m, 2 H); m/z = 484.2 [M + H] + .
Figure S11 Ex‐vivo measurement of ATX activity in plasma and homogenates from un‐perfused and perfused livers. Heparin plasma and un‐perfused and perfused livers rat livers were collected and immediately frozen at −20°C until analysis. 100 mg liver were homogenized in 500 μL PBS buffer using a dry iced cooled Precyllis Evolution (Bertin Technologies). The cell debris were pelleted by centrifugation at 10000 rpm, 10 min and 4°C. 400 μL of the supernatants or 35 μL plasma were supplemented with 10 μM 18:2 LPC and samples were incubated at 37°C. After 1 h the reaction was stopped by the addition of 40 mM disodiumhydrogenphosphate buffer containing 30 mM citric acid (pH 4) and 1 μM 17:0 LPA (internal standard). Subsequently, LPA was extracted by liquid–liquid extraction using 500 μL butanol as described above. Data represent individual values and mean ± SD, n = 3.
Figure S12A Individual relative plasma LPA species in CCl4 model. LPA levels were determined as described in the materials and methods section. Data represent mean ± SEM, n = 10 (control) and n = 13 (CCl4‐treated), *P < 0.05, n.s. = not significant. Levels were below detection limit in several samples of the vehicle and CCl4/Ex_31 groups for 16:0 LPA and in the CCl4/Ex_31 group for 18:0 LPA and 18:1 LPA. Thus statistical power calculations are not applicable for these groups (NA).
Figure S12B Individual relative liver LPA species in CCl4 model. LPA levels were determined as described in the materials and methods section. Data represent mean ± SEM, n = 10 (control) and n = 13 (CCl4‐treated), *P < 0.05, n.s. = not significant.
Figure S12C Individual relative plasma LPA species in CDAA model. LPA levels were determined as described in the materials and methods section. Data represent mean ± SEM, n = 12, *P < 0.05, n.s. = not significant. Levels were below detection limit in several samples of the CDAA/Ex_31 groups for 16:0 LPA, 18:0 LPA and 18:1 LPA. Thus statistical power calculations are not applicable for these groups (NA).
Figure S12D Individual relative liver LPA species in CDAA model. LPA levels were determined as described in the materials and methods section. Data represent mean ± SEM, n = 12, *P < 0.05, n.s. = not significant.
Acknowledgement
We thank the corresponding scientists at Gubra (Denkmark) for excellent interaction during the phase and execution of the CDAA study.
Baader, M. , Bretschneider, T. , Broermann, A. , Rippmann, J. F. , Stierstorfer, B. , Kuttruff, C. A. , and Mark, M. (2018) Characterization of the properties of a selective, orally bioavailable autotaxin inhibitor in preclinical models of advanced stages of liver fibrosis. British Journal of Pharmacology, 175: 693–707. doi: 10.1111/bph.14118.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174 (Suppl 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain G, Shannon KE, Huang F, Darlington J, Goulet L, Prodanovich P et al (2017). Selective inhibition of autotaxin is efficacious in mouse models of liver fibrosis. J Pharmacol Exp Ther 360: 1–13. [DOI] [PubMed] [Google Scholar]
- Baker DL, Desiderio DM, Miller DD, Tolley B, Tigyi GJ (2001). Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography‐mass spectrometry. Anal Biochem 292: 287–295. [DOI] [PubMed] [Google Scholar]
- Barbayianni E, Kaffe E, Aidinis V, Kokotos G (2015). Autotaxin, a secreted lysophospholipase D, as a promising therapeutic target in chronic inflammation and cancer. Prog Lipid Res 58: 76–96. [DOI] [PubMed] [Google Scholar]
- Bektas M, Payne SG, Liu H, Goparaju S, Milstien S, Spiegel S (2005). A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J Cell Biol 169: 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretschneider T, Luippold AH, Romig H, Bischoff D, Klinder K, Nicklin P et al (2017). Ultrafast and predictive mass spectrometry‐based autotaxin assays for label‐free potency screening. SLAS Discov 22: 425–432. [DOI] [PubMed] [Google Scholar]
- Castagna D, Budd DC, Macdonald SJ, Jamieson C, Watson AJ (2016). Development of autotaxin inhibitors: an overview of the patent and primary literature. J Med Chem 59: 5604–5621. [DOI] [PubMed] [Google Scholar]
- Chu X, Wei X, Lu S, He P (2015). Autotaxin‐LPA receptor axis in the pathogenesis of lung diseases. Int J Clin Exp Med 8: 17117–17122. [PMC free article] [PubMed] [Google Scholar]
- Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J et al (2013). Diet‐induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol 305: G483–G495. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim SH, Hirsova P, Malhi H, Gores GJ (2016). Animal models of nonalcoholic steatohepatitis: eat, delete, and inflame. Dig Dis Sci 61: 1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffe E, Katsifa A, Xylourgidis N, Ninou I, Zannikou M, Harokopos V et al (2017). Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 65: 1369–1383. [DOI] [PubMed] [Google Scholar]
- Kalari S, Zhao Y, Spannhake EW, Berdyshev EV, Natarajan V (2009). Role of acylglycerol kinase in LPA‐induced IL‐8 secretion and transactivation of epidermal growth factor‐receptor in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 296: L328–L336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Ikeda H, Miyakawa S, Futakawa S, Nonaka Y, Fujiwara M et al (2016). Structural basis for specific inhibition of autotaxin by a DNA aptamer. Nat Struct Mol Biol 23: 395–401. [DOI] [PubMed] [Google Scholar]
- Khanna D, Denton CP, Jagerschmidt A, Jasson M, Distler O, Allanore Y (2014). SAR100842, an antagonist of lysophaphatidic acid receptor 1, as a potential treatment for patients with systemic sclerosis: results from a phase 2a study. In ACR/ARHP Annual Meeting. Boston, MA.
- Kihara Y, Maceyka M, Spiegel S, Chun J (2014). Lysophospholipid receptor nomenclature review: IUPHAR review 8. Br J Pharmacol 171: 3575–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW et al (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41: 1313–1321. [DOI] [PubMed] [Google Scholar]
- Kondo M, Ishizawa T, Enooku K, Tokuhara Y, Ohkawa R, Uranbileg B et al (2014). Increased serum autotaxin levels in hepatocellular carcinoma patients were caused by background liver fibrosis but not by carcinoma. Clin Chim Acta 433: 128–134. [DOI] [PubMed] [Google Scholar]
- Kristiansen MN, Veidal SS, Rigbolt KT, Tolbol KS, Roth JD, Jelsing J et al (2016). Obese diet‐induced mouse models of nonalcoholic steatohepatitis‐tracking disease by liver biopsy. World J Hepatol 8: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc R, Peyruchaud O (2015). New insights into the autotaxin/LPA axis in cancer development and metastasis. Exp Cell Res 333: 183–189. [DOI] [PubMed] [Google Scholar]
- Lee JI, Lee KS, Paik YH, Nyun Park Y, Han KH, Chon CY et al (2003). Apoptosis of hepatic stellate cells in carbon tetrachloride induced acute liver injury of the rat: analysis of isolated hepatic stellate cells. J Hepatol 39: 960–966. [DOI] [PubMed] [Google Scholar]
- Llona‐Minguez S, Ghassemian A, Helleday T (2015). Lysophosphatidic acid receptor (LPAR) modulators: the current pharmacological toolbox. Prog Lipid Res 58: 51–75. [DOI] [PubMed] [Google Scholar]
- Luippold AH, Arnhold T, Jorg W, Kruger B, Sussmuth RD (2011). Application of a rapid and integrated analysis system (RIAS) as a high‐throughput processing tool for in vitro ADME samples by liquid chromatography/tandem mass spectrometry. J Biomol Screen 16: 370–377. [DOI] [PubMed] [Google Scholar]
- Maher JJ (2016). Pathogenesis of NAFLD and NASH In: Chalasani N, Szabo G. (eds). Alcoholic and Non‐Alcoholic Fatty Liver Disease: Bench to Bedside. Springer International Publishing: Switzerland, pp. 71–101. [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meeteren LA, Ruurs P, Stortelers C, Bouwman P, van Rooijen MA, Pradere JP et al (2006). Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol 26: 5015–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Ikeda H, Nakamura K, Ohkawa R, Masuzaki R, Tateishi R et al (2011). Autotaxin as a novel serum marker of liver fibrosis. Clin Chim Acta 412: 1201–1206. [DOI] [PubMed] [Google Scholar]
- Noel B, Rigou G, Degallaix N, Daix V, Cambula L, Roudot A et al (2015). Elafibranor (GFT505) prevents nash, hepatic fibrosis and hepatocarcinoma in a new disease model. In The Liver Meeting 2015 – AASLD. San Francisco, CA.
- Pages C, Simon MF, Valet P, Saulnier‐Blache JS (2001). Lysophosphatidic acid synthesis and release. Prostaglandins Other Lipid Mediat 64: 1–10. [DOI] [PubMed] [Google Scholar]
- Pasquinelli C (2013). Safety and efficacy of a lysophosphatidic acid receptor antagonist (bMS 986020) in idiopathic pulmonary fibrosis. In PFF Summit. San Diego, CA.
- Perrakis A, Moolenaar WH (2014). Autotaxin: structure‐function and signaling. J Lipid Res 55: 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleli T, Martin D, Kronenberger B, Brunner F, Koberle V, Grammatikos G et al (2014). Serum autotaxin is a parameter for the severity of liver cirrhosis and overall survival in patients with liver cirrhosis‐‐a prospective cohort study. PLoS One 9: e103532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savard C, Tartaglione EV, Kuver R, Haigh WG, Farrell GC, Subramanian S et al (2013). Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 57: 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer M, Schmitz G, Liebisch G (2009). High‐throughput analysis of sphingosine 1‐phosphate, sphinganine 1‐phosphate, and lysophosphatidic acid in plasma samples by liquid chromatography‐tandem mass spectrometry. Clin Chem 55: 1218–1222. [DOI] [PubMed] [Google Scholar]
- Sevastou I, Kaffe E, Mouratis MA, Aidinis V (2013). Lysoglycerophospholipids in chronic inflammatory disorders: the PLA(2)/LPC and ATX/LPA axes. Biochim Biophys Acta 1831: 42–60. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stortelers C, Kerkhoven R, Moolenaar WH (2008). Multiple actions of lysophosphatidic acid on fibroblasts revealed by transcriptional profiling. BMC Genomics 9: 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Okudaira S, Kishi Y, Ohkawa R, Iseki S, Ota M et al (2006). Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J Biol Chem 281: 25822–25830. [DOI] [PubMed] [Google Scholar]
- Tangkijvanich P, Melton AC, Chitapanarux T, Han J, Yee HF (2002). Platelet‐derived growth factor‐BB and lysophosphatidic acid distinctly regulate hepatic myofibroblast migration through focal adhesion kinase. Exp Cell Res 281: 140–147. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Ikeda H, Nakamura K, Ohkawa R, Kume Y, Tomiya T et al (2007). Plasma lysophosphatidic acid level and serum autotaxin activity are increased in liver injury in rats in relation to its severity. Life Sci 81: 1009–1015. [DOI] [PubMed] [Google Scholar]
- Xu F, Zhou D, Meng X, Wang X, Liu C, Huang C et al (2016). Smad2 increases the apoptosis of activated human hepatic stellate cells induced by TRAIL. Int Immunopharmacol 32: 76–86. [DOI] [PubMed] [Google Scholar]
- Yanase M, Ikeda H, Matsui A, Maekawa H, Noiri E, Tomiya T et al (2000). Lysophosphatidic acid enhances collagen gel contraction by hepatic stellate cells: association with rho‐kinase. Biochem Biophys Res Commun 277: 72–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Expression of LPA receptors (LPAR) in primary human hepatic stellate cells from 3 different donors. Data represent relative mRNA levels normalized versus levels of RNA‐polymerase II (RNAPol2). Dots represent three individual donors.
Figure S2 IC50 curves of EX_31 from the (A) biochemical and (B) rat whole blood assay. The potency of Ex_31 was determined as described recently (Bretschneider et al., 2017). Briefly, the reaction buffer consisted of 50 mM Tris (pH 8.0), 3 mM KCl, 1 mM CaCl2, 1 mM Mg Cl2, 0.14 mM NaCl and 0.1% bovine serum albumin, which was supplemented with 5 nM recombinant rat ATX. Ex_31 was added to the assay solution in a concentration range from 0.1 nM to 10 μM. To 2.5 μL of this solution, 2.5 μL of a10 μM 18:1 LPC substrate solution was added to initiate the biochemical reaction. After 2 h incubation at room temperature the samples were analysed by LC–MS. The whole blood potency was determined by incubation of 45 μL heparinized rat whole blood with a serial dilution of Ex_31 (0.12 to 100 μM). After 1 h at 37°C the reaction was stopped by addition of 100 μL 40 mM disodium hydrogen phosphate buffer containing 30 mM citric acid (pH 4) and 1 μM 17:0 LPA (internal standard). Subsequently, LPA was extracted by liquid–liquid extraction using 500 μL butanol. The samples were analysed by LC–MS. IC50 values were calculated by applying a sigmoidal Hill equation.
Table S3 Eurofin Cerep profile of Ex_31. Values represent relative activities in Ex_31 treated samples (10 μM) compared to untreated controls.
Table S4 In the CCl4 study, male Sprague Dawley rats were administered CCl4 (0.25 mL kg−1 in olive oil, twice weekly) for a total duration of 10 weeks. After 6 weeks, animals were stratified based on plasma collagen IV (Col. IV) and 18:1 LPA levels. Each row represents one individual animal, which was assigned to the Vehicle (columns 1 and 2) or Ex_31 (columns 3 and 4) group.
Table S5 Male Wistar rats were fed a CDAA diet for a total duration of 14 weeks. After 9 weeks, animals were stratified based on histology assessment of liver biopsies. Values represent quantification of fibrotic area based on collagen I‐stained liver sections. Each row represents one individual animal, which was assigned to the Vehicle (column 1) or Ex_31 (column 2) group.
Figure S6 Normalized plasma lysophosphytidylcholine (LPC) levels and lysophosphatidic acid to LPC ratios in choline‐deficient amino acid‐defined diet‐induced liver injury model. Data represent mean ± SEM, n = 12, **P < 0.01.
Table S7 Permeability of LPA species in cm ·sec−1.
Table S8 List of TaqMan assays and primers/probes for RT‐PCR.
Figure S9 Synthesis scheme for ATX inhibitor Ex_31 (Example 31(24)).
Figure S10 Analytical Data for Ex_31: Rf = 0.4 (10% MeOH:Dichloromethane); m.p.: 176–180°C; 1H–NMR (400 MHz, DMSO‐d6) δ ppm 12.01 (s, 1 H), 8.20 (dd, J = 4.7, 1.3 Hz, 1 H), 7.93–7.90 (m, 1 H), 7.50–7.46 (m, 1 H), 7.20–7.17 (m, 1 H), 7.11 (dd, J = 7.8, 4.8 Hz, 1 H), 6.90–6.83 (m, 1 H), 5.53–5.48 (m, 2 H), 4.66 (s, 2 H), 3.82–3.75 (m, 2 H), 2.79–2.66 (m, 2 H), 2.34–2.06 (m, 3 H), 1.90–1.65 (m, 5 H), 1.34–1.22 (m, 2 H), 1.05–0.86 (m, 2 H); m/z = 484.2 [M + H] + .
Figure S11 Ex‐vivo measurement of ATX activity in plasma and homogenates from un‐perfused and perfused livers. Heparin plasma and un‐perfused and perfused livers rat livers were collected and immediately frozen at −20°C until analysis. 100 mg liver were homogenized in 500 μL PBS buffer using a dry iced cooled Precyllis Evolution (Bertin Technologies). The cell debris were pelleted by centrifugation at 10000 rpm, 10 min and 4°C. 400 μL of the supernatants or 35 μL plasma were supplemented with 10 μM 18:2 LPC and samples were incubated at 37°C. After 1 h the reaction was stopped by the addition of 40 mM disodiumhydrogenphosphate buffer containing 30 mM citric acid (pH 4) and 1 μM 17:0 LPA (internal standard). Subsequently, LPA was extracted by liquid–liquid extraction using 500 μL butanol as described above. Data represent individual values and mean ± SD, n = 3.
Figure S12A Individual relative plasma LPA species in CCl4 model. LPA levels were determined as described in the materials and methods section. Data represent mean ± SEM, n = 10 (control) and n = 13 (CCl4‐treated), *P < 0.05, n.s. = not significant. Levels were below detection limit in several samples of the vehicle and CCl4/Ex_31 groups for 16:0 LPA and in the CCl4/Ex_31 group for 18:0 LPA and 18:1 LPA. Thus statistical power calculations are not applicable for these groups (NA).
Figure S12B Individual relative liver LPA species in CCl4 model. LPA levels were determined as described in the materials and methods section. Data represent mean ± SEM, n = 10 (control) and n = 13 (CCl4‐treated), *P < 0.05, n.s. = not significant.
Figure S12C Individual relative plasma LPA species in CDAA model. LPA levels were determined as described in the materials and methods section. Data represent mean ± SEM, n = 12, *P < 0.05, n.s. = not significant. Levels were below detection limit in several samples of the CDAA/Ex_31 groups for 16:0 LPA, 18:0 LPA and 18:1 LPA. Thus statistical power calculations are not applicable for these groups (NA).
Figure S12D Individual relative liver LPA species in CDAA model. LPA levels were determined as described in the materials and methods section. Data represent mean ± SEM, n = 12, *P < 0.05, n.s. = not significant.