

Abstract
Purpose
Electroretinograms (ERGs) are abnormal in diabetic retinas before the appearance of vascular lesions, providing a possible biomarker for diabetic vision loss. Previously, we reported that decreased retinal dopamine (DA) levels in diabetic rodents contributed to early visual and retinal dysfunction. In the current study, we examined whether oscillatory potentials (OPs) could serve as a potential marker for detecting early inner retinal dysfunction due to retinal DA deficiency.
Methods
Retinal function was tested with dark-adapted ERGs, taken at 3, 4, and 5 weeks after diabetes induction with streptozotocin. Electrical responses were analyzed and correlations were made with previously reported retinal DA levels. The effect of restoring systemic DA levels or removing DA from the retina in diabetic mice on OPs was assessed using L-3,4-dihydroxyphenylalanine (L-DOPA) treatments and retina-specific tyrosine hydroxylase (Th) knockout mice (rTHKO), respectively.
Results
Diabetic animals had significantly delayed OPs compared to control animals in response to dim, but not bright, flash stimuli. L-DOPA treatment preserved OP implicit time in diabetic mice. Diabetic rTHKO mice had further delayed OPs compared to diabetic mice with normal retinal Th, with L-DOPA treatment also providing benefit. Decreasing retinal DA levels significantly correlated with increasing OP delays mediated by rod pathways.
Conclusions
Our data suggest that inner retinal dysfunction in early-stage diabetes is mediated by rod-pathway deficits and DA deficiencies. OP delays may be used to determine the earliest functional deficits in diabetic retinopathy and to establish an early treatment window for DA therapies that may prevent progressive vision loss.
Keywords: diabetic retinopathy, dopamine, oscillatory potentials, electroretinogram
Diabetic retinopathy (DR) is a vision-threatening complication of diabetes mellitus (DM). After considering population growth and aging, DM in adults has nearly quadrupled in the last 30 years1 and is predicted to increase 54% by 2030.2 Concurrently, the incidence of vision loss due to DR is expected to nearly double between 2010 and 2050.1,3–5
A growing body of literature suggests that neuronal dysfunction in the retina is observable in early diabetes, before any vascular abnormalities are present in the eye.6–10 Changes in retinal thickness, alterations in color vision and contrast sensitivity, and decreased electroretinogram (ERG) responses have been detected in diabetic patients with no or minimal retinopathy.9,11–22 In diabetic animal models, altered glial cell activities,23–25 loss of retinal neurons, downregulation of synaptic proteins,26 and attenuated electrical responses of the inner retinal neurons8,27–33 have been found. Collectively, these studies strongly suggest negative effects of hyperglycemia in the sensory neurons of the retina, and emphasize the need to elucidate the underlying mechanisms to find novel treatment options to preserve vision earlier in the disease.
Chronic hyperglycemia appears to alter neuronal function in part by reducing the synthesis of dopamine (DA), a key neuromodulator in the retina and brain.34 Alterations to DA metabolism and synthesis in retinal neurons have been documented in streptozotocin (STZ)-induced hyperglycemic rats as early as 3 weeks post STZ.35,36 In addition, loss of dopaminergic amacrine cells was reported in animal models of both chemically and spontaneously induced diabetes 6 months after the onset of disease.37 Dopamine dysregulation in the diabetic rat retina has been shown to have functional consequences, including delayed b-wave latencies and reduced spatial frequency and contrast sensitivity thresholds in the early stages of disease.35 Further implicating DA's role in neural dysfunction during diabetes, visual function in these same animals could be partially preserved with the treatment of L-3,4-dihydroxyphenylalanine (L-DOPA), a precursor to DA.35
Oscillatory potentials (OPs), high-frequency oscillations on the leading edge of the ERG b-wave,38,39 have shown consistent delays in diabetic subjects6–8 and in diabetic animals even prior to development of vascular abnormalities.27,29–32 We have previously reported rod-mediated OP delays in hyperglycemic rats as early as 4 weeks post STZ.33 Since these small wavelet OPs are generated by the inner retina,38 these early deficits suggest a susceptibility of the inner retina to diabetic insult.
The aim of the current study was to determine whether the early changes in OP latency due to diabetes are attributable to DA deficiencies affecting inner retinal function. Here we examined the temporal appearance of OP delays in early-stage DR in STZ-induced diabetic mice and evaluated the effects of DA therapy using L-DOPA or DA deficiency using a retinal-specific tyrosine hydroxylase (Th) knockout mouse model to evaluate the contributions of DA signaling on OP delays.
Methods
Animals
Albino CD-1 and pigmented C57BL mice were obtained from Charles River Laboratories (Wilmington, MA, USA) and Jackson Laboratories (Bar Harbor, ME, USA), respectively, and housed in the animal facility at the Atlanta Veterans Affairs Medical Center (Decatur, GA) under a 12:12 (light:dark) hour cycle with food and water ad libitum. All procedures were approved by the Institutional Animal Care and Use Committee of the Atlanta Veterans Affairs Medical Center and performed in full accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
DM CD-1 mice: Diabetic mice were used to find ERG deficits with hyperglycemia. Diabetic mice (DM; n = 11) received STZ (200 mg/kg; Sigma-Aldrich Corp., St. Louis, MO, USA) while control mice (CTRL; n = 11) received equal volumes of citrate buffer. STZ was dissolved in citrate buffer mixed in 50% glucose (pH 4.0) and immediately injected intraperitoneally (IP). Blood glucose (BG) levels were measured using a handheld glucose meter (Freestyle; Abbott, Abbott Park, IL, USA) with blood collected from the tail vein. DM was confirmed with two successive measurements of BG greater than 200 mg/dL BG, and body weights (BW) were checked every other day for the first week and weekly thereafter until the end of the study. Upon two successive measurements of weight reduction in hyperglycemic animals, insulin (1 mL; Lantus Sanofi-Aventis, Bridgewater, NJ, USA) was injected intraperitoneally to prevent further weight loss. The low-dose insulin injection did not cause hypoglycemia. Retinas from a subset of these mice (CTRL, n = 8; DM, n = 6) were used for DA analysis, as described below.
L-DOPA-treated and Th-deficient mice: To restore retinal DA levels, diabetic mice (genotype information below) were given daily IP injections of vehicle only or L-DOPA (10 mg/kg; Sigma-Aldrich Corp.) dissolved in 0.1% ascorbic acid in saline. L-DOPA was made fresh daily with treatments beginning the week after STZ injections and lasting until 6 weeks post hyperglycemia. All injections occurred 4 to 8 hours after light onset. ERGs were measured 30 to 60 minutes after L-DOPA or vehicle injection.
Tyrosine hydroxylase (TH) is an enzyme critical for the conversion of tyrosine to L-DOPA for the synthesis of DA. To determine the effect of chronic reduction of retinal DA on inner retinal dysfunction in diabetes, we used retina-specific Th gene deletion. Commercially available Chx10-Cre mice (Jackson Laboratories) were crossed with mice expressing a floxed Th allele on C57BL background, as previously described.40 Offspring with Chx10-Cre:ThloxP/loxP genotype result in an 85% reduction in retinal DA,41 and were designated as rTHKO. Mice with the ThloxP/loxP genotype were referred to as THctrl. Diabetes was induced with five daily injections of low-dose STZ (50 mg/kg) (DM rTHKO + Veh, n = 8; DM THctrl + Veh, n = 15; THctrl, n = 15). The baseline BG levels of rTHKO mice were normal (181.8 ± 7.75 mg/dL, n = 17) versus THctrl (180.6 ± 6.5 mg/dL, n = 19; t-test P = nonsignificant).
To determine the effect of DA treatment on inner retinal function in rTHKO mice, a subset of mice (DM THctrl + L-DOPA, n = 15; DM rTHKO + L-DOPA, n = 8) were given injections of L-DOPA (IP, 10 mg/kg; Sigma-Aldrich Corp.) as described above. Half of the THctrl mice were given L-DOPA and half given vehicle, but there was no statistical difference between the groups so they were combined for analyses. Average BG and BW for these mice can be found in Aung et al.35
Animals used in the experiments were followed for 6 weeks post STZ. Mice involved in the DA study were killed by cervical dislocation in order to measure retinal DA using HPLC without interference of anesthesia, while all others were euthanized with an overdose of pentobarbital.
Measuring Retinal Function With Scotopic Full-Field ERG
Full-field scotopic ERGs were measured at baseline (data not shown) and 3, 4, and 5 weeks post STZ using a commercial ERG system (UTAS E-3000; LKC Technologies, Gaithersburg, MD, USA). After overnight dark adaptation, animals were anesthetized under red light with ketamine (80 mg/kg) and xylazine (16 mg/kg); corneas were anesthetized (0.5% tetracaine HCl), and pupils were dilated (1.0% cyclopentolate HCl and 1.0% tropicamide). Body temperature was maintained at 37°C with a heating pad. Custom-made DTL fiber electrodes42–45 were used as recording electrodes, contacting the cornea through a thin layer of methylcellulose (Refresh Celluvisc; Allergan, Irvine, CA, USA), while platinum needle electrodes (Grass Technologies, West Warwick, RI, USA) were inserted in the cheeks and tail as reference and ground, respectively. Ganzfeld flash stimuli ranging from −3.4 to 1.4 log cd s/m2, with interstimulus intervals ranging from 4 to 65 seconds, were used to elicit a retinal response. Responses were differentially amplified (1–1500 Hz) with a recording length of 250 ms and a sampling rate of 2000 Hz (UTAS Bigshot: LKC Technologies). After the recording, animals were given an IP injection of yohimbine (2.0 mg/kg; Lloyd Laboratories, Shenandoah, IA, USA) for recovery from anesthesia and prevention of corneal ulcers.46 STZ-treated mice were given a subcutaneous bolus of 0.9% saline solution for hydration after the recording.
ERG data analysis was performed on one eye of each animal. Four animals were excluded from the study, due to death (n = 1), absence of mixed rod- and cone-mediated ERG response (n = 2), and failed outlier test (n = 1). Amplitudes and implicit times of a- and b-wave and OPs were analyzed, as previously described.47 Inner retinal function was examined by extracting OPs from the ERG waveforms38,48,49 using a bandpass filter setting of 75 to 500 Hz (EM for Win version 8.1.2, LKC Technologies). Flashes within the rod-dominated dim luminance range8,33 of −3.02 and −1.8 log cd s/m2 were compared to a bright flash of 0.61 log cd s/m2 to compare the inner retinal function dominated by rod pathways versus mixed rod and cone pathways, respectively.50,51
Dopamine Analysis
After euthanasia at 6 weeks post STZ, retinas were taken from a subset of mice (n = 14) and DA levels were analyzed, as previously described.35 Retinas were flash-frozen and stored at −20°C. Ion-pair reverse-phase HPLC with coulometric detection was used to measure DA levels. Frozen retinas were homogenized in a 0.2 N HClO4 solution with 0.01% sodium meta-bisulfite and 25 ng/mL 3,4-dihydroxybenzylamine hydrobromide as an internal standard. Samples were then centrifuged, and supernatant fractions were separated on a HPLC column (Ultrasphere ODS; Hichrom, Berkshire, UK). DA was quantified based on a standard curve from 2 to 20 ng/mL, and measured as picograms per retina.
Statistical Analysis
Statistical analyses were performed with repeated-measures ANOVAs (Holm-Sidak post hoc analyses, SigmaStat 3.5; Systat Software, Chicago, IL, USA) when comparing the diabetic and control groups across time. One-way ANOVAs with Holm-Sidak post hoc analyses using the rough false discovery rate (RFDR) method for alpha level adjustment [calculated as p*(#tests+1)/(2*(#tests)] were used for analysis of L-DOPA–treated mice.35 All four OPs generated similar results, so OP2 was chosen for presentation here. Pearson's correlation test was used to calculate the correlation coefficient between the retinal DA level (pg/retina) and OP2 implicit time (ms) at dim (−3.02 log cd s/m2) and bright (0.61 log cd s/m2) flash stimuli. All figures show mean values with error bars representing SEM. Reported P values represent Holm-Sidak post hoc results.
Results
Blood Glucose and Body Weight of STZ-Injected CD-1 Mice
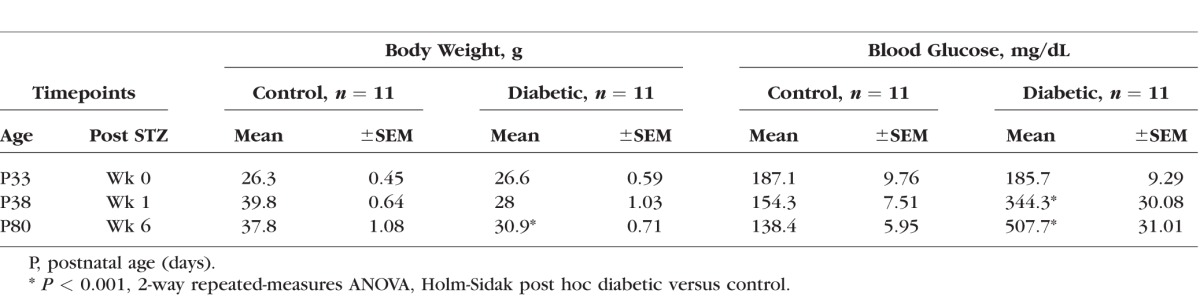
As expected, mice injected with STZ became hyperglycemic (344 ± 30 mg/dL) compared to controls (154 ± 7.5 mg/dL) within a week, and their BG levels remained elevated (507 ± 31.01 mg/dL) to the end of the study (Table, P < 0.001). Mice injected with STZ failed to gain weight over time, but remained relatively stable over the 5 weeks of hyperglycemia.
Table.
Blood Glucose and Weight Measurements in Diabetic and Control Mice
ERG Shows OP Delays in Diabetic CD-1 Mice After 4 to 5 Weeks Post STZ
Representative ERG recordings illustrated no discernable differences in a- and b-waveforms between the two groups at this early stage of diabetes (Fig. 1A). No significant differences were found in the a- and b-wave amplitudes (Fig. 1B) or implicit times (data not shown).
Figure 1.

No differences in ERG a- and b-wave responses at 5 weeks post STZ. (A) Representative raw ERG waveforms of control and diabetic animals at 5 weeks post STZ. (B) Mean (±SEM) amplitudes of a- and b-waves at 5 weeks post STZ plotted against increasing flash stimuli.
No differences were observed in any of the OP amplitudes at 3 or 4 weeks post STZ (data not shown). Similarly, at 5 weeks post STZ, OP1 and OP2 amplitudes were indistinguishable between the groups. However, OP3 and OP4 amplitudes were significantly higher in diabetic animals in response to brighter flash stimuli (Fig. 2C). OP3 showed a 31% increase in amplitude in response to 1.4 log cd s/m2 stimuli (P = 0.02). OP4 showed a 35% increase to −0.02 log cd s/m2 stimuli (P = 0.02), a 48% increase to −0.61 log cd s/m2 stimuli (P = 0.002), and a 65% increase to 1.4 log cd s/m2 stimuli (Fig. 2C, P = 0.004).
Figure 2.

Delayed implicit times of rod-driven OPs in diabetic animals. Representative OP waveforms from control and diabetic mice in response to a representative (A) dim (−3.02 log cd s/m2) or (B) bright flash (1.4 log cd s/m2). Flash onset was at 0 ms. The gray lines indicate OP2 for the control animal and the gray arrowhead indicates a delayed OP2. OP amplitudes (C) and implicit times (D) (mean ± SEM) of OP1, OP2, OP3, and OP4 at 5 weeks post STZ are plotted against flash stimuli. Asterisks represent Holm-Sidak post hoc analysis for individual steps: *P < 0.05, **P < 0.01, ***P < 0.001.
In contrast to differential changes in amplitudes across the OPs, implicit times of all four OPs were uniformly delayed by hyperglycemia at 5 weeks post STZ (Fig. 2D). OP implicit times were significantly delayed at 4 weeks (data not shown) and 5 weeks post STZ from 10% to 12% for OP1 through OP4 (Fig. 2D, P < 0.05 to P < 0.001). Note that the magnitude of the delay was greatest in response to the dimmest flash stimuli.
Rod-dominated signaling was the most impacted by diabetes. When diabetic versus control differences in all OP implicit times were averaged together and compared between representative dim (−3.02 log cd s/m2) and bright (0.61 log cd s/m2) stimuli at 3, 4, and 5 weeks post STZ, only responses to dim stimuli showed significant delays in the diabetic animals at week 4 (Fig. 3A, P < 0.001) and week 5 (Fig. 3A, P < 0.001).
Figure 3.

Rod-driven stimuli reveal selective delays in OP implicit time across diabetes progression, unlike mixed rod–cone stimuli. Mean (±SEM) OP1 through OP4 implicit time differences of diabetic relative to control animals over the duration of diabetes show progressive delays in response to (A) dim (−3.02 log cd s/m2), but not (B) bright (0.61 log cd s/m2) flash stimuli. Colored symbols represent each OP, and the solid line with black circles represents the average of all four OPs. Asterisks represent Holm-Sidak post hoc analysis for individual weeks, ***P < 0.001.
Diabetes-Induced OP Delays Are Ameliorated by L-DOPA Treatment
Similar to the data shown in Figure 2, DM THctrl + Veh animals showed an 11% delay in OP2 implicit times compared to THctrl animals in response to dim flash stimuli (Figs. 4A, 4C; −1.8 log cd s/m2, P = 0.008). Treatment with L-DOPA prevented the OP delay induced by hyperglycemia. DM THctrl + L-DOPA animals were statistically indistinguishable from THctrl mice (Figs. 4A, 4C, P = 0.5), as shown by the representative OP response (gray vertical line marks OP2 in Fig. 4A). OP2 implicit times for DM THctrl + Veh and DM THctrl + L-DOPA mice in response to bright flash stimuli did not reach statistical significance (Figs. 4B, 4D).
Figure 4.

L-DOPA treatment reverses the rod-driven inner retinal dysfunction in diabetic animals. (A, B) Representative raw waveforms of THctrl, DM THctrl + Veh, and DM THctrl + L-DOPA animals and their (C, D) OP2 implicit times (mean ± SEM) at 5 weeks post STZ in response to representative dim (−1.8 log cd s/m2; A, C) and bright (0.61 log cd s/m2; B, D) flash stimuli. Gray lines indicate the onset of OP2 in a representative control animal. The gray arrow indicates the onset of OP2 when it is significantly delayed in comparison to the control animal. (C) A significant delay (P = 0.008) is seen in OP2 implicit times of DM THctrl + Veh under dim flash stimulus. Treatment with L-DOPA in diabetic animals ameliorated the functional deficit to a level indistinguishable from the THctrl group. (D) Bright flash stimulus did not reveal any significant differences among the groups. Asterisks represent Holm-Sidak post hoc analysis with RFDR correction: **P < 0.01.
Retinal Dopamine Depletion Elicits OP Delays That Can Be Partially Rescued by L-DOPA
While the DM THctrl + Veh mice showed a selective delay in response to only dim flash stimuli (P = 0.02, similar to the CD-1 mice in Figs. 2 and 3), deletion of retinal Th had a deleterious effect on inner retinal function in DM mice in response to both dim and bright flashes. The implicit time of OP2 was 18% delayed in DM rTHKO + Veh mice compared to THctrl mice for dim flash stimuli (Fig. 5C, P = 0.003) and 20% delayed for bright flash stimuli (Fig. 5D, P < 0.004). For mixed rod- and cone-mediated responses, the Th knockout produced a delay more extreme compared to diabetes alone. The reduction in retinal Th produced 14% delays in OP2 implicit times for DM rTHKO + Veh animals compared to DM THctrl + Veh animals in response to bright flash stimuli (Fig. 5D, P < 0.03).
Figure 5.

Diabetic mice lacking retinal Th show larger inner retinal dysfunction, of which the rod-driven function can be ameliorated with L-DOPA. (A, B) Representative raw waveforms and (C, D) implicit time (mean ± SEM) of THctrl, DM THctrl + Veh, DM rTHKO + Veh, and DM rTHKO + L-DOPA animals at 5 weeks post STZ in response to (A, C) dim (−1.8 log cd s/m2) and (B, D) bright (0.61 log cd s/m2) flash stimuli. Suppression of retina-specific Th significantly increased OP2 implicit times compared to the control group in both dim (C; P = 0.003) and bright (D; P = 0.004) stimuli. However, L-DOPA treatment only protected the OP2 implicit time for the dim flash stimulus (C; P = 0.02). Gray lines indicate the onset of OP2 in a representative control animal. Gray arrows indicate the onset of OP2 whenever it is delayed in comparison to the control animal. Asterisks represent Holm-Sidak post hoc analysis with RFDR correction: *P < 0.03, **P < 0.01, ***P < 0.001. The waveforms and data for THctrl and DM THctrl + Veh are the same as for Figure 4.
L-DOPA treatment restored rod-driven function in DM rTHKO + L-DOPA mice (Fig. 5C) to the same level as the THctrl group (P = 0.54). Interestingly, the treatment did not rescue mixed rod- and cone-driven function. The treatment effect of L-DOPA in DM rTHKO + L-DOPA animals was not statistically significant under the bright flash stimulus (Fig. 5D).
Lower Retinal Dopamine Levels Correlate With Greater OP Delays
Next, we determined if decreased retinal DA was associated with inner retinal dysfunction, as measured with OP implicit time. A Pearson's correlation test was performed on the OP responses from control and diabetic animals with retinal DA levels from these same mice, as previously reported35 (Fig. 6). The correlation coefficient was found for the total sample size (inclusive of diabetic and control animals). In response to dim flash stimuli (−3.02 log cd s/m2), delays in OP2 implicit time significantly correlated with decreasing retinal DA level (R = −0.552, P = 0.04; Fig. 6A). In contrast, a nonsignificant correlation (R = −0.31; P = 0.28) was observed between the DA level and OP2 implicit times at brighter flash luminance (0.61 log cd s/m2; Fig. 6B).
Figure 6.

Dopamine levels are highly correlated with delays in inner retinal function in response to rod-driven stimuli. Scatter plot of OP2 implicit time at dim (−3.02 log cd s/m2; A) and bright (0.61 log cd s/m2; B) flash stimuli and corresponding retinal dopamine levels from the same mice at 5 weeks post STZ. Pearson correlation was performed on the total population (n = 14). Diabetic animals are shown with solid red circles; controls are shown with solid black diamonds. (A) There is a significant negative correlation (R = −0.552, P = 0.04) between the implicit time of OP2 and the retinal dopamine level in response to dim flash stimulus. Similar correlations for OP1, OP3, and OP4 delays and dopamine levels were found with the dim flash. In contrast, with bright flash stimulus (B), no correlations (P = 0.28) were observed between the two variables. OP1, OP3, and OP4 also did not show correlations with bright flash stimulus.
Discussion
This study showed that inner retinal function, as measured by OP latency, is impacted by diabetes early in the disease, in agreement with previous work.6–9 Significant reduction of retinal DA using diabetic rTHKO mice resulted in even greater OP delays. L-DOPA effectively ameliorated rod-dominated functional deficits of the inner retina seen with diabetes and the rTHKO model. The OP delay was mostly limited to dim flash stimuli and correlated with reduced retinal DA levels. Our results indicate that chronic hyperglycemia alters rod-driven inner retinal signal transmission, which is likely mediated by changes in retinal DA levels.
Inner Retinal Neurons Are Most Susceptible to Early Diabetes
The current study highlights the rapid onset of inner retinal changes with diabetes, which suggests greater vulnerability of inner retinal cell types to dysregulation by chronic hyperglycemia. Delayed OP implicit times have consistently been recognized as indicators of early inner retinal dysfunction in experimental diabetic animal models.27,29–32 Similarly, observations of altered OPs have been reported in diabetic patients without DR,6,7 in early DR without detectable vascular abnormalities,7–9 and in late DR.9,52 While reductions in OP amplitude7,9,52 and delays in implicit time6,7,9 have been identified as potential predictive markers for the progression of neuronal dysfunction in early diabetes, OP delays were the only ERG component that showed deficits at the time points studied here.
While the exact identity of OP generators remains unclear, pharmacologic studies have shown that selective blockage of dopaminergic amacrine cells can attenuate the activity of the first OP,53 and inhibition of second- and third-order interneurons of the depolarizing ON-pathway decreases the amplitudes of intermediate and late-appearing OPs.48 Since we observed similar delays in all OP wavelets, it would appear the inner retinal neurons generating all four OP peaks are affected by the diabetic insult. However, it should be noted that OP3 and OP4 showed larger than normal amplitudes in the current dataset, which may indicate alterations in inhibitory signals to the neurons generating these wavelets. Supporting the possibility that chronic hyperglycemia insults inner retinal neurons directly, elevated glucose induced changes in AMPA receptor subunit distribution and localization in cultured retinal neural cells.54 In addition, AII amacrine cells in diabetic rats after 3 weeks of STZ had reduced Ca(2+) permeability of extrasynaptic AMPA receptors.55
Systemic hyperglycemia has been reported to decrease amplitude and/or delay implicit time in second-order inner retinal neurons (bipolar cells, amacrine cells, ganglion cells) and the photoreceptors.24 However, in the present study at an early stage of diabetes, we did not detect abnormal electrical activities of the photoreceptors as measured by a-wave amplitudes (Figs. 1A, 1B) or implicit times (data not shown), in agreement with our previous results.33,35 While changes in photoreceptor structure and function have been reported that fit within the time course of the current experiment,56–58 others have demonstrated unaltered a-wave responses during the course of 6 to 12 weeks of hyperglycemia.25,27,28,30,32 Evidence is accumulating that diabetes elicits photoreceptor dysfunction at some point during the progression of retinopathy,56,59–63 but it may not be at a detectable level within the 5-week period tested in the current study.
Hyperglycemia Induces Dopamine-Mediated Neuronal Dysfunction
We found an association of hyperglycemia and the disruption of DA-mediated inner retinal function by employing rTHKO mice and L-DOPA treatments. Loss of retinal DA may explain the early inner retinal dysfunction in diabetes, as drastically lowering Th in the diabetic retina resulted in larger OP delays than diabetes alone and as L-DOPA was able to ameliorate the OP deficits in diabetic mice. Data reported here are in agreement with our previous findings of the therapeutic effect of L-DOPA on visual signals in early-stage diabetes.35 Moreover, the severity of retinal DA deficiency caused by diabetes may reflect the degree of inner retinal dysfunction, as suggested by our findings of worsening OP delays when retinal DA levels were drastically diminished in rTHKO mice and our correlational results in Figure 6.
Underlining DA's integral role in retinal function, we previously showed that diabetic rTHKO mice develop functional deficits beyond OP delays, including b-wave delays and optokinetic tracking impairments.35 Additionally, diabetic mice with normal retinal Th genes have shown a decrease in retinal DA at the early stage of 4 weeks post STZ, though Th transcript levels were unaltered, suggesting that the mechanisms which lead to eventual loss of DA synthesis may be multifaceted and disease-duration dependent.35
In addition, we found that most of the retinal functional deficits were observed in response to dim stimuli, suggesting that in early-stage diabetes, mainly rod-driven pathways are affected. Other studies have also reported increased susceptibility of rod pathways to hyperglycemia insult.8,33,64,65 However, only with more severe loss of DA in the rTHKO mice results in significantly delayed OPs in response to both dim and bright flash stimuli. This suggests that cells or circuits involved in cone-mediated signaling are less susceptible to DA depletion compared to rod-mediated signaling cells. Supporting the sensitivity of rod-driven pathways to DA, function was significantly improved with L-DOPA in rTHKO mice in response to dim flashes only.
While we have focused on the role of DA in altering inner retinal function in diabetes, we cannot rule out the involvement of GABAergic amacrine cells and the co-release of γ-aminobutyric acid (GABA; inhibitory) from the dopaminergic amacrine cells.66 GABA metabolism is altered in STZ-induced rats and partially credited to the reduction of OP amplitudes and delay in their implicit times as early as 6 weeks post STZ.67 Furthermore, STZ-induced diabetic mice have early deficits in inhibition in the rod pathway.65
Conclusions
The present study contributes to our understanding of one of the underlying mechanisms for altered signal transmission by inner retinal neurons in the presence of hyperglycemia. We have demonstrated that (1) early inner retinal dysfunction in diabetes can be partly attributed to DA deficiency; (2) restoration of DA levels using L-DOPA can ameliorate inner retinal deficits in early-stage diabetes; and (3) dim, rod-dominated flash stimuli have greater sensitivity for showing OP delays than bright, mixed rod–cone flashes, implicating increased susceptibility of rod-driven pathways to hyperglycemic insult. These findings suggest that rod-driven OP delays may provide a detection method for early-stage DR, hence revealing a potentially earlier treatment window, and that the retinal dopaminergic system could be a novel therapeutic target for early-stage diabetic retinopathy. Additional studies are needed to determine if slowing the early neuronal changes will prevent or slow the late-stage vision loss in DR.
Acknowledgments
Supported by Department of Veterans Affairs Rehabilitation Research and Development Service (Merit Award I01RX2615 [MTP] and Research Career Scientist Award C9257S [MTP]) and Department of Veterans Affairs Biological Laboratory Research and Development Service Merit Award (PMT), Juvenile Diabetes Research Foundation Innovative Award (PMT), Research to Prevent Blindness (Department of Ophthalmology, Emory University), and National Institutes of Health R01 EY004864, P30 EY006360.
Disclosure: M.K. Kim, None; M.H. Aung, P; L. Mees, None; D.E. Olson, None; N. Pozdeyev, None; P.M. Iuvone, P; P.M. Thule, None; M.T. Pardue, P
References
- 1. Ding J, Wong TY. . Current epidemiology of diabetic retinopathy and diabetic macular edema. Curr Diab Rep. 2012; 12: 346– 354. [DOI] [PubMed] [Google Scholar]
- 2. Rowley WR, Bezold C, Arikan Y, Byrne E, Krohe S. . Diabetes 2030: insights from yesterday, today, and future trends. Popul Health Manag. 2017; 20: 6– 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Danaei G, Friedman AB, Oza S, Murray CJ, Ezzati M. . Diabetes prevalence and diagnosis in US states: analysis of health surveys. Popul Health Metr. 2009; 7: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kollias AN, Ulbig MW. . Diabetic retinopathy: early diagnosis and effective treatment. Dtsch Arztebl Int. 2010; 107: 75– 83; quiz 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. National Eye Institute. United States Diabetic Retinopathy Statistics and Data. 2010. Available at: https://nei.nih.gov/eyedata/diabetic. Accessed December 26, 2017.
- 6. Lecleire-Collet A, Audo I, Aout M,et al. . Evaluation of retinal function and flicker light-induced retinal vascular response in normotensive patients with diabetes without retinopathy. Invest Ophthalmol Vis Sci. 2011; 52: 2861– 2867. [DOI] [PubMed] [Google Scholar]
- 7. Luu CD, Szental JA, Lee SY, Lavanya R, Wong TY. . Correlation between retinal oscillatory potentials and retinal vascular caliber in type 2 diabetes. Invest Ophthalmol Vis Sci. 2010; 51: 482– 486. [DOI] [PubMed] [Google Scholar]
- 8. Pardue MT, Barnes CS, Kim MK,et al. . Rodent hyperglycemia-induced inner retinal deficits are mirrored in human diabetes. Trans Vis Sci Tech. 2014; 3 3: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kizawa J, Machida S, Kobayashi T, Gotoh Y, Kurosaka D. . Changes of oscillatory potentials and photopic negative response in patients with early diabetic retinopathy. Jpn J Ophthalmol. 2006; 50: 367– 373. [DOI] [PubMed] [Google Scholar]
- 10. Heng LZ, Comyn O, Peto T,et al. . Diabetic retinopathy: pathogenesis, clinical grading, management and future developments. Diabet Med. 2013; 30: 640– 650. [DOI] [PubMed] [Google Scholar]
- 11. Asefzadeh B, Fisch BM, Parenteau CE, Cavallerano AA. . Macular thickness and systemic markers for diabetes in individuals with no or mild diabetic retinopathy. Clin Exp Ophthalmol. 2008; 36: 455– 463. [DOI] [PubMed] [Google Scholar]
- 12. Shimura M, Yasuda K, Nakazawa T, Tamai M. . Visual dysfunction after panretinal photocoagulation in patients with severe diabetic retinopathy and good vision. Am J Ophthalmol. 2005; 140: 8– 15. [DOI] [PubMed] [Google Scholar]
- 13. van Dijk HW, Verbraak FD, Kok PH,et al. . Early neurodegeneration in the retina of type 2 diabetic patients. Invest Ophthalmol Vis Sci. 2012; 53: 2715– 2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roy MS, Gunkel RD, Podgor MJ. . Color vision defects in early diabetic retinopathy. Arch Ophthalmol. 1986; 104: 225– 228. [DOI] [PubMed] [Google Scholar]
- 15. Sokol S, Moskowitz A, Skarf B, Evans R, Molitch M, Senior B. . Contrast sensitivity in diabetics with and without background retinopathy. Arch Ophthalmol. 1985; 103: 51– 54. [DOI] [PubMed] [Google Scholar]
- 16. Della Sala S, Bertoni G, Somazzi L, Stubbe F, Wilkins AJ. . Impaired contrast sensitivity in diabetic patients with and without retinopathy: a new technique for rapid assessment. Br J Ophthalmol. 1985; 69: 136– 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bresnick GH, Condit RS, Palta M, Korth K, Groo A, Syrjala S. . Association of hue discrimination loss and diabetic retinopathy. Arch Ophthalmol. 1985; 103: 1317– 1324. [DOI] [PubMed] [Google Scholar]
- 18. Cho NC, Poulsen GL, Ver Hoeve JN, Nork TM. . Selective loss of S-cones in diabetic retinopathy. Arch Ophthalmol. 2000; 118: 1393– 1400. [DOI] [PubMed] [Google Scholar]
- 19. Katz G, Levkovitch-Verbin H, Treister G, Belkin M, Ilany J, Polat U. . Mesopic foveal contrast sensitivity is impaired in diabetic patients without retinopathy. Graefes Arch Clin Exp Ophthalmol. 2010; 248: 1699– 1703. [DOI] [PubMed] [Google Scholar]
- 20. Ng JS, Bearse MA Jr, Schneck ME, Barez S, Adams AJ. . Local diabetic retinopathy prediction by multifocal ERG delays over 3 years. Invest Ophthalmol Vis Sci. 2008; 49: 1622– 1628. [DOI] [PubMed] [Google Scholar]
- 21. Harrison WW, Bearse MA Jr, Ng JS,et al. . Multifocal electroretinograms predict onset of diabetic retinopathy in adult patients with diabetes. Invest Ophthalmol Vis Sci. 2011; 52: 772– 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolff BE, Bearse MA Jr, Schneck ME, Barez S, Adams AJ. Multifocal VEP. (mfVEP) reveals abnormal neuronal delays in diabetes. Doc Ophthalmol. 2010; 121: 189– 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barber AJ, Antonetti DA, Kern TS,et al. . The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci. 2005; 46: 2210– 2218. [DOI] [PubMed] [Google Scholar]
- 24. Fletcher EL, Phipps JA, Ward MM, Puthussery T, Wilkinson-Berka JL. . Neuronal and glial cell abnormality as predictors of progression of diabetic retinopathy. Curr Pharm Des. 2007; 13: 2699– 2712. [DOI] [PubMed] [Google Scholar]
- 25. Ly A, Yee P, Vessey KA, Phipps JA, Jobling AI, Fletcher EL. . Early inner retinal astrocyte dysfunction during diabetes and development of hypoxia, retinal stress, and neuronal functional loss. Invest Ophthalmol Vis Sci. 2011; 52: 9316– 9326. [DOI] [PubMed] [Google Scholar]
- 26. VanGuilder HD, Brucklacher RM, Patel K, Ellis RW, Freeman WM, Barber AJ. . Diabetes downregulates presynaptic proteins and reduces basal synapsin I phosphorylation in rat retina. Eur J Neurosci. 2008; 28: 1– 11. [DOI] [PubMed] [Google Scholar]
- 27. Bui BV, Loeliger M, Thomas M,et al. . Investigating structural and biochemical correlates of ganglion cell dysfunction in streptozotocin-induced diabetic rats. Exp Eye Res. 2009; 88: 1076– 1083. [DOI] [PubMed] [Google Scholar]
- 28. Ramsey DJ, Ripps H, Qian H. . An electrophysiological study of retinal function in the diabetic female rat. Invest Ophthalmol Vis Sci. 2006; 47: 5116– 5124. [DOI] [PubMed] [Google Scholar]
- 29. Ozawa Y, Kurihara T, Sasaki M,et al. . Neural degeneration in the retina of the streptozotocin-induced type 1 diabetes model. Exp Diabetes Res. 2011; 2011: 108328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kohzaki K, Vingrys AJ, Bui BV. . Early inner retinal dysfunction in streptozotocin-induced diabetic rats. Invest Ophthalmol Vis Sci. 2008; 49: 3595– 3604. [DOI] [PubMed] [Google Scholar]
- 31. Kaneko M, Sugawara T, Tazawa Y. . Electrical responses from the inner retina of rats with streptozotocin-induced early diabetes mellitus [in Japanese]. Nippon Ganka Gakkai Zasshi. 2000; 104: 775– 778. [PubMed] [Google Scholar]
- 32. Shinoda K, Rejdak R, Schuettauf F,et al. . Early electroretinographic features of streptozotocin-induced diabetic retinopathy. Clin Exp Ophthalmol. 2007; 35: 847– 854. [DOI] [PubMed] [Google Scholar]
- 33. Aung MH, Kim MK, Olson DE, Thule PM, Pardue MT. . Early visual deficits in streptozotocin-induced diabetic long evans rats. Invest Ophthalmol Vis Sci. 2013; 54: 1370– 1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Witkovsky P. . Dopamine and retinal function. Doc Ophthalmol. 2004; 108: 17– 40. [DOI] [PubMed] [Google Scholar]
- 35. Aung MH, Park HN, Han MK,et al. . Dopamine deficiency contributes to early visual dysfunction in a rodent model of type 1 diabetes. J Neurosci. 2014; 34: 726– 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nishimura C, Kuriyama K. . Alterations in the retinal dopaminergic neuronal system in rats with streptozotocin-induced diabetes. J Neurochem. 1985; 45: 448– 455. [DOI] [PubMed] [Google Scholar]
- 37. Gastinger MJ, Singh RS, Barber AJ. . Loss of cholinergic and dopaminergic amacrine cells in streptozotocin-diabetic rat and Ins2Akita-diabetic mouse retinas. Invest Ophthalmol Vis Sci. 2006; 47: 3143– 3150. [DOI] [PubMed] [Google Scholar]
- 38. Wachtmeister L. . Oscillatory potentials in the retina: what do they reveal. Prog Retin Eye Res. 1998; 17: 485– 521. [DOI] [PubMed] [Google Scholar]
- 39. Wachtmeister L. . Basic research and clinical aspects of the oscillatory potentials of the electroretinogram. Doc Ophthalmol. 1987; 66: 187– 194. [DOI] [PubMed] [Google Scholar]
- 40. Jackson CR, Ruan GX, Aseem F,et al. . Retinal dopamine mediates multiple dimensions of light-adapted vision. J Neurosci. 2012; 32: 9359– 9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bergen MA, Park HN, Chakraborty R,et al. . Altered refractive development in mice with reduced levels of retinal dopamine. Invest Ophthalmol Vis Sci. 2016; 57: 4412– 4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ciavatta VT, Mocko JA, Kim MK, Pardue MT. . Subretinal electrical stimulation preserves inner retinal function in RCS rat retina. Mol Vis. 2013; 19: 995– 1005. [PMC free article] [PubMed] [Google Scholar]
- 43. DeMarco PJ Jr, Katagiri Y, Enzmann V, Kaplan HJ, McCall MA. . An adaptive ERG technique to measure normal and altered dark adaptation in the mouse. Doc Ophthalmol. 2007; 115: 155– 163. [DOI] [PubMed] [Google Scholar]
- 44. Sagdullaev BT, DeMarco PJ, McCall MA. . Improved contact lens electrode for corneal ERG recordings in mice. Doc Ophthalmol. 2004; 108: 181– 184. [DOI] [PubMed] [Google Scholar]
- 45. Mocko JA, Kim M, Faulkner AE, Cao Y, Ciavatta VT, Pardue MT. . Effects of subretinal electrical stimulation in mer-KO mice. Invest Ophthalmol Vis Sci. 2011; 52: 4223– 4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Turner PV, Albassam MA. . Susceptibility of rats to corneal lesions after injectable anesthesia. Comp Med. 2005; 55: 175– 182. [PubMed] [Google Scholar]
- 47. Boatright JH, Moring AG, McElroy C,et al. . Tool from ancient pharmacopoeia prevents vision loss. Mol Vis. 2006; 12: 1706– 1714. [PubMed] [Google Scholar]
- 48. Dong CJ, Agey P, Hare WA. . Origins of the electroretinogram oscillatory potentials in the rabbit retina. Vis Neurosci. 2004; 21: 533– 543. [DOI] [PubMed] [Google Scholar]
- 49. Hancock HA, Kraft TW. . Human oscillatory potentials: intensity-dependence of timing and amplitude. Doc Ophthalmol. 2008; 117: 215– 222. [DOI] [PubMed] [Google Scholar]
- 50. Jaissle GB, May CA, Reinhard J,et al. . Evaluation of the rhodopsin knockout mouse as a model of pure cone function. Invest Ophthalmol Vis Sci. 2001; 42: 506– 513. [PubMed] [Google Scholar]
- 51. Toda K, Bush RA, Humphries P, Sieving PA. . The electroretinogram of the rhodopsin knockout mouse. Vis Neurosci. 1999; 16: 391– 398. [DOI] [PubMed] [Google Scholar]
- 52. Bresnick GH, Palta M. . Oscillatory potential amplitudes. Relation to severity of diabetic retinopathy. Arch Ophthalmol. 1987; 105: 929– 933. [DOI] [PubMed] [Google Scholar]
- 53. Harnois C, Marcotte G, Bedard PJ. . Alteration of monkey retinal oscillatory potentials after MPTP injection. Doc Ophthalmol. 1987; 67: 363– 369. [DOI] [PubMed] [Google Scholar]
- 54. Castilho AF, Liberal JT, Baptista FI, Gaspar JM, Carvalho AL, Ambrosio AF. . Elevated glucose concentration changes the content and cellular localization of AMPA receptors in the retina but not in the hippocampus. Neuroscience. 2012; 219: 23– 32. [DOI] [PubMed] [Google Scholar]
- 55. Castilho A, Madsen E, Ambrosio AF, Veruki ML, Hartveit E. . Diabetic hyperglycemia reduces Ca2+ permeability of extrasynaptic AMPA receptors in AII amacrine cells. J Neurophysiol. 2015; 114: 1545– 1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kern TS, Berkowitz BA. . Photoreceptors in diabetic retinopathy. J Diabetes Investig. 2015; 6: 371– 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Park SH, Park JW, Park SJ,et al. . Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia. 2003; 46: 1260– 1268. [DOI] [PubMed] [Google Scholar]
- 58. Phipps JA, Fletcher EL, Vingrys AJ. . Paired-flash identification of rod and cone dysfunction in the diabetic rat. Invest Ophthalmol Vis Sci. 2004; 45: 4592– 4600. [DOI] [PubMed] [Google Scholar]
- 59. Du Y, Veenstra A, Palczewski K, Kern TS. . Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc Natl Acad Sci U S A. 2013; 110: 16586– 16591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arden GB. . The absence of diabetic retinopathy in patients with retinitis pigmentosa: implications for pathophysiology and possible treatment. Br J Ophthalmol. 2001; 85: 366– 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de Gooyer TE, Stevenson KA, Humphries P, Simpson DA, Gardiner TA, Stitt AW. . Retinopathy is reduced during experimental diabetes in a mouse model of outer retinal degeneration. Invest Ophthalmol Vis Sci. 2006; 47: 5561– 5568. [DOI] [PubMed] [Google Scholar]
- 62. Tonade D, Liu H, Kern TS. . Photoreceptor cells produce inflammatory mediators that contribute to endothelial cell death in diabetes. Invest Ophthalmol Vis Sci. 2016; 57: 4264– 4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu H, Tang J, Du Y,et al. . Photoreceptor cells influence retinal vascular degeneration in mouse models of retinal degeneration and diabetes. Invest Ophthalmol Vis Sci. 2016; 57: 4272– 4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Castilho A, Ambrosio AF, Hartveit E, Veruki ML. . Disruption of a neural microcircuit in the rod pathway of the mammalian retina by diabetes mellitus. J Neurosci. 2015; 35: 5422– 5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moore-Dotson JM, Beckman JJ, Mazade RE,et al. . Early retinal neuronal dysfunction in diabetic mice: reduced light-evoked inhibition increases rod pathway signaling. Invest Ophthalmol Vis Sci. 2016; 57: 1418– 1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hirasawa H, Betensky RA, Raviola E. . Corelease of dopamine and GABA by a retinal dopaminergic neuron. J Neurosci. 2012; 32: 13281– 13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ishikawa A, Ishiguro S, Tamai M. . Changes in GABA metabolism in streptozotocin-induced diabetic rat retinas. Curr Eye Res. 1996; 15: 63– 71. [DOI] [PubMed] [Google Scholar]