

Abstract
Anal squamous cell carcinomas (ASCC) are rare tumours in humans. The etiological role of HPV infection is now well established but little is known about the molecular landscape and signalling pathways involved in the pathogenesis of this cancer. Here we report the results from a whole exome sequencing of a homogeneous group of 20 treatment-naive ASCC. A total of 2422 somatic single nucleotide variations (SNV) were found, with an overall moderate rate of somatic mutations per tumour (median: 105 relevant SNV per tumour) but a high mutational load in 3 tumours. The mutational signatures associated with age and APOBEC were observed in 100% and 60% of tumours respectively. The most frequently mutated genes were PIK3CA (25%) followed by FBXW7 (15%), FAT1 (15%), and TRIP12 (15%), the two last ones having never been described in ASCC. The main copy number alterations were gains of chromosome 3q (affecting PIK3CA) and losses of chromosome 11q (affecting ATM). The combined analysis of somatic mutations and copy number alterations show that recurrent alterations of the PI3K/AKT/mTOR pathway are frequent (60%) in these tumours, as well as potentially targetable alterations of other signalling pathways that have never been described in ASCC such as chromatin remodelling (45%) and ubiquitin mediated proteolysis (35%). These results highlight the possible implication of these aberrant signalling pathways in anal carcinogenesis and suggest promising new therapeutic approaches in ASCC. The high somatic mutation burden found in some tumours, suggesting an elevated neoantigen load could also predict sensitivity of ASCC to immunotherapy.
Keywords: anal squamous cell carcinoma; whole exome sequencing; somatic mutation, copy number alteration; signalling pathway
INTRODUCTION
Anal squamous cell carcinoma (ASCC) is a rare tumour that accounts for less than 5% of all lower gastrointestinal tract malignancies in Europe [1], but its incidence has increased in the last two decades, especially in HIV men [2, 3]. Human papilloma virus (HPV) infection, predominantly by HPV-16, is the main risk factor of these tumours [4]. Other risk factors include anoreceptive intercourse, a high lifetime number of sexual partners, chronic immunosuppression and a history of smoking.
The majority of ASCC are diagnosed at a localized or locally advanced stage, where chemoradiotherapy (CRT) is the standard of care but is associated with a local failure or recurrence in approximately 30% of the patients [5]. A salvage abdominoperineal resection is then indicated but 30 to 60% of operated patients will experience a locoregional and/or metastatic recurrence [6, 7]. In these patients with an inoperable locally advanced or metastatic disease, very few treatments are available and their effectiveness is limited. New therapeutic approaches and predictive factors of outcome are required in this context.
A better understanding of molecular markers involved in anal carcinogenesis is required for the identification of new therapeutic targets as well as prognostic and predictive biomarkers. In contrast to other squamous cell carcinomas (SCC) and HPV-related cancers for which several recent large-scale studies have respectively defined the genetic and epigenetic processes involved in oncogenesis [8], the molecular landscape of ASCC is currently not completely characterized. Previously studies have revealed frequent alterations in the PIK3CA/AKT/mTOR pathway, high levels of EGFR expression but few KRAS or BRAF mutations [9–13]. Most of these analyses were performed on a limited number of genes or in tumours previously treated by CRT. Only two exome-wide mutational studies are available: the first study of Mouw et al. [14] evaluated the impact of CRT on genomic evolution in ASCC recurrences samples (after initial curative CRT) and the second study of Morris KW et al. evaluated the genomic evolution in metastatic ASCC samples [15].
To identify potential driver genes and to better understand molecular markers involved in anal squamous carcinogenesis, we performed a whole exome sequencing (WES) analysis focused on a homogeneous cohort of 20 frozen treatment-naive ASCC patients, and identified recurrent somatic alterations and common altered signalling pathways.
RESULTS
Patients and tumour characteristics
From a large previous reported cohort of ASCC [10], we identified and included in the present study 20 treatment-naive and fresh frozen tumour tissues for WES analysis. Patients and tumour characteristics were summarized in Table 1. All tumours were HPV positive, with HPV16 in 19 cases and HPV6-11 in the only HIV positive patient of the series. Most of the tumours (80%) were moderate or well differentiated. All patients except one were treated by CRT or radiotherapy (RT) preceded by surgery or tumour excision in 4 of them.
Table 1. Clinico-pathological features of the 20 treatment-naive ASCC patients.
| Clinico-pathological features | Number of patients, n |
|---|---|
| Total | 20 |
| Gender | |
| Female | 16 |
| Male | 4 |
| Concomitant HIV infection | |
| Yes | 1 |
| No | 19 |
| Tumour differentiation | |
| Moderate/Well | 16 |
| Poor | 4 |
| HPV status | |
| HPV positive | 20 |
| genotype 16 | 19 |
| genotype 6–11 | 1 |
| HPV negative | 0 |
| Initial tumour stage (AJCC-2010) | |
| I | 1 |
| II | 3 |
| IIIA | 8 |
| IIIB | 6 |
| IV* | 1 |
| ND | 1 |
| Initial therapy | |
| Surgery (tumour excision/APR): | 5 |
| alone | 1 |
| followed by radiotherapy | 3 |
| followed by chemoradiation | 1 |
| Radiation | 1 |
| Chemoradiation | 14 |
| with concomitant 5FU-CDDP | 11 |
| with concomitant 5FU-MMC | 1 |
| with other concomitant Chemo | 2 |
*liver metastasis
APR: abdominoperineal resection; CDDP: cisplatin; Chemo: chemotherapy; MMC: mitomycin C
Mutational spectrum
WES of 20 tumours and matched normal samples targeted 216320 exons in 23103 genes, for a total of 50.4 Mb of captured regions. On average, 62M reads were sequenced per sample and 96.2% aligned on the reference genome, among which 31.2% were identified as PCR duplicates. Overall, the mean depth of coverage of the targeted regions was 119X and 51% of the exome was covered by more than 100X. In the total of 20 treatment-naive anal carcinoma samples, WES identified 2396 somatic variants in 2049 genes: 660 (27.3%) synonymous variants, 1492 (61.6%) missense variants, 25 in-frame indels (1.0%) and 219 protein-truncating variants (9.0%), including 38 splicing variants, 61 frameshift indels, 116 nonsense variants and 4 stoploss variants. The average number of non-synonymous mutations was 75 per tumour, ranging from 25 to 182 variants per tumour. Mutational trinucleotide signature in the 20 ASCC as determined by approaches described in the Materials and Methods section is showed in Figure 1. The signature associated with age (signature 1A) was observed in all tested ASCC. After the signature associated with age, the more frequent mutational trinucleotide signature, observed in 12 out of 20 tested ASCC, was the signature associated with APOBEC (“apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like”) family (signature 2), followed by signatures associated with POLE mutations (5 cases) and UV light exposure (2 cases).
Figure 1. Mutational trinucleotide signatures in the series of 20 ASCCs.

Mutational trinucleotide signatures were determined by approaches described in the Materials and Methods section.18 Signature 1A (Age) is characterized by C>T in ACG, CCG, GCG or TCG; signature 2 (APOBEC) by C>T and C>G in TCA or TCT ; signature 7 (UV) by C>T in TCG; and signatures 10 (POLE mutation ) by C>T in TCG and C>A in TCT. Signature R2 is not validated.
Recurrent gene mutations
The 15 genes mutated in at least two tumours (≥10%) are described in Table 2. The majority of mutations were confirmed using Sanger analysis. PIK3CA mutations were found in 5 (25%) of the 20 ASCC, corresponding to the classical somatic activating hot-spot mutations described in the COSMIC database of somatic mutations in cancer [16]. The three other genes the more frequently mutated were FBXW7 (n = 3), FAT1 (n = 3), and TRIP12 (n = 3). It is noteworthy that the FBXW7 gene is a well-known gene frequently mutated in various histological types of cancer [17–19]. The three FBXW7 mutations (2 missenses and 1 nonsense) were all referenced as recurrent mutations in COSMIC database [16]. The FAT1 gene has been described as a tumour suppressor gene involved in Wnt/β-catenin signalling [20] and frequently mutated in SCCs [16, 21]. TRIP12 is an ubiquination-related gene less frequently reported as mutated in cancers. Mutations of this gene were essentially described in gastric and colorectal cancer with microsatellite instability, but also in the cervix carcinomas [16, 22]. The comparison of the observed mutation frequencies of the genes FAT1 and TRIP12 in various common cancers is indicated in the Supplementary Table 1. The 11 other genes mutated in 2 of the 20 tumours (10%) were PHLDA3, MCM4, TAGAP, ASXL1, MCC, CYLD, ZNF469, ZAK, NOTCH1, MLL3 and DDX5.
Table 2. Frequently mutated genes in the series of 20 ASCCs.
| Gene | Chromosomal location | Number of mutated samples | % | Mutations (n) |
|---|---|---|---|---|
|
PIK3CA NM_006218 |
3q26.3 | 5 | 25 | MS (5) exon10:c.1624G>A:p.E542K exon10:c.1624G>A:p.E542K exon10:c.1633G>A:p.E545K exon21:c.3140A>T:p.H1047L exon21:c.3062A>G:p.Y1021C |
|
FBXW7 NM_033632 |
4q31.3 | 3 | 15 | NS(1), MS(2) exon8:c.1177C>T:p.R393X exon10:c.1435C>G:p.R479G exon9:c.1322G>A:p.R441Q |
|
FAT1 NM_005245 |
4q35 | 3 | 15 | NS (2), MS (1) exon2:c.1882C>T :p.R628X exon11:c.9014C>G:p.S3005X exon2:c.440C>T:p.P147L |
|
TRIP12 NM_004238 |
2q36.3 | 3 | 15 | MS(1), FS(1), NS(1) exon33:c.4783C>T:p.R1595X exon37:c.5462_5469del:p.1821_1823del exon3:c.590C>T:p.S197F |
|
PHLDA3 NM_012396 |
1q31 | 2 | 10 | MS(2) exon1:c.315C>A:p.F105L exon1:c.110G>A:p.G37E |
|
MCM4 NM_005914 |
8q11.2 | 2 | 10 | FS(1), MS(1) exon8:c.977_978del:p.326_326del exon7:c.793G>A:p.A265T |
|
TAGAP NM_054114 |
6q25.3 | 2 | 10 | MS(1), NS (1) exon10:c.1130C>G:p.S377X exon4:c.118G>A:p.E40K |
|
ASXL1 NM_015338 |
20q11 | 2 | 10 | NS(2) exon11:c.1249C>T:p.R417X exon12:c.2593G>T:p.E865X |
|
MCC NM_002387 |
5q21 | 2 | 10 | MS(1), NS(1) exon9:c.1176G>T:p.K392N exon12:c.1469C>A:p.S490X |
|
CYLD NM_015247 |
16q12.1 | 2 | 10 | MS(1), NS(1) exon11:c.1616G>A:p.C539Y exon12:c.1794C>A:p.Y598X |
|
ZNF469 NM_001127464 |
16q24 | 2 | 10 | MS(1), NS(1) exon2:c.7453C>T:p.Q2485X exon2:c.4766C>T:p.P1589L |
|
ZAK NM_016653 |
2q24.2 | 2 | 10 | NS(1), MS(1) NM_133646:exon12:c.1052C>T:p.S351L NM_016653:exon13:c.1045C>T:p.Q349X |
|
NOTCH1 NM_017617 |
9q34.3 | 2 | 10 | MS(2) exon23:c.3859C>T:p.R1287C exon6:c.976G>A:p.G326S |
|
MLL3 NM_170606 |
7q36.1 | 2 | 10 | MS (1), NS(1) exon42:c.9638C>G:p.S3213X exon7:c.965A>T:p.H322L |
|
DDX5 NM_004396 |
17q21 | 2 | 10 | MS (2) exon12:c.1251C>G:p.D417E exon5:c.496G>A:p.D166N |
FS: frameshift indels, MS: missense variants, NS: nonsense variants
Microsatellite and chromosomal instability
No MSI was observed in the 20 ASCC. The Figure 2 illustrates the SCNA observed in the 20 tumours. The statistical analysis identified 6 regions with relevant gains and 8 regions with relevant losses of DNA copy number. Full arm chromosomal gains were observed in 1p (n = 4), 1q (n = 7), 3q (n = 7) and 8q (n = 3). In addition, focal gains were observed in 1p, 3q, 5p, 8q and 16p (Supplementary Table 2). The focal gain in 3q observed in 18 of 20 (90%) tumours affects the PIK3CA gene. MYC and TERT genes were also affected by focal gains in 8q and 5p observed in 55% and 40% of the tumours respectively (Figure 2 and Supplementary Table 2). In addition, 4 well-known oncogenes showed focal amplifications (characterized as described in Material and methods section): DDR2 (1q23.3; n = 1), CCND1 (11q13; n = 3), AKT2 (19q13.2; n = 2) and MDM2 (12q14.3; n = 1). We validated the focal status of gene amplification (size < 10Mb) for 2 tumours (focal amplification of CCND1 for tumour T8 and focal amplifications of DDR2, AKT2 and MDM2 for tumour T16) using array comparative genomic hybridization (Supplementary Figure 1). Full arm chromosomal losses were observed in 3p (n = 3), 4p (n = 4), 4q (n = 2), 16q (n = 4) and Xq (n = 6). Focal deletions were also identified in 2q, 3p, 4p, 4q, 10q, 13q, and 16q in 35% to 45% of the tumours (Supplementary Table 2). Among them, deletions in 4q (FAT1 and FBXW7) and 2q (TRIP12) affect genes that are frequently mutated in some cancer types, suggesting a tumour suppressor role of these ones (Table 2 and Supplementary Table 2). Allelic losses in 16q that contains the putative tumour suppressor gene CYLD were also observed in 9 of 20 (45%) tumours. A deletion of the PTEN locus in 10q was identified in 9 tumours, including the one harbouring a PTEN mutation. We observed biallelic inactivation (mutations + LOH) for 1 of the 3 FAT1 mutated tumors, for 3 of the 3 TRIP12 mutated tumors and for the 2 CYLD mutated tumors (Table 2). We suggested that other biallelic inactivation of FAT1, TRIP12 and CYLD (as well as PTEN) could occur through promoter methylation of these four genes in the tumours showing only monoallelic inactivation (only LOH without somatic mutation or only somatic mutation without LOH).
Figure 2. Somatic copy number alterations in the series of 20 ASCCs.
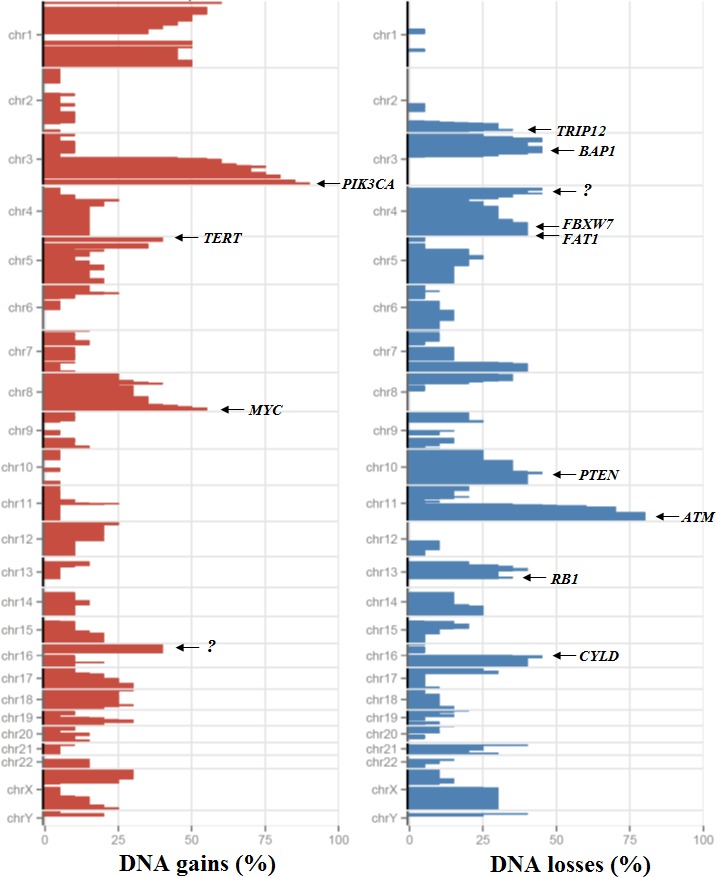
Frequency of copy number gains (red) and losses (blue) in x axis, chromosome position in y axis. “?” indicates the absence of well characterized driver gene.
Other focal deletions involve tumour suppressor genes in 3p (BAP1, FHIT, SETD2, MLH1, TGFBR2, CTNNB1 and PBRM1), 13q (RB1) and 11q22-q25 (ATM) without any mutations found in these genes. In addition, 5 homozygous deletions were identified for the following loci: EED (11q14.2-q22.3), CHL1 (3p26.1), STK11 (19p13.3), LDB1 (10q14) and RB1 (13q14.2).
DNA alterations in one primary tumour and its matched metastasis
For one primary tumour (T11), we performed WES in its matched pre-treated metachronous metastasis (M11). Similar somatic genomic alterations were observed between the primary tumour and the metastasis, including mutational load (124 mutations versus 142 mutations, with 40 mutations in common), mutational signature and SCNA profiles, with an EED homozygous deletion (Figure 3 and Figure 4). However, it is noteworthy that the metastasis M11 exhibited a mutation in the KRAS gene (G12S) that was not found in the primary tumour and a PIK3CA mutation (E542K) different from this observed in the primary tumour T11 (Y1021C). The mutational status (KRAS and PIK3CA genes) of the primary tumour (T11) and its matched metastasis (M11) was confirmed by Sanger analysis.
Figure 3. Common genetic alterations by signalling pathways in the series of 20 ASCCs.

Figure 4. DNA alterations in one primary tumour (T11) and its matched metastasis (M11).

(A) mutational load (number of mutations per tumour). (B) mutational signature as determined by approaches described in the Materials and Methods section. (C) Copy number analyses. The two different homologous chromosomes for each couple are showed using two different lines. Copy number gains are in red and copy number losses in blue.
Significant altered signalling pathways
We integrated mutation and SCNA data to characterize genomic alterations in the most frequently altered signalling pathways in SCCs [8]. Among the SCNAs, only the focal amplifications and the homozygous deletion have been considered relevant. The 5 most frequently altered pathways were the PI3K/AKT/mTOR pathway with 12 of 20 tumours (60%) harbouring focal amplifications and/or homozygous deletions and/or mutations, followed by the chromatin remodelling (45%) and ubiquitin mediated proteolysis (UBP) (35%) pathways and those involved in cell cycle regulation (35%) and squamous cell differentiation (30%) (Figure 3). No gene of the RAS/MAPK signalling pathway was altered.
Alterations in the PI3K/AKT/mTOR pathway included activating mutations of PIK3CA and MTOR, inactivating mutations (or homozygous deletions) of PIK3R1, PTEN and STK11 and focal amplification of AKT2. All these somatic events were mutually exclusive.
The UBP pathway was altered in 7 tumours with mutations in TRIP12, FBXW7, KEAP1 and CYLD. In this UBP pathway, two genes less well known to be involved in human carcinogenesis were identified: TRIP12 and CYLD. These two putative tumour suppressor genes were located in chromosome 2q and 16q respectively that were frequently deleted in our series (Figure 2).
The three other frequently altered pathways in our series were the squamous cell differentiation pathway, where the totality of somatic events (FAT1, NOTCH1 and HES5) were also mutually exclusive, the cell cycle regulation (CCND1, RB1, TP53, CDK4 and MDM2) and the chromatin remodelling pathways (EED, LDB1, MLL3, ASXL1, ARID1A, NSD1, KDM6A, SETD2 and HIST1H3B).
Correlation between altered pathways and clinico-histological parameters
No significant association was found between the five mainly altered pathways and tumour differentiation status or recurrence (Figure 3), which might be due to the small number of samples tested.
It is noteworthy that the only HPV16 negative sample (T20) did not show any alteration of the PI3K/AKT/mTOR pathway but harboured a mutation of TP53, a gene rarely mutated in ASCC [10, 23].
DISCUSSION
Genomic characterization of ASCC is crucial to better understand molecular mechanisms involved in anal carcinogenesis and identify new therapeutic targets and predictive biomarkers of response to chemoradiation. The integration of HPV DNA in the human genome is recognised as the main cause of ASCC [24], through the interaction of E6 and E7 oncoproteins with tumour suppressor genes, but other somatic genomic alterations could be involved in this process [25]. Recently 2 exome-wide mutational analysis interestingly showed a similar mutational profile in matched pre-, post-chemoradiotherapy and metastatic ASCC samples [14–15]. In order to complete the characterization of ASCC genomic landscape and avoid the identification of genetic alterations that could be induced by radiation or chemotherapy, we performed WES in a homogenous cohort of 20 treatment-naive ASCCs with corresponding complete clinico-pathological data. Our cohort is representative of the overall ASCC population, with 95% of HPV16 positive tumours and a 20% rate of tumour relapse after radiation-based therapy.
Among the most frequently mutated genes identified in this present study, PIK3CA and FBXW7 have been reported consistently with similar frequencies in previous studies, mostly limited to the analysis of a panel of genes and performed on heterogeneous ASCC samples [9–13]. As PIK3CA and FBXW7 genes are significantly mutated in other HPV associated squamous cell carcinomas, they could act as driver events in this histological type of cancers.
We identified recurrent somatic mutations in FAT1 (15%) and TRIP12 (15%) genes that have not been previously reported in ASCC. The FAT1 gene is implicated in the squamous cell differentiation and encodes a cadherin-like protein that is able to suppress cancer cell growth in vitro and in vivo by binding ß-catenin and antagonizing its nuclear localization. Recurrent somatic variants of FAT1 have been identified in several cancers and particularly in squamous cell carcinomas [21, 26, 27]. The inactivation of FAT1 via mutations has been demonstrated to activate Wnt signalling, tumorigenesis and cell invasiveness [21]. The second newly identified mutated gene TRIP12, codes for an E3 ubiquitin-protein ligase involved in ubiquitin mediated proteolysis (UBP) pathway and regulation of DNA repair. In normal cells, TRIP12 mediates ubiquitination and degradation of the isoform p19ARF/ARF of CDKN2A, a well-known tumour suppressor required for p53/TP53 activation under oncogenic stress. The alteration of TRIP12 promotes the degradation of ARF rendering cells unprotected from oncogenic stress [28]. Few mutations of TRIP12 have been described in cancers and in particular in HPV related SCCs as only 0.5% and 1.5% of head an neck squamous cell carcinomas (HNSCC) and cervical squamous cell carcinomas (CSCC) tested were found mutated in the COSMIC database [16]. However, it was recently demonstrated that TRIP12 is negatively regulated by HPV/p16-related protein and that the inhibition of TRIP12 expression, via repression of DNA damage repair pathway, was associated with a radiosensitization in HPV-positive HNSCC cells and xenografts. Moreover, overexpression of TRIP12 was associated with poor survival in 18 patients with HPV-positive HNSCC [29] and also be a predictive biomarker of response to chemoradiation in ASCC.
We confirmed, as previously reported [10, 12, 14, 15], frequent PI3K/AKT/mTOR pathway alterations, with at least one altered partner in 60% of ASCC samples but, additionally, our data suggest a complete mutual exclusion of the different alterations affecting this pathway, which has to be taken with caution considering the small sample size of the series. The majority of the altered partners in the PI3K/AKT/mTOR pathway were identical to those reported by Chung et al. [12] and concerned PIK3CA, PIK3R1, PTEN, STK11 and AKT2. This result is promising because PI3K/AKT/mTOR pathway alterations have been associated with clinical response to therapies targeting this pathway in other squamous cell or HPV-associated carcinomas [30–32].
Apart from PI3K/AKT/mTOR pathway, alterations of other pathways were observed in at least 30% of our cohort and concerned specifically chromatin remodelling (45%), cell cycle regulation (35%), UBP (35%), and squamous differentiation (30%) pathways. These alterations are common in many other SCCs and have been associated with potential clinical responses to targeted therapies. An alteration of other druggable targets involved in receptor tyrosine kinase signalling pathways was also observed in 15% of ASCC, concerning particularly FGFR3, DDR2 and FLT1 genes. These data, adding to those of Chung et al. [12] who described a number of alterations in genes involved in DNA damage pathway, chromatin remodeling, FGFR and ERBB signalling, suggest promising new targeted therapy approaches in ASCC.
It is noteworthy that the only HPV16 negative sample (T20) did not show any alteration of the PI3K/AKTmTOR pathway but harboured a TP53 mutation. These mutations are rare in ASCC but are correlated with HPV16 negative status, resistance to chemoradiotherapy and a poor prognosis [10, 23]. We reported a global moderate rate of somatic genetic mutations in ASCCs, which is known to correlate with the production of neoantigens [33]. This should make us consider the evaluation of immunomodulatory agents in the treatment of the ASCCs showing high rate of somatic genetic mutations: 3 out of 20 tested ASCC showed a mutational load >200 somatic mutations. It is noteworthy that these three patients did not show particular clinical characteristics, and did not show germline or somatic mutation either of the gene POLE, or of one of the genes involved in DNA mismatch repair (MMR) or in homologous recombination deficiency (HRD).
In this regard, promising results have been recently reported in metastatic ASCC with the programmed death-1 (PD-1) immune checkpoint inhibitor nivolumab, in a phase 2 study [34]. Moreover, Mouw et al. recently described a significant and durable response to the anti-PD-1 antibody pembrolizumab in a metastatic ASCC patient whose tumour harbored a high mutational burden and neoantigens.
Based on the classification of Alexandrov et al. [35], we identified in our cohort a recurrent mutational trinucleotide signature, observed in 100% of tested ASCC, which was reported to be associated with age. This result is concordant with our population of patients containing a majority of elderly females with a long-term history of HPV infection. The second more frequently signature (60%) was associated with APOBEC cytosine deaminase activity, which has recently emerged as a significant mutagenic factor in human cancer and more particularly in HPV-induced cancer [36].
The WES analysis of one primary tumour and matched metastasis (T11-M11) in our series showed similar mutational load, mutational signature and SCNA profiles but revealed a different gene mutation status between the primary tumour and the metastasis, suggesting a tumour heterogeneity as this phenomenon was already observed in samples from different tumour sites in a previous study [10].
In conclusion, the WES analysis performed in our study extends knowledge of the genomic landscape of ASCC highlights the high frequency of PIK3CA and FBXW7 mutations in non pre-treated primary tumours and reports the presence of new recurrently mutated genes that have not been previously described in ASCC, such as FAT1 and TRIP12. The combined analysis of somatic mutations and copy number alterations reveals the presence of recurrent alterations of the PI3K/AKT/mTOR pathway, including mutations, homozygous deletions or amplifications, in 60% of the tumours and many other altered genes involved in druggable signalling pathways. The high somatic mutation burden found in some tumours, suggesting an elevated neoantigen load could also predict sensitivity of ASCC to immunotherapy. These results are obviously limited by the small sample size of the study and need to be confirmed in a larger series but they suggest promising new therapeutic approaches and bring new insights in the treatment of ASCC.
MATERIALS AND METHODS
Sample collection
Human samples from ASCC and matched adjacent normal anal tissues were collected and frozen at diagnosis in the Institut Curie. The adjacent normal tissues were analysed and confirmed without dysplastic component by our referent pathologic team. All biopsy tissues were residual specimens collected after initial diagnosis (from anal tumour or peripheral lymph node tissue). No patient received previous treatment (treatment-naive tumour samples). All the tumour samples were macrodissected to achieve a maximal neoplastic cellularity (more than 50%). All adjacent control samples were confirmed to be free of tumour cells. For patients without adjacent normal adjacent tissue, a peripheral blood sample was collected to extract germline DNA. This retrospective study was reviewed and approved by the Ethics Committee of the Institut Curie (No. A10-024). According to French regulations, patients were informed of research performed with the biological specimens obtained during their treatment and did not express opposition. Clinical and biological data were collected for each patient. We also analysed an additional tumour sample corresponding to a metachronous (occurred 10 months after CRT) and distant peritoneal metastasis (M11) matched to the primary ASCC (T11), which was treated with systemic chemotherapy.
Whole exome sequencing analysis
The WES analysis was performed on paired DNA samples (tumour and matched normal control DNA). The coding sequences from individual libraries for each sample were captured using the SureSelect version 5.0 human All Exome kit (Agilent Technologies), according to the manufacturer’s protocols. Captured DNA libraries were then sequenced using the Illumina HiSeq 2500 Genome Analyzer, yielding 200 (2 × 100) base pairs from the final library fragments. Raw data were deposited on the NCBI SRA (Sequence Read Archive) database (https://www.ncbi.nlm.nih.gov/sra), under series accession number SRP119025.
Detection of somatic mutations
After a quality control of the FastQ data using FastQC (v0.10.1, Babraham Institute), sequenced reads were aligned to the hg19 human reference genome using Burrows-Wheeler Aligner BWA (v0.6.2, 1 mismatch allowed in the 22bp-seed, a maximum of 4 differences to the reference in the whole read and 1 alignment reported per read (Li Bioinformatics 2009) and PCR duplicates were quantified and removed using Picard Tools (v1.97, http://broadinstitute.github.io/picard). Finally, only alignment intersecting the targeted sequence (Agilent SureSelect V5) were and the overall quality of the targeted regions was assessed using BedTools (v2.17.0) (Quinlan Bioinformatics 2010).
VarScan2 (v2.3.6) was then used to call both Single Nucleotide Variation (SNVs) and small insertions/deletions (indels) predicted as present in the tumours and not in their matched normal control [37]. Only reads having a mapping quality higher than 20 and bases having a base quality higher than 17 were considered in the analysis. Somatic SNV was validated only when the following criteria were met: 1) it was identified by more than distinct pairs; 2) the number of distinct tags containing a particular mismatched base was higher than 5% of the total distinct tags in the tumour sample. The estimated neoplastic cell proportion was taken into account in the analysis. ANNOVAR (10/2013 version) was then used to annotate variants with the following databases: NCBI refGene, 1000 Genomes project frequencies, ESP project (Exome Sequencing Project) frequencies and COSMIC database [38]. Mutation function importance were predicted using SIFT, PolyPhen2 and MutationTaster. In order to prioritize the variants, only those present in the coding or splicing sites in the tumour have been considered relevant. The synonymous variants were analysed separately to identify any recurrence or impact on splicing assessed with MaxEntScan algorithm [39]. Because of the limited numbers of samples available for our study, a simple enumeration was realized to analyze the data for significantly mutated genes. Mutational trinucleotide signatures were obtained using the computational framework developed by Alexandrov et al. and both a homemade script and the R Package (R Core team, 2015) deconstructSigs [35, 40]. In order to best reconstruct the mutational profile of the tumours, an iterative approach was performed to determine the weights to assign to each signature. To this effect, raw trinucleotide counts were normalized by the number of times each trinucleotide context is observed in the human exome.
Sanger sequencing validation
A total of 34 relevant somatic SNVs and indels identified by WES were verified by Sanger sequencing.
Detection of somatic copy number alterations
From the WES analysis, somatic copy number alterations (SCNA) in the tumour compared to the normal sample were detected using the R package (R Core team, 2015) Sequenza (v2.1.0) [41]. Sequenza uses read depth as well as B-allele ratio (BAF) of variants found in both germline/tumour samples to estimate the allele-specific copy number variations and the optimal ploidy and tumour cellularity, after a GC percentage correction.
Homozygous deletion and focal amplification events were characterized using FACETS (v0.5.0) that uses default parameters and a number of 50 iterations per model to obtain convergence [42]. FACETS estimates subclonal events which allows for a more accurate detection of homozygous deletion or focal amplification by discriminating them from low cellular fraction events. The mean log2 ratio in a chromosome region between 0.30 and 1.0 was classified as genomic gain, more than 1.0 (with a size <10 Mb) as focal amplification, less than –0.30 as heterozygous deletion, and less than –1.0 (with a size <5 Mb) as homozygous deletion. Relevant genes affected by a focal gain or deletion of a chromosomal region were selected using the IntOGen database [43].
Assessment of microsatellite instability status
Microsatellite instability (MSI) status was performed on all samples subjected to WES using the MSI Analysis System (Promega, France), which is composed of 5-mononucleotide repeats (BAT-25, BAT-26, NR-21, NR-24 and MONO-27) to detect MSI and 2-pentanucleotide repeat loci to confirm identity between normal and tumour samples, following the manufacturer’s instructions.
Human papilloma virus genotyping
HPV status has been assessed in the Pathology Department of Institut Curie. Total DNA, isolated from formalin-fixed tissue blocks, was used for HPV typing. Real-time PCR using Sybr®Green and specific primers for HPV16 and 18, was performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA).
SUPPLEMENTARY MATERIALS FIGURE AND TABLES
Acknowledgments
We thank Emmanuelle Lappartient, Carine Tran-Perennou, Petros Tsantoulis and Xavier Sastre Garau for technical help.
Abbreviations
- APR
abdominoperineal resection
- ASCC
anal squamous cell carcinoma
- CGH
comparative genomic hybridization
- CNA
copy number alteration
- CSCC
cervical squamous cell carcinoma
- CRT
chemoradiotherapy
- HNSCC
head and neck squamous cell carcinoma
- HPV
human papilloma virus
- MSI
microsatellite instability
- RT
radiotherapy
- SCNA
somatic copy number alterations
- SNV
single nucleotide variation
- UBP
ubiquitin mediated proteolysis
- WES
whole exome sequencing
Footnotes
CONFLICTS OF INTEREST
The authors declare that they have no Conflicts of Interest.
FUNDING
High-throughput sequencing has been performed by the ICGex NGS platform of the Institut Curie supported by the grants ANR-10-EQPX-03 (Equipex) and ANR-10-INBS-09-08 (France Génomique Consortium) from the Agence Nationale de la Recherche (“Investissements d1Avenir” program), by the Canceropole Ile-de-France and by the SiRIC-Curie program -SiRIC Grant « INCa-DGOS- 4654.
REFERENCES
- 1.Glynne-Jones R, Nilsson PJ, Aschele C, Goh V, Peiffert D, Cervantes A, Arnold D, European Society for Medical O, European Society of Surgical O, European Society of Radiological Oncology Anal cancer: ESMO-ESSO-ESTRO clinical practice guidelines for diagnosis, treatment and follow-up. Eur J Surg Oncol. 2014;40:1165–1176. doi: 10.1016/j.ejso.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 2.Forman D, de Martel C, Lacey CJ, Soerjomataram I, Lortet-Tieulent J, Bruni L, Vignat J, Ferlay J, Bray F, Plummer M, Franceschi S. Global burden of human papillomavirus and related diseases. Vaccine. 2012;30:12–23. doi: 10.1016/j.vaccine.2012.07.055. [DOI] [PubMed] [Google Scholar]
- 3.Piketty C, Selinger-Leneman H, Bouvier AM, Belot A, Mary-Krause M, Duvivier C, Bonmarchand M, Abramowitz L, Costagliola D, Grabar S. Incidence of HIV-related anal cancer remains increased despite long-term combined antiretroviral treatment: results from the french hospital database on HIV. J Clin Oncol. 2012;30:4360–4366. doi: 10.1200/JCO.2012.44.5486. [DOI] [PubMed] [Google Scholar]
- 4.Baricevic I, He X, Chakrabarty B, Oliver AW, Bailey C, Summers J, Hampson L, Hampson I, Gilbert DC, Renehan AG. High-sensitivity human papilloma virus genotyping reveals near universal positivity in anal squamous cell carcinoma: different implications for vaccine prevention and prognosis. Eur J Cancer. 2015;51:776–785. doi: 10.1016/j.ejca.2015.01.058. [DOI] [PubMed] [Google Scholar]
- 5.James RD, Glynne-Jones R, Meadows HM, Cunningham D, Myint AS, Saunders MP, Maughan T, McDonald A, Essapen S, Leslie M, Falk S, Wilson C, Gollins S, et al. Mitomycin or cisplatin chemoradiation with or without maintenance chemotherapy for treatment of squamous-cell carcinoma of the anus (ACT II): a randomised, phase 3, open-label, 2 x 2 factorial trial. Lancet Oncol. 2013;14:516–524. doi: 10.1016/S1470-2045(13)70086-X. [DOI] [PubMed] [Google Scholar]
- 6.Lefevre JH, Corte H, Tiret E, Boccara D, Chaouat M, Touboul E, Svrcek M, Lefrancois M, Shields C, Parc Y. Abdominoperineal resection for squamous cell anal carcinoma: survival and risk factors for recurrence. Ann Surg Oncol. 2012;19:4186–4192. doi: 10.1245/s10434-012-2485-1. [DOI] [PubMed] [Google Scholar]
- 7.Mariani P, Ghanneme A, De la Rochefordiere A, Girodet J, Falcou MC, Salmon RJ. Abdominoperineal resection for anal cancer. Dis Colon Rectum. 2008;51:1495–1501. doi: 10.1007/s10350-008-9361-x. [DOI] [PubMed] [Google Scholar]
- 8.Dotto GP, Rustgi AK. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell. 2016;29:622–637. doi: 10.1016/j.ccell.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernardi MP, Ngan SY, Michael M, Lynch AC, Heriot AG, Ramsay RG, Phillips WA. Molecular biology of anal squamous cell carcinoma: implications for future research and clinical intervention. Lancet Oncol. 2015;16:611–621. doi: 10.1016/S1470-2045(15)00292-2. [DOI] [PubMed] [Google Scholar]
- 10.Cacheux W, Rouleau E, Briaux A, Tsantoulis P, Mariani P, Richard-Molard M, Buecher B, Dangles-Marie V, Richon S, Lazartigues J, Jeannot E, Farkhondeh F, Sastre-Garau X, et al. Mutational analysis of anal cancers demonstrates frequent PIK3CA mutations associated with poor outcome after salvage abdominoperineal resection. Br J Cancer. 2016;114:1387–1394. doi: 10.1038/bjc.2016.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casadei Gardini A, Capelli L, Ulivi P, Giannini M, Freier E, Tamberi S, Scarpi E, Passardi A, Zoli W, Ragazzini A, Amadori D, Frassineti GL. KRAS, BRAF and PIK3CA status in squamous cell anal carcinoma (SCAC) PLoS One. 2014;9:92071. doi: 10.1371/journal.pone.0092071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung JH, Sanford E, Johnson A, Klempner SJ, Schrock AB, Palma NA, Erlich RL, Frampton GM, Chalmers ZR, Vergilio J, Rubinson DA, Sun JX, Chmielecki J, et al. Comprehensive genomic profiling of anal squamous cell carcinoma reveals distinct genomically defined classes. Ann Oncol. 2016;27:1336–1341. doi: 10.1093/annonc/mdw152. [DOI] [PubMed] [Google Scholar]
- 13.Smaglo BG, Tesfaye A, Halfdanarson TR, Meyer JE, Wang J, Gatalica Z, Reddy S, Arguello D, Boland PM. Comprehensive multiplatform biomarker analysis of 199 anal squamous cell carcinomas. Oncotarget. 2015;6:43594–43604. doi: 10.18632/oncotarget.6202. https://doi.org/10.18632/oncotarget.6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mouw KW, Cleary JM, Reardon B, Pike J, Braunstein LZ, Kim J, Amin-Mansour A, Miao D, Damish A, Chin J, Ott PA, Fuchs CS, Martin NE, et al. Genomic Evolution after Chemoradiotherapy in Anal Squamous Cell Carcinoma. Clin Cancer Res. 2017;23:3214–3222. doi: 10.1158/1078-0432.CCR-16-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris VK, Rao X, Pickering C, Foo WC, Rashid A, Eterovic K, Kim T, Chen K, Wang J, Shaw K, Eng C. Comprehensive Genomic Profiling of Metastatic Squamous Cell Carcinoma of the Anal Canal. Mol Cancer Res. 2017 Aug 7 doi: 10.1158/1541-7786.MCR-17-0060. https://doi.org/10.1158/1541-7786.MCR-17-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45:777–783. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laforest A, Aparicio T, Zaanan A, Silva FP, Didelot A, Desbeaux A, Le Corre D, Benhaim L, Pallier K, Aust D, Pistorius S, Blons H, Svrcek M, et al. ERBB2 gene as a potential therapeutic target in small bowel adenocarcinoma. Eur J Cancer. 2014;50:1740–1746. doi: 10.1016/j.ejca.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio L, Cibulskis K, Bertelsen B, Romero-Cordoba S, Trevino V, Vazquez-Santillan K, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–375. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan S, Eng S, Kannan K, Zou Y, Peng L, Banuchi VE, Paty P, Zeng Z, et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet. 2013;45:253–261. doi: 10.1038/ng.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas N Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoo NJ, Park SW, Lee SH. Frameshift mutations of ubiquitination-related genes HERC2, HERC3, TRIP12, UBE2Q1 and UBE4B in gastric and colorectal carcinomas with microsatellite instability. Pathology. 2011;43:753–755. doi: 10.1097/PAT.0b013e32834c7e78. [DOI] [PubMed] [Google Scholar]
- 23.Meulendijks D, Tomasoa NB, Dewit L, Smits PH, Bakker R, van Velthuysen ML, Rosenberg EH, Beijnen JH, Schellens JH, Cats A. HPV-negative squamous cell carcinoma of the anal canal is unresponsive to standard treatment and frequently carries disruptive mutations in TP53. Br J Cancer. 2015;112:1358–1366. doi: 10.1038/bjc.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst. 2000;92:690–698. doi: 10.1093/jnci/92.9.690. [DOI] [PubMed] [Google Scholar]
- 25.Thomas M, Pim D, Banks L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene. 1999;18:7690–7700. doi: 10.1038/sj.onc.1202953. [DOI] [PubMed] [Google Scholar]
- 26.Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, Zhang F, Zhao ZR, Li ZT, Liu ZY, Zhao YD, Sun J, Zhou CC, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 27.Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, Tsai KY, Curry JL, Tetzlaff MT, Lai SY, Yu J, Muzny DM, Doddapaneni H, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. 2014;20:6582–6592. doi: 10.1158/1078-0432.CCR-14-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collado M, Serrano M. The TRIP from ULF to ARF. Cancer Cell. 2010;17:317–318. doi: 10.1016/j.ccr.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Zhang P, Molkentine DP, Chen C, Molkentine JM, Piao H, Raju U, Zhang J, Valdecanas DR, Tailor RC, Thames HD, Buchholz TA, Chen J, et al. TRIP12 as a mediator of human papillomavirus/p16-related radiation enhancement effects. Oncogene. 2016 doi: 10.1038/onc.2016.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dolly SO, Wagner AJ, Bendell JC, Kindler HL, Krug LM, Seiwert TY, Zauderer MG, Lolkema MP, Apt D, Yeh RF, Fredrickson JO, Spoerke JM, Koeppen H, et al. Phase I Study of Apitolisib (GDC-0980), Dual Phosphatidylinositol-3-Kinase and Mammalian Target of Rapamycin Kinase Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2016;22:2874–2884. doi: 10.1158/1078-0432.CCR-15-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou MM, Liu X, Wheler J, Naing A, Hong D, Coleman RL, Tsimberidou A, Janku F, Zinner R, Lu K, Kurzrock R, Fu S. Targeted PI3K/AKT/mTOR therapy for metastatic carcinomas of the cervix: A phase I clinical experience. Oncotarget. 2014;5:11168–11179. doi: 10.18632/oncotarget.2584. https://doi.org/10.18632/oncotarget.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, Tsimberidou AM, Stepanek VM, Moulder SL, Lee JJ, Luthra R, Zinner RG, Broaddus RR, et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014;6:377–387. doi: 10.1016/j.celrep.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris VK, Salem ME, Nimeiri H, Iqbal S, Singh P, Ciombor K, Polite B, Deming D, Chan E, Wade JL, Xiao L, Bekaii-Saab T, Vence L, et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2017;18:446–453. doi: 10.1016/S1470-2045(17)30104-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014;7:1833–1841. doi: 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 40.Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, Eklund AC. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol. 2015;26:64–70. doi: 10.1093/annonc/mdu479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A, Santos A, Lopez-Bigas N. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013;10:1081–1082. doi: 10.1038/nmeth.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.