

Key Points
Clinical response of SSc patients after AHSCT is associated with thymic and bone marrow rebounds.
Responder patients showed higher Treg and Breg counts and lower pre-/post-AHSCT TCR repertoire overlap than nonresponder patients.
Abstract
To evaluate the immunological mechanisms associated with clinical outcomes after autologous hematopoietic stem cell transplantation (AHSCT), focusing on regulatory T- (Treg) and B- (Breg) cell immune reconstitution, 31 systemic sclerosis (SSc) patients underwent simultaneous clinical and immunological evaluations over 36-month posttransplantation follow-up. Patients were retrospectively grouped into responders (n = 25) and nonresponders (n = 6), according to clinical response after AHSCT. Thymic function and B-cell neogenesis were respectively assessed by quantification of DNA excision circles generated during T- and B-cell receptor rearrangements. At the 1-year post-AHSCT evaluation of the total set of transplanted SSc patients, thymic rebound led to renewal of the immune system, with higher T-cell receptor (TCR) diversity, positive correlation between recent thymic emigrant and Treg counts, and higher expression of CTLA-4 and GITR on Tregs, when compared with pretransplant levels. In parallel, increased bone marrow output of newly generated naive B-cells, starting at 6 months after AHSCT, renovated the B-cell populations in peripheral blood. At 6 and 12 months after AHSCT, Bregs increased and produced higher interleukin-10 levels than before transplant. When the nonresponder patients were evaluated separately, Treg and Breg counts did not increase after AHSCT, and high TCR repertoire overlap between pre- and posttransplant periods indicated maintenance of underlying disease mechanisms. These data suggest that clinical improvement of SSc patients is related to increased counts of newly generated Tregs and Bregs after AHSCT as a result of coordinated thymic and bone marrow rebound.
Visual Abstract
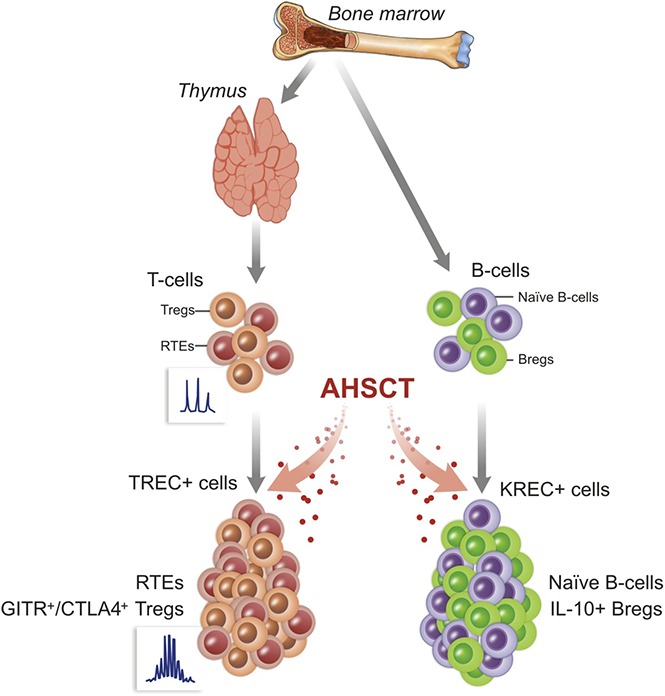
Introduction
Systemic sclerosis (SSc) is an autoimmune disease characterized by microvascular damage and progressive fibrosis within the skin and internal organs.1,2 Conventional therapy has limited benefit on disease control and modest impact on mortality.3-6 Three randomized studies have shown that autologous hematopoietic stem cell transplantation (AHSCT) has superior efficacy when compared with conventional therapy for SSc.7-10 Nevertheless, clinical guidelines and immune monitoring studies after AHSCT aim to further improve patient care and transplant outcomes.11-13
In SSc, decreased regulatory T-cell (Treg) counts and impaired immunosuppressive function have been associated with loss of self-tolerance, correlating with disease severity.14-17 Diminished thymopoiesis and abnormalities of T-cell receptor (TCR) repertoire, with fewer polyclonal families, overexpression of skewed families, and reduced overall TCR diversity were described.18,19 The role of B cells in the pathogenesis of SSc has been investigated,20 with reports of B-cell hyperactivation,21-23 autoantibody production,24 decreased regulatory B-cell (Breg) counts, and impaired interleukin-10 (IL-10) production.25,26
AHSCT aims to deplete the autoimmune repertoire and generate a new immune system, thereby reestablishing a state of autotolerance, already shown in multiple sclerosis,27,28 systemic lupus erythematosus,29,30 juvenile arthritis,31 Crohn disease,32 and SSc.19,33 In SSc, we previously showed how posttransplant CD4 T-cell reconstitution correlates with long-term clinical response to AHSCT.19,33 However, recovery of specific lymphocyte subpopulations, including those with regulatory functions, as well as thymic and bone marrow functions, and how they may be associated with clinical outcomes remain to be assessed. Here, we analyzed the immunological profile and T- and B-cells immune reconstitution of SSc patients that underwent AHSCT.
Methods
Study design
We prospectively analyzed and compared the determinants of immunological and clinical response in a group of 31 severe and rapidly progressive SSc patients who underwent AHSCT from 2010 to 2015, at the Ribeirão Preto Medical School University Hospital (Brazil). All patients met the 1980 American College of Rheumatology (ACR) and/or 2013 ACR/European League against Rheumatism classification criteria for SSc.34 The transplantation protocol and inclusion and exclusion criteria were previously published.35 Briefly, autologous hematopoietic stem cells were mobilized from the bone marrow with 2 g/m2 of cyclophosphamide plus granulocyte colony-stimulating factor (10 μg/kg/d, subcutaneous) and subsequently harvested from the peripheral blood by leukoapheresis. Then, patients were treated with total dose of 200 mg/kg cyclophosphamide plus 4.5 mg/kg rabbit antithymocyte globulin in 4 days, followed by infusion of nonmanipulated, previously cryopreserved autologous hematopoietic stem cells.
Sixteen nontransplanted severe SSc patients prospectively followed and clinically monitored at the Hôpital Saint-Louis, APHP (France), who were part of the control group of the ASTIS trial8 or for whom AHSCT was refused or unfeasible due to contraindications, were evaluated as control group for quantification of thymic and bone marrow functions (supplemental Table 1).
This study has been approved by the institutional review boards of both Brazilian and French centers, where the patients were enrolled, and complied with country-specific regulations. The study was conducted according to the Declaration of Helsinki and Good Practice Guidelines. All patients read and signed informed consents, which are available at each research site.
Immune monitoring and clinical follow-up
Clinical follow-up before and after AHSCT was performed as previously described during the first year19 and semiannually thereafter, until 36 months. The same observer assessed clinical response using repeated functional and physical examination of organ involvement. Clinical evaluations included assessment of modified Rodnan’s skin score (mRSS), lung, kidney, gastrointestinal, and heart function, and quantification of antitopoisomerase (anti-Scl-70) autoantibodies and C-reactive protein. Relapsing disease post-AHSCT was defined by 1 of the following criteria: increase of the mRSS by 25% from best improvement, or decline in forced vital capacity by 10%, renal crisis, start of total parenteral nutrition, or restarting of immune suppressive or modulating medication, as previously described.33,35 According to clinical response at long-term post-AHSCT, SSc patients were retrospectively classified as “responders” and “nonresponders.” According to the European Group for Blood and Marrow Transplantation guidelines,13 peripheral blood samples were collected at baseline and every 6 months until 36 months after transplantation for immune monitoring.
Flow cytometry analysis
Whole blood was collected into EDTA-containing tubes and immunophenotyped with predetermined optimal antibody concentrations to quantify lymphocyte subsets and regulatory molecule expressions.36-38 Antihuman monoclonal antibodies (mAbs) included the following: CD3 (UCHT1), CD4 (RPA-T4), CD8 (RPA-T8), CD19 (HIB19), CD31 (WM59), CD45RA (HI100), CD45RO (UCHL1), CD27 (L128), CD25 (2A3), immunoglobulin D (IgD) (IA6-2), CD38 (HIT2), CD24 (ML5), and CTLA-4 (BNI3) from BD Pharmingen (San Diego, CA), and GITR (eBioAITR) and FoxP3 (PCH101) from eBioscience (San Diego, CA). Cells were acquired in FACSCalibur (BD Biosciences) cytometer and analyzed with Flow Jo (TreeStar) software. All analyses were performed on fresh blood.
Cell isolation
Heparinized peripheral blood samples were collected, and peripheral blood mononuclear cells (PBMCs) were isolated through Ficoll-Hypaque (Amersham-Pharmacia, Uppsala, Sweden) density-gradient separation. PBMCs were cryopreserved in 10% dimethyl sulfoxide and stored in liquid nitrogen or lysed and stored in TRIzol reagent (Invitrogen, Carlsbad, CA) at −80°C.
Quantification of thymic (T-cell) and bone marrow (B-cell) functions
Genomic DNA was extracted with TRIzol according to the manufacturer’s instructions. Signal-joint or β-chain TCR (TCRβ) excision circles (sjTREC or βTREC), as well as coding and signal-joint K-chain recombination excision circles (Cj and sjKREC), were quantified by real-time quantitative polymerase chain reaction (RT-PCR) as previously described.39-41 Primers and probes were obtained from Eurogentec (Paris, France) (supplemental Table 2).
Multiplex preamplification.
A first PCR reaction was carried out in multiplex with different outer primer mixes (supplemental Table 2), with 1 to 2 μg of genomic DNA, 200 µM of each 2′-deoxynucleoside 5'-triphosphate, 2.5 mM MgSO4, 1× buffer, and 1.25 unit of Platinum Taq DNA pol High Fidelity (Thermo Fisher Scientific, Courtaboeuf, France) in 50 μL (3 minutes at 95°C, then 18 cycles of 95°C for 15 seconds, 60°C for 30 seconds, and 68°C for 30 seconds). Samples were stored at −20°C.
Quantification of sjTREC, βTREC, Cj, and sjKREC.
Final quantification was made on a ViiA7 Real-Time PCR System (Applied Biosystems, Foster City, CA), in duplicate with a second multiplex reaction containing 4 μL of 1/200 or 1/2000 dilution of the first PCR product, primers and probes for albumin gene, sjTREC, Cj, sjKREC, or 1 of the Dβ-Jβ segments and 2× Takyon Low Rox Probe MM (Eurogentec) in 10 μL (5 minutes at 95°C, then 40 cycles of 95°C for 15 seconds, and 60°C for 1 minute). The sum of the 10 Dβ-Jβ segments were finally multiplied by 1.3 to take into account the 3 Dβ-Jβ that were not quantified (Dβ2-Jβ 2.5, 2.6, and 2.7). sjTREC, βTREC, Cj, and sjKREC were normalized to 150 000 cells (∼1 µg of DNA) using albumin gene quantification. Data were expressed as Log10/150 000 PBMC.
Analysis of IL-10 production
PBMCs were thawed rapidly in preheated media, washed, and seeded in 96-well U-bottom plates at a concentration of 1 × 106 cells per well in fresh RPMI 1640 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% glutamate (Sigma-Aldrich). Cell viability was determined by Trypan blue exclusion and Annexin/PI (BD Pharmingen) staining and exceeded 95%.
T-cell IL-10 production.
Cells were either mock treated or cultured in the presence of anti-CD3/CD28 Dynabeads (Life Technologies) following the manufacturer's instructions (bead-to-cell ratio = 1:1). Phorbol myristate acetate (50 ng/mL; Sigma-Aldrich), ionomycin (1 μg/mL; Life Technologies, Waltham, MA), and brefeldin A (BFA, 10 μg/mL; Sigma-Aldrich) were added during the last 6 hours of culture. All 3 stimulants were added together, here cited as PIB. Cells were harvested, washed, and stained for CD4 and CD25 surface marker and then permeabilized with fluorescence-activated cell sorter (FACS) permeabilization solution (BD Bioscience) following the manufacturer's instructions. IL-10 was detected with anti–IL-10 antibody (JES3-9D7) from eBioscience or by an isotype-matched control at the same time as FoxP3 intracellular staining. FoxP3+IL-10+ gate positioning was determined in the mock-treated cells and cultured with BFA only during the last 6 hours. FACS determined relative lymphocyte percentages.
B-cell IL-10 production.
To evaluate the immunosuppressive capacity of Bregs, IL-10 production was assessed by 2 different in vitro methods (cytosine guanine dinucleotide [CpG] ± CD40L).25,42,43 Cells were cultured in the presence of 10 μg/mL CpG control (InvivoGen, San Diego, CA), 10 μg/mL CpG-B (ODN 2006), or CpG-B and recombinant human CD40L (1 μg/mL; R&D Systems, Minneapolis, MN) for 24 hours, as previously published.25,42 PIB was added in the last 6 hours. Cells were harvested, washed, stained for CD19 surface marker, and permeabilized. IL-10 was detected by anti–IL-10 or isotype-matched control. IL-10+ gate positioning was determined in cells cultured with CpG control, and BFA (instead of PIB) was added to the cultures in the last 6 hours. FACS determined relative lymphocyte percentages.
Analysis of TCRβ repertoire by NGS
According to the manufacturer’s instructions, total RNA was extracted from PBMCs using TRIzol. TCR new generation sequencing (NGS) protocol was adapted from Mamedov et al.44 First-strand complementary DNA (cDNA) was synthesized for 1 hour using Smartscribe RT (Clontech) with primers bc1R (specific to both constant regions of the human TCRβ; for primer sequences, see supplemental Table 3) and the adapter primer Smart NNN (containing bar-coding sequence) and 1.5 µg of total RNA. A first PCR amplification used 1 µL of cDNA, Taq platinum high fidelity (Invitrogen), and primers Smart20 and BC2R for 15 cycles (95°C 20 seconds, 65°C 20 seconds, and 72°C 50 seconds). The PCR product was purified using QiaQuick MinElute (Qiagen). A second PCR used Mix Step1 and Mix Hum bcj primers identically to PCR1 but for 10 cycles only. The library was prepared using Nextera XT index kit (Illumina) and Taq platinum high fidelity according to manufacturer’s instructions before purification using AMPure XP beads. Denatured library was paired-end (2 × 250) sequenced on a MiSeq (Illumina). Unique molecular identifiers were treated using pRESTO,45 and CDR3 clonotypes were assembled using MiTCR software.46 Postanalysis of TCR repertoire diversity, segment usage, spectratyping, clonotype tracking, and repertoire overlap were performed by VDJTools.47 Raw sequencing data were deposited in the NCBI SRA database (SRP106516).
Statistical analysis
Patient characteristics are described as mean ± standard error (SE). Nonparametric 2-tailed Wilcoxon's signed-rank test was performed to compare pre- and posttransplant values. Continuous variables in 2 different groups were compared by nonparametric 2-tailed Mann-Whitney U test, and results were expressed as median ± interquartile range (IQR). Correlations were assessed using nonparametric Spearman test. When indicated, logarithmic transformation was performed on skewed data prior to correlation. SPSS Statistics 20 (IBM, Armonk, NY), GraphPad 7 (La Jolla, CA) or R Project Package in VDJTools environment were used for figures and analyses.47 Significance was set at 0.05.
Results
SSc patient clinical characteristics
Thirty-one transplanted patients (26 women) with a median (range) age of 34 (19-58) years and disease duration of 27 (8-340) months were included in the study (supplemental Table 1). Sixteen nontransplanted severe SSc patients (14 women), median (range) age of 45 (30-63) years and disease duration of 16 (2-295) months, were included as control group for thymic and bone marrow function evaluations (supplemental Table 1). Transplanted patients showed significant improvement in the mRSS (P < .01) from 6 to at least 24 months of follow-up and stabilized pulmonary function (P > .05) when compared with pretransplant scores. There was also significant decrease in the antitopoisomerase (anti-Scl-70, P = .01) and C-reactive protein serum (P = .03) levels, starting at 6 months, until 18 and 24 months after AHSCT, respectively (supplemental Figure 1). Twenty-five patients were classified as responders, because they remained stable or improved at clinical evaluations, and 6 were classified as nonresponders (Table 1), and underwent subgroup analyses. One responder patient died at 7 months post-AHSCT because of pulmonary embolism.
Table 1.
Nonresponder patient characteristics
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | |
|---|---|---|---|---|---|---|
| Patient characteristics at inclusion | ||||||
| Age, y | 27 | 30 | 29 | 59 | 29 | 41 |
| Sex | M | F | F | F | F | F |
| Disease duration, mo | 24 | 25 | 31 | 86 | 14 | 33 |
| Previous treatments | MTX, CST | CYC, MTX, MMF | CYC, MTX, CST | CYC, MTX, CST, AZA | CYC, MTX, CST | CYC, CST, AZA |
| Modified Rodnan’s skin score | 14 | 30 | 24 | 20 | 41 | 29 |
| C-reactive protein, mg/dL | 4.43 | 0.25 | 1.99 | 0.76 | 0.9 | 0.34 |
| Anti-Scl-70 positivity | + | + | − | + | + | + |
| Patient characteristics after transplant | ||||||
| Months of follow-up after AHSCT, at disease reactivation | 36 | 36 | 24 | 24 | 6 | 12 |
| Disease-reactivation details | Developed RA | Digital ulcerations | Lung function worsening | Lung function worsening | Skin worsening | Skin worsening |
| Immunosuppression after disease reactivation* | MTX | RTX | MMF | RTX | MTX | MTX, CST |
AZA, azathioprine; CST, corticosteroids; CYC, cyclophosphamide intravenous; F, female; M, male; MMF, mycophenolate mofetil; MTX, methotrexate; RA, rheumatoid arthritis; RTX, rituximab.
Immunosuppressive therapy was started at the moment disease reactivation was detected, and maintained until the end of follow-up.
Thymic function rebound after AHSCT
sjTREC and βTREC values reflect generation of thymic-derived cells, whereas the sj/βTREC ratio allows estimation of intrathymic thymocyte proliferation rates (supplemental Figure 2).48 At 6 months after AHSCT, both sjTREC and βTREC values were low, when compared with pretransplant and nontransplanted SSc group levels. At 1 year after AHSCT, sjTREC reached pretransplant levels, progressively increasing after the 18 months’ time point and becoming higher than pretransplant levels at 3 years (Figure 1A). Similarly, βTREC values increased at 1-year post-AHSCT, presenting higher levels than the nontransplanted patients (Figure 1B). Nontransplanted patients did not change sjTREC and βTREC levels along the entire follow-up. For both transplanted and nontransplanted patients, no change in intrathymic thymocyte proliferation rates was observed (Figure 1C).
Figure 1.

Exportation of thymic-derived newly generated naive T cells correlates with posttransplantation thymic recovery. Median (± IQR) of (A) sjTREC and (B) βTREC values as measured by quantitative RT-PCR analysis on PBMC genomic DNA at baseline (0 months, pretransplant) and at the following time points in transplanted (AHSCT, n = 26 patients at baseline, n = 15 at 6 and 12 months, and n = 11 at 18, 24, and >24 months) and nontransplanted (non-AHSCT, n = 14 patients at baseline and at 6 months, n = 13 at 12 months, and n = 8 at 18, 24, and >24 months) SSc patients. The results were expressed by log10 in 150 000 PBMCs. (C) Intrathymic T-cell division (n) as calculated using following formula: n = LOG(sjTREC/βTREC)/LOG2. *P < .05, AHSCT vs non-AHSCT (Mann-Whitney U test). §P < .05, §§P < .01 comparing posttransplant values to baseline (Wilcoxon’s). Panels D-H include transplanted patients only. (D) Percentage of CD45RA and CD31 coexpression by CD3+CD4+ T cells immunophenotyped by flow cytometry. The boundaries of the boxes indicate the 25th and 75th percentiles; the lines within the boxes indicate the median, and the whiskers mark the 10th and the 90th percentiles. (E) Mean (± SE) of RTEs absolute values (cells per microliter). *P < .05; **P < .01 comparing posttransplant values to baseline (Wilcoxon’s). (F) Correlation between absolute values of CD3+CD4+CD31+CD45+ T cells and sjTREC values (Spearman’s). Mean (± SE) percentage of Naive (CD27+CD45RO−), Central Memory (CD27+CD45RO+), Effector Memory (CD27-CD45RO+), and Effector (CD27−CD45RO−) (G) CD4+ and (H) CD8+ T cells.
Thymic reactivation was assessed after AHSCT through CD45RA/CD31 coexpressions by naive T cells.49 After a decline at 6 months post-AHSCT, both the percentages and the absolute numbers of recent thymic emigrant (RTE) increased to pretransplant levels from 12-month post-AHSCT until the end of follow-up (Figure 1D-E), correlating (rs = 0.68, P < .0001) with sjTREC values (Figure 1F). These findings confirm the posttransplant thymic function rebound, with the exportation of newly generated thymic-derived naive T cells. Responder and nonresponder groups did not differ for sjTREC, βTREC, or RTE quantifications (supplemental Figure 3).
The effect of AHSCT on the distribution of naive and memory T cells was assessed by CD27 and CD45RO expressions.50 CD27+CD45RO− naive and CD27+CD45RO+ central-memory cells were depleted at 6 months and started to increase afterward, whereas CD27−CD45RO− effector cells presented the opposite profile (Figure 1G-H). The ratio between CD4+ naive/central-memory T cells increased from 0.25 ± 0.05 at 6 months to 0.64 ± 0.11 (P = .004) at 12 months, suggesting that thymic reactivation induces phenotypic rejuvenation of the CD4+ repertoire despite slow reconstitution of CD4+ T cells (supplemental Figure 4).
Low clonotype overlap and high TCR diversity are related to clinical response after AHSCT
Normalized unique clonotype counts were used to estimate the TCR diversity in 5 responder and 3 nonresponder transplanted patients (Table 2).47 Minimal transplant-induced changes in the TCR repertoire and clonotype specificities were assessed by NGS.28,51 At 1-year posttransplantation, the TCR diversity had significantly increased from pretransplant levels, indicating the generation of a new immune system (Figure 2A). These data were further confirmed by an increase of Chao1 diversity estimation52 at 1-year post-AHSCT (supplemental Figure 5A), with higher frequency of singletons (clonotypes with single occurrence in the sample) (supplemental Figure 5B).
Table 2.
Description of patients included for TCR NGS
| Patient characteristics | Age, y | Sex | Disease duration, mo | Previous treatments | mRSS | CRP | Anti-Scl-70 | Disease status after AHSCT (time point of reactivation) | Posttransplant IS |
|---|---|---|---|---|---|---|---|---|---|
| Responders | |||||||||
| P1 | 44 | M | 36 | CYC, CST, AZA | 29 | 0.35 | + | Remission | None |
| P2 | 35 | F | 51 | CYC, MTX, MMF, CST | 26 | 0.08 | + | Remission | None |
| P6 | 21 | M | 12 | MMF, CST | 26 | 0.48 | + | Remission | None |
| P7 | 45 | F | 22 | CYC, CST | 12 | 0.62 | + | Remission | None |
| P8 | 29 | F | 64 | CYC, MTX, CST | 18 | 0.92 | + | Remission | None |
| Non-responders | |||||||||
| P3 | 29 | F | 14 | CYC, MTX, CST | 41 | 0.9 | + | Skin worsening (6 mo) | MTX |
| P4 | 30 | F | 25 | CYC, MTX, MMF | 30 | 0.25 | + | Digital ulcerations (3 y) | RTX |
| P5 | 27 | M | 24 | MTX, CST | 14 | 4.43 | + | Developed RA (1 y) | MTX |
CRP, C-reactive protein (mg/dL); IS, immunosuppression.
Figure 2.
Low clonotype overlap and high TCR diversity are related to favorable clinical response to AHSCT. (A) Comparisons of the observed TCR repertoire diversity based on unique clonotypes (n = 8 transplanted SSc patients at baseline, n = 5 at 6 months, n = 4 at 12 months, and n = 5 at 24 months). The boundaries of the boxes indicate the 25th and 75th percentiles; the lines within the boxes indicate the median, and the whiskers mark the 10th and the 90th percentiles. *P < .05 comparing posttransplant values with baseline (Wilcoxon’s). (B) Rarefaction analysis of repertoire samples from a representative responder (patient P6, left) and nonresponder (patient P5, right) patient. Number of unique clonotypes in a subsample is plotted against its size (number of TCR cDNA molecules). Solid and dashed lines mark interpolated and extrapolated regions of rarefaction curves, respectively, and points mark exact sample size and diversity. Shaded areas mark 95% confidence intervals. (C) Representative spectratype profile of a responder (patient P8, left) and nonresponder (patient P3, right) patient at baseline (upper panels) and at 2 years after AHSCT (lower panels). Panels display distribution of clonotype frequency by CDR3 length. Most abundant clonotypes are explicitly shown. The nonresponder patient did not achieve a Gaussian distribution even at later periods after transplant. (D) Clonotype tracking stackplot shows details for highly frequent clonotypes shared between baseline and posttransplant time points. Overlapping clonotype shows average frequencies of a responder (patient P6, left) and a nonresponder (patient P5, right) patient. Clonotypes are colored by the peak position of their abundance profile. Other low-frequency clonotypes that were observed in both samples are marked as “Not-shown” and the remaining clonotypes are marked as “Non-overlapping.” (E) Representative joint clonotype abundance scatter plot of a responder (patient P1, upper panels) and nonresponder (patient P5, lower panels) SSc patient, showing the overlap between baseline and 6 months (left panels) as well as between baseline and 2 years (right panels). The main frame contains a scatter plot of clonotype abundances (overlapping clonotypes only) and a linear regression. Point size is scaled to the geometric mean of clonotype frequency in both samples. Scatter plot axes represent log10 clonotype frequencies in each sample. R2 represents squared Pearson’s correlation coefficient. (F) Clonotype tracking heat map of a responder (patient P6, left) and nonresponder (patient P5, right) patient showing joint clonotype abundances. For the responder patients, the most frequent clonotypes at baseline disappear at 2 years posttransplant.


Estimation of TCR diversity showed that, for responder patients, overall specificities increased following thymic rebound at 1 and 2 years (Figure 2B). Nonresponder patients failed to achieve higher TCR diversity, with reduced frequencies of singletons (supplemental Figure 5B).
Both clonotype and variable segment spectratyping analyses depicted a skewed TCRβ CDR3 distribution at baseline (Figure 2C; supplemental Figures 6-8), as observed for an autoimmune profile.19,27,28,30 After transplant, responder patients achieved a Gaussian distribution for clonotype and V-segment spectratyping at 2 years (Figure 2C; supplemental Figures 6-8). Nonresponder patients maintained a skewed and oligoclonally expanded profile, demonstrating a persistent autoimmune repertoire.
The overlapping and shared clonotype frequencies increased from before to the 6-month posttransplant time point in both responder and nonresponder patient groups (Figure 2D). Six months post-AHSCT, shared clonotype frequencies met only 25% of the total TCR repertoire in responder patients, whereas it was 60% of the repertoire in nonresponders (Figure 2D). Two years after transplantation, reflecting thymic reactivation, these shared clonotype frequencies comprised ∼10% of the overall TCR repertoire in responders, whereas still 50% in nonresponders (Figure 2D; supplemental Figure 9).
Next, the complete TCR repertoire overlap was analyzed to compare clonotype distribution and abundance. Weak correlations between pre- and post-AHSCT clonotypes indicate low repertoire overlap and high renewal of the immune system.47 Individual patient scatter plots show that at 6-months post-AHSCT, repertoire overlaps were lower in the responder (representative patient 1, r2 < 0.09) than in the nonresponder patients (representative patient 5, r2 > 0.51) (Figure 2E; supplemental Figure 10). At 2-years post-AHSCT, TCR overlap correlation rates were even lower in responder (representative patient 1, r2 = 0.0183), when compared with nonresponder patients (representative patient 5, r2 = 0.202) (supplemental Figure 10). Clonotype tracking heat map confirmed these changes, evidencing that for responder patients the most frequent clonotypes at baseline disappeared at 2-years posttransplantation, whereas remaining high in nonresponders patients after AHSCT (Figure 2F; supplemental Figure 11).
Increased natural Tregs after AHSCT correlate with thymic function
In transplanted patients, higher percentages of Tregs were detected at 6 (P = .01), 12 (P < .01), and 18 (P = .03) months after AHSCT when compared with pretransplant levels (Figure 3A). Absolute Treg numbers increased significantly (P = .01) at 12 months posttransplantation (Figure 3B), concurrent with thymic rebound. Following AHSCT, there was positive correlation between RTE and Treg counts (rs = 0.61, P < .0001; Figure 3C), and between Treg and sjTREC levels (rs = 0.45, P < .001; supplemental Figure 12).
Figure 3.

Increased natural Tregs after AHSCT correlate with thymic function and are associated with clinical response. (A) Percentage of CD4+CD25highFoxP3+ Tregs within CD3+CD4+ T cells and (B) Treg absolute values at baseline (0 months, pretransplant) and following time points in the transplanted patients immunophenotyped by flow cytometry. N = 26 transplanted patients at baseline, n = 15 at 6 and 12 months, and n = 11 at 18, 24, and >24 months. The boundaries of the boxes indicate the 25th and 75th percentiles; lines within the boxes indicate the median, and the whiskers mark the 10th and the 90th percentiles. Plots show mean ± SE. *P < .05; **P < .01 comparing posttransplant values to baseline (Wilcoxon’s). (C) Correlation between the absolute values of RTEs and Tregs (Spearman’s). (D) GITR (left) and CTLA-4 (right) median of fluorescence intensity (MFI) expression by CD4+CD25high Tregs at baseline (Pre-Tx) and 12 months posttransplant. *P < .05; **P < .01 comparing posttransplant values to baseline (Wilcoxon’s). (E) Median (± IQR) baseline percentage of (left) CD4+CD25hiFoxP3+ Tregs and (right) FoxP3 expression by CD4+CD25hi Tregs in nonresponder patients (red) after AHSCT or in responder patients (blue). *P < .05 comparing groups (Mann-Whitney U test). (F) Median (± IQR) baseline and 12-month Treg counts and GITR/CTLA-4 expressions by CD4+CD25hi Tregs in nonresponder patients after AHSCT or in responder patients. *P < .05 comparing groups (Mann-Whitney U test) and *P < .05 comparing posttransplant values to baseline (Wilcoxon’s).
To evaluate the Treg anti-inflammatory phenotype, GITR and CTLA-4 expressions and IL-10 production were assessed before and after AHSCT. At 12 months posttransplant, Tregs presented significantly increased CTLA-4 (P < .01) and GITR (P = .02) expressions, and a trend toward higher IL-10 production, from 15.75 ± 11 to 17.0 ± 1.4 (P > .05), when compared with pretransplant levels (Figure 3D).
When compared with values before transplant, responder patients presented higher CD4+CD25highFoxP3+ Treg percentages (P = .02), higher FoxP3 expression by CD4+CD25high Tregs (P = .02), and higher absolute CD4+CD25highFoxP3+ Treg counts (P = .04) than nonresponders (Figure 3E). At 12-months post-AHSCT, only the responder patients presented increased counts of Tregs (P = .03) and increased expression of GITR and CTLA-4 by CD4+CD25hi Tregs (P = .02), compared with baseline (Figure 3F).
B-cell ontogeny after AHSCT
B-cell ontogenesis was assessed in transplanted and nontransplanted patients by multiplex evaluation of sjKREC and CjKREC values (supplemental Figure 2). sjKREC reflects newly generated B cells, whereas CjKREC is representative of total B cells.40,53 When compared with pretransplant values and to those from nontransplanted SSc patients, sjKREC values progressively increased (P < .01) from 12 to 36 months post-AHSCT (Figure 4A). Cj values, on the other hand, transiently increased (P = .01) from 12 to 18 months posttransplant (Figure 4B) and total CD19+ B cells increased (P = .02) only at 12-months posttransplant, as compared with pretransplant values (Figure 4C). In transplanted patients, there was positive correlation (rs = 0.54, P < .0001) between Cj values and B-cell counts (Figure 4D).
Figure 4.

Increased output of newly generated B cells result in reduced B-cell replication in the periphery. Median (± IQR) of (A) sjKREC and (B) Cj values as measured by quantitative RT-PCR analysis on PBMC genomic DNA at baseline (0 months, pretransplant) and at the following time points in transplanted (AHSCT, n = 26 patients at baseline, n = 15 at 6 and 12 months, and n = 11 at 18, 24, and >24 months) and nontransplanted (non-AHSCT, n = 14 patients at baseline and 6 months, n = 13 at 12 months, and n = 8 at 18, 24, and >24 months) SSc patients. The results were expressed by log10 in 150 000 PBMCs. Panels C and D include transplanted patients only. (C) Quantification of CD19+ B cells by FACS. The boundaries of the boxes indicate the 25th and 75th percentiles; the lines within the boxes indicate the median, and the whiskers mark the 10th and the 90th percentiles. *P < .05 comparing posttransplant values to baseline (Wilcoxon’s). (D) Spearman’s correlation between B-cells count and Cj values. (E) Median (± IQR) number of B-cells division in the peripheral blood (n) as calculated using following formula: n = LOG(Cj/sjKREC)/LOG2. *P < .05, AHSCT vs non-AHSCT (Mann-Whitney U test). §P < .05; §§P < .01 comparing posttransplant values to baseline (Wilcoxon’s).
The ratio between Cj and sjKREC values reflects the mature B-cell replicative history division in the peripheral blood (supplemental Figure 2).40,53 Transplanted patients had significant decrease (P < .01) in B-cell replication starting at 6-months posttransplant, persisting until the end of follow-up (Figure 4E). No difference in sjKREC, Cj, or B-cell replication was observed between responder and nonresponder patients (supplemental Figure 13). Nontransplanted patients did not experience any change in B-cell proliferation.
Increased output of naive B-cells after AHSCT
Increasing sjKREC values and lower B-cell replication in the peripheral blood indicate that there is a continuous supply of more quiescent B cells after AHSCT. Except for transient decline in switched memory cell counts at 6 months post-AHSCT, no changes were observed in plasma cell, unswitched memory, or double-negative B-cell absolute values, as compared with pretransplant (Figure 5A-B). In contrast, naive B-cell counts increased (P = .01) from 6 months until the last time point of 36 months. After AHSCT, naive B-cell counts positively correlated (rs = 0.46, P = .002) with sjKREC values (Figure 5C).
Figure 5.

Increased output of bone marrow-derived naive B cells after AHSCT. (A) Mean (± SE) frequency of CD27brightIgD− plasma cells, CD27+IgD− switched memory, CD27+IgD+ nonswitched memory, CD27−IgD+ naive, and CD27−IgD− double-negative B cells immunophenotyped by flow cytometry at baseline (0 months, pretransplant) and following time points. (B) Quantification of the B-cell subpopulations absolute values. *P < .05 comparing posttransplant values to baseline (Wilcoxon’s). (C) Correlation between naive B-cells count and sjKREC values (Spearman’s). (D) Mean (± SE) frequency of CD38−IgD+ Bm1, CD38lowIgD+ Bm2, CD38highIgD+ Bm2′, CD38highIgD− Bm3+4, CD38lowIgD− early Bm5, and CD38−IgD− late Bm5 B cells immunophenotyped by flow cytometry at baseline and following time points. (E) Quantification of the B-cell subpopulations absolute values. *P < .05; **P < .01 comparing posttransplant values to baseline (Wilcoxon’s). (F) Correlation between Bm2 B-cells count and sjKREC values (Spearman’s). N = 18 transplanted patients at baseline, n = 14 at 6 and 12 months, n = 9 at 18 months, n = 12 at 24 months, and n = 7 at >24 months.
Through IgD and CD38 expressions, CD19+ B cells were classified into Bm1 to Bm5 subsets, reflecting several steps of B-cell differentiation.54 After AHSCT, although Bm1 and Bm5 cell counts transiently decreased (P = .01) at 6 months and Bm2′ cell numbers increased (P = .02) from 6 to 12 months, activated naive Bm2 cell counts progressively increased (P = .03), starting at 6 months until end of follow-up (Figure 5D-E) and positively correlated (rs = 0.47, P = .001) with sjKREC values (Figure 5F).
Increased output of IL-10–producing Bregs after AHSCT
Percentage and absolute numbers of CD24highCD38high Bregs increased significantly at 6 (P < .01 and P = .02, respectively) and 12 (P = .02 and P < .01, respectively) months post-AHSCT (Figure 6A-C). Higher Breg/unswitched memory (P = .01) (Figure 6D) and Breg/switched memory (P = .01) (Figure 6E) ratios were observed starting at 6-months posttransplantation, until the end of follow-up. Breg counts positively correlated (rs = 0.51, P < .0001) with sjKREC values after AHSCT (Figure 6F). Following CpG stimulation, IL-10–producing Breg frequency increased at 6 (P = .03) and 12 (P < .01) months after AHSCT as compared with pretransplant values. Similar increase (P = .02) was observed using CpG plus CD40L stimulation (Figure 6G), therefore confirming increased IL-10 production posttransplantation.
Figure 6.
Increased output of IL-10–producing Bregs after AHSCT. (A) Gating strategy of 1 representative patient showing the frequency of CD24hiCD38−, CD24intCD38int, CD24−CD38hi memory, and CD24hiCD38hi Bregs immunophenotyped by flow cytometry at baseline (0 months, pretransplant) and following time points. Quantification of the B-cell subpopulations frequency (B) and absolute values (C) are shown in panel A. The boundaries of the boxes indicate the 25th and 75th percentiles; the lines within the boxes indicate the median, and the whiskers mark the 10th and the 90th percentiles. Plots show mean ± SE. *P < .05; **P < .01 comparing posttransplant values with baseline (Wilcoxon’s). Mean ± SE changes on (D) Breg/CD19+CD27+IgD+ Unswitched memory and on (E) Breg/CD19+CD27+IgD− Switched memory ratios. *P < .05 comparing posttransplant values to baseline (Wilcoxon’s). N = 17 transplanted patients at baseline, n = 14 at 6 and 12 months, and n = 9 at 18, 24, and >24 months. (F) Correlation between Bregs and sjKREC values (Spearman’s). (G) Whole PBMCs from AHSCT patients at baseline, 6 months, and 12 months posttransplant were cultured for 18 hours with CpG or CpG and rhCD40L followed by restimulation with phorbol myristate acetate + ionomycin + BFA (PIB) in the last 6 hours of culture, fixed, permeabilized, and intracellular IL-10 assessed in CD19+ B cells by flow cytometry. The position of all gates was determined using isotype-matched control mAb staining. Negative controls consisted of PBMCs cultured in the presence of CpG control and BFA. These data are representative of those obtained in 6 independent experiments, with numbers representing the frequency of IL-10–producing B10 cells. Quantifications (mean ± SE) are expressed in parentheses. *P < .05; **P < .01 comparing posttransplant values with baseline (Wilcoxon’s). (H) Correlation between CD19+CD24hiCD38hi Bregs and the C-reactive protein levels (Spearman’s). (I) Responder patients after AHSCT presented higher Breg percentage at 12 months after transplant than nonresponder patients. *P < .05 comparing groups with each other (Mann-Whitney U test). (J) Correlation between sjKREC and sjTREC (Spearman’s).


Among the 31 SSc patients studied before and after AHSCT, negative correlation was observed between Breg percentages and C-reactive protein serum levels (Figure 6H). Responder subjects presented significantly higher (P = .01) frequencies of Bregs than nonresponders (Figure 6I). sjTREC and sjKREC levels positively correlated (rs = 0.38, P = .008) (Figure 6J) as well as Treg and Breg values (rs = 0.27, P = .02) (supplemental Figure 14), indicating simultaneous reestablishment of immunoregulatory processes after AHSCT as the result of immune rejuvenation.
Discussion
Mechanistic studies have shown profound transplant-induced changes in the immune system of SSc patients, some of which are disease specific, whereas others may be more widely observed across different autoimmune diseases.55-57 Persistent increase of Th1/Th2 ratios,58 recovery of Treg function,14 changes in thymic output,19,33,59 and lower profibrotic serum cytokine levels60 have been described. Several questions still remain, especially concerning the immunological determinants of clinical outcomes. Here, we investigated the immune reconstitution process in SSc patients after AHSCT, focusing on thymic and bone marrow function, searching for potential biomarkers of response. Complementary immunological and molecular approaches have enabled us to show unprecedented evidence in SSc that thymic rebound, as well as increased bone marrow output of newly generated naive B cells, are exclusive of the posttransplant setting and are not observed in SSc patients receiving conventional treatment.
Although AHSCT includes high-dose immunosuppression, potentially pathogenic T cells are not completely depleted, because specific T-cell clones can still be detected post-AHSCT.27,61 In responder patients, thymic reactivation led to replacement of the previous immune system, and an important TCR repertoire diversification was evidenced by the low overlap rates and reduced number of shared clonotypes, similar to what was previously reported in multiple sclerosis patients.28 In the nonresponder patients after AHSCT, conversely, high clonotype overlap was observed even after periods that correspond to thymic rebound, indicating that residual autoreactive cells may be associated with clinical reactivation.19,33 Indeed, the noticeable presence of overlapping clones in nonresponder patients, early after transplant, suggests that further immunosuppression after engraftment, or perhaps CD34+ graft selection, may be beneficial for this subgroup.62,63
Expansion of the Treg compartment following transplantation starts with lymphopenia-induced proliferation, followed by thymic generation of natural Tregs.64 This mechanism appears to be non–disease-specific and has been reported in other autoimmune diseases.14,30,31,36-38,65-67 Indeed, as already shown in multiple sclerosis,37 our group of responder SSc patients experienced significantly increased expression of regulatory molecules after AHSCT, when compared with nonresponders. Of note, nonresponder SSc patients presented lower FoxP3 expression before transplantation and consequently lower Treg percentage when compared with responders, indicating a possible selection bias or perhaps a potential biomarker to predict response to AHSCT.
We were able to identify sustained bone marrow output of newly generated B cells posttransplantation, evidencing bone marrow rebound as a contributing mechanism to the renewal of the immune system. Increasing sjKREC levels and reduced B-cell proliferation rates were detected in the peripheral blood over the entire follow-up after transplant. This indicates that in SSc patients, starting early after AHSCT, the reconstitution of the peripheral B-cell compartment is mostly due to the high output of bone marrow newly generated naive B cells rather than to clones that may have resisted the procedure and undergone homeostatic expansion. High rate of B-cell divisions is related to a B-cell hyperactivation state in vivo,40 and conventional therapies have failed to reset defective B-cell tolerance checkpoints.68 We show that transplanted SSc patients present reduced B-cell division rates long term, suggestive of a more tolerant immune status over time, probably by an immunological balance established by tolerant B cells.
CD19+CD24hiCD38hi B-cell populations are well-characterized Bregs in humans, playing an important role in the control of autoreactivity.43 Their decreased numbers and function have been reported in several autoimmune diseases.69-71 In SSc patients, low levels of Bregs are associated with higher disease activity.26 To date, there are no available reports on Breg levels after AHSCT. Here, we show that Breg frequencies transiently increased after AHSCT, tending to remain higher than pretransplant values for at least 2 years. These cells may be involved in the reestablishment of autotolerance after AHSCT, as suggested by persistently increased Breg/memory B-cell ratio, as well as higher IL-10 production.
Although Breg ontogeny is still not completely understood,43 Breg counts positively correlated with sjKREC values, indicating that Bregs could be bone marrow–derived, increasing after AHSCT due to higher output of newly generated B cells.56 In this scenario, studies have shown that donor-derived Bregs are important for suppression of murine sclerodermatous chronic graft-versus-host disease.72 In the present study, we found a correlation between favorable clinical outcomes and Breg levels after AHSCT in SSc patients.
The low number of nonresponders and possible influence of posttransplant immunosuppressive therapy on immunological outcomes limit the conclusions of our study. However, we learned that in addition to established clinical AHSCT guidelines,11,12 transplant protocols may still be improved at specific points. Future studies are required to evaluate T- and B-cell profile in the autografts and possible immunological effects of posttransplant immunization and immunosuppression. Furthermore, adjuvant therapies aiming to enhance immunoregulatory function or adoptive Treg and/or Breg therapy may improve rates of clinical remission.
In conclusion, we describe the coordinated recovery of Treg and Breg compartments after AHSCT in SSc patients. Here, both thymus and bone marrow proved important for successful immune reconstitution and clinical responsiveness to AHSCT. Their improved function after AHSCT in responder SSc patients contributes to an efficient reset of both the Treg and Breg populations.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Julien Pelé from Technological Platform of the Institut Universitaire d'Hématologie for the TCR-NGS technical support, Sandra Navarro Bresciani for the illustrations and graphic design, and Patrícia Vianna Bonini Palma for the flow cytometry data acquisition. The authors also thank the multidisciplinary team of the Bone Marrow Unit (Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto–Universidade de São Paulo) and Hemotherapy Regional Blood Center of Ribeirão Preto staff as well as D. Farge and all of the multidisciplinary team of the Internal Medicine Unit at St. Louis Hospital, for patient care and collection of clinical data and biological samples.
This study was supported by research funding from CNPq, INSERM, Programme hospitalier de recherche clinique–AOM 97 030 (Autologous Stem Cell Transplantation International Scleroderma trial) and São Paulo Research Foundation (grants 13/08135-2 [D.T.C.], 13/18678-3 [L.C.M.A.], and 14/20922-2 [L.C.M.A.]).
Footnotes
The data reported in this article have been deposited in the National Center for Biotechnology Information database (accession number SRP106516).
Authorship
Contribution: L.C.M.A., K.C.R.M., A.T., and M.C.O. designed the study; L.C.M.A., J.R.L.-J., C.D., I.F., and E.C. performed the experiments; L.C.M.A., P.L., J.R.L.-J., J.B.E.D., D.A.M., B.P.S., and H.M.-T. collected the data and performed data analysis; E.C., C.D., and A.J.A. prepared the NGS library; E.C. and L.C.M.A. performed the sequencing analyses; D.T.C. provided essential funding to the development of this work; L.C.M.A., A.T., E.C., and M.C.O. wrote the final report; and all authors contributed to the editing of the final report and agreed to all of the content of the submitted manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Antoine Toubert, Laboratoire Alloimmunité, Autoimmunité, Transplantation, Hôpital Saint-Louis, Université Paris Diderot, Sorbonne Paris Cité, INSERM UMR-1160, Institut Universitaire d'Hématologie, 1 Ave Claude Vellefaux, 75010 Paris, France; e-mail: antoine.toubert@univ-paris-diderot.fr; and Maria Carolina Oliveira, Divisão de Imunologia Clínica, Departamento de Clínica Médica, Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto, Avenida dos Bandeirantes 3900, Ribeirão Preto, São Paulo 14048-900, Brazil; e-mail: mcarolor@usp.br.
References
- 1.Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390(10103):1685-1699. [DOI] [PubMed] [Google Scholar]
- 2.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360(19):1989-2003. [DOI] [PubMed] [Google Scholar]
- 3.Nannini C, West CP, Erwin PJ, Matteson EL. Effects of cyclophosphamide on pulmonary function in patients with scleroderma and interstitial lung disease: a systematic review and meta-analysis of randomized controlled trials and observational prospective cohort studies. Arthritis Res Ther. 2008;10(5):R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tashkin DP, Elashoff R, Clements PJ, et al. ; Scleroderma Lung Study Research Group. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655-2666. [DOI] [PubMed] [Google Scholar]
- 5.Tashkin DP, Roth MD, Clements PJ, et al. ; Sclerodema Lung Study II Investigators. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4(9):708-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volkmann ER, Tashkin DP, Li N, et al. . Mycophenolate mofetil versus placebo for systemic sclerosis-related interstitial lung disease: an analysis of scleroderma lung studies I and II. Arthritis Rheumatol. 2017;69(7):1451-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burt RK, Shah SJ, Dill K, et al. . Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet. 2011;378(9790):498-506. [DOI] [PubMed] [Google Scholar]
- 8.van Laar JM, Farge D, Sont JK, et al. ; EBMT/EULAR Scleroderma Study Group. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. 2014;311(24):2490-2498. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan KM, Shah A, Sarantopoulos S, Furst DE. Review: hematopoietic stem cell transplantation for scleroderma: effective immunomodulatory therapy for patients with pulmonary involvement. Arthritis Rheumatol. 2016;68(10):2361-2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sullivan K, Keyes-Elstein L, McSweeney P, et al. . Myeloablative autologous transplantation of CD34+ selected hematopoietic stem cells (HSCT) vs monthly intravenous cyclophosphamide (CY) for severe scleroderma with internal organ involvement: outcomes of a randomized North American clinical trial. Biol Blood Marrow Transplant. 2017;23(3):S23-S24. [Google Scholar]
- 11.Farge D, Burt RK, Oliveira M, et al. . Cardiopulmonary assessment of patients with systemic sclerosis for hematopoietic stem cell transplantation : recommendations from the European Society for Blood and Marrow Transplantation Autoimmune Diseases Working Party and collaborating partners. Bone Marrow Transpl. 2017;52(11):1495-1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snowden JA, Saccardi R, Allez M, et al. ; Paediatric Diseases Working Party (PDWP). Haematopoietic SCT in severe autoimmune diseases: updated guidelines of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 2012;47(6):770-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexander T, Bondanza A, Muraro PA, et al. . SCT for severe autoimmune diseases: consensus guidelines of the European Society for Blood and Marrow Transplantation for immune monitoring and biobanking. Bone Marrow Transplant. 2015;50(2):173-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baraut J, Grigore EI, Jean-Louis F, et al. . Peripheral blood regulatory T cells in patients with diffuse systemic sclerosis (SSc) before and after autologous hematopoietic SCT: a pilot study. Bone Marrow Transplant. 2014;49(3):349-354. [DOI] [PubMed] [Google Scholar]
- 15.Papp G, Horvath IF, Barath S, et al. . Altered T-cell and regulatory cell repertoire in patients with diffuse cutaneous systemic sclerosis. Scand J Rheumatol. 2011;40(3):205-210. [DOI] [PubMed] [Google Scholar]
- 16.Ugor E, Simon D, Almanzar G, et al. . Increased proportions of functionally impaired regulatory T cell subsets in systemic sclerosis. Clin Immunol. 2017;184(11):54-62. [DOI] [PubMed] [Google Scholar]
- 17.Klein S, Kretz CC, Krammer PH, Kuhn A. CD127(low/-) and FoxP3(+) expression levels characterize different regulatory T-cell populations in human peripheral blood. J Invest Dermatol. 2010;130(2):492-499. [DOI] [PubMed] [Google Scholar]
- 18.Reiff A, Krogstad P, Moore S, et al. . Study of thymic size and function in children and adolescents with treatment refractory systemic sclerosis eligible for immunoablative therapy. Clin Immunol. 2009;133(3):295-302. [DOI] [PubMed] [Google Scholar]
- 19.Farge D, Henegar C, Carmagnat M, et al. . Analysis of immune reconstitution after autologous bone marrow transplantation in systemic sclerosis. Arthritis Rheum. 2005;52(5):1555-1563. [DOI] [PubMed] [Google Scholar]
- 20.Sakkas LI, Bogdanos DP. Systemic sclerosis: New evidence re-enforces the role of B cells. Autoimmun Rev. 2016;15(2):155-161. [DOI] [PubMed] [Google Scholar]
- 21.Sato S, Fujimoto M, Hasegawa M, Takehara K. Altered blood B lymphocyte homeostasis in systemic sclerosis: expanded naive B cells and diminished but activated memory B cells. Arthritis Rheum. 2004;50(6):1918-1927. [DOI] [PubMed] [Google Scholar]
- 22.Soto L, Ferrier A, Aravena O, et al. . Systemic sclerosis patients present alterations in the expression of molecules involved in B-cell regulation. Front Immunol. 2015;6:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simon D, Balogh P, Bognár A, et al. . Reduced non-switched memory B cell subsets cause imbalance in B cell repertoire in systemic sclerosis. Clin Exp Rheumatol. 2016;34(5 Suppl 100):30-36. [PubMed] [Google Scholar]
- 24.Allanore Y, Simms R, Distler O, et al. . Systemic sclerosis. Nat Rev Dis Primers. 2015;1(4):15002. [DOI] [PubMed] [Google Scholar]
- 25.Mavropoulos A, Simopoulou T, Varna A, et al. . Breg cells are numerically decreased and functionally impaired in patients with systemic sclerosis. Arthritis Rheumatol. 2016;68(2):494-504. [DOI] [PubMed] [Google Scholar]
- 26.Matsushita T, Hamaguchi Y, Hasegawa M, Takehara K, Fujimoto M. Decreased levels of regulatory B cells in patients with systemic sclerosis: association with autoantibody production and disease activity. Rheumatology (Oxford). 2016;55(2):263-267. [DOI] [PubMed] [Google Scholar]
- 27.Muraro PA, Douek DC, Packer A, et al. . Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med. 2005;201(5):805-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muraro PA, Robins H, Malhotra S, et al. . T cell repertoire following autologous stem cell transplantation for multiple sclerosis. J Clin Invest. 2014;124(3):1168-1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L, Bertucci AM, Ramsey-Goldman R, Burt RK, Datta SK. Regulatory T cell (Treg) subsets return in patients with refractory lupus following stem cell transplantation, and TGF-beta-producing CD8+ Treg cells are associated with immunological remission of lupus. J Immunol. 2009;183(10):6346-6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexander T, Thiel A, Rosen O, et al. . Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long-term remission through de novo generation of a juvenile and tolerant immune system. Blood. 2009;113(1):214-223. [DOI] [PubMed] [Google Scholar]
- 31.de Kleer I, Vastert B, Klein M, et al. . Autologous stem cell transplantation for autoimmunity induces immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4+CD25+ immune regulatory network. Blood. 2006;107(4):1696-1702. [DOI] [PubMed] [Google Scholar]
- 32.Clerici M, Cassinotti A, Onida F, et al. . Immunomodulatory effects of unselected haematopoietic stem cells autotransplantation in refractory Crohn’s disease. Dig Liver Dis. 2011;43(12):946-952. [DOI] [PubMed] [Google Scholar]
- 33.Farge D, Arruda LCM, Brigant F, et al. . Long-term immune reconstitution and T cell repertoire analysis after autologous hematopoietic stem cell transplantation in systemic sclerosis patients. J Hematol Oncol. 2017;10(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van den Hoogen F, Khanna D, Fransen J, et al. . 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burt RK, Oliveira MC, Shah SJ, et al. . Cardiac involvement and treatment-related mortality after non-myeloablative haemopoietic stem-cell transplantation with unselected autologous peripheral blood for patients with systemic sclerosis: a retrospective analysis. Lancet. 2013;381(9872):1116-1124. [DOI] [PubMed] [Google Scholar]
- 36.Arruda LCM, Lorenzi JCC, Sousa APA, et al. . Autologous hematopoietic SCT normalizes miR-16, -155 and -142-3p expression in multiple sclerosis patients. Bone Marrow Transplant. 2015;50(3):380-389. [DOI] [PubMed] [Google Scholar]
- 37.Arruda LCM, de Azevedo JTC, de Oliveira GLV, et al. . Immunological correlates of favorable long-term clinical outcome in multiple sclerosis patients after autologous hematopoietic stem cell transplantation. Clin Immunol. 2016;169:47-57. [DOI] [PubMed] [Google Scholar]
- 38.Malmegrim KCR, de Azevedo JTC, Arruda LCM, et al. . Immunological balance is associated with clinical outcome after autologous hematopoietic stem cell transplantation in type 1 diabetes. Front Immunol. 2017;8:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clave E, Lisini D, Douay C, et al. . Thymic function recovery after unrelated donor cord blood or T-cell depleted HLA-haploidentical stem cell transplantation correlates with leukemia relapse. Front Immunol. 2013;4:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glauzy S, Soret J, Fournier I, et al. . Impact of acute and chronic graft-versus-host disease on human B-cell generation and replication. Blood. 2014;124(15):2459-2462. [DOI] [PubMed] [Google Scholar]
- 41.Clave E, Busson M, Douay C, et al. . Acute graft-versus-host disease transiently impairs thymic output in young patients after allogeneic hematopoietic stem cell transplantation. Blood. 2009;113(25):6477-6484. [DOI] [PubMed] [Google Scholar]
- 42.de Masson A, Bouaziz J-D, Le Buanec H, et al. . CD24(hi)CD27+ and plasmablast-like regulatory B cells in human chronic graft-versus-host disease. Blood. 2015;125(11):1830-1839. [DOI] [PubMed] [Google Scholar]
- 43.Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221-241. [DOI] [PubMed] [Google Scholar]
- 44.Mamedov IZ, Britanova OV, Zvyagin IV, et al. . Preparing unbiased T-cell receptor and antibody cDNA libraries for the deep next generation sequencing profiling. Front Immunol. 2013;4:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vander Heiden JA, Yaari G, Uduman M, et al. . pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics. 2014;30(13):1930-1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolotin DA, Shugay M, Mamedov IZ, et al. . MiTCR: software for T-cell receptor sequencing data analysis. Nat Methods. 2013;10(9):813-814. [DOI] [PubMed] [Google Scholar]
- 47.Shugay M, Bagaev DV, Turchaninova MA, et al. . VDJtools: unifying post-analysis of T cell receptor repertoires. PLOS Comput Biol. 2015;11(11):e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toubert A, Glauzy S, Douay C, Clave E. Thymus and immune reconstitution after allogeneic hematopoietic stem cell transplantation in humans: never say never again. Tissue Antigens. 2012;79(2):83-89. [DOI] [PubMed] [Google Scholar]
- 49.Kimmig S, Przybylski GK, Schmidt CA, et al. . Two subsets of naive T helper cells with distinct T cell receptor excision circle content in human adult peripheral blood. J Exp Med. 2002;195(6):789-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22(1):745-763. [DOI] [PubMed] [Google Scholar]
- 51.Mamedov IZ, Britanova OV, Bolotin DA, et al. . Quantitative tracking of T cell clones after haematopoietic stem cell transplantation. EMBO Mol Med. 2011;3(4):201-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colwell RK, Chao A, Gotelli NJ, et al. . Models and estimators linking individual-based and sample-based rarefaction, extrapolation and comparison of assemblages. J Plant Ecol. 2012;5(1):3-21. [Google Scholar]
- 53.van Zelm MC, Szczepanski T, van der Burg M, van Dongen JJM. Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J Exp Med. 2007;204(3):645-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bohnhorst JO, Bjørgan MB, Thoen JE, Natvig JB, Thompson KM. Bm1-Bm5 classification of peripheral blood B cells reveals circulating germinal center founder cells in healthy individuals and disturbance in the B cell subpopulations in patients with primary Sjögren’s syndrome. J Immunol. 2001;167(7):3610-3618. [DOI] [PubMed] [Google Scholar]
- 55.Arruda LCM, Clave E, Moins-Teisserenc H, Douay C, Farge D, Toubert A. Resetting the immune response after autologous hematopoietic stem cell transplantation for autoimmune diseases. Curr Res Transl Med. 2016;64(2):107-113. [DOI] [PubMed] [Google Scholar]
- 56.Muraro PA, Martin R, Mancardi GL, Nicholas R, Sormani MP, Saccardi R. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis. Nat Rev Neurol. 2017;13(7):391-405. [DOI] [PubMed] [Google Scholar]
- 57.Swart JF, Delemarre EM, van Wijk F, et al. . Haematopoietic stem cell transplantation for autoimmune diseases. Nat Rev Rheumatol. 2017;13(4):244-256. [DOI] [PubMed] [Google Scholar]
- 58.Tsukamoto H, Nagafuji K, Horiuchi T, et al. . Analysis of immune reconstitution after autologous CD34+ stem/progenitor cell transplantation for systemic sclerosis: predominant reconstitution of Th1 CD4+ T cells. Rheumatology (Oxford). 2011;50(5):944-952. [DOI] [PubMed] [Google Scholar]
- 59.Bohgaki T, Atsumi T, Bohgaki M, et al. . Immunological reconstitution after autologous hematopoietic stem cell transplantation in patients with systemic sclerosis: relationship between clinical benefits and intensity of immunosuppression. J Rheumatol. 2009;36(6):1240-1248. [DOI] [PubMed] [Google Scholar]
- 60.Michel L, Farge D, Baraut J, et al. . Evolution of serum cytokine profile after hematopoietic stem cell transplantation in systemic sclerosis patients. Bone Marrow Transplant. 2016;51(8):1146-1149. [DOI] [PubMed] [Google Scholar]
- 61.Dubinsky AN, Burt RK, Martin R, Muraro PA. T-cell clones persisting in the circulation after autologous hematopoietic SCT are undetectable in the peripheral CD34+ selected graft. Bone Marrow Transplant. 2010;45(2):325-331. [DOI] [PubMed] [Google Scholar]
- 62.Farge D, Marolleau JP, Zohar S, et al. ; Intensification et Autogreffe dans les Maladies Auto Immunes Resistantes (ISAMAIR) Study Group. Autologous bone marrow transplantation in the treatment of refractory systemic sclerosis: early results from a French multicentre phase I-II study. Br J Haematol. 2002;119(3):726-739. [DOI] [PubMed] [Google Scholar]
- 63.Oliveira MC, Labopin M, Henes J, et al. . Does ex vivo CD34+ positive selection influence outcome after autologous hematopoietic stem cell transplantation in systemic sclerosis patients? Bone Marrow Transplant. 2016;51(4):501-505. [DOI] [PubMed] [Google Scholar]
- 64.Delemarre EM, van den Broek T, Mijnheer G, et al. . Autologous stem cell transplantation aids autoimmune patients by functional renewal and TCR diversification of regulatory T cells. Blood. 2016;127(1):91-101. [DOI] [PubMed] [Google Scholar]
- 65.Abrahamsson SV, Angelini DF, Dubinsky AN, et al. . Non-myeloablative autologous haematopoietic stem cell transplantation expands regulatory cells and depletes IL-17 producing mucosal-associated invariant T cells in multiple sclerosis. Brain. 2013;136(Pt 9):2888-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burt RK, Craig RM, Milanetti F, et al. . Autologous nonmyeloablative hematopoietic stem cell transplantation in patients with severe anti-TNF refractory Crohn disease: long-term follow-up. Blood. 2010;116(26):6123-6132. [DOI] [PubMed] [Google Scholar]
- 67.Darlington PJ, Touil T, Doucet JS, et al. ; Canadian MS/BMT Study Group. Diminished Th17 (not Th1) responses underlie multiple sclerosis disease abrogation after hematopoietic stem cell transplantation. Ann Neurol. 2013;73(3):341-354. [DOI] [PubMed] [Google Scholar]
- 68.Chamberlain N, Massad C, Oe T, Cantaert T, Herold KC, Meffre E. Rituximab does not reset defective early B cell tolerance checkpoints. J Clin Invest. 2016;126(1):282-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Flores-Borja F, Bosma A, Ng D, et al. . CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med. 2013;5(173):173ra23. [DOI] [PubMed] [Google Scholar]
- 70.Menon M, Blair PA, Isenberg DA, Mauri C. A regulatory feedback between plasmacytoid dendritic cells and regulatory B cells is aberrant in systemic lupus erythematosus. Immunity. 2016;44(3):683-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blair PA, Noreña LY, Flores-Borja F, et al. . CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity. 2010;32(1):129-140. [DOI] [PubMed] [Google Scholar]
- 72.Le Huu D, Matsushita T, Jin G, et al. . Donor-derived regulatory B cells are important for suppression of murine sclerodermatous chronic graft-versus-host disease. Blood. 2013;121(16):3274-3283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.