

Abstract
Defective nucleotide modifications of mitochondrial tRNAs have been associated with several human diseases, but their pathophysiology remains poorly understood. In this report, we investigated the pathogenic molecular mechanism underlying a hypertension-associated 4435A→G mutation in mitochondrial tRNAMet. The m.4435A→G mutation affected a highly conserved adenosine at position 37, 3′ adjacent to the tRNA's anticodon, which is important for the fidelity of codon recognition and stabilization. We hypothesized that the m.4435A→G mutation introduced an m1G37 modification of tRNAMet, altering its structure and function. Primer extension and methylation activity assays indeed confirmed that the m.4435A→G mutation created a tRNA methyltransferase 5 (TRMT5)–catalyzed m1G37 modification of tRNAMet. We found that this mutation altered the tRNAMet structure, indicated by an increased melting temperature and electrophoretic mobility of the mutated tRNA compared with the wildtype molecule. We demonstrated that cybrid cell lines carrying the m.4435A→G mutation exhibited significantly decreased efficiency in aminoacylation and steady-state levels of tRNAMet, as compared with those of control cybrids. The aberrant tRNAMet metabolism resulted in variable decreases in mitochondrial DNA (mtDNA)-encoded polypeptides in the mutant cybrids. Furthermore, we found that the m.4435A→G mutation caused respiratory deficiency, markedly diminished mitochondrial ATP levels and membrane potential, and increased the production of reactive oxygen species in mutant cybrids. These results demonstrated that an aberrant m1G37 modification of mitochondrial tRNAMet affected the structure and function of its tRNA and consequently altered mitochondrial function. Our findings provide critical insights into the pathophysiology of maternally inherited hypertension, which is manifested by the deficient tRNA nucleotide modification.
Keywords: mutation, nucleotide modification, oxidative phosphorylation, oxygen consumption rates, translation
Introduction
Defects in nucleotide modifications of mitochondrial tRNAs have been associated with several clinical abnormalities including cancer, diabetes, neurological disorders, deafness, and hypertension (1–4). These nucleotide modifications of human 22 mitochondrial tRNAs encoded by its own genome were catalyzed by tRNA-modifying enzymes encoded by nuclear genome (5–8). To date, 15 types of modifications have been identified in 118 positions in different mammalian mitochondrial tRNAs (9, 10). These nucleotide modifications play a vital role in the structure and function of tRNAs (11–14). Core modifications including pseudouridinylation at position 55 at the TΨC loop primarily contributed to structural stability of tRNAs and in some cases, may affect the aminoacylation (2, 3). These were exemplified by our recent discovery that the loss of pseudouridinylation at position 55 at the TΨC loop of tRNAGlu caused the maternally inherited deafness and diabetes (4). Indeed, the anticodon loop modifications including nucleotides at positions 34 and 37 regulate the stabilization of anticodon structure, fidelity, and efficiency of translation (2, 3, 11, 15–17). The defective 5-taurinomethyluridine (τm5U) at U34 of tRNALeu(UUR) was associated with mitochondrial encephalopathy lactic acidosis and stroke-like episodes, whereas the lack of 5-taurinomethyl-2-thiouridine (τm5s2U) at U34 of tRNALys was responsible for myoclonus epilepsy associated with ragged red fibers (MERRF) (18–20). Furthermore, mutations in tRNA modifying enzymes TRMU, MTO1, GTPBP3, and NSUN3 involved in nucleotide modifications at position 34 of mitochondrial tRNAs have been associated several clinical phenotypes including deafness (21–25).
The nucleotides at position 37 (A or G) of 17 mammalian mitochondrial tRNAs carry the diverse species of modifications, whereas no nucleotide modification at this position was detected in 5 tRNAs, such as tRNAMet and tRNAAsp (9). In fact, modifications of nucleotides A37 and G37 in human mitochondrial tRNAs were catalyzed by the modifying enzymes TRIT1 and TRMT5 encoded by nuclear genes, respectively (8, 26, 27). The modifications at position 37 contributed to the high fidelity of codon recognition and to the structural formation and stabilization of functional tRNAs (11, 15, 26, 27). The defective i6A37 or m1G37 modifications caused by mutations in the TRIT1 or TRMT5 were responsible for mitochondrial dysfunction leading to clinical phenotypes (26, 27). Mutations in the nucleotides at position 37 including the tRNAIle 4295A→G, tRNAAsp 7551A→G, and tRNAMet 4435A→G mutations were associated with hypertension, diabetes, visual loss, and deafness (28–33). In particular, the tRNAMet 4435A→G mutation was identified in four genetically unrelated Chinese families with maternal transmission of hypertension (31–33). Therefore, it was hypothesized that the substitution of A with G at position 37 of the tRNAMet may introduce the m1G37 modification catalyzed by TRMT5, thereby altering the structure and function of tRNAMet. In particular, the m.4435A→G mutation may affect the aminoacylation capacity and stability of tRNAMet and then impair mitochondrial translation. It was also anticipated that defective mitochondrial translation caused by the m.4435A→G mutation alters the respiration, production of ATP and reactive oxygen species (ROS).3 To further investigate the pathogenic mechanism of the m.4435A→G mutation, cybrid cell lines were generated by transferring mitochondria from lymphoblastoid cell lines derived from an hypertensive matrilineal relative carrying the mutation and from a control subject lacking the mtDNA mutation but belonging to the same mtDNA haplogroup, into human mtDNA-less (ρ°) cells (34, 35). Using these cell lines, we investigated if the m.4435A→G mutation introduced the m1G37 modification of tRNAMet by primer extension and methylation activity assays. These cell lines were then assessed for the effects of the m.4435A→G mutation on the stability and aminoacylation capacity of tRNAMet, mitochondrial translation, enzymatic activities of electron transport chain complexes, the rate of O2 consumption, ATP, and oxidative reactive species (ROS) as well as mitochondrial membrane potential.
Results
The m.4435A→G mutation created the m1G37 modification of tRNAMet
Neither i6A37 nor t6A37 modification was detected in the mammalian mitochondrial tRNAMet (9). To investigate if the m.4435A→G mutation introduced the m1G37 modification of tRNAMet, we subjected mitochondrial RNAs from mutant and control cybrid cell lines to the reverse transcription with a digoxigenin (DIG)-labeled oligonucleotide probe specific for tRNAMet (Fig. 1A). This resulted in a stop band one residue 3′ to the methylation on 15% polyacrylamide gel. As shown in Fig. 1B, the m1G37 modification was present in tRNAMet derived from three mutant cell lines. However, the m1G37 modification was not detected in the tRNAMet derived from three control cell lines.
Figure 1.
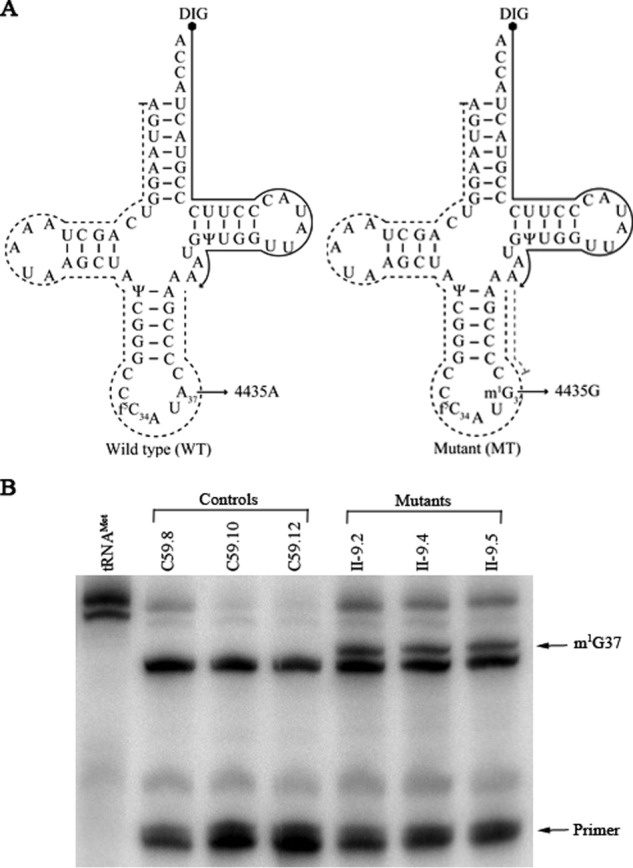
The m.4435A→G mutation created the m1G37 modification of tRNAMet. A, cloverleaf structure of human mitochondrial tRNAMet. Arrows indicate the location of the m.4435A→G mutation. Solid lines represent the DIG-labeled oligonucleotide probe specific for tRNAMet. Broken lines represented the potential stops of primer extension caused by the m1G37 modification. B, primer extension demonstrated the creation of m1G37 in the tRNAMet carrying the m.4435A→G mutation. The primer extension termination products caused by m1G37 modification are showed.
Methanocaldococcus jannaschii Trm5 (Mj-Trm5) catalyzed the m1G37 modification in mutant tRNAMet transcripts
Trm5 is one of the tRNA (m1G37)-methyltransferases that catalyzes the identical tRNA modification, m1G37 (36, 37). To further examine if the m.4435A→G mutation introduced the m1G37 modification of tRNAMet, we prepared wildtype and mutant tRNAs by in vitro transcription and evaluated the methylation activity catalyzed by the recombinant M. jannaschii Trm5 (Mj-Trm5) (36, 37). As shown in Fig. 2A, the mutant tRNAMet transcripts (G37) were modified with m1G37 modification by the Mj-Trm5 but as less efficiently as cytoplasmic tRNALeu(CAG) transcripts (G37) (10). In contrast, the modification was not detected in the wildtype tRNAMet transcripts (A37) in the presence of Mj-Trm5. As controls, the human cytoplasmic tRNALeu(CAG) transcripts (G37) were modified by the Mj-Trm5, whereas human cytoplasmic tRNAThr transcripts (A37) were not modified in the presence of Mj-Trm5. These results indicated that the substation of A to G at position 37 introduced the m1G37 modification of tRNAMet.
Figure 2.

Methylation activity assays. The unmodified human mitochondrial wildtype (A37) and mutant (G37) tRNAMet, cytosolic tRNALeu(CAG), and tRNAThr were generated from in vitro transcription. A, analysis for the m1G37 modification of tRNAMet. The unmodified tRNA transcripts were incubated with M. jannaschii (Mj-Trm5) in the presence of S-adenosyl-l-methionine. Samples were withdrawn and stopped after 2, 4, 6, or 8 min, respectively. The relative modification efficiency was calculated from the initial phase of the reaction. The calculations were based on three independent determinations. Graph shows the results of a representative experiment. B, electrophoretic mobility shift assay. The unmodified mitochondrial wildtype (A37) and mutant (G37) tRNAMet were incubated with various concentrations of enzymes Mj-Trm5. These samples were electrophoresed through 6% polyacrylamide gel and stained with ethidium bromide.
We then analyzed the binding affinities of tRNAMet with Mj-Trm5 using an electrophoretic mobility shift assay. The shift representing the Trm5–tRNA complex occurred at an enzyme concentration of 0.25 μm. As shown in Fig. 2B, the mutant and wildtype tRNAMet transcripts were fully shifted to the Mj-Trm5–tRNA complex at enzyme concentrations of 1.5 and 4.0 μm, with the calculated Kd value of 1.105 ± 0.028 and 1.822 ± 0.025 μm, respectively. These data provided further evidence that the m.4435A→G mutation introduced the m1G37 modification of tRNAMet.
Altered conformation and stability of tRNAMet
It was anticipated that the m.44535A→G mutation caused structural alteration and the instability of tRNAMet. To test if the m.4435A→G mutation affected the conformation of tRNAMet, total RNAs from mitochondria isolated from mutant and control cell lines were electrophoresed through a 10% native gel and then electroblotted onto a positively charged nylon membrane for hybridization analysis with oligodeoxynucleotide probes for tRNAMet and tRNAThr, tRNALeu(CUN) and tRNASer(AGY), respectively. As shown in Fig. 3A, electrophoretic patterns showed that the tRNAMet in three mutant cybrid cell lines carrying the m.4435A→G mutation migrated faster than those of control cybrid cell lines lacking this mutation.
Figure 3.

The analysis of the stability of tRNAMet. A, Northern blot analysis of tRNA under native conditions. Two μg of total mitochondrial RNA from various cell lines were electrophoresed through native polyacrylamide gels, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for the tRNAMet, tRNAThr, tRNALeu(CUN), and tRNASer(AGY), respectively. B, thermal stability of wildtype (A37) and mutant (G37) tRNAMet. ΔTm indicates the difference of the Tm value between wildtype (A37) and mutant (G37) tRNAMet. The calculations were based on three independent experiments.
We then examined the stability of the wildtype and mutant tRNAMet transcripts through the measurement of the melting temperatures (Tm) by calculating the derivatives of the absorbance against a temperature curve. As shown in Fig. 3B, the Tm values for wildtype (A37) and mutant (G37) transcripts were 43.52 ± 0.71 and 46.02 ± 0.41 °C, respectively. These results suggested that the m.4435A→G mutation affected the stability of tRNAMet.
Marked decrease in the steady-state levels of tRNAMet
To assess if the m.4435A→G mutation ablated the metabolism of tRNAMet, we subjected mitochondrial RNAs from mutant and control cybrid cell lines to Northern blots and hybridized them with DIG-labeled oligodeoxynucleotide probes for tRNAMet, tRNALys, tRNAThr, and tRNALeu(CUN) derived from the heavy (H)-strand transcription unit and tRNASer(UCN) and tRNAGlu derived from the light (L)-strand transcription unit (38, 39). As shown in Fig. 4A, the steady-state level of tRNAMet in the mutant cells was markedly decreased compared with those in control cells. For comparison, the average levels of tRNAMet in the mutant cybrid cell lines were among ∼74.4, 69.5, 73.3, 64.9, and 75.3% of average values of three control cybrids after normalization to tRNALys, tRNAThr, tRNALeu(CUN), tRNASer(UCN), and tRNAGlu (p = 0.0001 to 0.0003), respectively.
Figure 4.

Northern blot analysis of tRNA under a denaturing condition. A, 2 μg of total mitochondrial RNA from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for the tRNAMet, tRNALys, tRNASer(UCN), tRNAThr, tRNALeu(CUN), and tRNAGlu, respectively. B, quantification of tRNA levels. Average relative tRNAMet content per cell, normalized to the average content per cell of tRNALys, tRNASer(UCN), tRNAThr, tRNALeu(CUN), and tRNAGlu in three cybrid cell lines derived from one affected subject (II-9) and three cybrid cell lines derived from one Chinese control subject (C59). The values for the latter are expressed as percentages of the average values for the control cell lines. The calculations were based on three independent experiments. The error bars indicate mean ± 2 S.D. P indicates the significance, according to the t test, of the differences between mutant and control cell lines.
Deficient aminoacylation of tRNAMet
To evaluate if the m.4435A→G mutation affects aminoacylation of tRNA, we examined the aminoacylation level of tRNAMet as well as tRNALeu(UUR), tRNALeu(CUN), tRNAIle, and tRNAHis by the use of electrophoresis in an acidic urea PAGE system to separate uncharged tRNA species from the corresponding charged tRNA, electroblotting and hybridizing with the tRNA probes described above. As shown in Fig. 5A, the upper band represents the charged tRNA, and the lower band represents uncharged tRNA. There were no obvious differences in electrophoretic mobility between the control and mutant cell lines. To further distinguish nonaminoacylated tRNA from aminoacylated tRNA, samples of tRNAs were deacylated by being heated for 10 min at 60 °C (pH 8.3) and then run in parallel. As shown in Fig. 5B, only one band (uncharged tRNA) was present in both mutant and control cell lines after deacylating. However, the efficiencies of aminoacylated tRNAMet in the mutant cell lines varied from 61.6 to 83.2%, with the average of 69%, relative to the average values of control cell lines (p = 0.034) (Fig. 5C). However, the levels of aminoacylation in tRNALeu(UUR), tRNALeu(CUN), tRNAIle, and tRNAHis in mutant cell lines were comparable with those in the control cell lines.
Figure 5.

In vivo aminoacylation assays. A, 2 μg of total mitochondrial RNA purified from six cell lines under acid conditions were electrophoresed at 4 °C through an acid (pH 4.5) 10% polyacrylamide, 8 m urea gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for the tRNAMet, tRNALeu(UUR), tRNALeu(CUN), tRNAIle, and tRNAHis, respectively. B, the samples from one control (C59–8) and one mutant (II-9.2) cell lines were deacylated (DA) by heating for 10 min at 60 °C (pH 8.3), electrophoresed, and hybridized with DIG-labeled oligonucleotide probes specific for tRNAMet and tRNALeu(UUR). C, quantification of aminoacylated proportions of tRNAMet in the mutant and controls. The calculations were based on three independent experiments. Graph details and symbols are explained in the legend to Fig. 4.
Decreases in the levels of mitochondrial proteins
To investigate whether the aberrant tRNA metabolism caused by the m.4435A→G mutation impaired mitochondrial translation, a Western blot analysis was carried out to examine the steady-state levels of seven oxidative phosphorylation subunits of (encoded by mtDNA) in mutant (5) and control cells lines, whereas Tom20 (encoded by nuclear gene) as a loading control. As shown in Fig. 6A, the overall levels of seven mitochondrial translation products in the mutant cell lines ranged from ∼29.6 to 97.5%, with an average of 67.2% (p < 0.0001), relative to the mean value measured in the control cell lines. However, there were variable reductions in levels of ND3, ND4, and ND5, subunits 3, 4, and 5 of NADH dehydrogenase; ATP6, ATP8, subunits 6 and 8 of the H+-ATPase; CYTB, apocytochrome b; CO2, subunit II of cytochrome c oxidase in mutant cell lines. In particular, mutant cell lines exhibited marked reductions (49.4 to 70.4%) in the levels of ND3, ATP6, and CYTB harboring 3.9 to 7.0% methionine codons, whereas relatively mild reductions (2.5 to 22.9%) of the levels of ND4, ND5, ATP8, and CO2 containing 4.3 to 8.7% methionine codons were observed in mutant cell lines. However, the levels of polypeptide synthesis in mutant cells, relative to those in control cells, showed no significant correlation with either the number of codons or the proportion of methionine (Table S1).
Figure 6.

Western blot analysis of mitochondrial proteins. A, 20 μg of total cellular proteins from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with 7 polypeptides (mtDNA-encoded subunits of respiratory complexes) and with Tom20 as a loading control. B, quantification of total mitochondrial protein levels. The levels of mitochondrial proteins in 3 mutant cell lines and 3 control cell lines were determined as described elsewhere (53, 54). C, quantification of 7 polypeptides. The levels of ND3, ND4, ND5, CO2, CYTB, ATP6, and ATP8 in three mutant cell lines and three control cell lines were determined as described elsewhere (53, 54). Graph details and symbols are explained in the legend to Fig. 4.
Reduced activities of respiratory chain complexes I, III, IV, and V
To evaluate the effect of the m.4435A→G mutation on the oxidative phosphorylation, we measured the activities of respiratory complexes by the use of isolating mitochondria from three mutant and three control cell lines. The activity of Complex I (NADH ubiquinone oxidoreductase) was determined through the oxidation of NADH with ubiquinone as the electron acceptor (40–42). Complex II (succinate ubiquinone oxidoreductase) was the only respiratory complex that was encoded by the nuclear DNA, so we examined the activity of complex II through the artificial electron acceptor dichlorophenolindophenol (41, 42). The activity of complex III (ubiquinone cytochrome c oxidoreductase) was measured through the reduction of cytochrome c (III) by using d-ubiquinol-2 as the electron donor. The activity of complex IV (cytochrome c oxidase) was monitored through the oxidation of cytochrome c (II). The activity of complex V (F1-ATP synthase) was explored through the NADH oxidation via conversion of phosphoenolpyruvate to lactate by a two-step reaction (41). As shown in Fig. 7, the activity of complexes I, III, IV, and V in the mutant cells carrying the m.4435A→G mutation were 78.8 (p = 0.013), 71.6 (p < 0.001), 81.1 (p = 0.003), and 73.0% (p = 0.018) of the mean value measured in three control cell lines, respectively, whereas the activity of complex II in the mutant cells carrying the m.4435A→G mutation was 99.5% of the mean value measured in three control cell lines (p = 0.868), which was similar to the activity of complex II in the wildtype cell lines as expected.
Figure 7.

Enzymatic activities of respiratory chain complexes. The activities of respiratory complexes were investigated by enzymatic assay on complexes I, II, III, IV, and V in mitochondria isolated from various cell lines. The calculations were based on 4 independent experiments. Graph details and symbols are explained in the legend to Fig. 4.
Respiration deficiency
To evaluate if the m.4435A→G mutation alters cellular bioenergetics, we examined the oxygen consumption rates (OCR) of three mutant cell lines carrying the m.4435A→G mutation and three control cell lines (43). As shown in Fig. 8, the basal OCR in mutant cell lines was 57.8% (p < 0.001) relative to the mean value measured in the control cell lines. To investigate which of the enzyme complexes of the respiratory chain were affected in the mutant cell lines, oligomycin (to inhibit the ATP synthase), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (to uncouple the mitochondrial inner membrane and allow for maximum electron flux through the ETC), rotenone (to inhibit complex I), and antimycin A (to inhibit complex III) were added sequentially while measuring OCR. The difference between the basal OCR and the drug-insensitive OCR yields the amount of ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and non-mitochondrial OCR. As shown in Fig. 8, the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and non-mitochondrial OCR in mutant cell lines were ∼53.1 (p < 0.001), 79.7 (p = 0.122), 48.6 (p < 0.001), 40.1 (p < 0.001), and 99.5% (p = 0.880), relative to the mean value measured in the control cell lines, respectively.
Figure 8.

Respiration assays. A, an analysis of O2 consumption in the various cell lines using different inhibitors. The rates of O2 (OCR) were first measured on 2 × 104 cells of each cell line under basal conditions and then sequentially added to oligomycin (1.5 μm), FCCP (0.5 μm), rotenone (1.0 μm), and antimycin A (1.0 μm) at the indicated times to determine different parameters of mitochondrial functions. B, graphs presented the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and non-mitochondrial OCR in mutant and control cell lines. Non-mitochondrial OCR was determined as the OCR after rotenone/antimycin A treatment. Basal OCR was determined as OCR before oligomycin minus OCR after rotenone/antimycin A. ATP-lined OCR was determined as OCR before oligomycin minus OCR after oligomycin. Proton leak was determined as basal OCR minus ATP-linked OCR. Maximal was determined as the OCR after FCCP minus non-mitochondrial OCR. Reserve Capacity was defined as the difference between Maximal OCR after FCCP minus Basal OCR. The average values of 3–5 independent experiments for each cell line are shown, the horizontal dashed lines represent the average value for each group. Graph details and symbols are explained in the legend to Fig. 4.
Reduced level in mitochondrial ATP production
We used the luciferin/luciferase assay to examine the capacity of oxidative phosphorylation in mutant and wildtype cell lines. Populations of cells were incubated in the media in the presence of glucose, and 2-deoxy-d-glucose with pyruvate (44). As shown in Fig. 9A, the levels of ATP production in mutant cell lines in the presence of glucose (total cellular levels of ATP) were comparable with those measured in control cell lines. In contrast, as shown in Fig. 9B, the levels of ATP production in mutant cell lines, in the presence of 2-deoxy-d-glucose and pyruvate to inhibit the glycolysis (mitochondrial levels of ATP), varied from 66.0 to 73.1%, with an average of 69.8% relative to the mean value measured in the control cell lines (p < 0.001).
Figure 9.
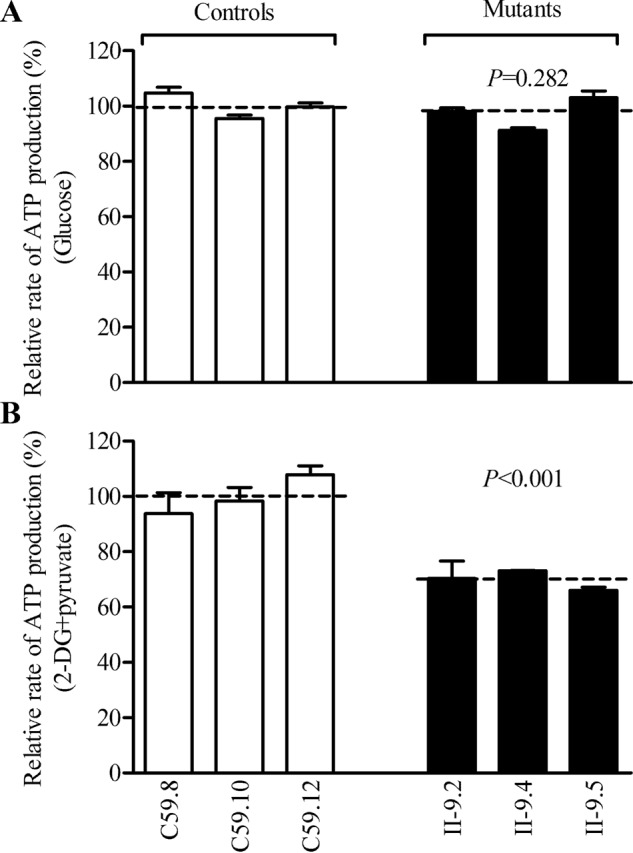
Measurement of cellular and mitochondrial ATP levels using bioluminescence assay. Cells were incubated with 10 mm glucose or 5 mm 2-deoxy-d-glucose plus 5 mm pyruvate to determine ATP generation under mitochondrial ATP synthesis. Average rates of ATP level per cell line and are shown: A, ATP level in total cells. B, ATP level in mitochondria. Three independent experiments were made for each cell line. Graph details and symbols are explained in the legend to Fig. 4.
Decrease in mitochondrial membrane potential
The mitochondrial membrane potentials (ΔΨm) were measured through the fluorescence probe JC-10 assay system in three mutant and three control cell lines (45). The ratio of fluorescence intensities excitation/emission = 490/590 and 490/530 nm (FL590/FL530) were recorded to delineate the ΔΨm of each sample. The relative ratios of FL590/FL530 geometric mean between mutant and control cell lines were calculated to represent the level of ΔΨm, as described elsewhere (44). As shown in Fig. 10, the ΔΨm of three mutant cell lines carrying the m.4435A→G mutation ranged from 62.8 to 64.0%, with an average 63.4% (p < 0.001) of the mean value measured in three control cell lines. In contrast, the levels of ΔΨm in mutant cell lines in the presence of FCCP were comparable with those of control cell lines (p = 0.290).
Figure 10.

Mitochondrial membrane potential analysis. ΔΨm was measured in three mutant and three control cell lines using a fluorescence probe JC-10 assay system. The ratio of fluorescence intensities excitation/emission = 490/590 and 490/530 nm (FL590/FL530) were recorded to delineate the ΔΨm level of each sample. Represented flow cytometry images of cell lines II-9.2 and C59.8 with (A) and without (B) 10 μm FCCP. Relative ratio of JC-10 fluorescence intensities at excitation/emission = 490/530 and 490/590 nm in the absence (C) and presence (D) of 10 μm FCCP. The average of three to five determinations for each cell line are shown. Graph details and symbols are explained in the legend to Fig. 4.
The increase of ROS production
The levels of mitochondrial ROS among the cybrids derived from three mutant cybrid cell lines carrying the m.4435A→G mutation and three control cybrid cell lines lacking the mutation were determined using MitoSOX assay via flow cytometry under normal conditions and then following H2O2 stimulation (46–48). Geometric mean intensity was recorded to measure the production rate of ROS of each sample. As shown in Fig. 11, A and B, the levels of ROS generation in the mutant cell lines carrying the m.4435A→G mutation ranged from 140.0 to 157.4%, with an average 147.5% (p < 0.001) of the mean value measured in three control cell lines under unstimulated conditions. As illustrated in Fig. 11, C and D, the levels of ROS generation in the mutant cell lines varied from 157.1 to 170.9%, with an average 165.6% (p < 0.001) of the mean value measured in the control cell lines under stimulation conditions.
Figure 11.

Measurement of ROS. Ratio of geometric mean intensity between levels of the ROS generation in the vital cells with or without H2O2 stimulation. The rates of production in ROS from three mutant cell lines and three control cell lines were analyzed by BD-LSR II flow cytometer system using MitoSox (5 μm) either without (A) or with (C) H2O2 stimulation. The relative ratio of intensity (stimulated or unstimulated with H2O2) was calculated. B and D, the average of three independent determinations for each cell lines is shown. Graph details and symbols are explained in the legend to Fig. 4.
Discussion
In the present study, we investigated the pathogenic mechanism of the hypertension-associated m.4435A→G mutation in the tRNAMet gene. The occurrences of the m.4435A→G mutation in four genetically unrelated Chinese families affected by hypertension strongly indicate that this mutation is involved in the pathogenesis of this disease (31–33). The m.4435A→G mutation affected a highly conserved adenosine (A37), adjacent to the 3′ end of the anticodon of tRNAMet (3, 7). The nucleotides at position 37 (A or G) of tRNAs are often modified, by such processes as thiolation and methylthiolation (3, 15). However, nucleotides at this position in 5 of 22 mammalian mitochondrial tRNAs including tRNAMet and tRNAAsp were not modified (9, 28). In particular, neither i6A37 nor t6A37 modification was detected in the mammalian mitochondrial tRNAMet (9). Therefore, it was hypothesized that the substitution of A37 with G37 caused by the m.4435A→G mutation created the m1G37 modification of tRNAMet, catalyzed by TRMT5. In vitro assays showed that the m.4435A→G mutation impaired the f5C formation mediated by NSUN3 (49). In this study, the primer extension experiment revealed that the m.4435A→G mutation introduced the m1G37 modification of tRNAMet. This hypothesis was further supported by the fact that an archaea M. jannaschii Trm5 catalyzed the m1G37 modification in the unmodified mutant (G37) but not wildtype (A37) tRNAMet transcripts. Furthermore, an electrophoretic mobility shift assay showed that M. jannaschii Trm5 had higher affinity with the mutant tRNAMet transcripts (G37) than the wildtype tRNAMet transcript (A37). These data demonstrated that the m.4435A→G mutation created the m1G37 modification of tRNAMet, catalyzed by TRMT5.
Modification at position 37 contributes to the high fidelity of codon recognition and the structural formation and stabilization of functional tRNAs (3, 15, 50, 51). In Escherichia coli, the deficient modification of A37 decreased the activity of the corresponding tRNA (52) and increased +1 frameshifts for tRNAPhe (53), whereas the A to G substitution at position 37 led to a 10-fold reduction in the section of tRNAs at the A-site (54). Therefore, the m1G37 modification introduced by the m.4435A→G mutation altered the structure and function of tRNAMet, as in the case of m.7551A→G mutation in the tRNAAsp (28). Here, the altered structure of tRNAMet caused by the m.4435A→G mutation was evidenced by the increased melting temperature and electrophoretic mobility of mutated tRNA with respect to the wildtype molecule. Furthermore, the m.4435A→G mutation caused 31% reduction in aminoacylated efficiency of tRNAMet in mutant cell lines, in contrast to the increasing efficiency of aminoacylation in several tRNAs caused by TRMU mutation (22). Both altered structure and improper aminoacylation of tRNAMet made the mutant tRNAMet to be metabolically less stable and more subject to degradation, thereby lowering the level of this tRNA, as in the case of tRNAAsp 7551A→G mutation (28). In the present study, 40% reductions in the steady-state level of tRNAMet observed in mutant cell lines were consistent with the previous observations in the lymphoblastoid cell lines bearing the m.4435A→G mutation (30, 31). However, the reduced levels of tRNAMet in mutant cells harboring the m.4435A→G mutation were indeed above the proposed threshold, which is 30% of the control levels of tRNA, to support the normal rate of mitochondrial translation (39, 44, 55, 56), indicating that the m.4435A→G mutation itself is insufficient to produce a clinical phenotype.
The inefficient aminoacylation and shortage of tRNAMet, or the faulty interaction between mutant tRNAMet and the translational machinery, contributed to the defective mitochondrial translation (57, 58). In the present study, reduced levels of mitochondrial proteins (an average decrease of ∼33%) were comparable with the reduced rate of mitochondrial translation observed in cell lines carrying the tRNAAsp 7551A→G, tRNAIle 4263A→G, tRNAGlu 14692A→G, and tRNAAla 5655A→G mutations (4, 28, 35, 56). The variable decreases in the levels of 7 mtDNA-encoded polypeptides were observed in the mutant cell lines. As shown in Table S2, mutant cell lines carrying the m.4435A→G mutation exhibited marked reductions (49.4 to 70.4%) in the levels of 3 polypeptides (ND3, ATP6, and CYTB), and relative mild reductions (2.5 to 23%) in the levels of ND4, ND5, CO2, and ATP8. In contrast to what was previously shown in cells carrying the tRNALys 8344A→G and tRNASer(UCN) 7445A→G mutations (39, 58), the reduced levels of these polypeptides in mutant cell lines did not significantly correlate with the number or proportion of methionine codons. The impaired synthesis of these polypeptides was responsible for the reduced activities of complexes I, III, IV, and complex V. Furthermore, the impairment of mitochondrial translation resulted in the reduced rates in the basal OCR, or ATP-linked OCR, reserve capacity, and maximal OCR in the mutant cell lines. These observations highlighted an important role of tRNAMet metabolic failure in producing their respiratory phenotypes, as in the cases of cell lines harboring the tRNAAsp 7551A→G and tRNAGlu 14692T→C mutations (4, 28).
The respiratory deficiency caused by the m.4435A→G mutation may cause uncoupling of the oxidative pathway for ATP synthesis, oxidative stress, and subsequent failure of the cellular energetic process (59). In this investigation, 32% decreases in mitochondrial ATP production in the cell lines carrying the m.4435A→G mutation, which may be caused by the defective activity of complexes I, III, IV, and V, was comparable with those in cell lines carrying the tRNAGlu 14692T→C, tRNAAsp 7551A→G, and tRNAAla 5655A→G mutations (4, 28, 35). Furthermore, the deficient oxidative phosphorylation often affected mitochondrial membrane potentials, which reflect the pumping of hydrogen ions across the inner membrane during the process of electron transport and oxidative phosphorylation (44, 46). In this study, 37% reduction in mitochondrial membrane potential in cybrid cell lines carrying the m.4435A→G mutation was comparable with those in cell lines carrying the tRNAGlu 14692T→C and tRNAAsp 7551A→G (4, 28). The abnormal oxidative phosphorylation and mitochondrial membrane potential enhanced the production of reactive oxygen species and the subsequent failure of cellular energetic processes in mutant cells carrying the m.4435A→G mutation. In particular, the overproduction of ROS can establish a vicious cycle of oxidative stress in the mitochondria, thereby damaging mitochondrial and cellular proteins, lipids, and nucleic acids and increasing apoptotic signaling (60, 61). The skeletal and vascular smooth muscles may be preferentially involved because they were exquisitely sensitive to inefficient metabolism, subtle imbalance in cellular redox state, and increased level of free radicals (62, 63). An inefficient metabolism caused by the mitochondrial dysfunction in skeletal and vascular smooth muscles may lead to the elevation of systolic blood pressure and therefore be involved in hypertension. The relative mild mitochondrial dysfunction caused by the m.4435A→G mutation suggested that the mutation is an inherited risk factor necessary for the development of hypertension but may by itself be insufficient to produce a clinical phenotype. The nuclear genetic or epigenetic factors and lifestyles may contribute to the development of clinical phenotypes in subjects bearing the m.4435A→G mutation (64, 65). In particular, the tissue-specific effect of this tRNA mutation may be attributed to the tissue-specific RNA metabolism or the involvement of nuclear modifier genes (66–69).
In summary, our findings suggested the pathogenic mechanism leading to an impaired oxidative phosphorylation in cells carrying the hypertension-associated m.4435A→G mutation in the tRNAMet gene. The m.4435A→G mutation altered the structure and function of tRNAMet. The aberrant tRNA metabolism resulted in the defects in mitochondrial translation, respiratory deficiency, decreasing membrane potentials and ATP production, and finally increasing ROS production. As a result, mitochondrial dysfunction caused by the m.4435A→G mutation manifests hypertension. However, the tissue specificity of this pathogenic mtDNA mutation is likely due to the involvement of nuclear modifier genes or tissue-specific differences in tRNA metabolism. Thus, our findings may provide new insights into the pathophysiology of hypertension, which was manifested by the deficient modification of mitochondrial tRNAMet.
Experimental procedures
Cell lines and culture conditions
Immortalized lymphoblastoid cell lines were generated from one hypertensive matrilineal relative (II-9) of a Chinese family carrying the m.4435A→G mutation (33) and one genetically unrelated Chinese control individual (C59) belonging to the same mtDNA haplogroup B5 but lacking the mutation (Table S2) (70). These cell lines were grown in RPMI 1640 medium with 10% fetal bovine serum (FBS). The bromodeoxyuridine (BrdU)-resistant 143B.TK− cell line was grown in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) (containing 4.5 mg of glucose and 0.11 mg of pyruvate/ml), supplemented with 100 μg of BrdU/ml and 5% FBS. The mtDNA-less ρ°206 cell line, derived from 143B.TK− (34, 35, 71) was grown under the same conditions as the parental line, except for the addition of 50 μg of uridine/ml. Transformation by cytoplasts of mtDNA-less ρo206 cells using one affected subject (II-9) carrying the m.4435A→G mutation and one control individual (C59) was performed as described elsewhere (34, 35, 71). All cybrid cell lines constructed with enucleated lymphoblastoid cell lines were maintained in the same medium as the 143B.TK− cell line. An analysis for the presence and level of the m.4435A→G mutation was carried out as described previously (Fig. S1) (31). Quantification of the mtDNA copy number from different cybrids was performed as detailed elsewhere (47). Three mutant cybrids (II-9.2, II-9.4, and II-9.5) carrying the m.4435A→G mutation and three control cybrids (C59.8, C59.10, and C59.12) lacking the mutation with similar mtDNA copy numbers and the same karyotype were used for the biochemical characterization described below.
Primer extension assay
A primer extension experiment to analyze the m1G37 modification of tRNAMet was carried out by a modified procedure, as described elsewhere (72, 73). Total mitochondrial RNAs were obtained from mitochondria isolated from mutant and control cell lines (∼2.0 × 108 cells) using the TOTALLY RNATM kit (Ambion), as described previously (74). A DNA primer (5′-TGGTAGTACGGGAAGGGTATAACCAACATT-3′) complementary to the 3′ end of the tRNAMet was 5′ end labeled with DIG. Two μg of total mitochondrial RNA as templates and 1 μm DIG-labeled oligodeoxynucleotide probe specific for the tRNAMet were used in the PrimeScript II 1st Strand cDNA Synthesis Kit (TAKARA) for reverse transcription. Extension reactions were carried out as detailed previously (72, 73). The samples were applied onto 15% PAGE with 8 m urea in Tris borate-EDTA buffer and electroblotted onto a positively charged nylon membrane (Roche).
Methylation activity assays
The fragments spanning tRNAMet (corresponding to mtDNA at positions 4402–4469) were PCR-amplified genomic DNAs from one hypertensive subject (II-9) carrying the m.4435A→G mutation and control subject (C59) and cloned into the HindIII/BamHI sites of pUC19. The plasmid carrying the human cytoplasmic tRNALeu(CAG) and tRNAThr genes were the gifts from Dr. En-Duo Wang (75, 76). The unmodified tRNAs were generated by in vitro transcription using T7 RNA polymerase, as detailed elsewhere (77). The purifications of archaeal Trm5 (aTrm5) from M. jannaschii (Mj-Trm5) was performed as detailed previously (36, 37). The methylation reaction contained 200 μm [3H]SAM, 50 mm Tris-HCl (pH 7.0), 100 mm KCl, 10 mm MgCl2, 100 μg/ml of bovine serum albumin (BSA), 5 mm DTT, and 5 μm transcribed wildtype or mutant tRNAMet. The reaction was initiated by the addition of 2 μm Mj-Trm5. Reactions were performed under identical conditions at 37 °C, at time intervals ranging between 2 and 8 min, aliquots of 5 μl were removed, absorbed on paper discs, and precipitated in trichloroacetic acid.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed as described elsewhere (78, 79). A range of 0.25–8 μm Mj-Trm5 was incubated with 100 nm tRNA in the presence of a 30-μl reaction volume including 50 mm Tris-HCl (pH 8.0), 100 mm KCl, 5 mm MgCl2, and 20% glycerol at 37 °C for 10 min. After incubation, each sample was loaded onto a 6% polyacrylamide native gel immediately after adding loading buffer. The gel was stained with ethidium bromide.
UV melting assays
UV melting assays were carried out as previously described (4, 80). The wildtype and mutant tRNAMet transcripts were generated as described above. The tRNAMet transcripts were diluted in the buffer including 50 mm sodium phosphate (pH 7.0), 50 mm NaCl, 5 mm MgCl2, and 0.1 mm EDTA. Absorbance against temperature melting curves were measured at 260 nm with a heating rate of 0.5 °C/min from 25 to 95 °C through Agilent Cary 100 UV Spectrophotometer.
Mitochondrial tRNA analysis
For the tRNA Northern blot analysis, 2 μg of total mitochondrial RNAs were electrophoresed through a 10% polyacrylamide gel without (native gel) or with (denature gel) 8 m urea. The gels were then electroblotted onto a positively charged nylon membrane (Roche) for the hybridization analysis with specific DIG-labeled oligodeoxynucleotide tRNAMet probe (21, 31, 81). After stripping, the same blots were hybridized with five DIG-labeled oligodeoxynucleotide tRNA probes in the following order: tRNALys, tRNASer(UCN), tRNAThr, tRNALeu(CUN), and tRNAGlu, respectively. After hybridization, the membranes were washed with Washing buffer (0.1 m maleic acid, 0.15 m NaCl, 0.3% (v/v) Tween 20 (pH 7.5)), incubated for 30 min in Blocking solution (dilute blocking reagent (Roche) in 0.1 m maleic acid, 0.15 m NaCl (pH 7.5)), and then incubated for 30 min in Antibody solution (dilute anti-digoxigenin-AP Fab fragments (Roche), 1:10,000, in blocking solution). Anti-digoxigenin-AP Fab fragments for binding to the hybridized probes were used for the immunological detection. After incubation, the membranes were washed with 20 ml of washing buffer twice and equilibrated with detection buffer (0.1 m Tris-HCl, 0.1 m NaCl (pH 9.5)). CDP-Star, ready-to-use solution (Roche) was used to soak the membrane evenly before exposure and signals were detected using the ECL system (CWBIO). After exposure, the membranes were washed with sterile diethyl pyrocarbonate-treated distilled water, incubated for 2 × 60 min at 80 °C in stripping buffer (50 mm Tris-HCl (pH 7.5), 50% formamide, 5% SDS), then washed for 2 × 5 min in 2 × SSC (300 mm NaCl, 30 mm sodium citrate (pH 7.0)) before prehybridization and hybridization with the next probe. To reduce the loss of tRNA during stripping, fresh stripping buffers were used for each time under sterile RNase-free conditions. The DIG-labeled oligodeoxynucleotides were generated by using a DIG oligonucleotide tailing kit (Roche). The hybridization and quantification of density in each band were performed as detailed previously (21, 31).
Oligodeoxynucleotides specific for tRNAMet, tRNALys, tRNASer(UCN), tRNAThr, tRNALeu(CUN), and tRNAGlu were as follows: 5′-TGGTAGTACGGGAAGGGTATAACCAACATT-3′ (tRNAMet), 5′-AAAGAGGTGTTGGTTCTCTTAATCTTTAAC-3′ (tRNALys), 5′-AAAGGAAGGAATCGAACCCCCCAAAGCTGG-3′ (tRNASer(UCN)), 5′-TGTCCTTGGAAAAAGGTTTTCATCTCCGG-3′ (tRNAThr), 5′-ACTTTTATTTGGAGTTGCACCAAAATTTTT-3′ (tRNALeu(CUN)) and 5′-ATTCTCGCACGGACTACAACCACGACCAAT-3′ (tRNAGlu) (GenBankTM accession no. NC_001807) (5).
For the aminoacylation assays, total mitochondrial RNAs were isolated under acid conditions, and 2 μg of total mitochondrial RNAs was electrophoresed at 4 °C through an acid (pH 5.0) 10% PAGE with 8 m urea in 100 mm sodium acetate buffer to separate the charged and uncharged tRNA as detailed elsewhere (82, 83). To further distinguish nonaminoacylated tRNA from aminoacylated tRNA, total RNAs were treated with being heated for 10 min at 60 °C (pH 8.3) and then run in parallel (82, 83). The gels were then electroblotted as described above. Quantification of density in each band was performed as detailed previously (82, 83).
Western blot analysis
Western blotting analysis was performed as detailed elsewhere (44, 46). Antibodies were obtained from different companies including ND4 (sc-20499-R) from Santa Cruz Biotechnology, ATP6 (55313-1-AP), ATP8 (26723-1-AP), and CYTB (55090-1-AP) from Proteintech, CO2 (ab110258), ND5 (ab92624), ND3 (ab170681), and Tom20 (ab56783) from Abcam. Peroxidase Affini Pure goat anti-mouse IgG and goat anti-rabbit IgG (Jackson) were used as a secondary antibody and protein signals were detected using the ECL system (CWBIO). Quantification of density in each band was performed as detailed previously (44, 46).
Assays of activities of respiratory complexes
The enzymatic activities of complexes I, II, III, IV, and V were assayed as detailed before (40, 41).
Measurements of oxygen consumption
The OCR in cybrid cell lines were measured with a Seahorse Bioscience XF-96 extracellular flux analyzer (Seahorse Bioscience), as detailed previously (43, 44).
ATP measurements
The Cell Titer-Glo® Luminescent Cell Viability Assay kit (Promega) was used for the measurement of cellular and mitochondrial ATP levels, following the modified manufacturer's instructions (44, 46).
Assessment of mitochondrial membrane potential
Mitochondrial membrane potential was assessed with a JC-10 Assay Kit-Microplate (Abcam) according to the general manufacturer's recommendations with some modifications, as detailed elsewhere (44, 45).
Measurement of ROS production
ROS measurements were performed as detailed previously (29, 35, 47, 48).
Computer analysis
Statistical analysis was carried out using the unpaired, two-tailed Student's t test contained in the Microsoft-Excel program or Macintosh (version 2007). Differences were considered significant at p < 0.05.
Author contributions
M. Z., L. X., Y. C., H. L., Q. H., B. W., F. M., and M.-X. G. data curation; M. Z., L. X., and M.-X. G. formal analysis; M. Z. and M.-X. G. funding acquisition; M. Z., B. W., M. W., and M.-X. G. validation; M. Z., L. X., and Y. C. methodology; M. Z. and M.-X. G. writing-original draft; H. L. and M.-X. G. resources; Q. H. and M.-X. G. investigation; M. W. software; M.-X. G. conceptualization; M.-X.G. supervision; M.-X. G. project administration; M.-X. G. writing-review and editing.
Supplementary Material
Acknowledgments
We are grateful to Dr. En-Duo Wang (Shanghai Institute of Biochemistry and Cell Biology) for the pTrc99B-tRNALeu(CAG) and pTrc99B-tRNAThr plasmids and Dr. Shigeyuki Yokoyama (RIKEN Structural Biology Laboratory) for the pET26b-aTrm5 plasmid. We thank Dr. Gilbert Eriani (Institut de Biologie Moléculaire et Cellulaire) for the critical comments of this manuscript.
This work was supported by National Basic Research Priorities Program of China Grant 2014CB541704 (to M. X. G.), National Natural Science Foundation of China Grant 81600326 (to M. Z.), Postdoctoral Science Foundation of China Grant 2016M591987 (to M. Z.), and Fundamental Research Funds for the Central Universities Grant 2017QNA7026 (to M. Z.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1 and Tables S1 and S2.
- ROS
- reactive oxygen species
- DIG
- digoxigenin
- OCR
- oxygen consumption rate
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- ΔΨm
- mitochondrial membrane potentials.
References
- 1. Zhang X., Cozen A. E., Liu Y., Chen Q., and Lowe T. M. (2016) Small RNA modifications: integral to function and disease. Trends Mol. Med. 22, 1025–1034 10.1016/j.molmed.2016.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bohnsack M. T., and Sloan K. E. (2017) The mitochondrial epitranscriptome: the roles of RNA modifications in mitochondrial translation and human disease. Cell Mol. Life Sci. 75, 241–260 10.1007/s00018-017-2598-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Suzuki T., Nagao A., and Suzuki T. (2011) Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 45, 299–329 10.1146/annurev-genet-110410-132531 [DOI] [PubMed] [Google Scholar]
- 4. Wang M., Liu H., Zheng J., Chen B., Zhou M., Fan W., Wang H., Liang X., Zhou X., Eriani G., Jiang P., and Guan M. X. (2016) A deafness- and diabetes-associated trna mutation causes deficient pseudouridinylation at position 55 in tRNAGlu and mitochondrial dysfunction. J. Biol. Chem. 291, 21029–21041 10.1074/jbc.M116.739482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Andrews R. M., Kubacka I., Chinnery P. F., Lightowlers R. N., Turnbull D. M., and Howell N. (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147 10.1038/13779 [DOI] [PubMed] [Google Scholar]
- 6. Phizicky E. M., and Hopper A. K. (2010) tRNA biology charges to the front. Genes Dev. 24, 1832–1860 10.1101/gad.1956510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. El Yacoubi B., Bailly M., and de Crécy-Lagard V. (2012) Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 46, 69–95 10.1146/annurev-genet-110711-155641 [DOI] [PubMed] [Google Scholar]
- 8. Hori H. (2014) Methylated nucleosides in tRNA and tRNA methyltransferases. Front. Genet. 5, 144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki T., and Suzuki T. (2014) A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res. 42, 7346–7357 10.1093/nar/gku390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Machnicka M. A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K. M., et al. (2013) MODOMICS: a database of RNA modification pathways: 2013 update. Nucleic Acids Res. 41, D262–D267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bjork G. R. (1995) Genetic dissection of synthesis and function of modified nucleosides in bacterial transfer RNA. Prog. Nucleic Acids Res. Mol. Biol. 50, 263–338 10.1016/S0079-6603(08)60817-X [DOI] [PubMed] [Google Scholar]
- 12. Väre V. Y., Eruysal E. R., Narendran A., Sarachan K. L., and Agris P. F. (2017) Chemical and conformational diversity of modified nucleosides affects tRNA structure and function. Biomolecules 7, pii: E29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Helm M., Brulé H., Degoul F., Cepanec C., Leroux J. P., Giegé R., and Florentz C. (1998) The presence of modified nucleotides is required for cloverleaf folding of a human mitochondrial tRNA. Nucleic Acids Res. 26, 1636–1643 10.1093/nar/26.7.1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gingold H., and Pilpel Y. (2011) Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 7, 481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Allnér O., and Nilsson L. (2011) Nucleotide modifications and tRNA anticodon-mRNA codon interactions on the ribosome. RNA 17, 2177–2188 10.1261/rna.029231.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Drummond D. A., and Wilke C. O. (2008) Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134, 341–352 10.1016/j.cell.2008.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johansson M. J., Esberg A., Huang B., Björk G. R., and Byström A. S. (2008) Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol. Cell. Biol. 28, 3301–3312 10.1128/MCB.01542-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kirino Y., Goto Y., Campos Y., Arenas J., and Suzuki T. (2005) Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc. Natl. Acad. Sci. U.S.A. 102, 7127–7132 10.1073/pnas.0500563102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yasukawa T., Suzuki T., Ueda T., Ohta S., and Watanabe K. (2000) Modification defect at anticodon wobble nucleotide of mitochondrial tRNALeu(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J. Biol. Chem. 275, 4251–4257 10.1074/jbc.275.6.4251 [DOI] [PubMed] [Google Scholar]
- 20. Yasukawa T., Suzuki T., Ishii N., Ohta S., and Watanabe K. (2001) Wobble modification defect in tRNA disturbs codon-anticodon interaction in a mitochondrial disease. EMBO J. 20, 4794–4802 10.1093/emboj/20.17.4794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guan M. X., Yan Q., Li X., Bykhovskaya Y., Gallo-Teran J., Hajek P., Umeda N., Zhao H., Garrido G., Mengesha E., et al. (2006) Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 79, 291–302 10.1086/506389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meng F., Cang X., Peng Y., Li R., Zhang Z., Li F., Fan Q., Guan A. S., Fischel-Ghosian N., Zhao X., and Guan M. X. (2017) Biochemical evidence for a nuclear modifier allele (A10S) in TRMU (Methylaminomethyl-2-thiouridylate-methyltransferase) related to mitochondrial tRNA modification in the phenotypic manifestation of deafness-associated 12S rRNA mutation. J. Biol. Chem. 292, 2881–2892 10.1074/jbc.M116.749374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kopajtich R., Nicholls T. J., Rorbach J., Metodiev M. D., Freisinger P., Mandel H., Vanlander A., Ghezzi D., Carrozzo R., Taylor R. W., Marquard K., et al. (2014) Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am. J. Hum. Genet. 95, 708–720 10.1016/j.ajhg.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghezzi D., Baruffini E., Haack T. B., Invernizzi F., Melchionda L., Dallabona C., Strom T. M., Parini R., Burlina A. B., Meitinger T.,Prokisch H., Ferrero I., and Zeviani M. (2012) Mutations of the mitochondrial-tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am. J. Hum. Genet. 90, 1079–1087 10.1016/j.ajhg.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van Haute L., Dietmann S., Kremer L., Hussain S., Pearce S. F., Powell C. A., Rorbach J., Lantaff R., Blanco S., Sauer S., et al. (2016) Deficient methylation and formylation of mt-tRNAMet wobble cytosine in a patient carrying mutations in NSUN3. Nat. Commun. 7, 12039 10.1038/ncomms12039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yarham J. W., Lamichhane T. N., Pyle A., Mattijssen S., Baruffini E., Bruni F., Donnini C., Vassilev A., He L., Blakely E. L., et al. (2014) Defective i6A37 modification of mitochondrial and cytosolic tRNAs results from pathogenic mutations in TRIT1 and its substrate tRNA. PLoS Genet. 10, e1004424 10.1371/journal.pgen.1004424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Powell C. A., Kopajtich R., D'Souza A. R., Rorbach J., Kremer L. S., Husain R. A., Dallabona C., Donnini C., Alston C. L., Griffin H., et al. (2015) TRMT5 mutations cause a defect in post-transcriptional modification of mitochondrial tRNA associated with multiple respiratory-chain deficiencies. Am. J. Hum. Genet. 97, 319–328 10.1016/j.ajhg.2015.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang M., Peng Y., Zheng J., Zheng B., Jin X., Liu H., Wang Y., Tang X., Huang T., Jiang P., and Guan M. X. (2016) A deafness-associated tRNAAsp mutation alters the m1G37 modification, aminoacylation and stability of tRNAAsp and mitochondrial function. Nucleic Acids Res. 44, 10974–10985 10.1093/nar/gkw726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Merante F., Myint T., Tein I., Benson L., and Robinson B. H. (1986) An additional mitochondrial tRNAIle point mutation (A-to-G at nucleotide 4295) causing hypertrophic cardiomyopathy. Hum. Mutat. 8, 216–222 [DOI] [PubMed] [Google Scholar]
- 30. Qu J., Li R., Tong Y., Lu F., Qian Y., Hu Y., Mo J. Q., West C. E., and Guan M. X. (2006) The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation. Invest. Ophthalmol. Vis. Sci. 47, 475–483 [DOI] [PubMed] [Google Scholar]
- 31. Liu Y., Li R., Li Z., Wang X. J., Yang L., Wang S., and Guan M. X. (2009) Mitochondrial transfer RNAMet 4435A>G mutation is associated with maternally inherited hypertension in a Chinese pedigree. Hypertension. 53, 1083–1090 10.1161/HYPERTENSIONAHA.109.128702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu Z., Chen H., Meng Y., Wang Y., Xue L., Zhi S., Qiu Q., Yang L., Mo J. Q., and Guan M. X. (2011) The tRNAMet 4435A>G mutation in the mitochondrial haplogroup G2a1 is responsible for maternally inherited hypertension in a Chinese pedigree. Eur. J. Hum. Genet. 19, 1181–1186 10.1038/ejhg.2011.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xue L., Wang M., Li H., Wang H., Jiang F., Hou L., Geng J., Lin Z., Peng Y., Zhou H.,Yu H., Jiang P., Mo J. Q., and Guan M. X. (2016) Mitochondrial tRNA mutations in 2070 Chinese Han subjects with hypertension. Mitochondrion. 30, 208–221 10.1016/j.mito.2016.08.008 [DOI] [PubMed] [Google Scholar]
- 34. King M. P., and Attadi G. (1996) Mitochondria-mediated transformation of human rho(0) cells. Methods Enzymol. 264, 313–334 10.1016/S0076-6879(96)64030-0 [DOI] [PubMed] [Google Scholar]
- 35. Jiang P., Wang M., Xue L., Xiao Y., Yu J., Wang H., Yao J., Liu H., Peng Y., Liu H., Li H., Chen Y., and Guan M. X. (2016) A hypertension-associated tRNAAla mutation alters tRNA metabolism and mitochondrial function. Mol. Cell. Biol. 36, 1920–1930 10.1128/MCB.00199-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goto-Ito S., Ito T., Ishii R., Muto Y., Bessho Y., and Yokoyama S. (2008) Crystal structure of archaeal tRNA m1G37 methyltransferase aTrm5. Proteins 72, 1274–1289 10.1002/prot.22019 [DOI] [PubMed] [Google Scholar]
- 37. Goto-Ito S., Ito T., and Yokoyama S. (2017) Trm5 and TrmD: two enzymes from distinct origins catalyze the identical tRNA modification, m1G37. Biomolecules 7, E32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ojala D., Montoya J., and Attardi G. (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474 10.1038/290470a0 [DOI] [PubMed] [Google Scholar]
- 39. Guan M. X., Enriquez J. A., Fischel-Ghodsian N., Puranam R. S., Lin C. P., Maw M. A., and Attardi G. (1998) The deafness-associated mitochondrial DNA mutation at position 7445, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase subunit ND6 gene expression. Mol. Cell. Biol. 18, 5868–5879 10.1128/MCB.18.10.5868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Y., D'Aurelio M., Deng J. H., Park J. S., Manfredi G., Hu P., Lu J., and Bai Y. (2007) An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. J. Biol. Chem. 282, 17557–17562 10.1074/jbc.M701056200 [DOI] [PubMed] [Google Scholar]
- 41. Thorburn D. R., Chow C. W., and Kirby D. M. (2004) Respiratory chain enzyme analysis in muscle and liver. Mitochondrion 4, 363–375 10.1016/j.mito.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 42. Scheffler I. E. (2015) Mitochondrial disease associated with complex I (NADH-CoQ oxidoreductase) deficiency. J. Inherit. Metab. Dis. 38, 405–415 10.1007/s10545-014-9768-6 [DOI] [PubMed] [Google Scholar]
- 43. Dranka B. P., Benavides G. A., Diers A. R., Giordano S., Zelickson B. R., Reily C., Zou L., Chatham J. C., Hill B. G., Zhang J., Landar A., and Darley-Usmar V. M. (2011) Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 51, 1621–1635 10.1016/j.freeradbiomed.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gong S., Peng Y., Jiang P., Wang M., Fan M., Wang X., Zhou H., Li H., Yan Q., Huang T., and Guan M. X. (2014) A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 42, 8039–8048 10.1093/nar/gku466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reers M., Smiley S. T., Mottola-Hartshorn C., Chen A., Lin M., and Chen L. B. (1995) Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 260, 406–417 10.1016/0076-6879(95)60154-6 [DOI] [PubMed] [Google Scholar]
- 46. Jiang P., Jin X., Peng Y., Wang M., Liu H., Liu X., Zhang Z., Ji Y., Zhang J., Liang M., et al. (2016) The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 25, 584–596 10.1093/hmg/ddv498 [DOI] [PubMed] [Google Scholar]
- 47. Zhang J., Liu X., Liang X., Lu Y., Zhu L., Fu R., Ji Y., Fan W., Chen J., Lin B., et al. (2017) A novel ADOA-associated OPA1 mutation alters the mitochondrial function, membrane potential, ROS production and apoptosis. Sci. Rep. 7, 5704 10.1038/s41598-017-05571-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mahfouz R., Sharma R., Lackner J., Aziz N., and Agarwal A. (2009) Evaluation of chemiluminescence and flow cytometry as tools in assessing production of hydrogen peroxide and superoxide anion in human spermatozoa. Fertil. Steril. 92, 819–827 10.1016/j.fertnstert.2008.05.087 [DOI] [PubMed] [Google Scholar]
- 49. Nakano S., Suzuki T., Kawarada L., Iwata H., Asano K., and Suzuki T. (2016) NSUN3 methylase initiates 5-formylcytidine biogenesis in human mitochondrial tRNAMet. Nat. Chem. Biol. 12, 546–551 10.1038/nchembio.2099 [DOI] [PubMed] [Google Scholar]
- 50. Bjork G. R., and Hagervall T. G. (2014) Transfer RNA modification: presence, synthesis, and function. EcoSal Plus 6, 10.1128/ecosalplus [DOI] [PubMed] [Google Scholar]
- 51. Florentz C., Sohm B., Tryoen-Tóoth P., Pütz J., and Sissler M. (2003) Human mitochondrial tRNAs in health and disease. Cell Mol. Life Sci. 60, 1356–1375 10.1007/s00018-003-2343-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buck M., and Griffiths E. (1982) Iron mediated methylthiolation of tRNA as a regulator of operon expression in Escherichia coli. Nucleic Acids Res. 10, 2609–2624 10.1093/nar/10.8.2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Urbonavicius J., Qian Q., Durand J. M., Hagervall T. G., and Björk G. R. (2001) Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J. 20, 4863–4873 10.1093/emboj/20.17.4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yarus M., Cline S. W., Wier P., Breeden L., and Thompson R. C. (1986) Actions of the anticodon arm in translation on the phenotypes of RNA mutants. J. Mol. Biol. 192, 235–255 10.1016/0022-2836(86)90362-1 [DOI] [PubMed] [Google Scholar]
- 55. Li X., Fischel-Ghodsian N., Schwartz F., Yan Q., Friedman R. A., and Guan M. X. (2004) Biochemical characterization of the mitochondrial tRNASer(UCN) T7511C mutation associated with nonsyndromic deafness. Nucleic Acids Res. 32, 867–877 10.1093/nar/gkh226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang S., Li R., Fettermann A., Li Z., Qian Y., Liu Y., Wang X., Zhou A., Mo J. Q., Yang L., Jiang P., Tascherner A., Rossmanith W., and Guan M. X. (2011) Maternally inherited essential hypertension is associated with the novel 4263A>G mutation in the mitochondrial tRNAIle gene in a large Han Chinese family. Circ. Res. 108, 862–870 10.1161/CIRCRESAHA.110.231811 [DOI] [PubMed] [Google Scholar]
- 57. Masucci J. P., Davidson M., Koga Y., Schon E. A., and King M. P. (1995) In vitro analysis of mutations causing myoclonus epilepsy with ragged-red fibers in the mitochondrial tRNALys gene: two genotypes produce similar phenotypes. Mol. Cell. Biol. 15, 2872–2881 10.1128/MCB.15.5.2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Enriquez J. A., Chomyn A., and Attardi G. (1995) MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNALys and premature translation termination. Nat. Genet., 10, 47–55 10.1038/ng0595-47 [DOI] [PubMed] [Google Scholar]
- 59. Wallace D. C. (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407 10.1146/annurev.genet.39.110304.095751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hayashi G., and Cortopassi G. (2015) Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 88, 10–17 10.1016/j.freeradbiomed.2015.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schieber M., and Chandel N. S. (2014) ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 10.1016/j.cub.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Addabbo F., Montagnani M., and Goligorsky M. S. (2009) Mitochondria and reactive oxygen species. Hypertension 53, 885–892 10.1161/HYPERTENSIONAHA.109.130054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Eirin A., Lerman A., and Lerman L. O. (2015) Mitochondria: a pathogenic paradigm in hypertensive renal disease. Hypertension 65, 264–270 10.1161/HYPERTENSIONAHA.114.04598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vasan R.S., Beiser A., Seshadri S., Larson M.G., Kannel W.B., D'Agostino R.B., and Levy D. (2002) Residual lifetime risk for developing hypertension in middle-aged women and men: The Framingham Heart Study. JAMA 287, 1003–1010 [DOI] [PubMed] [Google Scholar]
- 65. Archer S. L., Marsboom G., Kim G. H., Zhang H. J., Toth P. T., Svensson E. C., Dyck J. R., Gomberg-Maitland M., Thébaud B., Husain A. N., Cipriani N., and Rehman J. (2010) Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121, 2661–2671 10.1161/CIRCULATIONAHA.109.916098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dittmar K. A., Goodenbour J. M., and Pan T. (2006) Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2, e221 10.1371/journal.pgen.0020221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tischner C., Hofer A., Wulff V., Stepek J., Dumitru I., Becker L., Haack T., Kremer L., Datta A. N., Sperl W., et al. (2015) MTO1 mediates tissue specificity of OXPHOS defects via tRNA modification and translation optimization, which can be bypassed by dietary intervention. Hum. Mol. Genet. 24, 2247–2266 10.1093/hmg/ddu743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Powell C. A., Nicholls T. J., and Minczuk M. (2015) Nuclear-encoded factors involved in post-transcriptional processing and modification of mitochondrial tRNAs in human disease. Front. Genet. 6, 79 10.3389/fgene.2015.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen D., Li F., Yang Q., Tian M., Zhang Z., Zhang Q., Chen Y., and Guan M. X. (2016) The defective expression of gtpbp3 related to tRNA modification alters the mitochondrial function and development of zebrafish. Int. J. Biochem. Cell Biol. 77, 1–9 10.1016/j.biocel.2016.05.012 [DOI] [PubMed] [Google Scholar]
- 70. Miller G., and Lipman M. (1973) Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc. Natl. Acad. Sci. U.S.A. 70, 190–194 10.1073/pnas.70.1.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. King M. P., and Attardi G. (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503 10.1126/science.2814477 [DOI] [PubMed] [Google Scholar]
- 72. Ofengand J., Del Campo M., and Kaya Y. (2001) Mapping pseudouridines in RNA molecules. Methods 25, 365–373 10.1006/meth.2001.1249 [DOI] [PubMed] [Google Scholar]
- 73. Qian Y., and Guan M. X. (2009) Interaction of aminoglycosides with human mitochondrial 12S rRNA carrying the deafness-associated mutation. Antimicrob. Agents Chemother. 53, 4612–4618 10.1128/AAC.00965-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. King M. P., and Attardi G. (1993) Post-transcriptional regulation of the steady-state levels of mitochondrial tRNAs in HeLa cells. J. Biol. Chem. 268, 10228–10237 [PubMed] [Google Scholar]
- 75. Long T., Li J., Li H., Zhou M., Zhou X. L., Liu R. J., and Wang E. D. (2016) Sequence-specific and shape-selective RNA recognition by the human RNA 5-methylcytosine methyltransferase NSun6. J. Biol. Chem. 291, 24293–24303 10.1074/jbc.M116.742569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li Y., Chen J., Wang E., and Wang Y. (1999) T7 RNA polymerase transcription of Escherichia coli isoacceptors tRNALeu. Sci. China Ser. C 42, 185–190 10.1007/BF02875516,10.1007/BF02880055 [DOI] [PubMed] [Google Scholar]
- 77. Bonnefond L., Fender A., Rudinger-Thirion J., Giegé R., Florentz C., and Sissler M. (2005) Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: characterization of AspRS and TyrRS. Biochemistry 44, 4805–4816 10.1021/bi047527z [DOI] [PubMed] [Google Scholar]
- 78. Watanabe K., Nureki O., Fukai S., Ishii R., Okamoto H., Yokoyama S., Endo Y., and Hori H. (2005) Roles of conserved amino acid sequence motifs in the SpoU (TrmH) RNA methyltransferase family. J. Biol. Chem. 280, 10368–10377 10.1074/jbc.M411209200 [DOI] [PubMed] [Google Scholar]
- 79. Zhou M., Long T., Fang Z. P., Zhou X. L., Liu R. J., and Wang E. D. (2015) Identification of determinants for tRNA substrate recognition by Escherichia coli C/U34 2′-O-methyltransferase. RNA Biol. 12, 900–911 10.1080/15476286.2015.1050576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhou M., Wang M., Xue L., Lin Z., He Q., Shi W., Chen Y., Jin X., Li H., Jiang P., and Guan M. X. (2017) A hypertension-associated mitochondrial DNA mutation alters the tertiary interaction and function oftRNALeu(UUR). J. Biol. Chem. 292, 13934–13946 10.1074/jbc.M117.787028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang X., Lu J., Zhu Y., Yang A., Yang L., Li R., Chen B., Qian Y., Tang X., Wang J.,Zhang X., and Guan M. X. (2008) Mitochondrial tRNAThr G15927A mutation may modulate the phenotypic manifestation of ototoxic 12S rRNA A1555G mutation in four Chinese families. Pharmacogenet. Genomics 18, 1059–1070 10.1097/FPC.0b013e3283131661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Enríquez J. A., and Attardi G. (1996) Analysis of aminoacylation of human mitochondrial tRNAs. Methods Enzymol. 264, 183–196 10.1016/S0076-6879(96)64019-1 [DOI] [PubMed] [Google Scholar]
- 83. Li R., and Guan M. X. (2010) Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol. Cell. Biol. 30, 2147–2154 10.1128/MCB.01614-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.