

Abstract
In cystic fibrosis, deletion of phenylalanine 508 (F508del) in the cystic fibrosis transmembrane conductance regulator (CFTR) anion channel causes misfolding and premature degradation. One possible approach to reducing the detrimental health effects of cystic fibrosis could be the identification of proteins whose suppression rescues F508del-CFTR function in bronchial epithelial cells. However, searches for these potential targets have not yet been conducted, particularly in a relevant airway background using a functional readout. To identify proteins associated with F508del-CFTR processing, we used a high-throughput functional assay to screen an siRNA library targeting 6,650 different cellular proteins. We identified 37 proteins whose silencing significantly rescued F508del-CFTR activity, as indicated by enhanced anion transport through the plasma membrane. These proteins included FAU, UBE2I, UBA52, MLLT6, UBA2, CHD4, PLXNA1, and TRIM24, among others. We focused our attention on FAU, a poorly characterized protein with unknown function. FAU knockdown increased the plasma membrane targeting and function of F508del-CFTR, but not of wild-type CFTR. Investigation into the mechanism of action revealed a preferential physical interaction of FAU with mutant CFTR, leading to its degradation. FAU and other proteins identified in our screening may offer a therapeutically relevant panel of drug targets to correct basic defects in F508del-CFTR processing.
Keywords: chloride channel, cystic fibrosis, cystic fibrosis transmembrane conductance regulator (CFTR), high-throughput screening (HTS), small interfering RNA (siRNA), interactome
Introduction
Cystic fibrosis (CF)2 is one of the most common and severe genetic diseases in the Caucasian population (1 affected in every ∼3,000 births) that causes meconium ileus, deterioration of lung function, pancreatic insufficiency, and male infertility (1). The basic defect in CF derives from loss-of-function mutations in the gene encoding the CF transmembrane conductance regulator (CFTR), a cAMP-regulated chloride channel expressed at the apical membrane of many types of epithelial cells. CFTR belongs to the ATP-binding cassette transporter superfamily (2) and consists of five distinct domains: two membrane-spanning domains (MSDs), two nucleotide-binding domains (NBDs), and a regulatory (R) domain. The CFTR channel is opened when the two NBDs bind to each other, resulting in the formation of two interaction sites for ATP (3). The cAMP-dependent phosphorylation of the R domain is the process that controls CFTR activity. Indeed, in the unphosphorylated state, the R domain physically blocks the oligomerization of NBDs (3).
The most common CF mutation (∼60% of all CF alleles worldwide with relevant ethnic variability) is the deletion of phenylalanine 508 (F508del). The mutation causes two distinct defects, namely a processing defect and a gating defect. Indeed, F508del mutation reduces the intrinsic stability of NBD1 and perturbs the interactions between NBD1 and the MSDs. Such abnormalities cause retention of F508del-CFTR at the endoplasmic reticulum (ER) and premature degradation by the ubiquitin/proteasome system (processing defect) (4, 5). In addition, gating of the mutant CFTR channel is significantly altered because of reduced open channel probability (6).
Druggability of F508del-CFTR has been demonstrated by several studies, and at present, two molecules are commercially available for CF patients. Ivacaftor, previously known as VX-770, is a potentiator, i.e. a molecule that increases channel activity and is therefore particularly suited for CFTR mutants having a gating defect (7). At the moment, it has been approved for the use on CF patients bearing at least one of the following mutations: R117H, G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, or G1349D. Recently, a second drug has been approved for the use on CF patients homozygous for the F508del mutation. This drug, named Orkambi, is a combo drug containing the potentiator ivacaftor plus the corrector lumacaftor, previously known as VX-809, i.e. a molecule that improves maturation and trafficking of F508del-CFTR (8).
Although potentiators are generally believed to directly bind mutant CFTR, improving channel gating, correctors may act in at least two different ways: as pharmacological chaperones or as proteostasis regulators (9, 10). Pharmacological chaperones are thought to act directly on mutant CFTR, and they have been grouped into three classes (11). Class 1 includes compounds (like VX-809) able to improve interactions at the interfaces between NBD1 and MSD1 and NBD1 and MSD2. Class 2 compounds stabilize NBD2 domain. Class 3 correctors are theoretical compounds that should stabilize NBD1 domain, i.e. the same mechanism of action displayed by chemical chaperones like glycerol. However, no drug-like small molecules belonging to class 3 have been yet identified. Proteostasis regulators are instead molecules able to modify the proteostasis environment leading to beneficial effects on CFTR maturation and trafficking to the plasma membrane. Proteostasis regulators could either modulate activity/expression of other proteins interacting with CFTR (such as proteins involved in quality control or degradation of CFTR in the ER or plasma membrane), or they could also act in a more general and indirect way, by modulating proteins (and pathways) that are not directly involved in CFTR biogenesis, but whose modulation results in increased CFTR processing (9, 10). Several proteins have already been identified that could represent useful drug targets for a CF therapy based on proteostasis modulation (12).
Regardless of the mechanism of action of known correctors, it has been shown that the use of a single compound is not sufficient to promote a therapeutically relevant F508del-CFTR rescue (11). This finding suggests that drug therapy for CF will require combinations of correctors exploiting different mechanisms of action, i.e. the use of pharmacological chaperones combined together or with a proteostasis regulator. For example, it has been shown that genetic suppression of the ubiquitin ligase RNF5/RMA1 in vivo leads to an attenuation of pathological phenotypes in CF mice homozygous for the F508del mutation (13). In addition, in vitro experiments on F508del human bronchial cells demonstrate a strong additive effect between RNF5/RMA1 suppression and treatment with corrector VX-809 (13).
An extensive search for other potential targets, useful for a combination therapy for CF, has not been undertaken yet, particularly in a relevant airway/epithelial background and using functional readouts. Here, by means of a functional genomics approach based on the screening of a druggable-genome library of siRNA molecules, we report the identification of novel proteins, whose suppression results in a significant F508del-CFTR rescue in bronchial epithelial cells of human origin. The identified targets include proteins associated to F508del-CFTR degradation, transcription factors and proteins with unknown function. Particularly interesting is FAU, a yet-uncharacterized fusion protein consisting of the ubiquitin-like protein FUBI at the N terminus and ribosomal protein S30 at the C terminus. These findings expand the panel of putative drug targets available for novel therapy strategies addressing F508del-CFTR mistrafficking and premature degradation.
Results
High-throughput screening of a siRNA library targeting 6,650 human genes to identify CFTR regulators
To identify proteins playing a role in CFTR biogenesis, we utilized the MISSION siRNA Human Druggable Genome Library (Sigma–Aldrich), composed of 6,650 therapeutically valuable gene targets and designed to specifically support drug screening programs. As depicted in Fig. 1A, CFBE41o− bronchial epithelial cells stably co-expressing F508del-CFTR and the halide-sensitive yellow fluorescent protein (HS-YFP) were transfected with each siRNA triplet separately. After 48 h, F508del-CFTR activity in the plasma membrane was assessed by measuring the rate of HS-YFP quenching caused by iodide influx into cells. As a positive control, we treated cells with the approved drug corrector VX-809.
Figure 1.
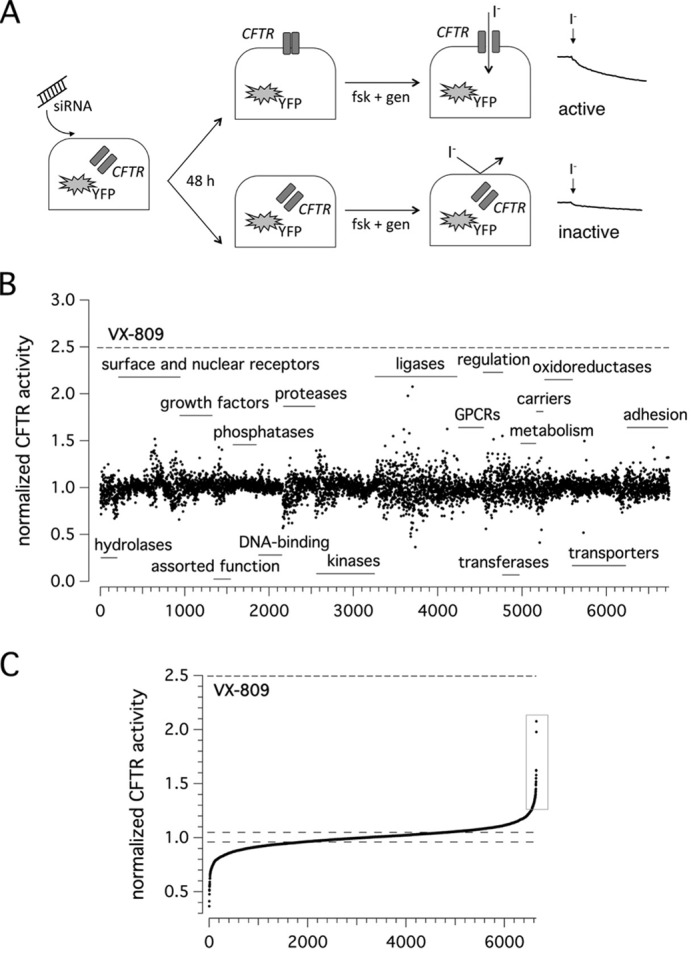
F508del-CFTR screening assay. A, screening procedure. CFBE41o− cells co-expressing F508del-CFTR and a halide-sensitive YFP were reverse-transfected with siRNAs (final concentration, 30 nm). The cells were incubated at 37 °C prior to analysis. The assay was carried out 48 h after transfection, using a plate reader, and activity was estimated according to YFP fluorescence quenching by iodide in the presence of forskolin (20 μm) plus genistein (50 μm) (fsk + gen). Representative traces showing iodide influx in the presence or absence of F508del-CFTR function rescue are shown on the right. B, results of siRNA library screening. Each dot represents CFTR activity following silencing of a single gene. Genes are grouped in functional classes. C, graph reporting the ordered distribution of CFTR activity scores displayed in B.
Results of the siRNA library screening are displayed in Fig. 1B. Targets are grouped in functional protein classes. For each individually silenced gene target, we evaluated the resulting CFTR activity, normalized to the activity measured in controls (cells treated with non-targeting siRNAs). CFTR activity scores were then put into an ordered distribution (Fig. 1C). Given the high reproducibility and robustness of the assay (normalized CFTR activity in controls, 1.00 ± 0.02), we chose to set a threshold at 1.25 so that gene targets giving CFTR activity of >1.25 were considered as positive. The primary screening highlighted 109 putative hits, i.e. proteins encoded by human genes whose genetic inhibition led to significant F508del-CFTR rescue and increased anion transport based on assessment of HS-YFP quenching. Of 109 putative hits, 95 underwent a validation step using siRNA molecules from Life Technologies (Stealth) or, for one target (SUMO2), from Riboxx (iBONI). Fourteen targets were instead not pursued, because siRNA molecules (from vendors other than Sigma–Aldrich) were not available for those targets.
Validation of putative hits identified by the primary screening
Fig. 2 shows results from the validation step, which confirmed CFTR rescue following knockdown of 37 of 95 primary hits, with a confirmation rate of ∼40%. In detail, activity was confirmed by 3 siRNAs for 2 hits, 2 siRNAs for 11 hits, and 1 siRNA for 24 hits. The results are summarized in Table 1.
Figure 2.

Validation of putative hits derived from primary screening. The bar graph shows F508del-CFTR activity in CFBE41o− cells based on the YFP assay. The cells were previously transfected with siRNAs (final concentration, 30 nm) against indicated targets (three siRNAs molecules per gene, corresponding to black, white, and gray bars) or with control, NT-siRNA. The assay was carried out 48 h after transfection. The activity measured upon treatments was normalized for the activity detected under control condition (NT-siRNA + DMSO). For each gene, the cells transfected with NT-siRNAs were also treated separately with VX-809 corrector (1 μm) for 24 h. The extent of rescue obtained with VX-809 is indicated by a solid line above each set of bars. The data are expressed as means ± S.E. (n = 8). Statistical significance was tested by parametric ANOVA followed by the Dunnet multiple comparisons test (all groups against the control group). Asterisks indicate statistical significance versus NT-siRNA: ***, p < 0.001; **, p < 0.01; and *, p < 0.05.
Table 1.
Validated hits deriving from primary screening
| HIT | Positive siRNA no. | Statistical significancea | Normalized F508del-CFTR activity |
Uniprot accession no. | |
|---|---|---|---|---|---|
| Mean | S.E. | ||||
| ALOX5AP | 1 | * | 1.302 | 0.04 | P20292 |
| ANXA11 | 1 | * | 1.307 | 0.031 | P50995 |
| CDK6 | 1 | ** | 1.342 | 0.065 | Q00534 |
| CHD4 | 3 | *** | 1.787 | 0.095 | Q14839 |
| CNTN1 | 1 | * | 1.309 | 0.026 | Q12860 |
| CSNK2A1 | 2 | * | 1.395 | 0.021 | P68400 |
| FAU | 2 | *** | 1.621 | 0.066 | P62861/P35544 |
| FRAG1 | 2 | * | 1.382 | 0.034 | Q9UHJ9 |
| GALK1 | 1 | * | 1.304 | 0.022 | P51570 |
| GJA5 | 1 | ** | 1.346 | 0.019 | P36382 |
| GJB4 | 1 | * | 1.293 | 0.029 | Q9NTQ9 |
| GPR149 | 1 | *** | 1.445 | 0.078 | Q86SP6 |
| KCTD8 | 1 | *** | 1.4 | 0.036 | Q6ZWB6 |
| LRRC59 | 1 | ** | 1.37 | 0.039 | Q96AG4 |
| MLLT6 | 1 | *** | 1.501 | 0.076 | P55198 |
| NR2E3 | 1 | *** | 1.419 | 0.029 | Q9Y5X4 |
| P2RX1 | 1 | * | 1.291 | 0.038 | P51575 |
| PHF12 | 2 | *** | 1.49 | 0.074 | Q96QT6 |
| PLXNA1 | 2 | *** | 1.484 | 0.041 | Q9UIW2 |
| PLXND1 | 1 | * | 1.295 | 0.021 | Q9Y4D7 |
| PRPS1 | 1 | * | 1.322 | 0.023 | P60891 |
| PTCH1 | 1 | *** | 1.425 | 0.029 | Q13635 |
| RARA | 1 | * | 1.31 | 0.022 | P10276 |
| RBBP6 | 1 | *** | 1.472 | 0.033 | Q7Z6E9 |
| RNF207 | 1 | *** | 1.449 | 0.056 | Q6ZRF8 |
| RORB | 1 | * | 1.328 | 0.023 | Q92753 |
| SEC22B | 1 | ** | 1.365 | 0.014 | O75396 |
| SGK223 | 1 | *** | 1.411 | 0.037 | Q86YV5 |
| SLC25A1 | 2 | * | 1.303 | 0.021 | P53007 |
| SNRNP25 | 1 | *** | 1.519 | 0.072 | Q9BV90 |
| TLR7 | 2 | * | 1.376 | 0.032 | Q9NYK1 |
| TRIM24 | 2 | *** | 1.739 | 0.089 | O15164 |
| UBA2 | 2 | *** | 1.653 | 0.029 | Q9UBT2 |
| UBA52 | 3 | *** | 1.922 | 0.162 | P62987 |
| UBE2C | 2 | * | 1.357 | 0.044 | O00762 |
| UBE2I | 2 | * | 1.639 | 0.047 | P63279 |
| UBXN6 | 1 | ** | 1.366 | 0.022 | Q9BZV1 |
a Asterisks indicate statistical significance versus respective NT-siRNA:
***, p < 0.001; **, p < 0.01; and *, p < 0.05.
To account for possible effect of target silencing on CFTR transcription, for the 37 validated hits, we evaluated CFTR mRNA level by means of real-time quantitative PCR. We found that only when UBA52 was silenced, there was a significant 2–3-fold up-regulation of CFTR mRNA level. Confirmed hits were analyzed by DAVID bioinformatics tool for functional annotation, which revealed the BIOCARTA “Basic Mechanisms of SUMOylation” pathway as the most significantly enriched with three genes (p = 0.037).
Effectiveness of silencing of positive hits was verified by evaluating target mRNA level using real-time quantitative PCR. The most effective targets were UBA52, CHD4, TRIM24, UBA2, UBE2I, and FAU. Silencing (95%) of UBA52 elicited a 90–95% increase in F508del-CFTR function over control-transfected cells. UBA52, ubiquitin A-52 residue ribosomal protein fusion product 1, is one of the four mammalian ubiquitin precursors (the others being UBA80, UBB, and UBC), and it comprises a single ubiquitin molecule C-terminally fused to a ribosomal protein, L40 (14). Knockdown (75%) of CHD4, the chromodomain–helicase–DNA-binding protein 4, caused 80% increase in mutant CFTR activity. CHD4 represents the main component of the nucleosome remodeling and deacetylase complex and plays an important role in transcriptional regulation (15).
We also observed significant rescue (75% increase in CFTR activity relative to control cells) following transfection with siRNAs against TRIM24 (silencing achieved 85%), previously known as transcriptional intermediary factor 1α. TRIM24 mediates transcriptional control by interaction with the activation function 2 region of several nuclear receptors, including the estrogen, retinoic acid, and vitamin D3 receptors. In addition, it also functions as E3 ubiquitin ligase: for example, it is responsible for ubiquitylation of p53. This activity is exploited via its RING domain (16).
Among promising targets, we found two proteins functioning in the sumoylation pathway: UBA2 and UBE2I. Indeed, silencing of UBE2I (which encodes for UBC9, the primary SUMO E2-conjugating enzyme; silencing achieved 80%) significantly rescued F508del-CFTR protein (65% increase in CFTR activity relative to control cells). Knockdown of UBA2 (85%), the SUMO-activating enzyme subunit 2 (also known as ubiquitin-like 1-activating enzyme E1B, UBLE1B), caused a 65% increase in mutant CFTR activity. These data are in agreement with our previous findings, demonstrating that inhibition of sumoylation may improve F508del-CFTR processing (13).
The most intriguing target that came to our attention was FAU, since there is little information available about this protein. Indeed, silencing (85%) of FAU caused a 65% increase in F508del-CFTR trafficking to plasma membrane. The FAU (for FBR-MuSV associated ubiquitously expressed) gene is the cellular homolog of the fox sequence in the Finkel–Biskis–Reilly murine sarcoma virus (FBR-MuSV). It encodes a ubiquitous, highly expressed fusion protein of 133 amino acids consisting of the ubiquitin-like protein FUBI at the N terminus and ribosomal protein S30 at the C terminus (17, 18). It has been proposed that the fusion protein is post-translationally processed to generate free FUBI and free ribosomal protein S30. The function of FUBI is currently unknown. Although FUBI has 37% amino acid sequence identity (57% sequence similarity) to ubiquitin and retains the C-terminal G-G dipeptide motif that participates in isopeptide bond formation between ubiquitin and lysines of target proteins, it lacks internal lysine residues, which serve as sites of polyubiquitin chain formation, indicating that the biological function of FUBI is distinct from that of ubiquitin (17, 18).
Functional and biochemical evaluation of additive effects between rescue maneuvers
Various studies demonstrated that combinations of rescue maneuvers (e.g. pairs of correctors or pairs of revertant mutations having different mechanism of action) are required to generate therapeutically relevant corrective effects on F508del-CFTR activity (11, 19, 20). Thus, we evaluated whether combining target silencing with treatment with the corrector VX-809 resulted in additive/synergistic effects. Therefore, we transfected CFBE41o− cells with siRNAs against selected targets, and the following day, we treated silenced cells with either vehicle (DMSO) or the corrector VX-809. After additional 24 h, F508del-CFTR activity in the plasma membrane was measured using the YFP assay. As shown in Fig. 3A, rescue of F508del-CFTR activity by VX-809 treatment was further enhanced (60–75%) by knockdown of specific targets, namely UBA52, KCTD8, UBA2, UBE2I, TRIM24, PLXNA1, and FAU. In agreement with previous findings from our group (13), we also observed increased mutant CFTR activity when corrector VX-809 was combined with down-regulation of the sumoylation pathway (55 or 75% increase in CFTR activity upon silencing of UBA2 or UBE2I, respectively).
Figure 3.

Prioritization of validated hits. A, additivity of target silencing with VX-809. The bar graph shows results of combined treatment with vehicle alone (DMSO, white bar) or VX-809 (1 μm; gray bars) on cells transfected with indicated siRNAs. The dashed line indicates CFTR activity in cells silenced with NT-siRNA and treated with VX-809. The data are expressed as means ± S.E. (n = 3). B, biochemical analysis of the F508del-CFTR expression pattern. The figures show the electrophoretic mobility of F508del-CFTR in CFBE41o− cells after transfection with indicated siRNAs (final concentration, 30 nm) in combination with treatment with vehicle alone (DMSO) or VX-809 (1 μm). The cells were reverse-transfected with siRNA, cultured for 24 h at 37 °C, and then treated with DMSO or VX-809 for an additional 24 h. Arrows indicate complex-glycosylated (band C) and core-glycosylated (band B) forms of CFTR protein. C, quantification of CFTR bands. The data are expressed as relative abundance of band C (upper panels) or C band/B band ratio (lower panels) in cells transfected with indicated siRNAs and treated with DMSO alone (left panels) or with VX-809 (1 μm; right panels) normalized for abundance of band B in cells treated with NT-siRNA + DMSO. The data are expressed as means ± S.E. (n = 3–5 independent experiments). Statistical significance was tested by parametric ANOVA followed by the Dunnet multiple comparisons test (all groups against the control group). Asterisks indicate statistical significance versus respective NT-siRNA band C: **, p < 0.01; and *, p < 0.05. D, additivity of targets silencing. The bar graph shows results of combined silencing of two targets on cells transfected with indicated siRNAs. The dashed line indicates CFTR activity in respective controls (cells transfected with NT-siRNA (top graph), anti-RNF5 (second graph), anti-UBE2I (third graph), or anti-FAU (fourth graph) siRNA).
We also evaluated rescue of mutant phenotypes biochemically by observing the electrophoretic mobility of CFTR protein. In Western blotting, CFTR protein is detected as two bands, named B and C, of ∼150 and 170 kDa, respectively. Band B corresponds to partially glycosylated CFTR residing in the ER. Band C is instead the mature fully processed CFTR that has passed through the Golgi. The prevalent form in cells expressing wild-type CFTR is band C. Lysates of cells expressing F508del-CFTR show primarily band B, consistent with the severe trafficking defect caused by the mutation (Fig. 3B). To evaluate the effect of target knockdown on CFTR electrophoretic mobility, we transfected CFBE41o− cells with siRNAs, and after 24 h we treated silenced cells with VX-809 (or vehicle). The following day, cells were lysed, and lysates were subjected to SDS-PAGE followed by Western blotting. Western blotting images were analyzed with ImageJ software. For each lane, CFTR bands, analyzed as regions of interest, were quantified after normalization for GAPDH to account for total protein loading. Treatment of F508del-CFTR cells with siRNAs against FAU, KCTD8, LRRC59, MLLT6, NR2E3, PHF12, PLXNA1, TRIM24, UBA2, or UBA52 significantly enhanced expression of mature CFTR (band C). However, only MLLT6 knockdown changed significantly the relative ratio of band C to band B (Fig. 3C). Incubation of cells for 24 h with VX-809 also elicited a nearly 2-fold increase in C/B band ratio. Combining VX-809 treatment with FAU, LRRC59, NR2E3, PHF12, PLXNA1, TRIM24, or UBA52 silencing further increased the intensity of band C, whereas only when VX-809 treatment was combined with UBA52 silencing did we observe a significant increase in C/B ratio (Fig. 3, B and C).
We wondered whether combined suppression of more than one target could result in an additive effect on mutant CFTR rescue. To this aim, we selected three main proteins, RNF5, UBE2I, and FAU, and evaluated the effect of their co-silencing together with other targets. As shown in Fig. 3D, rescue of F508del-CFTR function was significantly increased with specific combinations, the most effective one being the combined knockdown of UBE2I and FAU. Increased rescue was also observed with combined knockdown of RNF5 and KCTD8 or, to a lower extent, of RNF5 together with LRRC59 or TRIM24.
Characterization of FAU protein and the effect of its modulation on CFTR biogenesis
In an attempt to characterize the molecular mechanisms by which FAU suppression leads to F508del-CFTR rescue, we analyzed in more details this protein, and in parallel, we optimized experimental conditions for FAU silencing upon transfection of siRNA molecules. We initially evaluated the FAU expression pattern in various cell lines with or without stable expression of either wild-type or F508del-CFTR (Fig. 4A). In Western blot analysis, FAU protein was detected as two bands of ∼14 and 17 kDa, the latter one being the prevalent form (Fig. 4A). In none of the samples could we identify bands having lower molecular mass (∼6.5 and 7 kDa) corresponding to free FUBI and free ribosomal protein S30. To be noted that, in CFBE41o− cells, treatment with siRNAs targeting FAU (knockdown achieved 90%) caused a partial decrease in the intensity of the 17-kDa band, with negligible changes in the 14-kDa band appearance (Fig. 4A). This finding suggested that the 14-kDa band could be a nonspecific band detected by the anti-FAU antibody. However, the 14-kDa band was also detected by other anti-FAU antibodies targeting specifically the C terminus of FUBI, and the whole S30 protein (Fig. 4B), suggesting that the 14-kDa band is specific for FAU. To understand whether the 14-kDa band corresponds to a form of the protein characterized by a longer half-life, we assessed at the protein level the effect of FAU knockdown 96 h (instead of 48 h) after siRNA transfection. Disappointingly, we consistently observed only the partial reduction of the 17-kDa band, although at the mRNA level, the knockdown was >95% (Fig. S1).
Figure 4.

Analysis of FAU expression and effect of FAU knockdown on CFTR expression. A, biochemical analysis of FAU expression pattern. The pictures show the electrophoretic mobility of FAU in different cell lines, under control condition or, where indicated, following transfection with anti-FAU siRNA (final concentration, 30 nm). B, immunodetection of FAU protein by Western blotting using three different antibodies targeting different domains of the protein. C, biochemical analysis of CFTR and FAU expression pattern in lysates of primary bronchial epithelial cells from healthy donors or CF patients homozygous for the F508del mutation. D, electrophoretic mobility of F508del-CFTR in three different preparations of CFBE41o− cells after transfection with anti-FAU siRNA (final concentration, 30 nm) alone or in combination with VX-809 (1 μm) treatment. E, confocal microscopy images showing immunolocalization of F508del-CFTR protein in CFBE41o− cells after transfection with indicated siRNAs (final concentration, 30 nm) and treatment with DMSO alone or with VX-809 (1 μm). F, confocal microscopy images showing immunolocalization of wt-CFTR protein in CFBE41o− cells after transfection with indicated siRNAs (final concentration, 30 nm) and treatment with DMSO alone or with VX-809 (1 μm). Scale bars, 30 μm.
We also evaluated FAU expression by Western blotting performed on whole lysates from primary human bronchial epithelial cells derived from non-CF or CF patients (homozygous for the F508del mutation). We found that, similar to what was observed for the cell lines, the FAU prevalent form is the one migrating at 17 kDa (Fig. 4C).
We analyzed in more details the effect of FAU knockdown on cultures of CFBE41o− cells stably expressing mutant CFTR. Fig. 4D shows the results of three separate experiments. Upon FAU knockdown, we consistently observed a marked increase of both immature and mature CFTR bands (2.2- and 1.9-fold increase, respectively, in band intensity compared with control condition), with additive effects with VX-809 treatment. These results were in agreement with the functional rescue measured by the HS-YFP assay under similar conditions. Subsequently, we used immunofluorescence combined with confocal microscopy to establish the expression of wt- and F508del-CFTR in CFBE41o− cells. Silencing of FAU markedly increased the signal of F508del-CFTR, although the protein appeared to be largely localized in intracellular compartments (Fig. 4E). A similar but weaker effect was observed in cells treated with VX-809. Importantly, the expression of wt-CFTR was also enhanced by FAU knockdown (Fig. 4F).
We asked whether and how modulation of FAU levels affects maturation and function of membrane proteins other than mutant CFTR. Therefore, we evaluated in parallel the effect of FAU silencing on wild-type CFTR and on TMEM16A protein, a Ca2+-dependent Cl− channel (unrelated to CFTR) that is expressed, similarly to CFTR, on epithelial cell membrane. We transfected CFBE41o− cells stably expressing wt-CFTR with scrambled siRNAs or siRNAs against FAU or CFTR (as positive control). After 48 h, CFTR activity in the plasma membrane was measured using the YFP assay. As shown in Fig. 5A (left panel), cAMP-dependent ion transport measured under these conditions was severely reduced upon CFTR knockdown. On the contrary, FAU suppression determined a slight but statistically significant increase in the transport activity (10% increase relative to control cells). Similarly, we measured Ca2+-dependent ion transport in CFBE41o− cells, which endogenously express TMEM16A (21), by using the YFP assay. We found that anion transport elicited by Ca2+ stimulation was significantly reduced when TMEM16A was silenced; however, no apparent effect was seen upon FAU knockdown (Fig. 5A, right panel). We confirmed these results biochemically by analyzing the electrophoretic mobility of CFTR and TMEM16A proteins under control conditions and following FAU silencing. Therefore, CFBE41o− cells were transfected with siRNAs, and after 48 h, cells were lysed, and Western blotting was performed. As shown in Fig. 5B, FAU suppression caused a 40–60% increase in CFTR band C but had no effect on TMEM16A protein.
Figure 5.

Effect of FAU knockdown on wild-type CFTR and TMEM16A function and expression. A, the scatter dot plots show wt-CFTR (left panel) and TMEM16A (right panel) activity in CFBE41o− cells based on the YFP assay after transfection with indicated siRNAs (final concentration, 30 nm). The activity measured upon treatments was normalized for the activity detected under control condition (NT-siRNA). B, electrophoretic mobility of wt-CFTR (upper panel) and TMEM16A (lower panel) proteins in three different preparations of CFBE41o− cells after transfection with anti-FAU siRNA (final concentration, 30 nm). C, detection by cell surface biotinylation of CFTR forms expressed at the plasma membrane. Immunoblot detection of CFTR and control proteins in the biotinylated fraction (left panel) and in total lysates (right panel) is shown. Absence of the cytosolic proteins calnexin (CNX) and 14-3-3 in the biotinylated fraction confirms surface protein-specific labeling in each experiment.
We carried out cell surface biotinylation experiments to assess the changes elicited by FAU silencing on CFTR expression at the plasma membrane. In cells expressing wild-type CFTR, the mature form was prevalent (Fig. 5C), whereas cells expressing F508del-CFTR expressed significant levels of immature CFTR on their surface. This is not surprising, because it has been demonstrated that immature CFTR can traffic to the plasma membrane through the unconventional secretion route (13, 22). Importantly, the levels of both immature and mature CFTR available for biotinylation were markedly increased following FAU knockdown in F508del-CFTR–expressing cells. On the contrary, cells expressing the wild-type protein showed only a very modest increase in the total amount of CFTR expressed at the plasma membrane (less than 20%; see Fig. 5C), which was consistent with the slight increase in function observed upon FAU silencing (Fig. 5A).
Evaluation of the mechanism of action of FAU modulation as a mutant CFTR rescue maneuver
To investigate the mechanisms associated with enhanced cell surface expression of mutant CFTR protein, we assessed the potential role of the unconventional export pathway mediated by GRASP family proteins GRASP55 and GRASP65, which are implicated in trafficking of the immature CFTR to the plasma membrane (13, 22). To do so, we used GRASP55 and GRASP65 knockdown by siRNA molecules to ask whether rescue of F508del trafficking defect caused by FAU silencing was mediated by the GRASP system. Notably, knockdown of neither GRASP65 (achieved silencing, 75%) nor GRASP55 (achieved silencing, 85%) affected FAU knockdown-dependent rescue of F508del-CFTR function as assessed by halide transport measurements (Fig. S2). On the contrary, as previously reported (13), F508del-CFTR rescue because of RNF5 silencing was severely reduced upon GRASP65 knockdown (Fig. S2).
To evaluate whether the effect of FAU knockdown on CFTR trafficking and targeting to the membrane was mediated by a direct interaction between FAU and CFTR, we transfected CFBE41o− cells with two different FAU constructs, one having a triple hemagglutinin tag at the N terminus of FAU protein (3xHA-FAU), and the second one having the triple hemagglutinin tag at the C terminus (FAU-3xHA). As negative controls, we transfected cells using a YFP expression vector. Briefly, parental CFBE41o− cells and CFBE41o− cell lines stably expressing mutant F508del- or wt-CFTR were seeded and, after 6 h, were transfected with the indicated plasmids. Two days after the transfection, the cells were lysed, and Western blotting was performed (Fig. 6A). In a previous study, we performed gene expression profiling on CFBE41o− cells (23). Gene expression profiles were determined by microarrays (Affymetrix GeneChip HG133A2) and processed using standard procedures. Gene expression profiles demonstrated that FAU is highly expressed in CFBE41o− cells (Table S1). Indeed, FAU expression was comparable with housekeeping/reference genes such as GAPDH and B2M. Accordingly, as evidenced by immunoblotting with the anti-FAU antibody, transfection of FAU-3xHA construct did not change the total amount of FAU proteins expressed by CFBE41o− cells, whereas for the 3xHA-FAU construct, we observed an increase in the 17-kDa band only (Fig. 6A). In addition, the anti-FAU antibody detected a band at 23 kDa that was present in CFBE41o− cells expressing F508del-CFTR and transfected with 3xHA-FAU (Fig. 6A). Using an anti-HA antibody, we detected two bands, corresponding to 17 and 21 kDa in cells transfected with FAU-3xHA. Two bands, of 14 and 23 kDa, were also detected in cells transfected with 3xHA-FAU construct (Fig. 6A). On the same cell lysates, we also carried out immunoprecipitation using an anti-CFTR antibody. The immunoprecipitates were then subjected to SDS-PAGE followed by Western blotting to evaluate the presence of FAU protein (Fig. 6B). Immunoblotting was done with the anti-FAU, the anti-HA, the anti-FUBI, and the anti-S30 antibodies, each one blotted on a separate membrane to avoid spurious signals caused by incomplete stripping. In the lanes containing immunoprecipitates derived from F508del-CFTR cells transfected with the FAU constructs, the antibodies detected bands corresponding to the ones detected in the whole lysates. The same bands were also present, although very faint, in the lanes containing immunoprecipitates derived from F508del-CFTR cells transfected with the YFP construct (Fig. 6B).
Figure 6.

Analysis of CFTR interaction with FAU. A, biochemical analysis of CFTR and FAU expression pattern in whole lysates from CFBE41o− cells after transfection with a vector coding for the YFP or vectors coding for two different FAU constructs having a triple HA tag at the N or C terminus (3xHA-FAU and FAU-3xHA, respectively). B, whole lysates used in A were immunoprecipitated using an anti-CFTR antibody. Immunoblot detection of FAU protein was performed using three different antibodies targeting different domains of the protein and an anti-HA antibody. C, upper panel, immunoblot detection of mutant CFTR in whole lysates derived from CFBE41o− cells transfected with indicated siRNA (final concentration, 30 nm) and at different time points following cycloheximide (CHX)-induced block of protein synthesis. Lower panels, quantification of mutant CFTR (band B and band C) half-life. D, representative traces and scatter dot plots summarizing data from Ussing chamber recordings of human primary bronchial epithelia from a homozygous F508del patient (BE86) following reverse transfection with 50 nm of indicated siRNA molecules. E, scatter dot plots summarizing data from Ussing chamber recordings of human primary bronchial epithelia from a homozygous F508del patient (BE91) following reverse transfection with 50 nm of indicated siRNA molecules.
To investigate the effect of interaction between FAU and mutant CFTR, we evaluated the degradation rate of mutant CFTR in the presence or in the absence of FAU protein. To this aim, we reverse-transfected F508del-CFTR CFBE41o− cells with non-targeting or anti-FAU siRNA molecules, and after 48 h, we blocked protein synthesis by adding cycloheximide to the medium. We then lysed cells at different time points, and cell lysates were subjected to SDS-PAGE followed by Western blotting to evaluate CFTR expression. As shown in Fig. 6C, the expression of mutant CFTR (both band B and C) decreases over time. However, when FAU was silenced, the half-life of mutant CFTR was significantly increased by 2-fold.
We thus set to confirm the therapeutic relevance of silencing of FAU using a native cell system. We therefore transfected with siRNA molecules (final concentration, 50 nm) primary cultures of human bronchial epithelial cells derived from a CF patient homozygous for the F508del mutation. Human bronchial cells were reverse-transfected during cell plating on Snapwell inserts (Corning Costar). We monitored epithelia development by checking transepithelial resistance (Rt) and potential difference. To avoid loss of silencing caused by degradation of siRNA molecules by cellular RNases, when epithelia reached Rt = 4 kΩ, and potential difference = −30 mV (8 days after plating), inserts were mounted in a vertical perfusion chamber for measurement of chloride secretion by short-circuit current analysis (Fig. 6D). After blocking Na+ current with amiloride, cells transfected with NT-siRNA vehicle alone showed little response to the membrane-permeable cAMP analog CPT-cAMP, VX-770, or selective CFTR inhibitor 172 (24). Epithelia transfected with FAU siRNA displayed increased CFTR-mediated chloride current, paralleled by a modest reduction in epithelial sodium channel (ENaC)-mediated current (Fig. 6D). For comparison, in epithelia transfected with siRNA against TRIM24, we observed no increase in the CFTR-mediated chloride secretion, whereas a significant increase of the amiloride-sensitive current occurred. We evaluated CFTR mRNA level of α and β subunits of the ENaC channel by means of real-time quantitative PCR. Interestingly we found that silencing of FAU or TRIM24 did not alter expression of ENaC subunits at the mRNA level. Indeed, relative ENaC α subunit mRNA content (relative to the transcript abundance of the reference β2-microglobulin gene) in bronchial epithelia transfected with NT-, FAU-, or TRIM24-siRNA was equal to 2.7 × 10−2, 5.0 × 10−2, and 4.1 × 10−2, respectively, whereas relative ENaC β subunit mRNA content was equal to 0.9 × 10−3, 1.1 × 10−3, and 1.6 × 10−3, respectively. Rescue of F508del-CFTR function and reduction of ENaC-mediated sodium absorption following FAU silencing were confirmed on epithelia derived from another CF patient homozygous for the F508del mutation (Fig. 6E).
Discussion
The deletion of phenylalanine 508 occurring in CFTR protein is present in ∼60% of CF alleles worldwide, and it is associated to a severe phenotype. Several drug discovery projects have been launched in the last two decades, aimed to identify correctors able to restore proper folding of mutant CFTR and to improve its targeting to plasma membrane. Until now, only one drug, Orkambi, has been approved to treat homozygous F508del-CFTR patients, which is actually a combo treatment containing one corrector and one potentiator. However, the efficacy of the corrector is very limited and hope resides in second and third generation correctors and possibly in the combination of correctors to maximize F508del-CFTR rescue. This outcome may indeed be achieved by using pharmacological chaperones that directly interact with mutant CFTR or by modulating the activity of proteins that control the trafficking and degradation of the protein (9, 10). Therefore, the identification of proteins playing key roles in CFTR biogenesis and maturation is a priority, because they may constitute novel drug targets for CF therapy. In this regard, our study represents the first genome-wide search undertaken to identify targets whose suppression results in functionally and biochemically proved rescue of mutant CFTR, in a cell background that is, as much as possible, close to the native one, the bronchial epithelial cells.
Our search highlighted 37 validated targets among which there are proteins associated to F508del-CFTR bioprocessing but also transcriptional regulators and proteins with unknown function. Regarding known CFTR regulators, we identified UBA2 and UBE2I, being a E2 and a E3 ligase, respectively, of the sumoylation pathway (13, 25). In addition, we identified UBXD1, also known as UBXN6, that is a VCP-interacting protein involved in ER-associated degradation (26). It has been demonstrated that depletion of endogenous UBXD1 protein by RNA interference results in a defect in CFTR degradation (26). Peculiarly, other known CFTR regulators were missed by the screening, possibly because of poor knockdown efficiency of siRNA molecules present in the library (as in the case of siRNAs targeting RNF5) or because they are not present in our library (for example, AHA1).
Regarding known transcriptional regulators, such as CHD4, TRIM24, and MLLT6 (15, 16, 27), additional studies will be required to unravel the mechanism of action (which might be quite indirect) through which their suppression results in mutant CFTR rescue. We can hypothesize that they act by reprogramming gene transcription and protein expression in a way that results in facilitated CFTR processing, that is the most general concept of proteostasis regulators (9, 10). However, the F508del-CFTR rescue observed following TRIM24 suppression could be also due to inhibition of its E3 ubiquitin ligase activity, p53 being one of its identified substrate (16).
MLLT6 is another target of particular interest. Indeed, MLLT6, also known as AF17, is a transcription factor that up-regulates the transcription of the ENaC genes (27). ENaC is a major player in salt and water reabsorption and epithelial surface hydration in a number of tissues, including airways. In cystic fibrosis, the defective CFTR not only leads to inappropriately low chloride transport and impaired hydration but also to enhanced activity of ENaC driving sodium absorption and further dehydration of the airways epithelial surface (28). In vivo, deletion of Af17 leads to reduced ENaC function (27). Although the role of ENaC and its possible hyperactivity in CF is still controversial, it is widely accepted that a decrease in sodium absorption could be beneficial in CF airway epithelia. Indeed, ENaC inhibition has already been proposed as a novel CF treatment (29–32). In this view, MLLT6/AF17 may represent a promising candidate, combining F508del-CFTR rescue to decreased ENaC activity.
Among the various targets identified in our screening, we focused our attention on FAU given its marked effect on F508del-CFTR rescue and the little information available. FAU is a poorly characterized protein that is generated in cells as a fusion between the ubiquitin-like protein FUBI (at the N terminus) and the ribosomal protein S30 (at the C terminus). FAU appears to be highly and constitutively expressed in CFBE41o− cells, being present as stable forms of different molecular mass. Silencing of FAU resulted in a significant rescue of F508del-CFTR as detected by functional and biochemical assays. Interestingly, the effect of FAU knockdown was additive with that of the pharmacological corrector VX-809, thus indicating separate but complementary rescue mechanisms. At the protein level, FAU silencing appeared to increase the overall expression of wt- and F508del-CFTR, as indicated by immunoblotting and immunofluorescence. In particular, for mutant CFTR the effect of FAU knockdown consisted of an increase of both band C and band B levels. Because we could exclude a less direct effect on CFTR mRNA levels, we can postulate that FAU controls the expression and processing of CFTR at a post-transcriptional step, possibly in the ER. Experiments of cell surface biotinylation demonstrated that FAU suppression has a preferential effect on mutant CFTR. Indeed, FAU silencing increased the amount of F508del-CFTR but not of wt-CFTR at the plasma membrane. Furthermore, co-immunoprecipitation experiments demonstrated a marked interaction of FAU with mutant CFTR compared with wild-type protein. In addition, we demonstrated that FAU silencing increases mutant CFTR half-life. We can postulate that FAU is part of a quality control checkpoint where it has a particular role in controlling the processing of F508del-CFTR. Therefore, silencing of FAU improves F508del-CFTR rescue by preventing its degradation and therefore promoting its trafficking to the plasma membrane as band C and band B forms. Importantly, we could confirm that FAU knockdown is effective in primary bronchial epithelial cells, thus indicating that the rescue of mutant CFTR is not limited to an overexpressing cell line but also present in a native cell system. In addition, the observed reduction in ENaC-mediated sodium absorption following FAU silencing increases the interest in this target.
In conclusion, our study has evidenced a panel of proteins that are important for the processing of mutant CFTR, paving the way for using these proteins as new therapeutic targets. In particular, the identification of FAU may unravel novel mechanisms underlying protein synthesis and processing and may offer new therapeutic avenues to rescue mutant CFTR function.
Experimental procedures
Cell culture
CFBE41o− cells stably expressing F508del-CFTR and the HS-YFP YFP-H148Q/I152L were generated as previously described (23). The culture medium was as follows: MEM supplemented with 10% fetal calf serum, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. For fluorescence assays of CFTR activity, CFBE41o− cells were plated (50,000 cells/well) on clear-bottomed 96-well black microplates (Corning Life Sciences, Acton, MA).
The isolation, culture, and differentiation methods of primary bronchial epithelial cells were previously described in detail (21). Briefly, epithelial cells were obtained from main stem human bronchi, derived from CF individuals undergoing lung transplant. For this study, cells were obtained from two CF patients (homozygous for F508del mutation). Cells were detached by overnight incubation of bronchi at 4 °C in a solution containing protease XIV. Epithelial cells were then cultured in a serum-free medium (LHC9 mixed with RPMI 1640, 1:1) supplemented with various hormones and supplements. This medium favors cell number amplification. For cells derived from CF patients, the culture medium contained in the first days a complex mixture of antibiotics (usually colistin, piperacillin, and tazobactam) to eradicate bacteria. The collection of bronchial epithelial cells and the study to investigate the mechanisms of transepithelial ion transport were specifically approved by the ethics committee of the Istituto Giannina Gaslini following the guidelines of the Italian Ministry of Health. Each patient provided informed consent to the study using a form that was also approved by the ethics committee.
To obtain differentiated epithelia, cells were reverse-transfected with indicated siRNA molecules (final concentration, 50 nm) and seeded at high density on porous membranes (12 mm Snapwell inserts; Corning, code 3801). After 24 h, the serum-free medium was replaced with DMEM/Ham's F12 containing 2% fetal bovine serum plus hormones and supplements. Differentiation of cells into a tight epithelium was checked by measuring transepithelial electrical resistance and potential difference with an epithelial voltohmmeter (EVOM1; World Precision Instruments). The medium was replaced daily on both sides of permeable supports up to 8–10 days (liquid–liquid culture).
Fluorescence assay for CFTR or TMEM16A activity
At the time of the assay, the cells were washed with PBS containing 137 in mm NaCl, 2.7 in mm KCl, 8.1 in mm Na2HPO4, 1.5 in mm KH2PO4, 1 in mm CaCl2, and 0.5 in mm MgCl2. The cells were then incubated for 25 min with 60 μl of PBS plus forskolin (20 μm) and either genistein (50 μm) or VX-770 (1 μm) to maximally stimulate F508del-CFTR. Genistein was used for the primary screening of the siRNA library, whereas VX-770 was used for all other functional assays. For evaluation of wt-CFTR, the incubation solution contained forskolin alone. For TMEM16A, forskolin was also omitted. The cells were then transferred to a microplate reader (FluoStar Galaxy; BMG Labtech, Offenburg, Germany) for CFTR or TMEM16A activity determination. The plate reader was equipped with high-quality excitation (HQ500/20X: 500 ± 10 nm) and emission (HQ535/30M: 535 ± 15 nm) filters for YFP (Chroma Technology). Each assay consisted of a continuous 14-s fluorescence reading with 2 s before and 12 s after injection of 165 μl of an iodide-containing solution (PBS with Cl− replaced by I−; final I− concentration, 100 mm). In the case of TMEM16A, the injected solution was supplemented with 1 μm ionomycin to increase intracellular calcium and fully stimulate TMEM16A. The data were normalized to the initial background-subtracted fluorescence. To determine the I− influx rate, the final 11 s of the data for each well were fitted with an exponential function to extrapolate initial slope (dF/dt).
siRNA library screening
The protocol for high-throughput siRNA transfection in a 96-well format was previously established (13). Briefly, the conditions were as follows: CFBE41o− cells expressing F508del-CFTR and the HS-YFP were reverse-transfected with 30 nm (final concentration) siRNAs using Lipofectamine 2000 as transfection agent. 24 h after transfection and plating, the medium was changed, and the cells were incubated at 37 °C for additional 24 h, prior to proceeding with the functional HS-YFP-based assay.
For the primary screening, we used the MISSION siRNA Human Druggable Genome Library targeting 6,650 genes, with three siRNA molecules per gene. For each gene, the three siRNAs were pooled together and tested. For the validation step, we utilized Stealth siRNA molecules from Life Technologies (three duplexes per target) or iBONI siRNAs from Riboxx (four duplexes per target). Sequences and/or catalog numbers of siRNAs will be provided upon request. Target silencing was confirmed by evaluating expression of the target mRNA.
Evaluation of target mRNA level
To evaluate CFBE41o− cell mRNAs, we extracted total RNA using both TRIzol reagent (Gibco-BRL) and an RNeasy mini kit (Qiagen), both following the manufacturers' instructions. One μg of spectrophotometer-quantified RNA was retrotranscribed using an iScript RT kit (Bio-Rad). Real-time quantitative PCR was carried out using inventoried Assays-on-Demand provided by Applied Biosystems. β2-Microglobulin (Hs00187842_m1) served as reference gene to normalize transcript abundance. Real-time quantitative PCR was performed using an IQ5 Realtime PCR detection system (Bio-Rad). Cycling conditions were: 3-min hot start at 95 °C, followed by 40 cycles of denaturation at 95 °C for 30 s, and annealing and extension at 60 °C for 30 s. mRNA was quantified using the comparative CT Method. Each sample was run in triplicate, and the data were analyzed using IQ5 Optical System software (Bio-Rad). Changes in transcript levels were quantified using the comparative CT Method (Sequence Detection System Chemistry Guide; Applied Biosystems).
Antibodies and plasmids
The following antibodies were used: mouse monoclonal anti-CFTR (596; Cystic Fibrosis Foundation Therapeutics, University of North Carolina, Chapel Hill); rabbit polyclonal anti-CFTR (H182; Santa Cruz Biotechnology); mouse monoclonal anti Na+/K+ ATPase α1 (cl. C464.6; Millipore); rabbit monoclonal anti-calnexin antibody (ab22595; Abcam); rabbit polyclonal anti-14-3-3 ζ antibody (ab51129; Abcam); rabbit polyclonal anti-FAU (ab135765; Abcam; identified in the text as anti-FAU); rabbit polyclonal anti-FAU (LS-C97817; LifeSpan Biosciences; identified in the text as anti-FUBI); rabbit polyclonal anti-FAU (ab81442; Abcam; identified in the text as anti-S30); mouse monoclonal anti-HA 1.1 epitope tag antibody (Biolegend); rabbit monoclonal anti-TMEM16A (SP31; Abcam); mouse monoclonal anti-GAPDH (cl.6C5; Santa Cruz Biotechnology, Inc); HRP-conjugated anti-mouse IgG (Abcam); or HRP-conjugated anti-rabbit IgG (DAKO).
The following vectors were used to transiently transfect CFBE41o− cells: OmicsLink Expression Clone EX-A2953-M06 (3xHA-FAU) and OmicsLink Expression Clone EX-A2953-M07 (FAU-3xHA) (Genecopoeia). pcDNA3.1 expressed HS-YFP.
Cell surface biotinylation assay
Parental CFBE41o− cells, CFBE41o− cells expressing wt-CFTR, or CFBE41o− cells expressing F508del CFTR were treated as previously described (13). Briefly, cells were seeded on 100-mm dishes and reverse-transfected with 30 nm (final concentration) NT control siRNAs or siRNAs against selected targets. The day after, the cells were incubated with vehicle alone (DMSO) or with VX-809 (1 μm). A cell surface biotinylation assay was performed 24 h later. Briefly, the cells were washed twice with ice-cold PBS and incubated twice with biotin (0.35 mg/ml in PBS) for 25 min each time on a shaker at 4 °C. After three washes in PBS, biotin was quenched with two washes in NH4Cl solution (50 mm in PBS, 15 min each) on a shaker at 4 °C. The cells were then washed three times in PBS without Ca2+ and Mg2+ and then scraped into lysis buffer (50 mm Hepes, pH 7, 150 mm NaCl, 1% glycerol, 1% Triton X-100, 1.5 mm MgCl2, 5 mm EGTA). Cell lysates were collected in an Eppendorf tube and rocked for 30 min at 4 °C. Nuclei were then pelleted by centrifugation at 10,000 rpm at 4 °C for 20 min. Supernatant protein concentration was calculated using the BCA assay (Euroclone) following the manufacturer's instructions. Then an aliquot of supernatants corresponding to 600 μg of proteins was precipitated by rotating 6 h at 4 °C with high capacity streptavidin-agarose resin (Thermo Fischer Scientific), following the manufacturer's recommendation. The resin was then washed with the following solutions: once with lysis buffer, twice with buffer 1 (150 mm NaCl, 20 mm Tris-HCl, pH 8, 5 mm EDTA, 1% Triton X-100, 0.2% BSA), once with buffer 3 (150 mm NaCl, 20 mm Tris-HCl, pH 8, 5 mm EDTA, 0.5% Triton X-100), and once with buffer 4 (50 mm Tris-HCl, pH 8). Biotinylated proteins were eluted from the resin with reducing sample buffer 4X, and 30 μl of each sample were separated on a 4–15% or 4–20% gradient Criterion TGX gel (Bio-Rad) and analyzed by Western blotting.
Western blotting
Cells silenced with indicated siRNAs (final concentration, 30 nm) were grown to confluence on 60-mm diameter dishes and lysed in radioimmune precipitation assay buffer containing a complete protease inhibitor (Roche). Cell lysates were subjected to centrifugation at 12,000 rpm at 4 °C for 10 min. Supernatant protein concentration was calculated using the BCA assay (Euroclone) following the manufacturer's instructions. Equal amounts of protein (10 μg to detect CFTR and GAPDH and 50 μg to detect Fau and HA tag) were separated onto gradient (4–15% or 4–20% depending on target protein molecular mass) Criterion TGX Precast gels (Bio-Rad), transferred to nitrocellulose membrane with Trans-Blot Turbo system (Bio-Rad) and analyzed by Western blotting. Proteins were detected using antibodies indicated in the dedicated methods section and subsequently visualized by chemiluminescence using the SuperSignal West Femto Substrate (Thermo Scientific). Chemiluminescence was monitored using the Molecular Imager ChemiDoc XRS System. Images were analyzed with ImageJ software (National Institutes of Health). Bands were analyzed as regions of interest, normalized against the GAPDH loading control. The data are presented as means ± S.E. of independent experiments.
To evaluate F508del-CFTR half-life, CFBE41o− cells, silenced with indicated siRNAs (final concentration, 30 nm), were treated with cycloheximide (150 μg/ml) (Sigma–Aldrich) 48 h after siRNA transfection. At different time points (0, 2, 4, and 6 h), the cells were then lysed in radioimmune precipitation assay buffer 1× and subjected to SDS-PAGE as previously described.
CFTR-FAU co-IP assay
Cells were seeded at 80% confluence onto 60-mm diameter dishes and after 6 h were transiently transfected with vectors encoding FAU with a triple HA tag (Genecopoeia) or a vector encoding the YFP protein (as control vector) using Lipofectamine 2000 as transfection agent. 48 h after seeding cells were rinsed twice with ice-cold PBS without Ca2+/Mg2+ and then lysed with IP lysis buffer (#87788; Thermo Scientific) containing complete protease inhibitor (Roche). Nuclei were pelletted by centrifugation at 12,000 rpm at 4 °C for 10 min. Supernatant protein concentration was calculated using the BCA assay (Euroclone) following the manufacturer's instructions. An aliquot of supernatant corresponding to 500 μg of protein was incubated for 1 h with 2 μg/sample of rabbit polyclonal anti-CFTR H182 antibody (Santa Cruz Biotechnology), rocking at room temperature. Then ATP-Mg (Sigma) was added to each sample (final concentration, 2 mm). Antibody-antigen mixture was precipitated with 25 μl/sample of Pierce protein A/G magnetic beads (Thermo Scientific) for 1 h rocking at room temperature, following supplier instructions. Prior to IP, magnetic beads were saturated in lysis buffer with 5% BSA (Sigma) rocking at room temperature for 1 h. The beads were then washed twice with lysis buffer without BSA. Co-immunoprecipitated proteins were eluted from the resin under reducing conditions with 100 μl of Laemmli sample buffer 1×, at room temperature. Equal amounts of co-IP products were analyzed by Western blotting (25 μl were used to detect HA tag or FAU, and 10 μl were used to detect CFTR).
Immunofluorescence: Confocal microscopy
CFBE41o− cells expressing native CFTR or CFBE41o− cells expressing F508del CFTR were reverse-transfected as previously described and seeded on multiwell microscope slides (IBIDI). 24 h after transfection and plating, the medium was changed, and the cells were incubated at 37 °C for additional 24 h with DMSO or VX-809 1 μm prior to fixation.
The cells were washed with PBS and then fixed 5 min with Bouin's solution. After extensive washing with PBS, the cells were permeabilized with PBS and Triton X-100 (0.2%) and saturated in PBS and BSA (1%) for 2 h. Primary antibodies, mouse IgG1 anti-CFTR (ab570; J. R. Riordan, University of North Carolina at Chapel Hill, and Cystic Fibrosis Foundation Therapeutics) at 1:250 and polyclonal antibody anti-FAU 1:100 (ab135765; Abcam) were incubated overnight at 4 °C. Following incubation with primary antibody, the cells were rinsed three times in PBS and incubated with Alexa Fluor 488 goat anti-mouse IgG1 and Alexa Fluor 546 donkey anti-rabbit (Invitrogen) secondary antibodies diluted 1:200 in PBS-BSA 1% for 1 h in the dark. After three further washes in PBS, microscope slides were mounted with Fluoroshield with DAPI (Sigma–Aldrich) to stain cell nuclei. Confocal microscopy was performed using a laser scanning confocal microscope TCS SP8 (Leica Microsystems, Heidelberg, Germany) equipped with 63×/1.40 oil immersion objective.
Short-circuit current recordings
Snapwell inserts carrying differentiated bronchial epithelia were mounted in a vertical diffusion chamber resembling a Ussing chamber with internal fluid circulation. Both apical and basolateral hemichambers were filled with 5 ml of a solution containing 126 mm NaCl, 0.38 mm KH2PO4, 2.13 mm K2HPO4, 1 mm MgSO4, 1 mm CaCl2, 24 mm NaHCO3, and 10 mm glucose. Both sides were continuously bubbled with a gas mixture containing 5% CO2 and 95% air, and the temperature of the solution was kept at 37 °C. The transepithelial voltage was short-circuited with a voltage clamp (DVC-1000; World Precision Instruments) connected to the apical and basolateral chambers via Ag/AgCl electrodes and agar bridges (1 m KCl in 1% agar). The offset between voltage electrodes and the fluid resistance was adjusted to compensate parameters before experiments. The short-circuit current was recorded with a PowerLab 4/25 (ADInstruments) analogical to digital converter connected to a Macintosh computer.
Statistics
Because more than two groups were to be compared, the analysis of variance (ANOVA), followed by a post hoc test was used to avoid “multiple comparisons error.” In the case of normally distributed quantitative variables, a parametric ANOVA was performed, whereas when the quantitative variables were skewed, the non-parametric ANOVA (Kruskal–Wallis test) was applied. The Kolmogorov–Smirnov test was used to evaluate the assumption of normality.
Statistical significance of the effect of single siRNA treatments on CFTR activity or expression in CFBE41o− cells was tested by parametric one-way ANOVA followed by the Dunnet multiple comparisons test (all groups against the control group) as a post hoc test. In the case of combination of siRNAs against more than one target, statistical significance was verified by ANOVA followed by the Tukey test (for multiple comparisons) as a post hoc test.
Normally distributed data are expressed as means ± S.E., whereas skewed distributed data are expressed as median (min-max), and significances are two-sided. Differences were considered statistically significant when p < 0.05.
Author contributions
N. P. and E. P. performed the siRNA screening; V. T. performed all the biochemical and functional experiments, with the help of E. S. and E. P.; M. M. and E.C. performed target evaluation on primary bronchial epithelial cells; V. T. performed the immunofluorescence experiments that were analyzed at the confocal microscope by P. S.; F. A. and G. C. developed cell models; N. P. analyzed functional and biochemical data; R. R., N. P., and L. J. V. G. planned the study; and N. P. wrote the manuscript aided by R. R., G. C., and L. J. V. G.
Supplementary Material
Acknowledgments
We thank Dr. John Riordan (University of North Carolina, Chapel Hill), and Cystic Fibrosis Foundation Therapeutics for kindly providing us with anti-CFTR antibodies. We are grateful to Giuseppe Nicora and Carlo Pedemonte for valuable assistance with Ussing chamber equipment.
This work was supported by Ricerca Corrente from the Italian Ministry of Health (Linea1), by Italian Cystic Fibrosis Foundation Grants FFC 3/2009 (with the contribution of “Anna Iacomini, Gruppo di Sostegno FFC Rita Verona, Delegazione FFC di Como”) and FFC 5/2012 (with the contribution of “Danone S.p.A.”) (to N. P.), by Italian Ministry of Health Grant GR-2008-1141326 (to N. P.), and by Telethon Grant TMLGCBX16TT (to L. J. V. G.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Figs. S1 and S2.
- CF
- cystic fibrosis
- CFTR
- cystic fibrosis transmembrane conductance regulator
- MSD
- membrane-spanning domain
- NBD
- nucleotide-binding domain
- R
- regulatory
- ER
- endoplasmic reticulum
- HS-YFP
- halide-sensitive yellow fluorescent protein
- ENaC
- epithelial sodium channel
- NT
- non-targeting
- ANOVA
- analysis of variance
- IP
- immunoprecipitation.
References
- 1. Pilewski J. M., and Frizzell R. A. (1999) Role of CFTR in airway disease. Physiol. Rev. 79, S215–S255 [DOI] [PubMed] [Google Scholar]
- 2. Hyde S. C., Emsley P., Hartshorn M. J., Mimmack M. M., Gileadi U., Pearce S. R., Gallagher M. P., Gill D. R., Hubbard R. E., and Higgins C. F. (1990) Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature 346, 362–365 10.1038/346362a0 [DOI] [PubMed] [Google Scholar]
- 3. Liu F., Zhang Z., Csanády L., Gadsby D. C., and Chen J. (2017) Molecular structure of the human CFTR ion channel. Cell 169, 85–95 10.1016/j.cell.2017.02.024 [DOI] [PubMed] [Google Scholar]
- 4. Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., and Welsh M. J. (1992) Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358, 761–764 10.1038/358761a0 [DOI] [PubMed] [Google Scholar]
- 5. Lukacs G. L., Mohamed A., Kartner N., Chang X. B., Riordan J. R., and Grinstein S. (1994) Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 13, 6076–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Welsh M. J., and Smith A. E. (1993) Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73, 1251–1254 10.1016/0092-8674(93)90353-R [DOI] [PubMed] [Google Scholar]
- 7. Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Cao D., Neuberger T., Turnbull A., Singh A., Joubran J., Hazlewood A., Zhou J., McCartney J., Arumugam V., Decker C., Yang J., et al. (2009) Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U.S.A. 106, 18825–18830 10.1073/pnas.0904709106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wainwright C. E., Elborn J. S., Ramsey B. W., Marigowda G., Huang X., Cipolli M., Colombo C., Davies J. C., De Boeck K., Flume P. A., Konstan M. W., McColley S. A., McCoy K., McKone E. F., Munck A., et al. (2015) Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del CFTR. N. Engl. J. Med. 373, 1783–1784 10.1056/NEJMc1510466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mu T. W., Ong D. S., Wang Y. J., Balch W. E., Yates J. R. 3rd, Segatori L., and Kelly J. W. (2008) Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 134, 769–781 10.1016/j.cell.2008.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Balch W. E., Morimoto R. I., Dillin A., and Kelly J. W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 10.1126/science.1141448 [DOI] [PubMed] [Google Scholar]
- 11. Okiyoneda T., Veit G., Dekkers J. F., Bagdany M., Soya N., Xu H., Roldan A., Verkman A. S., Kurth M., Simon A., Hegedus T., Beekman J. M., and Lukacs G. L. (2013) Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol. 9, 444–454 10.1038/nchembio.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sondo E., Pesce E., Tomati V., Marini M., and Pedemonte N. (2017) RNF5, DAB2 and friends: novel drug targets for cystic fibrosis. Curr. Pharm. Des. 23, 176–186 [DOI] [PubMed] [Google Scholar]
- 13. Tomati V., Sondo E., Armirotti A., Caci E., Pesce E., Marini M., Gianotti A., Jeon Y. J., Cilli M., Pistorio A., Mastracci L., Ravazzolo R., Scholte B., Ronai Z., Galietta L. J., et al. (2015) Genetic inhibition of the ubiquitin ligase Rnf5 attenuates phenotypes associated to F508del cystic fibrosis mutation. Sci. Rep. 5, 12138 10.1038/srep12138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baker R. T., and Board P. G. (1991) The human ubiquitin-52 amino acid fusion protein gene shares several structural features with mammalian ribosomal protein genes. Nucleic Acids Res. 19, 1035–1040 10.1093/nar/19.5.1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O'Shaughnessy A., and Hendrich B. (2013) CHD4 in the DNA-damage response and cell cycle progression: not so NuRDy now. Biochem. Soc. Trans. 41, 777–782 10.1042/BST20130027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jain A. K., and Barton M. C. (2009) Regulation of p53: TRIM24 enters the RING. Cell Cycle 8, 3668–3674 10.4161/cc.8.22.9979 [DOI] [PubMed] [Google Scholar]
- 17. Kas K., Michiels L., and Merregaert J. (1992) Genomic structure and expression of the human fau gene: encoding the ribosomal protein S30 fused to a ubiquitin-like protein. Biochem. Biophys. Res. Commun. 187, 927–933 10.1016/0006-291X(92)91286-Y [DOI] [PubMed] [Google Scholar]
- 18. Michiels L., Van der Rauwelaert E., Van Hasselt F., Kas K., and Merregaert J. (1993) fau cDNA encodes a ubiquitin-like-S30 fusion protein and is expressed as an antisense sequence in the Finkel–Biskis–Reilly murine sarcoma virus. Oncogene 8, 2537–2546 [PubMed] [Google Scholar]
- 19. Pedemonte N., Tomati V., Sondo E., and Galietta L. J. (2010) Influence of cell background on pharmacological rescue of mutant CFTR. Am. J. Physiol. Cell Physiol. 298, C866–C874 10.1152/ajpcell.00404.2009 [DOI] [PubMed] [Google Scholar]
- 20. Lopes-Pacheco M., Sabirzhanova I., Rapino D., Morales M. M., Guggino W. B., and Cebotaru L. (2016) Correctors rescue CFTR mutations in nucleotide-binding domain 1 (NBD1) by modulating proteostasis. Chembiochem. 17, 493–505 10.1002/cbic.201500620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scudieri P., Caci E., Bruno S., Ferrera L., Schiavon M., Sondo E., Tomati V., Gianotti A., Zegarra-Moran O., Pedemonte N., Rea F., Ravazzolo R., and Galietta L. J. (2012) Association of TMEM16A chloride channel overexpression with airway goblet cell metaplasia. J. Physiol. 590, 6141–6155 10.1113/jphysiol.2012.240838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gee H. Y., Noh S. H., Tang B. L., Kim K. H., and Lee M. G. (2011) Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 146, 746–760 10.1016/j.cell.2011.07.021 [DOI] [PubMed] [Google Scholar]
- 23. Sondo E., Tomati V., Caci E., Esposito A. I., Pfeffer U., Pedemonte N., and Galietta L. J. (2011) Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am. J. Physiol. Cell Physiol. 301, C872–C885 10.1152/ajpcell.00507.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma T., Thiagarajah J. R., Yang H., Sonawane N. D., Folli C., Galietta L. J., and Verkman A. S. (2002) Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 110, 1651–1658 10.1172/JCI0216112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ahner A., Gong X., Schmidt B. Z., Peters K. W., Rabeh W. M., Thibodeau P. H., Lukacs G. L., and Frizzell R. A. (2013) Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol. Biol. Cell 24, 74–84 10.1091/mbc.E12-09-0678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nagahama M., Ohnishi M., Kawate Y., Matsui T., Miyake H., Yuasa K., Tani K., Tagaya M., and Tsuji A. (2009) UBXD1 is a VCP-interacting protein that is involved in ER-associated degradation. Biochem. Biophys. Res. Commun. 382, 303–308 10.1016/j.bbrc.2009.03.012 [DOI] [PubMed] [Google Scholar]
- 27. Chen L., Wu H., Pochynyuk O. M., Reisenauer M. R., Zhang Z., Huang L., Zaika O. L., Mamenko M., Zhang W., Zhou Q., Liu M., Xia Y., and Zhang W. (2011) Af17 deficiency increases sodium excretion and decreases blood pressure. J. Am. Soc. Nephrol. 22, 1076–1086 10.1681/ASN.2010121270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boucher R. C. (2007) Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med. 261, 5–16 10.1111/j.1365-2796.2006.01744.x [DOI] [PubMed] [Google Scholar]
- 29. Caci E., Melani R., Pedemonte N., Yueksekdag G., Ravazzolo R., Rosenecker J., Galietta L. J., and Zegarra-Moran O. (2009) Epithelial sodium channel inhibition in primary human bronchial epithelia by transfected siRNA. Am. J. Respir. Cell Mol. Biol. 40, 211–216 10.1165/rcmb.2007-0456OC [DOI] [PubMed] [Google Scholar]
- 30. Almaça J., Faria D., Sousa M., Uliyakina I., Conrad C., Sirianant L., Clarke L. A., Martins J. P., Santos M., Heriché J. K., Huber W., Schreiber R., Pepperkok R., Kunzelmann K., and Amaral M. D. (2013) High-content siRNA screen reveals global ENaC regulators and potential cystic fibrosis therapy targets. Cell 154, 1390–1400 10.1016/j.cell.2013.08.045 [DOI] [PubMed] [Google Scholar]
- 31. Clark K. L., Hughes S. A., Bulsara P., Coates J., Moores K., Parry J., Carr M., Mayer R. J., Wilson P., Gruenloh C., Levin D., Darton J., Weber W. M., Sobczak K., Gill D. R., et al. (2013) Pharmacological characterization of a novel ENaCα siRNA (GSK2225745) with potential for the treatment of cystic fibrosis. Mol. Ther. Nucleic Acids 2, e65 10.1038/mtna.2012.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gianotti A., Melani R., Caci E., Sondo E., Ravazzolo R., Galietta L. J., and Zegarra-Moran O. (2013) Epithelial sodium channel silencing as a strategy to correct the airway surface fluid deficit in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 49, 445–452 10.1165/rcmb.2012-0408OC [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.