

Overview
Sarcomas constitute a heterogeneous group of rare solid tumors of mesenchymal cell origin with distinct clinical and pathological features, and are usually divided into 2 broad categories: sarcomas of soft tissues (including fat, muscle, nerve and nerve sheath, blood vessels, and other connective tissues) and sarcomas of bone.
Soft tissue sarcomas (STS) are the most frequent sarcomas; the annual incidence in the United States for 2009 is estimated to be approximately 10,660 cases, with an overall mortality rate of approximately 3820 cases per year, including adults and children.1 The true incidence of sarcoma is underestimated, especially because a large proportion of patients with gastrointestinal stromal tumors (GIST) may not have been counted in tumor registry databases before 2001. GIST is expected to have an incidence of at least 5000 new cases per year in the United States.2,3
Collectively, sarcomas account for approximately 1% of all adult and 15% of pediatric malignancies. Prior radiation therapy (RT) to the affected area given generally some years before the development of the sarcoma is a risk factor for STS.4 More than 50 different histologic subtypes of STS have been identified, with pleomorphic sarcoma (also known as malignant fibrous histiocytoma), GIST, liposarcoma, leiomyosarcoma, synovial sarcoma, and malignant peripheral nerve sheath tumors the most common.5 Rhabdomyosarcoma is the most common STS of childhood. The most common primary sites are the extremities (60%), trunk (19%), retroperitoneum (15%), and head and neck (9%).6 The anatomic site of the primary disease is an important variable influencing treatment and outcome. STS most commonly metastasize to the lungs; tumors arising in the abdominal cavity commonly metastasize to the liver and peritoneum.
NCCN encompasses institutions with extensive experience in managing sarcomas using multidisciplinary care and they function as referral centers of consultative support for community-based practitioners. The expertise of these institutions lends their extensive experience in defining the consensus guidelines for the management of patients with sarcomas. The guidelines appearing in this issue of JNCCN address sarcoma management in adult patients from the perspective of 2 disease subtypes: STS of the extremity/trunk and GIST. Two additional subtypes, retroperitoneal or intra-abdominal STS and desmoid tumors, are addressed online, in these guidelines, at www.NCCN.org. These guidelines do not include the management of rhabdomyosarcoma, Ewing’s sarcoma, and desmoplastic small round cell tumor.
NOTE: This manuscript highlights only a portion of the NCCN Guidelines on Soft Tissue Sarcoma. Please refer to www.NCCN.org for the complete guidelines.
Pathology of STS
Biopsy
A pretreatment biopsy is highly preferred for diagnosing and grading sarcomas, and should be performed by an experienced surgeon or radiologist. Biopsy can be accomplished by core needle or open incisional techniques. Although fine needle aspiration (FNA) is a convenient technique, it can be difficult to make an accurate primary diagnosis with FNA alone.7 FNA may be acceptable in select institutions with clinical and pathologic expertise. Endoscopic or needle biopsy may be indicated for deep thoracic, abdominal, or pelvic sarcomas.
Principles of Pathologic Assessment
Pathologists with sarcoma expertise should review the pathologic assessment of biopsies and resected specimens, especially for initial histopathologic classification. Margins must be thoroughly evaluated in these specimens. Morphologic assessment based on microscopic examination of histologic sections remains the gold standard of sarcoma diagnosis. Differential diagnosis of a soft tissue mass includes malignant lesions (i.e., primary or metastatic carcinoma, melanoma, or lymphoma), desmoids, and benign lesions (i.e., lipomas, lymphangiomas, leiomyomas, and neuromas). Because identifying a histopathologic type of sarcoma is often difficult, several ancillary techniques, such as conventional cytogenetics, immunohistochemistry, and molecular genetic testing, are useful to support the morphologic diagnosis.
Pathologists should have access to optimal cytogenetic and molecular diagnostic techniques. Results of appropriate ancillary studies used as an adjunct to morphologic diagnosis should be included in the pathology report. The report should include specific details about the primary diagnosis (using standardized classification according to the WHO classification); organ; sarcoma site, depth, size, and histologic grade; presence or absence of necrosis; status of excision margins and lymph nodes; TNM stage; and additional features of the tumor, such as mitotic rate, presence or absence of vascular invasion, and the type and extent of inflammatory infiltration (see page 646). The size at presentation depends on the location—tumors in the proximal extremities and retroperitoneum are often large, whereas distal extremity tumors are often small.
Molecular Diagnosis of STS
Molecular genetic testing has emerged as a particularly useful ancillary test because many STS subtypes are associated with characteristic genetic aberrations, including single base-pair substitutions, deletions, amplifications, and translocations (see pages 647 and 648). STS can be divided into 2 major genetic groups: 1) sarcomas with specific genetic alterations, such as chromosomal translocations or point mutations, and usually simple karyotypes, and 2) sarcomas with nonspecific genetic alterations and complex unbalanced karyotypes.8
STS with recurrent chromosomal translocations can be classified into subtypes depending on the presence of fusion gene transcripts (e.g., EWS-ATF1 in clear cell sarcoma, TLS-CHOP in myxoid or round cell liposarcoma, SYT-SSX [SYT-SSX1 or SYT-SSX2] in synovial sarcoma, and PAX-FKHR [PAX3-FKHR or PAX7-FKHR] in alveolar rhabdomyosarcoma). The fusion genes resulting from chromosomal translocations can provide useful diagnostic and prognostic information.
Common techniques used in molecular diagnosis include conventional cytogenetic analysis, fluorescence in-situ hybridization, and polymerase chain reaction (PCR)–based methods.9 In a prospective study, Hill et al.10 found that PCR–based molecular analysis is a useful adjunct and more sensitive than conventional cytogenetics for the diagnosis of certain STS subtypes, including alveolar rhabdomyosarcoma, synovial sarcoma, and myxoid liposarcoma, which have variation in fusion gene partners.
The molecular heterogeneity of fusion transcripts has been suggested to predict prognosis in certain sarcoma subtypes. In patients with alveolar rhabdomyosarcoma presenting with metastatic disease, PAX7-FKHR was associated with a favorable prognosis compared with PAX3-FKHR.11 In those with synovial sarcoma, the prognostic impact of fusion gene transcripts SYT-SSX1 and SYT-SSX2 is less clear, with conflicting results in 2 large studies.12,13 In myxoid liposarcoma, the variability of fusion transcript has no effect on clinical outcome.14
Although molecular genetic testing appears promising, it involves highly complex techniques and the methods are not absolutely sensitive or provide specific results. In addition, technical limitations associated with molecular testing suggest that molecular evaluation be considered only as an ancillary technique. Molecular test results should therefore only be interpreted in the context of the morphologic features of a sarcoma.9
Staging
The American Joint Committee on Cancer (AJCC) STS staging system has historically used a 4-grade system, but within the STS staging groups this effectively functioned as a 2-tiered system (G1/G2 [low] and G3/G4 [high]). The 2 most widely used systems, the French Federation of Cancer Centers Sarcoma Group (FNCLCC) and National Cancer Institute system, are 3-tiered grading systems. The latter is based on the evaluation of tumor histology, location, and amount of tumor necrosis, whereas the former is based on tumor differentiation, mitosis count, and tumor necrosis.
In a comparative study of these 2 systems in 410 adult patients with STS, the FNCLCC system showed a slightly increased ability to predict distant metastasis development and tumor mortality.15 The 2002 AJCC staging system accommodated some of the 3- and 4-tiered systems for establishing the grade. The revised 2010 AJCC staging system incorporates a 3-tiered grading system (see the staging table, available online, in these guidelines, at www.NCCN.org [ST-1]). Because many clinicians prefer the 2-tiered system, this system is also used in the algorithm (see page 633).
Principles of Surgery
Because surgery is the standard primary treatment for most sarcomas, the panel has included a separate section on principles of sarcoma surgery (see page 649). If a patient cannot be surgically treated according to these principles, preoperative RT or chemotherapy should be considered as alternate treatment options. Because the risk for failure in the surgical bed can be high, many clinicians choose to augment surgery with RT and chemotherapy, either pre- or postoperatively.16,17 When appropriate, the guidelines incorporate those therapies that are supported by clinical trial data or extensive clinical experience.18
Sarcoma Surgery
The biopsy site should be excised en bloc with the definitive surgical specimen. Dissection should be through grossly normal tissue planes uncontaminated by the tumor. If it is close to or displaces major vessels or nerves, these do not need to be resected if the adventitia or perineurium is removed and the underlying neurovascular structures are not involved with gross tumor. Radical excision or entire anatomic compartment resection is not routinely necessary. If resections with microscopically or grossly positive margins are anticipated, surgical clips should be left in place to identify high-risk areas for recurrence, particularly for retroperitoneal or intra-abdominal sarcomas, to help guide future RT. If closed suction drainage is used, the drains should exit the skin close to the surgical incision edge (in case re-resection or RT is indicated).
Limb-sparing surgery is recommended for most patients with extremity STS to achieve local tumor control with minimal morbidity. Evaluation for postoperative rehabilitation is recommended for all patients with extremity sarcoma. If indicated, rehabilitation should be continued until maximum function is achieved.
Resection Margins
Resection with appropriately negative margins is recommended, although negative but closer margins may be effective in patients undergoing RT.19 Close margins may be necessary to preserve uninvolved critical neurovascular structures. Microscopically positive surgical margins are associated with a higher rate of local recurrence and lower rate of disease-free survival in patients with extremity sarcomas.20–22
Both the surgeon and pathologist should document surgical margins in evaluating a resected specimen. If surgical margins are positive on final pathology, re-resection to obtain negative margins should strongly be considered if it will not have a significant impact on functionality.23,24 Adjuvant RT should be considered after resections with close soft tissue margins (< 1 cm) or a microscopically positive margin on bone, major blood vessels, or nerve.
Amputation for Extremity Sarcoma
Before considering amputation, patients should be evaluated by a surgeon with expertise in the treatment of STS. Amputation should be considered for patient preference, or if the gross total resection of the tumor is expected to render the limb nonfunctional.25–27
Guidelines for RT
External-beam radiation therapy (XRT) can be administered as primary therapy, preoperatively, or postoperatively in STS. Advances in RT technology, such as brachytherapy, intensity-modulated radiation therapy (IMRT), and intraoperative radiation therapy (IORT), have led to improved treatment outcomes in patients with STS.28 Brachytherapy involves the direct application of radioactive seeds into the tumor bed through catheters placed during surgery. The main advantage of IMRT is its ability to more closely contour the high-dose radiation volume to the tumor while minimizing the volume of high-dose radiation to the surrounding tissues.29 IORT delivers radiation during surgery and is performed using different techniques, such as electron beam radiation or brachytherapy.
Preoperative RT
Preoperative RT has several advantages. First, the treatment volume is smaller because the need to cover the operative field is not present. Second, it may reduce seeding during surgical manipulation of the tumor. The tumor may or may not regress with preoperative RT, but the pseudocapsule may thicken and become acellular, easing resection and decreasing the risk for recurrence. However, the main disadvantage of preoperative RT is its effect on wound healing.30 A higher acute wound healing complication rate has been observed when primary closure is used. Therefore, involvement of a plastic surgeon may be necessary to reduce wound complications when preoperative radiation is contemplated. After preoperative radiation, a 3- to 6-week interval is necessary before resection to allow acute radiation reactions to subside and decrease the risk for wound complications. Very long intervals between resection and postoperative radiation are not recommended because of the development of late fibrosis.
The usual dose of preoperative RT is 50 Gy. If wide margins are obtained, additional radiation may not be needed. Radiation boost with brachytherapy, IORT, or XRT is recommended for positive or close margins (see page 650).31 Often, margins are close because of their proximity to major neurovascular bundles or bone. Brachytherapy boosts should be delivered several days after surgery, through catheters placed at operation, with doses of 12 to 20 Gy based on margin status. Alternatively, a single intraoperative dose of 10 to 16 Gy, based on margin status, can be delivered immediately after resection with exposure of the area at risk, avoiding uninvolved organs. XRT boosts may be an alternative to brachytherapy or IORT. Recommended doses are 10 to 14 Gy for close margins, 16 to 20 Gy for microscopically positive margins, and 20 to 26 Gy for grossly positive margins. Many institutions are no longer giving a boost after preoperative radiation to patients who have widely negative margins, based on local control rates that approach 95% with preoperative radiation at 50 Gy and negative margins.
Postoperative RT
Postoperative RT has been shown to improve local control in patients with high-grade extremity STS with positive surgical margins.32 In a recent report from Memorial Sloan-Kettering Cancer Center, in patients with extremity STS treated with limb-sparing surgery and a pathologically negative re-resection without RT, patients of elderly age and/or with stage III disease had a higher rate of local recurrence, even though the 5-year overall local recurrence rate was 9% with a median follow-up of 82 months.33 Therefore, treatment decisions regarding the use of postoperative RT should be individualized and not solely based on the finding of margin-negative re-resection.
When surgical resection is the initial therapy, postoperative RT choices include brachytherapy, IORT, or XRT (see page 650). When XRT is used, sophisticated treatment planning with IMRT, tomotherapy, and/or proton therapy can be used to improve therapeutic effect. Most institutions include the entire operative bed within that radiation field. Total doses of RT should always be determined through normal tissue tolerance. RT is not a substitute for suboptimal surgical resection, and re-resection may be necessary. If the patient has not previously undergone RT, control of microscopic residual disease would be attempted with postoperative RT if re-resection is not feasible.
Brachytherapy alone has been used as an adjuvant treatment. Radiation delivered at 45 to 50 Gy at low-dose rate to the tumor bed has been shown to reduce recurrence without a significant effect on wound healing.34 However, brachytherapy-alone techniques require special expertise and significant experience. The panel recommends 45 Gy low dose-rate brachytherapy for patients with negative margins. Low dose-rate brachytherapy (16–20 Gy) or a high dose-rate equivalent is recommended for patients with positive margins followed by XRT. XRT is delivered to the target volume to a total dose of 50 Gy (45 Gy for retroperitoneal or intra-abdominal sarcomas) after surgical healing is complete (3–8 weeks).
Recent reports from a retrospective study suggest that IORT provides excellent local control to STS of the extremity.35 However, because IORT has not been proven superior, the guidelines recommend IORT (10–16 Gy) followed by a 50-Gy dose of XRT.
If no IORT or brachytherapy was used in the immediate operative or postoperative period, XRT is delivered to the target volume of a 50-Gy total dose (45 Gy for retroperitoneal or intra-abdominal sarcomas) after surgical healing is complete. An XRT boost should be used based on the margin status. For negative margins, an additional 10 to 16 Gy is recommended to the original tumor bed. For microscopically positive margins, an additional 16 to 20 Gy is recommended, and for grossly positive margins, an additional 20 to 26 Gy is suggested.
STS of the Extremities or Trunk
Surgery
Amputation was once considered the standard treatment to achieve local control in patients with extremity sarcomas.36 In recent years, technical advances in reconstructive surgical procedures, implementation of multimodality therapy, and improved selection of patients for adjuvant therapy have minimized the functional deficits in patients who might otherwise require amputation.
In 1982, a randomized controlled trial (43 patients) showed that limb-sparing surgery with RT was an effective treatment in patients with high-grade STS of the extremities, with a local recurrence rate of 15% and no difference in overall and disease-free survival compared with amputation.37 In another series of 77 patients treated with limb-sparing surgery without RT, the local recurrence rate was only 7% and resection margin status was a significant predictor of local recurrence.38 The local recurrence rate was 13% when the resection margin was 1 cm or less compared with 0% when the resection margin was 1 cm or more. In a retrospective study of 115 patients with an STS of hand or foot, radical amputation as an initial treatment did not decrease the probability of regional metastasis and also did not improve the disease-specific survival.39 These results suggest that limb-sparing surgery with or without adjuvant RT is an effective treatment option for extremity STS, and amputation should be reserved only for cases in which resection or re-resection with adequate margins cannot be performed without sacrificing the functional outcome.
RT
Randomized clinical trial data support the use adjunctive XRT in appropriately selected patients with STS of extremity.40–42 In a phase III randomized trial conducted by the Canadian Sarcoma Group,40 local control and progression-free survival rates were similar in patients undergoing either pre- or postoperative XRT for localized primary or recurrent extremity sarcoma. However, preoperative RT was associated with a greater incidence of acute wound complications (35% vs. 17% for postoperative XRT), especially in lower extremity tumors (43% vs. 5% for upper extremity tumors). Late treatment-related side effects were more common in patients undergoing postoperative radiation, which is believed to be related to the higher postoperative XRT dose (66 vs. 50 Gy for preoperative) and the larger treatment volume. Therefore, the risk for local recurrence versus the toxicity of postoperative XRT should be assessed before making a decision regarding radiation.
The efficacy of postoperative XRT was shown in a prospective randomized trial comparing limbsparing surgery alone and with adjuvant XRT. Postoperative XRT reduced the 10-year local recurrence rate in patients with high-grade sarcoma (none in patients who underwent surgery plus XRT vs. 22% in those who underwent surgery alone) and those with low-grade (5% for surgery plus XRT group vs. 32% for surgery alone).
In a prospective randomized trial, 164 patients with completely resected STS of the extremity or superficial trunk were randomized intraoperatively to receive either adjuvant brachytherapy or no brachytherapy.43 With a median follow-up time of 76 months, the 5-year local control rates were 82% and 69% in the brachytherapy and no brachytherapy groups, respectively. Patients with high-grade lesions who received brachytherapy had higher local control rates than those randomized to no brachytherapy (89% and 66%, respectively). However, brachytherapy had no impact on local control in patients with low-grade lesions. The 5-year freedom-from-distant-recurrence rates were 83% and 76%, respectively. Results of this trial showed that adjuvant brachytherapy improves local control after complete resection of STS in patients with high-grade lesions.
Postoperative IMRT after limb-sparing surgery is associated with excellent local control in selected patients with high-risk features. In a retrospective analysis, the 5-year local control rate was 94% in patients with negative and positive or close margins.44 The risk for complications such as edema and joint stiffness were also favorable compared with conventional RT. Despite the excellent results of adjuvant IMRT in patients with extremity sarcomas, its efficacy must be confirmed in larger cohorts of patients with longer follow-up.
Definitive RT entails the delivery of maximal local dose compatible with known tissue tolerance, typically ranging from 70 to 80 Gy, using sophisticated instrument planning techniques. In a single-institution study (112 patients, 43% STS of extremity), tumor size and the dose of RT influenced local control and survival in patients with unresectable STS.45 Local control rate was 51% for tumors less than 5 cm and 9% for tumors greater than 10 cm. Patients who received 63 Gy or more had better 5-year local control, disease-free survival, and overall survival rates (60%, 36%, and 52%, respectively) than patients treated with less than 63 Gy (22%, 10%, and 14%, respectively). Local control for patients receiving more than 63 Gy was 72% for lesions 5 cm or less, 42% for lesions 5 to 10 cm, and 25% for those greater than 10 cm.
Evaluation and Workup
All patients should be managed by a multidisciplinary team with expertise in STS.46 The differential diagnosis of STS of the extremities includes ruling out desmoids and other malignant and benign lesions previously discussed. An essential element of the workup is a history and physical examination (H&P). Laboratory tests have a limited role. Adequate and high-quality imaging studies are crucial to good clinical management of patients, because the presence of metastatic disease may change the management of the primary lesion and the overall approach to the patient’s disease management. Imaging studies should also provide details about tumor size and contiguity to nearby visceral structures and neurovascular landmarks. The propensities to spread to various locations vary among the subtypes of sarcoma. Therefore, imaging should be individualized based on the subtype of sarcoma.
MRI with or without CT is indicated for all lesions with a reasonable chance of being malignant (see page 632). MRI is preferred for extremity sarcomas, whereas CT is preferred for retroperitoneal sarcomas.47–49 CT angiogram may be useful in patients for whom MRI is not feasible. Plain radiograph of the primary lesion is optional. Given the risk for hematogenous spread from a high-grade sarcoma to the lungs, imaging of the chest is essential for accurate staging. Abdominal/pelvic CT should be considered for myxoid round cell liposarcoma, leiomyosarcoma, epithelioid sarcoma, or angiosarcoma, and MRI of the total spine should be considered for myxoid round cell liposarcomas because of the higher risk for metastasis to spine compared with other STS.50–52 Central nervous system imaging should be considered for patients with alveolar soft part sarcomas and angiosarcomas because alveolar soft part sarcomas have a relatively increased propensity to metastasize to the brain, especially in patients with stage IV disease in the presence of pulmonary metastases.53
PET scan may be useful for prognostication and grading, and to assess response to chemotherapy.54 Tumor metabolism data acquired by PET will be useful in accurate grading and prognostication in sarcoma.55 Recent reports in literature have shown the value of PET in evaluating response to preoperative chemotherapy in patients with high-grade extremity STS and for predicting outcome in patients with liposarcoma.56,57 A large prospective study is underway to evaluate the value of PET scan combined with CT scan in predicting disease-free survival in patients undergoing preoperative chemotherapy for STS (www.cancer.gov/clinicaltrials/UMN-2005LS080).
Based on the initial workup, the patients are assigned to 1 of the following categories:
Stage I
Stage II–III
Stage IV
Recurrent disease
Stage I
Surgery is the primary treatment for low-grade stage I tumors and is considered definitive if margins are greater than 1 cm or the fascia plane is intact.58,59 Retrospective studies have shown a local control rate of 90% or more for surgery alone.60 Long-term results of a prospective trial showed that selected patients with primary T1 STS of the extremity and trunk can be treated with surgery alone (R0 resection) with acceptable local control and excellent long-term survival.61 In the surgery alone arm, the cumulative incidence rates of local recurrence at 5 and 10 years were 7.9% and 10.6%, respectively, in patients who underwent R0 resection, and the 5-and 10-year sarcoma-specific death rates were 3.2%.
The panel recommends surgery alone as the primary treatment for low-grade stage I tumors (T1a–2b, N0, M0). If the final surgical margins are 1.0 cm or less, postoperative RT is included with a category 2B recommendation for T1a–b tumors and a category 1 recommendation for T2a–b tumors (see page 633). RT may not be necessary in patients with small lesions (≤ 5 cm), because these tumors are less frequently associated with local recurrence.
Stage II–III
Large high-grade extremity sarcomas (> 8–10 cm) at high risk for local recurrences and metastases should be considered for preoperative therapy. Preoperative chemotherapy or chemoradiation is used in many centers for downstaging large high-grade tumors to enable effective surgical resection, especially in the case of chemosensitive histologies.37,62–65 Concurrent chemoradiation with doxorubicin-based regimens has been shown to improve local control rates in patients with STS, although acute reactions must be considered.66 Available evidence, although underpowered, suggests that anthracycline-based postoperative chemotherapy would improve disease-free survival in selected patients who are at high risk for recurrence but otherwise have good performance status.67,68
The Sarcoma Meta-Analysis Collaboration performed a meta-analysis of 14 randomized trials (1568 patients) comparing adjuvant chemotherapy with follow-up, and in some cases RT after surgery, for treatment of various sarcomas.69 The result of the meta-analysis showed that doxorubicin-based chemotherapy prolongs relapse-free survival in adults with localized, resectable STS of the extremity, and was associated with decreased recurrence rates. However, adjuvant chemotherapy does not seem to improve overall survival.70 Another recent analysis of 674 patients with stage III STS (1984–1999) showed that clinical benefits from doxorubicin-based chemotherapy lasted for less than a year.71
In an Italian randomized cooperative trial, patients with high-grade or recurrent extremity sarcoma were randomized to undergo postoperative chemotherapy with epirubicin and ifosfamide or observation alone. After a median follow-up of 59 months, median disease-free (48 vs. 16 months) and overall survivals (75 vs. 46 months) were significantly better in the treatment group.72,73
Remarkably little data have been generated in the adjuvant setting regarding the combination of aggressively dosed ifosfamide plus doxorubicin with growth factor support. EORTC-62931 is a completed phase III randomized study assessing the efficacy of adjuvant chemotherapy after definitive surgery in patients with excised high-grade STS at any site. Patients with macroscopically resected grade II and III tumors with no metastases were randomized to observation or chemotherapy with ifosfamide and doxorubicin with lenograstim. A planned interim analysis of this study showed no survival advantage for adjuvant chemotherapy with ifosfamide and doxorubicin in patients with resected high-grade STS.74 The estimated relapse-free survival rate was 52% in both arms. Further analysis of this study is needed to make a detailed assessment of the role of adjuvant chemotherapy in resected STS.
Treatment options for stage II or III high-grade tumors should be decided by a multidisciplinary team, based on the performance status; comorbid factors, including age, tumor location, and histologic subtype of the tumor; and institutional experience.
Resectable Tumors
Surgery followed by RT with or without adjuvant chemotherapy or surgery alone (for small tumors that can be resected with wider surgical margins) is the primary treatment for resectable high-grade sarcomas with acceptable functional outcomes (see page 633).75 The guidelines have also included preoperative RT, chemotherapy, or chemoradiation before surgery as alternative options for patients with resectable tumors with acceptable functional outcomes and for potentially resectable tumors with associated concerns for adverse functional outcomes. The panel included preoperative chemotherapy or chemoradiation for resectable disease with acceptable functional outcomes with a category 2B recommendation.
Postoperative RT boost for residual gross disease or microscopically positive margins or adjuvant chemotherapy alone can be considered for patients who have undergone preoperative RT or chemoradiation, whereas postoperative RT with or without adjuvant chemotherapy is recommended for those who underwent preoperative chemotherapy (see page 633). Because limited and conflicting data are available for adjuvant chemotherapy in patients with stage II or III disease, adjuvant chemotherapy for stage II or III tumors is included as a category 2B recommendation for all patients with resectable tumors, irrespective of the functional outcomes.
Unresectable Tumors
Unresectable tumors can be treated primarily with preoperative RT, chemoradiation, or chemotherapy. Tumors that become resectable after preoperative treatment can be treated with surgery (see page 634). Postoperative treatment options for these patients are similar to those described on page 633 of the algorithm for stage II or III resectable tumors.
Definitive RT (7000–8000 cGy) can be considered for select patients with unresectable tumors after preoperative treatment (see page 634). Observation is an option for asymptomatic patients whose tumors are not believed to be amenable to local control with definitive radiation. For symptomatic patients, the panel recommends moving directly to a palliative approach, defined broadly as chemotherapy, palliative surgery, or best supportive care.
Stage IV (Metastatic Disease)
Single agents (doxorubicin, ifosfamide, or dacarbazine) or anthracycline-based combination regimens (doxorubicin or epirubicin with ifosfamide and/or dacarbazine) have been widely used to treat metastatic disease.76–83 Liposomal anthracyclines were found to be active as first-line treatment for advanced sarcomas with a better toxicity profile than doxorubicin.84,85 Other chemotherapeutic agents have also been tested in clinical trials. Combined gemcitabine and docetaxel was found to be highly active in patients with predominantly uterine leiomyosarcomas who experienced no response to or for medical reasons could not tolerate ifosfamide plus doxorubicin.86 In a randomized phase II study, progression-free survival (6.2 vs. 3.0 months, respectively) and overall survival (17.9 vs. 11.5 months, respectively) were superior to gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic STS.87 In a separate report published after this study, this combination was found to be active in various histologic subtypes of sarcoma.88
In a retrospective study conducted by the French Sarcoma Group in 133 patients with unresectable or metastatic STS, the gemcitabine and docetaxel combination was tolerable and showed better response and survival rates in patients with leiomyosarcoma.89 Another phase II trial (MSKCC-99027) is evaluating the activity of gemcitabine, docetaxel, and filgrastim in patients with recurrent or persistent unresectable leiomyosarcoma or other STS that cannot be removed with surgery. In a phase II study, combination gemcitabine and vinorelbine was associated with clinically meaningful rates of disease control in patients with advanced STS.90 Clinical benefit (complete response, partial response, or stable disease at ≥ 4 months) was seen in 25% of patients. Temozolomide as a single agent is also active in patients with advanced pretreated STS, especially those with unresectable or metastatic leiomyosarcoma of both uterine and nonuterine origin.91,92
Ecteinascidin 743 (ET-743, also known as trabectedin or Yondelis), is a marine-derived antitumor agent, which has shown objective responses in phase II trials of patients with progressive STS refractory to chemotherapy.93–97 NCT00210665 is an ongoing multicenter, open label, single-arm study providing access to treatment with trabectedin for patients with persistent or recurrent STS and who are not expected to benefit from currently available treatments.
Interim overall survival data are encouraging from an ongoing phase III trial (EORTC-62961) of regional hyperthermia (RHT) versus chemotherapy with EIA (etoposide, ifosfamide, and adriamycin) alone for patients with locally advanced high-risk STS, especially those with extremity sarcomas.98 After a median follow-up of 24.9 months, disease-free survival (31.7 vs. 6.2 months, respectively), local progression-free survival (84% vs. 64%, respectively, for extremity sarcomas and 57% vs. 39%, respectively, for body wall and abdominal sarcomas), and overall response rate (28.7% vs. 12.6%, respectively) were significantly superior for patients treated with EIA plus RHT compared with those treated with EIA alone.99
Isolated limb perfusion (ILP) has been used in Europe as a limb-sparing treatment for unresectable intermediate or high-grade extremity STS.100 In European clinical trials, melphalan in combination with tumor necrosis factor-α (TNF-α) resulted in better response rates and limb-salvage rates than ILP with melphalan alone.101 Recombinant TNF-α–1A and melphalan has been approved in Europe for ILP in patients with locally advanced high-grade STS of the extremities.
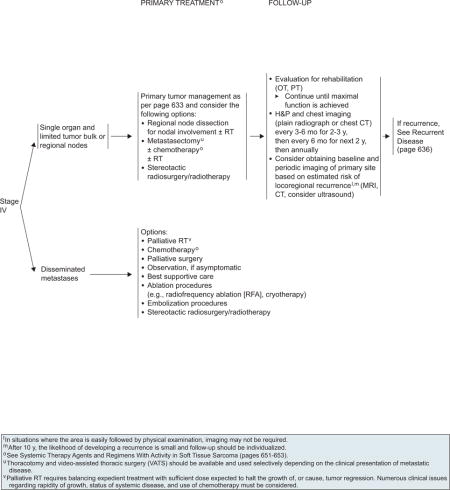
Limited Metastases
Patients with limited metastasis confined to a single organ and limited tumor bulk or regional lymph node involvement should undergo primary tumor management as described for stage II or III tumors on page 633. Another option is to consider regional node dissection for nodal involvement with or without RT or metastasectomy with or without chemotherapy with or without RT (see page 635). The guidelines do not specify rules governing metastasectomy, which remains controversial for many cancers, including sarcoma. Several variables influence the decision to use metastasectomy, including the disease-free interval from original diagnosis to detection of the metastases, the patient’s performance status, and the amount of prior therapy. Thoracotomy and video-assisted thoracic surgery should be used selectively depending on the clinical presentation of metastatic disease. In addition, patients can also undergo radiofrequency ablation or embolization procedures as an alternate method for control of metastatic lesions.
Disseminated Metastases
In the guidelines, a subsequent distinction is made between asymptomatic and symptomatic patients for those who present with disseminated disease (see page 635). One reasonable management option for asymptomatic patients is to offer close observation with a “watchful waiting” strategy, especially for patients who had a very long disease-free interval and have only a minimal burden of metastases (e.g., subcentimeter pulmonary nodules). Alternatively, patients can also be treated with palliative approaches, such as palliative RT, chemotherapy, or palliative surgery. Palliative RT involves expedient treatment with sufficient dose to halt tumor growth or cause tumor regression. The outcome of this approach depends on the rapidity of growth and the status of systemic disease.
In addition, the guidelines have included ablation procedures (e.g., radiofrequency ablation or cryotherapy), embolization procedures, or stereotactic radiosurgery/radiotherapy as options for symptomatic patients with disseminated metastases. The guidelines are intentionally nonspecific about this group of options, because many different factors impact this decision (e.g., patient performance status, patient preferences, specific clinical problems from the metastases, treatment availability) and specific details are best left to clinical judgment.
Surveillance
Surveillance is deemed important to detect recurrences that might still be potentially curable. However, limited data are available in the literature on effective surveillance strategies.102–104 The guidelines outline a prudent follow-up schedule that avoids excessive testing. Higher-grade and larger tumors have a higher risk for dissemination; therefore, the surveillance recommendations for patients with these tumors are somewhat more intensive, particularly for the first 3 years after resection. Periodic imaging (MRI, CT, or ultrasound) of the primary site should be performed based on the estimated risk for locoregional recurrence. However, when the area can be easily followed by physical examination, imaging may not be required. After 10 years, the likelihood of developing a recurrence is small and follow-up should be individualized.
Stage I tumors are routinely followed with H&P every 3 to 6 months for 2 to 3 years, then annually (see page 633). Chest imaging should also be considered every 6 to 12 months. For stage II through IV disease, H&P and chest imaging should be performed every 3 to 6 months for 2 to 3 years, then every 6 months for the next 2 years, and then annually (see pages 633–635). Because these patients’ risk never returns to zero, long-term follow-up is indicated, including consideration of MRI or CT scanning.105 No study has ever proved that the use of more sensitive CT scans in routine surveillance would improve clinical outcomes. According to the reported data from MD Anderson Cancer Center, routine use of chest CT adds little clinical benefit when risk for pulmonary metastases is low.106 However, in certain subsets of patients for whom chest radiographs are difficult to interpret because of anatomic considerations (e.g., scarring, emphysema), chest CT surveillance may be indicated.
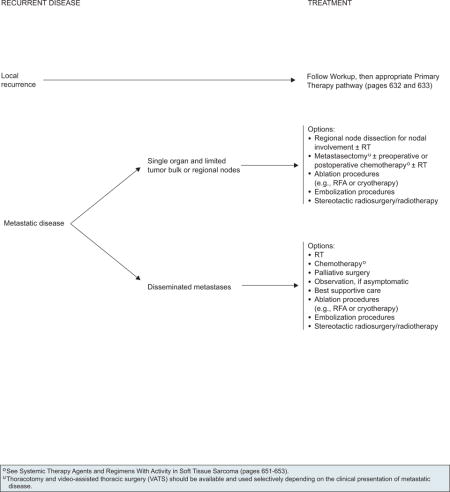
Recurrent Disease
The management of recurrent disease encompasses a heterogeneous group of patients and clinical scenarios (see page 636). For a patient with a local recurrence, treatment decisions should be made using the same algorithm as for patients with a new primary lesion (see page 633).107 For patients who present with metastatic recurrences, the guidelines distinguish between widely disseminated metastases and limited metastases confined to a single organ and the treatment options are similar to those described for stage IV disease at presentation (see page 635).
GISTs
GISTs are the most common mesenchymal neoplasms of the gastrointestinal tract, resulting from activating mutations in one of the receptor protein tyrosine kinases (KIT, also called CD117).128 Most GISTs (95%) are KIT positive. Approximately 5% of GISTs have mutations in the platelet-derived growth factor-alpha (PDGFRA) genes and express little or no KIT.129,130 Therefore, the diagnosis of GIST for a tumor that is otherwise morphologically typical is not precluded by an absence of KIT staining.
GISTs can arise anywhere along the gastrointestinal tract, but the stomach (60%) and small intestine (30%) are the most common primary sites.131,132 Gastric GISTs have a more favorable prognosis than the intestinal ones. Patients with a suspected GIST may present with various symptoms, which may include early satiety, abdominal discomfort from pain or swelling, intraperitoneal hemorrhage, gastrointestinal bleeding, or anemia-related fatigue. Liver metastases and/or dissemination within the abdominal cavity are the most common clinical manifestations of malignancy. Lymph node metastases are extremely rare. Metastases in the lungs and other extra-abdominal locations are observed only in advanced cases.
Targeted Therapy for Patients With GIST
GIST tumors have been documented to be resistant to conventional chemotherapies. Because KIT activation occurs in most GIST cases, KIT-inhibition has emerged as the primary therapeutic treatment along with surgery.
Imatinib Mesylate
Imatinib mesylate, a selective inhibitor of the KIT protein tyrosine kinase, has produced durable clinical benefit and objective antitumor responses in most patients with GIST. Multiple clinical trials worldwide have consistently shown the efficacy of imatinib for patients with GIST. Phase II and III studies have shown high overall response rates, exceptionally good progression-free survival, and objective responses in more than 50% for patients with unresectable and/or metastatic GIST treated with imatinib.133–136 In February 2002, the FDA approved imatinib mesylate for the treatment of patients with KIT-positive unresectable and/or metastatic malignant GIST.137
The presence and type of KIT or PDGFRA mutation status are predictive of response to imatinib therapy in patients with advanced or metastatic GISTs. KIT juxtamembrane domain (exon 11) mutations are the most common mutations in GISTs of all sites, whereas KIT extracellular domain (exon 9) mutations are specific for intestinal GISTs. PDGFRA mutations are common in gastric GISTs, with most affecting exon 18 in the tyrosine kinase domain 2.138
In randomized clinical trials, patients with KIT exon 11 mutations had better response rates and longer progression-free and overall survival than those with KIT exon 9 mutations or no KIT or PDGFRA mutation. In the U.S.-Finnish B2222 phase II trial, the partial response rate was 83.5% for patients with exon 11 mutation compared with 47.8% for those with exon 9 mutations.139 The EORTC-Italian Sarcoma Group-Australasian GI Trials Group phase III trial (EORTC-62005)140 and the North American phase III study SWOG S0033/CALGB 150105141 also confirmed the findings from the B2222 study: that the KIT exon 11 genotype is associated with favorable outcome in patients with advanced GIST compared with KIT exon 9 genotype or wild-type (WT)-GIST.
Two separate phase III trials have assessed the efficacy of imatinib mesylate at 2 initial dose levels (400 mg daily vs. 800 mg daily, given as 400 mg twice a day) in patients with metastatic or unresectable GIST.135,136 Both studies showed equivalent response rates and overall survival for both dose levels; however, higher dose was associated with more side effects in both studies. The EORTC 62005 trial documented an earlier time-to-tumor progression for patients treated with 400 mg daily.135 At a median follow-up of 760 days, 56% of patients assigned to imatinib once daily had experienced progression compared with 50% of those assigned to treatment twice daily. The S0033/CALGB 150105 study reported identical response rates (40% vs. 42%, respectively) at a median follow-up of 4.5 years, and no statistical differences were seen in progression-free survival (18 months for low-dose arm vs. 40 months for higher-dose arm) and median overall survival (55 and 51 months, respectively).136 After progression on 400 mg daily, 33% of patients who crossed over to the higher dose showed objective response rates and stable disease. However, the small advantage in progression-free survival observed for high-dose imatinib in the EORTC 62005 trial was not corroborated by the S0033/CALGB 150105 trial.
Available data confirm the safety and efficacy of imatinib at 400 mg/d as the initial standard dose to achieve response induction. Dose escalation to 800 mg/d is a reasonable option for patients progressing on 400 mg/d. Recent data support the use of imatinib at 800 mg/d in patients with exon 9 mutations and advanced GIST.142 In a randomized EORTC phase III trial, patients whose tumors expressed an exon 9 KIT mutation treated with high-dose imatinib (800 mg/d) had a significantly superior progression-free survival (P = .0013), with a reduction of the relative risk of 61%.143 In the North American Intergroup phase III trial (CALGB 150105), patients with exon 9 mutations treated with 800 mg of imatinib experienced improved response rates compared with those treated with 400 mg (67% vs. 17%, respectively).141 However, the progression-free survival advantage observed in EORTC-62005 among patients with exon 9 KIT mutations treated with high-dose imatinib was not confirmed in the S0033/CALGB 150105 study.
A meta-analysis of 1640 patients from both of these trials showed that treatment with high-dose imatinib (400 mg, twice daily) results in small but significant progression-free survival advantage compared with standard-dose imatinib (400 mg/d).144 This meta-analysis also showed a progression-free survival benefit in patients with KIT exon 9 mutations treated with 800 mg of imatinib.
Preoperative Imatinib
Two randomized phase II studies evaluated the safety and efficacy of preoperative imatinib in patients with primary GISTs or resectable metastatic disease. The RTOG 0132/ACRIN 6665 trial evaluated the efficacy of preoperative imatinib (600 mg/d) in patients with potentially resectable primary disease (30 patients) or potentially resectable recurrent or metastatic disease (22 patients).145 The response rates were 7% partial and 83% stable disease in patients with primary GIST, and 4.5% and 91%, respectively, in those with recurrent or metastatic disease. The estimated overall survival rates for these patients were 93% and 91%, respectively, and the 2-year progression-free survival rates were 83% and 77%, respectively.
In a randomized trial conducted at the MD Anderson Cancer Center, 19 patients undergoing surgical resection were randomized to receive 3, 5, or 7 days of preoperative imatinib (600 mg/d).146 The response rates assessed using FDG-PET and dynamic CT were 69% and 71%, respectively. Median disease-free survival of patients treated with surgery and imatinib was 46 months. Tumor size was a predictor of recurrence after postoperative imatinib.
Although the results of these 2 trials showed the safety and efficacy of preoperative imatinib in patients undergoing surgical resection, survival benefit could not be determined because all patients in both trials received imatinib postoperatively for 2 years. Currently, the decision to use preoperative therapy for patients with resectable primary or locally advanced GIST should be made on an individual basis.
Postoperative Imatinib
Surgery does not routinely cure GIST. Complete resection is possible in approximately 85% of patients with primary tumors. At least 50% of these patients will develop recurrence or metastasis after complete resection, and the 5-year survival rate is approximately 50%.147–149 Median time to recurrence after resection of primary high-risk GIST is approximately 2 years.
The American College of Surgeons Oncology Group (ACOSOG) first evaluated the efficacy of postoperative imatinib in a single-arm, multicenter, phase II intergroup trial involving 106 evaluable patients with primary GIST at high risk for recurrence based on clinicopathologic factors. Patients were treated with 1 year of imatinib at 400 mg/d. In this trial, postoperative imatinib prolonged relapse-free survival after complete resection and was also associated with improved overall survival compared with historical controls.150 In 2002, ACOSOG undertook a phase III, double-blind, randomized trial (Z9001) of postoperative imatinib after resection of primary localized GISTs. In the recently published analysis, 713 patients from 230 sites were randomized either to 400 mg of imatinib (359 patients) or to placebo (354 patients) for 1 year after surgical resection. Median follow-up was 19.7 months, and 67% of patients completed 1 year of adjuvant imatinib treatment. Interim analysis showed that postoperative imatinib after resection of primary GIST improved relapse-free survival,151 which at 1 year was 98% in the imatinib arm vs. 83% in the placebo arm and was statistically different. Overall survival was not different in both arms.
Although the trial was not designed to assess patient subsets, subset analysis showed that relapse-free survival was statistically in favor of the imatinib arm (96% for imatinib vs. 67%–86% for placebo) in patients with high-risk tumors (> 6 cm). However, at this point, the trial results are not conclusive regarding the appropriate duration of treatment, and regarding the effect of imatinib resistance and genetic mutations on the outcome of adjuvant imatinib. Long-term follow-up is ongoing.
Based on the results of ACOSOG Z9001 trial, in December 2008, the FDA approved imatinib for postoperative treatment of adult patients after resection of KIT-positive GIST. Optimum duration of postoperative treatment has not yet been determined.
Management of Toxicities Caused by Imatinib Mesylate
The most common side effects of imatinib include fluid retention, diarrhea, nausea, fatigue, muscle cramps, abdominal pain, and rash. The side-effect profile may improve with prolonged therapy.152 Serious side effects (e.g., lung toxicity, liver function test [LFT] abnormalities, low blood counts, gastrointestinal bleeding) have been reported rarely and often improve after imatinib is withheld. LFT abnormalities are seen in fewer than 5% of patients. Leukopenia is rare and imatinib has been associated with neutropenic fever only rarely. The side-effect profile may improve with prolonged therapy and can be managed with appropriate supportive care measures. If life-threatening side effects occur with imatinib that cannot be managed with maximum supportive treatment, then sunitinib should be considered after imatinib is discontinued.
A recent report described congestive heart failure as a potential side effect of imatinib. However, in a retrospective analysis of 219 consecutive patients treated with imatinib, grade 3 or 4 cardiotoxic occurred in 8.2% of patients, were manageable with medical therapy, and infrequently required dose reduction or discontinuation of imatinib.153 Arrhythmias, acute coronary syndromes, and heart failure were uncommon, occurring in fewer than 1% of treated patients. The authors concluded that imatinib is an uncommon cause of cardiotoxicity, and that the cardiovascular adverse events that occur are manageable when recognized and treated. However, patients on imatinib who present with significant fluid retention should be evaluated carefully.
Imatinib Mesylate Resistance
Imatinib benefits most patients with advanced GIST. However, some patients develop resistance to the drug. Primary resistance is defined as evidence of clinical progression developing during the first 6 months of imatinib therapy and is most commonly seen in patients with KIT exon 9, PDGFRA exon 18, or WT-GIST.154 Secondary resistance seems to be related to the acquisition of new kinase mutations.155 Patients who have been on imatinib for more than 6 months who experienced an initial response and then progression are categorized as having secondary resistance, which develops predominantly in patients who have secondary mutations in KIT exon 11.156 Imatinib resistance can be managed either with dose escalation or switching to sunitinib.
Sunitinib Malate
Sunitinib malate (previously known as SU11248) is a multi-targeted tyrosine kinase inhibitor (TKI) that can induce objective responses and control progressive disease in patients with imatinib-resistant GIST.
In a recent randomized placebo-controlled phase III trial, sunitinib produced significant and sustained clinical benefit in patients with imatinib-resistant or -intolerant GIST.157 In patients with imatinib-resistant GIST, sunitinib was associated with a significant improvement in median time to progression (27.3 vs. 6.4 weeks) and significantly greater estimated overall survival. Sunitinib treatment induced partial response in 14 patients (6.8%) and stable disease (≥ 22 weeks) in 36 patients (17.4%), compared with no partial responses and stable disease in 2 patients (1.9%) on placebo. In the imatinib-intolerant group, 4 of 9 patients randomized to sunitinib experienced partial response, with progressive disease in only one. In contrast, 3 of the 4 patients randomized to placebo had progressive disease at analysis and no partial response was observed. Sunitinib therapy was generally well tolerated. In January 2006, sunitinib malate received FDA approval for the treatment of GIST in patients who experience disease progression on or are intolerant to imatinib mesylate.
The safety and efficacy of sunitinib on a continuous daily dosing schedule at 37.5 mg was evaluated in an open-label, multicenter, randomized phase II study in patients with advanced GIST after imatinib failure.158 Patients were randomized (1:1) to receive continuous daily sunitinib (37.5 mg/d) in either the morning or evening for 28 days (1 cycle). The primary end point was the clinical benefit rate (CBR), defined as the percentage of patients with complete responses, partial responses, or stable disease for 24 weeks or more based on RECIST.
The overall CBR was 53% (13% had partial responses and 40% had stable disease). Median progression-free and overall survivals were 34 and 107 weeks, respectively. The most commonly reported treatment-related adverse events (diarrhea, fatigue, and nausea) were consistent with those known to be associated with intermittent dosing of sunitinib. Treatment-related hypertension and hypothyroidism experienced by 28% and 12% of patients, respectively, were successfully managed with appropriate supportive care measures. These adverse events have also been associated with long-term use of sunitinib at intermittent dosing. Results of this study suggest that continuous daily dosing seems to be an effective alternative dosing strategy with acceptable safety for patients with imatinib-resistant/intolerant GIST.
Management of Toxicities Caused by Sunitinib Malate
Sunitinib-related toxicities can often be managed with dose interruptions or reductions. Fatigue, nausea, and vomiting were dose-limiting toxicities for sunitinib in clinical trials. Other common toxicities include hematologic toxicities (anemia, neutropenia), diarrhea, abdominal pain, mucositis, anorexia, and skin discoloration. Sunitinib is associated with a significant risk for developing hand–foot skin reaction (HFSR).159 Early detection and proper management of HFSR is vital during treatment with sunitinib. HFSR can be prevented with routine application of emollient lotions, but if significant, interruption of therapy is indicated, and if severe, dose reduction should be considered.
Hypertension is a common side effect reported in clinical trials, because sunitinib targets vascular endothelial growth factor receptor. However, the risk is higher in patients with renal cell carcinoma than in those with non–renal cell carcinoma.160 Recent reports have shown that sunitinib is also associated with cardiotoxicity and hypothyroidism.161,162 In a retrospective analysis of the data from a phase I/II trial, 11% of patients experienced an adverse cardiovascular event, including congestive heart failure in 8% of patients and absolute reduction in the left ventricular ejection fraction in 28%.161 In a prospective, observational cohort study, abnormal serum thyroid stimulating hormone concentrations were documented in 62% of patients, and the risk for hypothyroidism increased with the duration of therapy.162
Close monitoring for hypertension and left ventricular ejection fraction is essential in patients receiving sunitinib, especially those with a history of heart disease or cardiac risk factors. Routine monitoring (every 3–6 months) of thyroid stimulating hormone concentration is indicated. If hypothyroidism is suggested, patients should receive thyroid hormone replacement therapy. Patients should monitor their blood pressure closely, and those who experience an increase in blood pressure should be treated with antihypertensives.
Principles of Biopsy and Pathologic Assessment
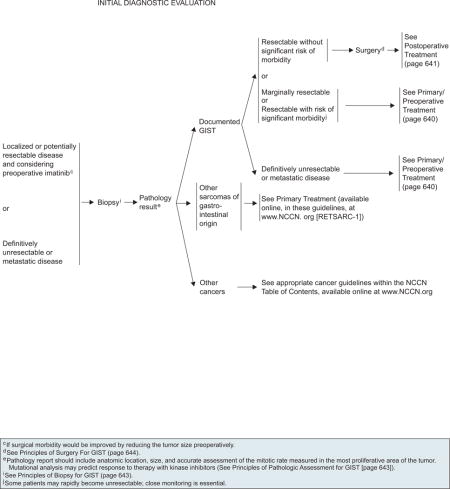
GISTs are soft and fragile, and biopsy may cause tumor hemorrhage and possibly increased risk for tumor dissemination. The decision to obtain a biopsy should be based on the extent of disease and the clinician’s degree of suspicion of other malignancies. Biopsy may not be necessary if the tumor is easily resectable and preoperative therapy is not required. However, biopsy should be performed if preoperative therapy is being considered for unresectable or marginally resectable tumors (see page 643). Endoscopic ultrasound (EUS) biopsy is preferred over percutaneous. Recent reports have suggested that definitive diagnosis of GIST requires tissue acquisition through EUS-guided FNA.163
Morphologic diagnosis based on careful microscopic examination of adequate tumor tissue is essential to confirm the diagnosis of GIST. The pathology report should include anatomic location, size, and an accurate assessment of the mitotic rate measured in the most proliferative area of the tumor and reported as the number of mitoses in 50 high power fields (see page 643). The differential diagnosis of GIST should be considered for any gastrointestinal sarcoma and for any other intra-abdominal sarcoma. The panel recommends referral to centers with expertise in sarcomas for cases with complex or unusual histopathologic features. Immunohistochemical staining for KIT and molecular genetic testing to identify mutations in the KIT or PDGFRA genes are useful in diagnosing GIST (page 643). However, 10% to 15% of GISTs have no detectable KIT or PDGFRA mutations (WT) GIST. The absence of mutations does not exclude the diagnosis of GIST. Results of a phase III study showed that patients with CD117-negative GIST have similar time-to-tumor progression but inferior overall survival compared with those with CD117-positive GIST, suggesting that patients with CD117-negative GIST may benefit from imatinib therapy.139 Therefore, it is rational to offer patients with KIT-negative GIST a therapeutic trial of imatinib mesylate with close evaluation and follow-up.
Principles of Surgery for GIST
Surgery is the preferred primary treatment for patients with localized or potentially resectable GIST lesions. Although imatinib is the primary therapy for patients with metastatic GIST, surgery may be indicated for locally advanced or previously unresectable disease after a favorable response to preoperative imatinib and for limited disease progression on systemic therapy (see page 644).
GISTs are fragile and should be handled with care to avoid tumor rupture. The goal is to achieve complete gross resection of the tumor with an intact pseudocapsule. After removal of any suspected GIST, postoperative pathology assessment is essential to confirm the diagnosis. Segmented or wedge resection to obtain histologically negative margins is often appropriate (see page 644). Lymphadenectomy is usually not required given the low incidences of nodal metastases. Resection should be accomplished with minimal morbidity, and complex multivisceral resection should be avoided. If the surgeon believes a complex surgical procedure is required, then a multidisciplinary consultation on the use of preoperative imatinib is recommended. Sphincter- and esophagus-sparing surgery should be considered for rectal and gastroesophageal junction GISTs, respectively. If abdominoperineal resection would be necessary to achieve a negative margin, then preoperative imatinib should be considered.
The role of laparoscopy in the resection of GISTs continues to expand. Although prospective trials are lacking, literature reports based on small series of patients and retrospective analyses have shown that laparoscopic or laparoscopic-assisted resections are not only possible, but are also associated with low recurrence rates, short hospital stay, and low morbidity. Laparoscopic approach may be considered for selected GISTs in favorable anatomic locations, such as anterior wall of the stomach, jejunum, and ileum. The same surgical principles of complete macroscopic resection, including preservation of the pseudocapsule and avoidance of tumor rupture, should be followed during laparoscopy (page 644). Resection specimen should be removed from the abdomen in a plastic bag to avoid spillage or seeding of port sites. Laparoscopic surgery could be feasible in other anatomic sites, such as smaller rectal GISTs. However, data on laparoscopic resection of GISTs at other sites are limited.
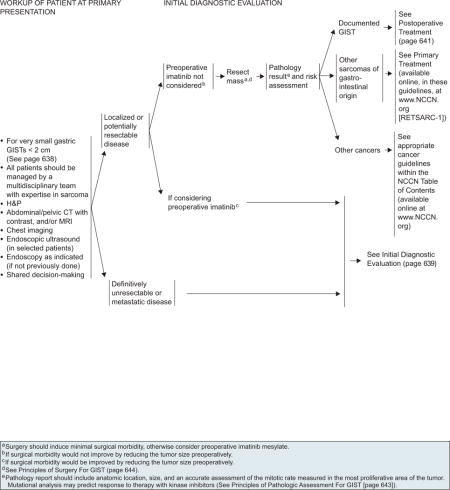
Initial Evaluation and Workup
All patients should be managed by a multidisciplinary team with expertise in sarcoma. Essential elements of the workup include the H&P, abdominal/pelvic CT scan with contrast and/or MRI, chest imaging, EUS in selected patients, endoscopy as indicated (if not performed previously), and surgical assessment (see page 637).
In patients with GIST, imaging is used for diagnosis, initial staging, restaging, monitoring response to therapy, and performing follow-up surveillance of possible recurrence. Imaging studies can include CT, MRI, and PET. Contrast-enhanced CT is the preferred imaging modality to characterize an abdominal mass and to evaluate its extent and the presence or absence of metastasis at the initial staging workup for biopsy-proven GIST. PET scan helps differentiate active tumor from necrotic or inactive scar tissue, malignant from benign tissue, and recurrent tumor from nondescript benign changes. PET provides significant value to the standard CT images, because changes in the metabolic activity of tumors often precede anatomic changes on CT. However, PET scan is not a substitute for CT. PET scans may be used to clarify ambiguous findings seen on CT or MRI. Many imaging centers are also equipped with combined PET-CT scanners, which may facilitate both anatomic and functional tumor evaluation in one step.164 If clinicians consider using PET scan to monitor therapy, a baseline PET should be obtained before the start of therapy.
Resectable Disease
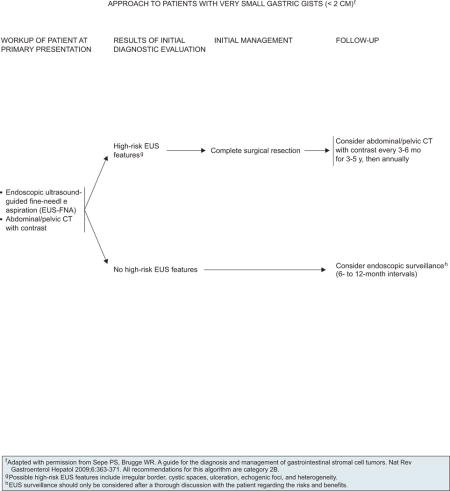
Surgery is the primary treatment for all patients with resectable GISTs of 2 cm or greater who have no significant risk for morbidity. However, the management of incidentally encountered small GISTs smaller than 2 cm remains controversial. Currently, data are insufficient to guide the management of very small GISTs (< 2 cm) discovered incidentally on endoscopy, and the usefulness of regular EUS surveillance remains unestablished. Complete surgical resection is the mainstay of treatment in symptomatic patients. For a subset of patients with very small gastric GISTs (< 2 cm) with no high-risk EUS features (irregular extraluminal border, heterogeneous echo pattern, presence of cystic spaces, and echogenic foci), endoscopic surveillance at 6- to 12-month intervals may be considered (see page 638). The panel included this approach with a category 2B recommendation.
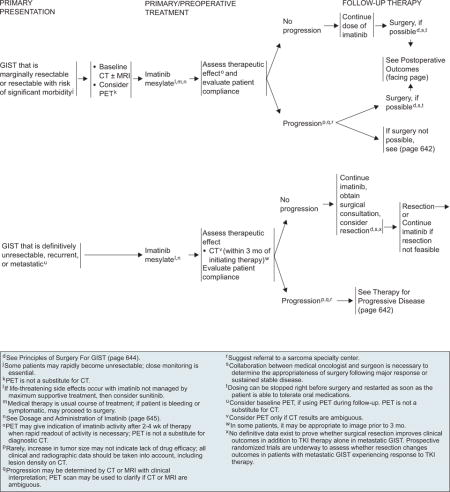
Patients with marginally resectable or resectable GIST with a significant risk for morbidity should be considered for preoperative imatinib before resection, if surgical morbidity would be improved by reducing the size of the tumor. However, close monitoring is essential, because some patients may become unresectable rapidly. Surgery is recommended if bleeding and/or symptoms are present. Baseline CT with or without MRI is recommended before the start of preoperative imatinib (see page 640). Because the optimal duration of preoperative therapy remains unknown, imatinib should be continued in patients experiencing response to therapy until maximal response (defined as no further improvement between 2 successive CT scans, which can take as long as 6–12 months). However, waiting for a maximal response before performing surgery is not always necessary. If no progression occurs, resection should be considered, if possible. If progression occurs, confirmed with CT scan, surgery is recommended after discontinuing imatinib (see page 640). Collaboration between the medical oncologist and surgeon is necessary to determine the appropriateness of surgery after major response or stable disease.
Metastatic, Unresectable, or Recurrent Disease
Advanced, unresectable, or metastatic GIST has a very high likelihood of clinical benefit and positive response after treatment with imatinib. Patients with a documented unresectable GIST, who would be at risk for severe functional deficit after resection, or who have widespread metastatic disease should be treated with imatinib mesylate in the preoperative setting (see page 640). Patients should be assessed within 3 months of initiating therapy to determine if their GIST has become resectable. In selected patients, imaging can be done before 3 months. If no progression occurs, resection can be considered after surgical consultation. Several studies have evaluated the impact of cytoreductive surgery on survival in patients with advanced GIST after treatment with imatinib. No definitive data exist to prove whether surgical resection improves clinical outcome in addition to TKI therapy for patients with resectable metastatic GIST. Prospective phase III trials are underway to assess whether resection changes outcome in patients with unresectable metastatic GIST responding to TKI therapy.
Imatinib therapy should be continued if resection is not feasible. Currently, continuous use of imatinib is recommended for metastatic GIST until progression. Patients should be maintained on the same dose, with no increase if they remain stable without objective progression of the disease. Termination of imatinib therapy in patients with GIST that is refractory to imatinib has been shown to result in a flare phenomenon, which in turn indicates that even in patients with progressive disease on imatinib therapy, imatinib may still be effective for some tumor cells.165 Updated results from a randomized phase III trial by a French sarcoma group showed a significant increase in the rate of progressive disease when imatinib therapy was interrupted in patients with advanced GIST that was stable or responding to treatment.166
Recurrence after complete resection should be managed as described for unresectable or metastatic disease, because recurrent disease represents locoregional metastatic or infiltrative spread of the malignancy and has essentially the same prognosis as distant metastases overall.
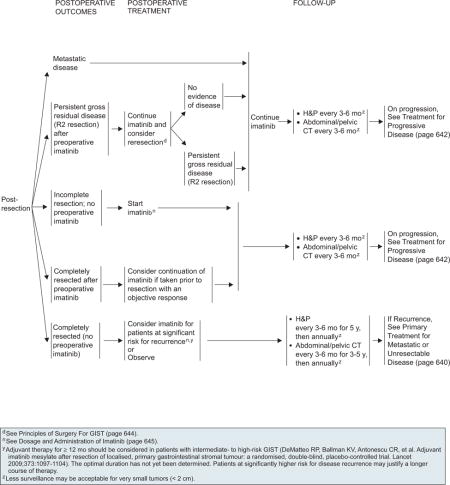
Postoperative Treatment
In patients taking preoperative imatinib, dosing can be stopped immediately before surgery and resumed as soon as the patient is able to tolerate oral medications after surgery regardless of surgical margins. If persistent gross disease is present after resection, additional resection may be considered to remove residual disease. Imatinib treatment should be continued after re-resection regardless of surgical margins until progression occurs. Postoperative imatinib should be initiated after resection in patients who did not undergo prior imatinib therapy.
The panel has included postoperative imatinib as an alternative to observation for patients at significant risk for recurrence who have undergone complete resection for primary GIST (see page 641). Optimum duration of postoperative treatment has not yet been determined. Postoperative imatinib is recommended for at least 12 months in patients with intermediate- to high-risk GIST. Higher-risk patients may require longer treatment.
Risk stratification after surgical resection should be based on tumor mitotic rate, size, and location.167 Gold et al.168 developed a nomogram that takes into account tumor size, site, and mitotic index for predicting relapse-free survival after resection of localized primary GIST. This nomogram accurately predicts relapse-free survival after resection of localized primary GIST and might be useful for patient care, interpretation of trial results, and selection of patients for postoperative imatinib therapy.
Progressive Disease
Progression is defined as the appearance of a new lesion or an increase in tumor size. It may be determined using CT or MRI with clinical interpretation; PET may be used if the results are ambiguous (see page 642). For patients with limited progressive disease or with widespread systemic disease and good performance status (0–2), options include continuation of imatinib at the same dose, dose escalation as tolerated, or switching to sunitinib. Patients with limited progression should not be switched to sunitinib if most of the disease is still controlled by imatinib. Before dose escalation, all clinical and radiologic data, including lesion density on CT, should be taken into account. Patient compliance to imatinib therapy at standard dose should be assessed before altering the dose of imatinib or switching to sunitinib. For limited progressive disease that is potentially easily resectable, surgical resection should be considered.169 Other treatment options include radiofrequency ablation or embolization (category 2B). RT (category 2B) for palliation can be considered for rare patients with bone metastases.
Options are limited for patients progressing on imatinib and sunitinib. Second-generation TKIs, such as sorafenib, dasatinib, and nilotinib, have shown activity in patients with imatinib- and sunitinib-resistant GIST. In a multicenter, ongoing phase II study involving patients with unresectable, KIT-positive GIST that had progressed on imatinib and sunitinib, 58% of patients who received sorafenib had stable disease.170 Median progression-free survival was 5.3 months, and the estimated 1-year survival rate was 62%.
In another phase I trial, nilotinib alone and in combination with imatinib showed significant activity in patients with GIST resistant to prior treatment with TKIs.171 In a phase I dose-escalation study, 3 of 19 patients with refractory GIST had stable disease, which lasted more than 3 months in 1 patient.172 The Sarcoma Alliance for Research through Collaboration is completing a phase II multi-arm study of dasatinib in patients with imatinib- and sunitinib-refractory GIST. The efficacy and safety of nilotinib as a third-line therapy for GIST are being studied in an ongoing phase III trial.
The guidelines include sorafenib, dasatinib, or nilotinib as options for patients who are no longer experiencing clinical benefit from imatinib or sunitinib (see pages 651–653). Any patient who experiences progression of GIST despite prior therapy or who experiences a recurrence regardless of presentation should be considered for enrollment in a clinical trial, if an appropriate trial is available. Recent data reported by Fumagalli et al.173 support rechallenging patients with imatinib after standard and investigational therapeutic options fail. In patients with progressive disease no longer receiving benefit from current TKI therapy, consider reintroduction of previously tolerated and effective TKI therapy for palliation of symptoms. The panel also feels that continuation of TKI therapy life-long for palliation of symptoms should be an essential component of best supportive care.
Surveillance
Every patient with a resected localized GIST should have a thorough H&P every 3 to 6 months; these patients should also have an abdominopelvic CT scan every 3 to 6 months. An identical schedule is used for patients who have persistent gross residual disease that is unresectable or for completely resected disease.
Guidelines Online
Information on retroperitoneal intra-abdominal soft tissue sarcomas and desmoid tumors (fibromatoses) can be found in the full soft tissue sarcoma guidelines, available online, at www.NCCN.org.
Acknowledgments
At the beginning of each NCCN Guidelines panel meeting, panel members disclosed any financial support they have received from industry. Through 2008, this information was published in an aggregate statement in JNCCN and online. Furthering NCCN’s commitment to public transparency, this disclosure process has now been expanded by listing all potential conflicts of interest respective to each individual expert panel member.
NCCN Categories of Evidence and Consensus
Category 1: The recommendation is based on high-level evidence (e.g., randomized controlled trials) and there is uniform NCCN consensus.
Category 2A: The recommendation is based on lower-level evidence and there is uniform NCCN consensus.
Category 2B: The recommendation is based on lower-level evidence and there is nonuniform NCCN consensus (but no major disagreement).
Category 3: The recommendation is based on any level of evidence but reflects major disagreement.
All recommendations are category 2A unless otherwise noted.
The full NCCN Clinical Practice Guidelines in Oncology: Soft Tissue Sarcoma are not printed in this issue of JNCCN, but can be accessed online at www.NCCN.org.
Clinical trials: The NCCN believes that the best management for any cancer patient is in a clinical trial. Participation in clinical trials is especially encouraged.
Please Note
The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines™) are a statement of consensus of the authors regarding their views of currently accepted approaches to treatment. Any clinician seeking to apply or consult these guidelines is expected to use independent medical judgment in the context of individual clinical circumstances to determine any patient’s care or treatment. The National Comprehensive Cancer Network® (NCCN®) makes no representation or warranties of any kind regarding their content, use, or application and disclaims any responsibility for their applications or use in any way.
Disclosures for the NCCN Soft Tissue Sarcoma Guidelines Panel
Individual disclosures for the NCCN Soft Tissue Sarcoma Guidelines Panel members can be found on page 674. (The most recent version of these guidelines and accompanying disclosures, including levels of compensation, are available on the NCCN Web site at www.NCCN.org.)
These guidelines are also available on the Internet. For the latest update, please visit www.NCCN.org.
NCCN Soft Tissue Sarcoma Panel Members
*George D. Demetri, MD/Chair†
Dana-Farber Cancer Institute|Harvard Cancer Center
Scott Antonia, MD, PhD†
H. Lee Moffitt Cancer Center & Research Institute
Robert S. Benjamin, MD†
The University of Texas MD Anderson Cancer Center
Marilyn M. Bui, MD, PhD≠
H. Lee Moffitt Cancer Center & Research Institute
Ephraim S. Casper, MD†Þ
Memorial Sloan-Kettering Cancer Center
Ernest U. Conrad III, MD¶π
University of Washington/Seattle Cancer Care Alliance
*Thomas F. DeLaney, MD§
Massachusetts General Hospital Cancer Center
Kristen N. Ganjoo, MD†
Stanford Comprehensive Cancer Center
Martin J. Heslin, MD¶
University of Alabama at Birmingham, Comprehensive Cancer Center
Raymond J. Hutchinson, MD€ξ
University of Michigan Comprehensive Cancer Center
*John M. Kane III, MD¶
Roswell Park Cancer Institute
G. Douglas Letson, MD¶
H. Lee Moffitt Cancer Center & Research Institute
Sean V. McGarry, MDπ
UNMC Eppley Cancer Center at The Nebraska Medical Center
Richard J. O’Donnell, MD¶
UCSF Helen Diller Family Comprehensive Cancer Center
I. Benjamin Paz, MD¶
City of Hope Comprehensive Cancer Center
John D. Pfeifer, MD, PhD≠
Siteman Cancer Center at Barnes-Jewish Hospital and Washington University School of Medicine
Raphael E. Pollock, MD¶
The University of Texas MD Anderson Cancer Center
R. Lor Randall, MD¶
Huntsman Cancer Institute at the University of Utah
Richard F. Riedel, MD†
Duke Comprehensive Cancer Center
Karen D. Schupak, MD§
Memorial Sloan-Kettering Cancer Center
Herbert S. Schwartz, MD¶
Vanderbilt-Ingram Cancer Center
Katherine Thornton, MD†
The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
*Margaret von Mehren, MD†
Fox Chase Cancer Center
Jeffrey Wayne, MD¶
Robert H. Lurie Comprehensive Cancer Center of Northwestern University
KEY:
*Writing Committee Member
Specialties: †Medical Oncology; ≠Pathology; ÞInternal Medicine; ¶Surgery/Surgical Oncology; πOrthopedics/Orthopedic Oncology; §Radiotherapy/Radiation Oncology; €Pediatric Oncology; ξBone Marrow Transplantation




PRINCIPLES OF BIOPSY FOR GIST
GISTs are soft, fragile tumors. Biopsy may cause tumor hemorrhage and possibly increased risk for tumor dissemination.
Consideration of biopsy should be based upon the extent of disease and suspicion of a given histologic subtype (e.g., lymphoma). EUS biopsy is preferred over percutaneous biopsy.
Biopsy is generally necessary when planning preoperative therapy for primary GIST.
Diagnosis is based on the Principles of Pathologic Assessment of Sarcoma Specimens (page 646); referrals to centers with expertise in sarcoma diagnosis is recommended for cases with complex or unusual histopathologic features.
PRINCIPLES OF PATHOLOGIC ASSESSMENT FOR GIST
Pathologic assessment should follow the guidelines outlined on page 646.
Morphologic diagnosis based on microscopic examination of histologic sections is the standard for GIST diagnosis. Several ancillary techniques are useful in support of GIST diagnosis, including immunohistochemistry (95% express CD117 and 80% express CD34) and molecular genetic testing (for mutations in KIT or PDGFRA). Referral to centers with expertise in sarcoma diagnosis is recommended for cases with complex or unusual histopathologic features.
Tumor size and mitotic rate are used as guides to predict the malignant potential of GISTs, although it is notoriously difficult to predict the biologic potential of individual cases. The mitotic rate should be measured in the most proliferative area of the tumor, and reported as the number of mitoses in 50 high power (400x total magnification) fields.
Approximately 80% of GISTs have a mutation in the gene encoding the KIT receptor tyrosine kinase; another 5% to 10% of GISTs have a mutation in the gene encoding the related PDGFRA receptor tyrosine kinase. Because approximately 10% to 15% of GISTs have no detectable KIT or PDGFRA mutation, the absence of a mutation does not exclude the diagnosis of GIST. The presence and type of KIT and PDGFRA mutations are not strongly correlated with prognosis.
The mutations in KIT and PDGFRA in GIST result in expression of mutant proteins with constitutive tyrosine kinase activity.
PRINCIPLES OF SURGERY FOR GIST
Primary (Resectable) GIST
The surgical procedure performed should aim to resect the tumor with histologically negative margins.
Given the limited intramural extension, extended anatomic resections (such as total gastrectomy) are rarely indicated. Segmental or wedge resection to obtain negative margins is often appropriate.
Lymphadenectomy is usually not required given the low incidence of nodal metastases.
As GIST tends to be very friable, every effort should be made not to violate the pseudocapsule of the tumor.
Re-resection is generally not indicated for microscopically positive margins on final pathology.
Resection should be accomplished with minimal morbidity and, in general, complex multi-visceral resection should be avoided. If the surgeon feels that a multi-visceral resection may be required, then multidisciplinary consultation is indicated regarding a course of preoperative imatinib therapy. Similarly, rectal GIST should be approached using a sphincter-sparing approach. If abdominoperineal resection (APR) would be necessary to achieve a negative margin resection, then preoperative imatinib therapy should be considered.
A laparoscopic approach may be considered for select GISTs in favorable anatomic locations (greater curvature or anterior wall of the stomach, jejunum, and ileum) by surgeons with appropriate laparoscopic experience.
All oncologic principles of GIST resection must still be followed, including preservation of the pseudocapsule and avoidance of tumor spillage.
Resection specimens should be removed from the abdomen in a plastic bag to prevent spillage or seeding of port sites.
Metastatic GIST
Imatinib is the primary therapy for metastatic GIST. Surgery may be indicated for:
Limited disease progression refractory to systemic therapy.
Locally advanced or previously unresectable tumors after a favorable response to preoperative imatinib.
If persistent metastatic or residual tumor remains after surgery, then imatinib should be continued as soon as the patient is able to tolerate oral intake.
DOSING AND ADMINISTRATION OF IMATINIB1
Unresectable and/or Metastatic GIST
Initiate dosing at 400 mg daily. Patients with documented mutations in KIT exon 9 may benefit from dose escalation up to 800 mg daily (given as 400 mg, twice daily), depending on tolerance.
IF PROGRESSION OF DISEASE IS DOCUMENTED: Imatinib dose increase up to 800 mg daily (given as 400 mg, twice daily) may be considered, as clinically tolerated, in patients showing objective signs of disease progression at a lower dose and in the absence of severe adverse drug reactions.
Adjuvant Treatment After Complete Gross Resection of GIST
400 mg daily. In the randomized clinical study ACOSOG Z9001, imatinib was administered for 1 year, and patients at highest risk for recurrence showed increased rate of recurrence after discontinuation of drug dosing. The optimal duration of adjuvant treatment is not known.
DOSING AND ADMINISTRATION OF SUNITINIB1
- The recommended dose of sunitinib is either:
-
➤37.5 mg orally once daily without interruption or
-
➤50 mg orally once daily on a schedule of 4 weeks on treatment followed by 2 weeks off (schedule 4/2).
-
➤
- In patients receiving sunitinib, selection of an alternate concomitant medication with no or minimal enzyme induction potential is recommended. Sunitinib dose modification is recommended in patients who must receive concomitant CYP3A4 inhibitors or inducers.
-
➤A dose reduction for sunitinib to a minimum of 37.5 mg daily should be considered if sunitinib must be coadministered with a strong CYP3A4 inhibitor.
-
➤A dose increase for sunitinib to a maximum of 87.5 mg daily should be considered if sunitinib must be co-administered with a CYP3A4 inducer. According to the package insert, in vitro studies indicate that sunitinib does not induce or inhibit major cytochrome enzymes.
-
➤
Sunitinib may be taken with or without food.
PRINCIPLES OF PATHOLOGIC ASSESSMENT OF SARCOMA SPECIMENS
Pathologic assessment of biopsies and resection specimens should be carried out by an experienced sarcoma pathologist.
Morphologic diagnosis based on microscopic examination of histologic sections remains the gold standard for sarcoma diagnosis. However, because several ancillary techniques are useful in support of morphologic diagnosis (including immunohistochemistry, classical cytogenetics, and molecular genetic testing), sarcoma diagnosis should be performed by pathologists who have access to these ancillary methods.1
- The pathologic assessment should include evaluation of the following features, all of which should be specifically addressed in the pathology report:
-
➤Organ, site, and operative procedure
-
➤Primary diagnosis (using standardized nomenclature, such as provided in the World Health Organization Classification of Soft Tissue Tumors2).
-
➤Depth of tumor
-
◊Superficial (tumor does not involve the superficial fascia)
-
◊Deep
-
◊
-
➤Size of tumor
-
➤Histologic grade (at the least, specify low or high grade, if applicable); ideally, grade using the French Federation of Cancer Centers Sarcoma Group or National Cancer Institute system
-
➤Necrosis
-
◊Present or absent
-
◊Microscopic or macroscopic
-
◊Approximate extent (percentage)
-
◊
-
➤Status of margins of excision
-
◊Uninvolved
-
◊Closer than 2 cm (state which margins and measured distance)
-
◊Involved (state which margins)
-
◊
-
➤Status of lymph nodes
-
◊Site
-
◊Number examined
-
◊Number positive
-
◊
-
➤Results of ancillary studies1
-
◊Type of testing (electron microscopy, immunohistochemistry, molecular genetic analysis)
-
◊Where performed
-
◊
-
➤Additional tumor features
-
◊Mitotic rate
-
◊Presence or absence of vascular invasion
-
◊Character of tumor margin (well circumscribed or infiltrative)
-
◊Inflammatory infiltrate (type and extent)
-
◊
-
➤TNM Stage (see the staging table, available online, in these guidelines, at www.NCCN.org [ST-1])
-
➤
PRINCIPLES OF ANCILLARY TECHNIQUES USEFUL IN THE DIAGNOSIS OF SARCOMAS
Morphologic diagnosis based on microscopic examination of histologic sections remains the gold standard for sarcoma diagnosis. However, several ancillary techniques are useful in support of morphologic diagnosis, including immunohistochemistry, classical cytogenetics, and molecular genetic testing. Molecular genetic testing has emerged as a particularly powerful ancillary testing approach because many sarcoma types harbor characteristic genetic aberrations, including single base-pair substitutions, deletions and amplifications, and translocations. Most molecular testing uses fluorescence in situ hybridization (FISH) approaches or polymerase chain reaction (PCR)-based methods.1 Recurrent genetic aberrations in sarcoma2 are listed below:
| TUMOR | ABERRATION | GENES INVOLVED |
|---|---|---|
| Malignant Round Cell Tumors | ||
| Ewing's sarcoma/peripheral neuroectodermal tumor | t(11;22)(q24;q12) t(21;22)(q22;q12) other rare variants |
EWS-FLI1 EWS-ERG various |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWS-WT1 |
| Embryonal rhabdomyosarcoma | Complex alterations | Unknown |
| Alveolar rhabdomyosarcoma | t(2;13)(q35;q14) t(1;13)(p36;q14) |
PAX3-FKHR PAX7-FKHR |
|
| ||
| Lipomatous Tumors | ||
| Myxoid/round cell liposarcoma | t(12;16)(q13;p11) | TLS-CHOP |
| Atypical lipomatous tumor/well-differentiated liposarcoma (ALT/WDLPS) | Supernumerary ring chromosomes; giant marker chromosomes | Amplification of region 12q14-15, including MDM2, CDK4, HMGA2, SAS, GL1 |
| Dedifferentiated liposarcoma | Same as for ALT/WDLPS | Same as for ALT/WDLPS |
| Pleomorphic liposarcoma | Complex alterations | Unknown |
|
| ||
| Other Sarcomas | ||
| Alveolar soft part sarcoma | der(17)(X;17)(p11;q25) | ASPL-TFE3 |
| Angiomatoid fibrous histiocytoma | t(2;22)(q33;q12) t(12;16)(q13;p11) |
EWS-CREB1 TLS-ATF1 |
| Clear cell sarcoma | t(12;22)(q13;q12) t(2;22)(q33;q12) |
EWS-ATF1 EWS-CREB1 |
| Congenital/infantile – fibrosarcoma | t(12;15)(p13;q25) | ETV6-NTRK3 |
| Dermatofibrosarcoma protuberans | t(17;22)(q22;q13) and derivative ring chromosomes | COL1A1-PDGFB |
| Desmoid fibromatosis | Trisomy 8 or 20; loss of 5q | CTNNB1 or APC mutations |
| Epithelioid sarcoma (proximal type) | Bi-allelic inactivation of 22q11.2 | INI1 |
| Extrarenal rhabdoid tumor | Bi-allelic inactivation of 22q11.2 | INI1 |
| Extraskeletal myxoid chondrosarcoma | Rearrangements of 9q22 | CHN |
| Sporadic GIST Familial GIST (Carney-Stratakis syndrome) |
Activating kinase
mutations KREBS cycle mutation |
KIT or
PDGFRA SDH subunit mutations |
| Inflammatory myofibroblastic tumor | Rearrangements of 2p23 | ALK |
| Leiomyosarcoma | Complex alterations | Unknown |
| Low-grade fibromyxoid sarcoma | t(7;16)(q34;p11) | TLS-BBF2H7 |
| Malignant peripheral nerve sheath tumor | Complex alterations | Unknown |
| Synovial sarcoma | t(X;18)(p11;q11) t(X;18)(p11;q11) |
SYT-SSX1 SYT-SSX2 |
| Tenosynovial giant cell tumor/pigmented villonodular synovitis (TGCT/PVNS) | t(1;2)(p13;q35) | CSF1 |
PRINCIPLES OF SURGERY
Biopsy of Sarcoma
A pretreatment biopsy to diagnose and grade a sarcoma is highly preferred. Biopsy should be performed by an experienced surgeon (or radiologist) and may be accomplished by open incisional or needle technique. Endoscopic or needle biopsy may be indicated for deep, thoracic, abdominal, or pelvic sarcomas.
Sarcoma Surgery
The surgical procedure necessary to resect the tumor with appropriately negative margins should be used. Close margins may be necessary to preserve uninvolved critical neurovascular structures, bones, joints, etc. Ideally, the biopsy site should be excised en bloc with the definitive surgical specimen. Dissection should be through grossly normal tissue planes uncontaminated by tumor. If the tumor is close to or displaces major vessels or nerves, these need not be resected if the adventitia or perineurium is removed and the underlying neurovascular structures are not involved with gross tumor. Radical excision/entire anatomic compartment resection is not routinely necessary. Surgical clips should be placed to mark the periphery of the surgical field and other relevant structures to help guide potential future radiation therapy. If closed suction drainage is used, the drains should exit the skin close to the edge of the surgical incision (in case re-resection or radiation is indicated).
Resection Margins
- Surgical margins should be documented by both the surgeon and pathologist when evaluating a resected specimen. If surgical resection margins are positive on final pathology (other than bone, nerve, or major blood vessels), surgical re-resection to obtain negative margins should be strongly considered if it will not have a significant impact on functionality. Consideration for adjuvant radiation therapy should be given for a close (< 1 cm) soft tissue margin or a microscopically positive margin on bone, major blood vessels, or a major nerve.
-
➤R0 resection - No residual microscopic disease
-
➤R1 resection - Microscopic residual disease
-
➤R2 resection - Gross residual disease
-
➤
Limb Salvage Surgery
For extremity sarcomas, the goal of surgery should be functional limb preservation, if possible, within the realm of an appropriate oncologic resection.
Amputation
Before considering amputation, patients should be evaluated by a surgeon with expertise in the treatment of soft tissue sarcomas. Amputation to treat an extremity sarcoma should be considered based on patient preference or if gross total resection of the tumor is expected to render the limb nonfunctional.
Evaluate postoperative rehabilitation (PT, OT) for patients with extremity sarcoma. Continue rehabilitation until maximal function is achieved.
GUIDELINES FOR RADIATION THERAPY1

| Extremity, Retroperitoneal, Intra-abdominal | Angiosarcoma | Desmoid Tumors (Fibromatosis) | GIST | |
|
Combination
regimens • AD (doxorubicin. dacarbazine)1,2 • AIM (doxorubicin. ifosfamide, mesna)3,4 • MAID (mesna, doxorubicin, ifosfamide, dacarbazine)5 • Ifosfamide, epirubicin, mesna6 • Gemcitabine and docetaxel7.8 • Gemcitabine and vinorelbine9 |
Single
agents • Doxorubicin10 • Ifosfamide6,11 • Epirubicin • Gemcitabine • Dacarbazine • Liposomal doxorubicin12 • Temozolomide13 |
• Paclitaxel14,15 • Docetaxel • Vinorelbine • Sorafenib16,17 • Sunitinib18 • Bevacizumab19 • All other systemic therapy options as per extremity sarcoma |
• Sulindac20 or other
non-steroidal anti-inflammatory druas (NSAIDs) including
celecoxibc • Tamoxifen21 • Toremifene22 • Methotrexate and vinblastine23 • Low-dose interferon24 • Doxorubicin-based regimens25,26 • Imatinib mesylate27 |
• Imatinib28,29 • Sunitinib30 • Sorafenib31 • Nilotinib32,33 • Dasatanib34 |
|
| ||||
|
Solitary
Fibrous
Tumor/Hemangiopericytoma • Bevacizumab and temozolomide35 • Sunitinib36,37 |
Pigmented
Villonodular Synovitis/Tenosynovial Giant Cell Tumor
(PVNS/TGCT) • Imatinib38 |
|||
|
| ||||
|
Alveolar
Soft Part Sarcoma (ASPS) • Sunitinib39,40 (category 2B) |
PEComa,
Recurrent Angiomyolipoma,
Lymphangioleiomyomatosis • Sirolimus41–45 |
|||
|
| ||||
| Chordoma (All recommendations are category 2B) | ||||
|
Combination
regimens • Erlotinib and cetuximab • Imatinib and cisplatin46 • Imatinib and sirolimus47 |
Single
agents • Erlotinib48 • Imatinib49,50 • Sunitinib37 |
|||
Alveolar soft part sarcoma and clear cell sarcomas are generally not sensitive to chemotherapy.
References for regimens, see pages 652 and 653.
The risk for cardiovascular events may be increased in patients receiving celecoxib. Physicians prescribing celecoxib should consider this emerging information when weighing the benefits against risks for individual patients (FDA Talk Paper T04-61, Dec 23, 2004).
Zalupski M, Metch B, Balcerzak S, et al. Phase III comparison of doxorubicin and dacarbazine given by bolus versus infusion in patients with soft-tissue sarcomas: a Southwest Oncology Group study. J Natl Cancer Inst 1991;83:926-932.
Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase Ill randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276-1285.
Grobmyer SR, Maki RG, Demetri GD, et al. Neo-adjuvant chemotherapy for primary high-grade extremity soft tissue sarcoma. Ann Oncol 2004;15:1667-1672.
Edmonson J, Ryan L, Blum R, et al. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269-1275.
Elias A, Ryan L, Sulkes A, et al. Response to mesna, doxorubicin, ifosfamide, and dacarbazine in 108 patients with metastatic or unresectable sarcoma and no prior chemotherapy. J Clin Oncol 1989;7:1208-1216.
Frustaci S, Gherlinzoni F, De Paoli A, et al. Adjuvant chemotherapy for soft tissue sarcomas of the extremities and girdles: results of the Italian Randomized Cooperative Trial. J Clin Oncol 2001;19:1238-1247.
Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;20:2824-2831.
Leu KM, Ostruszka U, Schewach D, et al. Laboratory and clinical evidence of synergistic cytotoxicity of sequential treatment with gemcitabine followed by docetaxel in the treatment of sarcoma. J Clin Oncol 2004;22:1706-1712.
Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863-1869.
Tierney JF. Adjuvant chemotherapy for localized resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647-1654.
Antman KH, Elias A. Dana-Farber Cancer Institute studies in advanced sarcoma. Semin Oncol 1990;1(Suppl 2):7-15.
Judson I, Radford J, Harris M, et al. Randomized phase II trial of pegylated liposomal doxorubicin versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: a study by the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 2001;37:870-877.
Talbot SM, Keohan ML, Hesdorffer M, et al. A phase II trial of temozolomide in patients with unresectable or metastatic soft tissue sarcoma. Cancer 2003;98:1942-1946.
Penel N, Bui BN, Bay JO, et al. Phase II trial of weekly paclitaxel for unresectable angiosarcoma: the ANGIOTAX study. J Clin Oncol 2008;26:5269-5274.
Schlemmer M, Reichardt P, Verweij J, et al. Paclitaxel in patients with advanced angiosarcomas of soft tissue: a retrospective study of the EORTC soft tissue and bone sarcoma group. Eur J Cancer 2008;44:2433-2436.
Maki RG, Keohan ML, Undevia SD, et al. Updated results of a phase II study of oral multi-kinase inhibitor sorafenib in sarcomas, CTEP study #7060 [abstract]. J Clin Oncol 2008;26(Suppl 1):Abstract 10531.
Maki RG, D'Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol 2009;27:3133-3140.
Keohan ML, Morgan JA, D'Adamo DR, et al. Continuous daily dosing (CDD) of sunitinib (SU) in patients with metastatic soft tissue sarcomas (STS) other than GIST: results of a phase II trial [abstract]. J Clin Oncol 2008;26(Suppl 1):Abstract 10533.
Agulnik M, Okuno SH, Von Mehren M, et al. An open-label multicenter phase II study of bevacizumab for the treatment of angiosarcoma [abstract]. J Clin Oncol 2009;27(Suppl 1):Abstract 10522.
Hansmann A, Adolph C, Vogel T, et al. High-dose tamoxifen and sulindac as first-line treatment for desmoid tumors. Cancer 2004;100:612-620.
Chao AS, Lai CH, Hsueh S, et al. Successful treatment of recurrent pelvic desmoid tumor with tamoxifen: case report. Hum Reprod 2000;15:311-313.
Benson JR, Mokbel MK, Baum M. Management of desmoid tumours including a case report of toremifene. Ann Oncol 1994;5:173-177.
Azzarelli A, Gronchi A, Bertulli R, et al. Low-dose chemotherapy with methotrexate and vinblastine for patients with advanced aggressive fibromatosis. Cancer 2001;92:1259-1264.
Leithner A, Schnack B, Katterschafka T, et al. Treatment of extra-abdominal desmoid tumors with interferon-alpha with or without tretinoin. J Surg Oncol 2000;73:21-25.
Seiter K, Kemeny N. Successful treatment of a desmoid tumor with doxorubicin. Cancer 1993;71:2242-2244.
Patel SR, Evans HL, Benjamin RS. Combination chemotherapy in adult desmoid tumors. Cancer 1993;72:3244-3247.
Chugh R, Maki RG, Thomas DG, et al. A SARC phase II multicenter trial of imatinib mesylate (IM) in patients with aggressive fibromatosis [abstract]. J Clin Oncol 2006;24(Suppl 1):Abstract 9515.
Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N EngI J Med 2002;347:472-480.
Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomized trial. Lancet 2004;364:1127-1134.
Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumor after failure of imatinib: a randomized controlled trial. Lancet 2006;368:1329-1338.
Wiebe L, Kasza KE, Maki RG, et al. Activity of sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitinib (SU)-resistant (RES) gastrointestinal stromal tumors (GIST): a phase II trial of the University of Chicago Phase II Consortium [abstract]. J Clin Oncol 2008;26(Suppl 1):Abstract 10502.
Blay JY, Casali PG, Reichardt P, et al. A phase I study of nilotinib alone and in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors (GIST): study update [abstract]. J Clin Oncol 2008;26(Suppl 1):Abstract 10553.
Montemurro M, Schoffski P, Reichardt P, et al. Nilotinib in advanced GIST: a retrospective analysis of nilotinib in compassionate use [abstract]. J Clin Oncol 2008;26(Suppl 1):Abstract 10523.
Demetri G, Lo Russo P, MacPherson RJ, et al. Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin Cancer Res 2009;15:6232-6240.
Park MS, Patel SR, Ludwig JA, et al. Combination therapy with temozolomide and bevacizumab in the treatment of hemangiopericytoma/malignant solitary fibrous tumor [abstract]. J Clin Oncol 2008;26(Suppl 1):Abstract 10512.
Casali PG, Stacchiotti S, Palassini E, et al. Evaluation of the antitumor activity of sunitinib malate (SM) in solitary fibrous tumor (SFT) [abstract]. J Clin Oncol 2009;27(Suppl 1):Abstract 10571.
George S, Merriam P, Maki RG, et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J Clin Oncol 2009;27:3154-3160.
Blay JY, El Sayadi H, Thiesse P, et al. Complete response to imatinib in relapsing pigmented villonodular synovitis/tenosynovial giant cell tumor (PVNS/TGCT). Ann Oncol 2008;19:821-822.
Stacchiotti S, Tamborini E, Bertulli R, et al. Response to sunitinib malate (SM) in alveolar soft part sarcoma (ASPS) [abstract]. J Clin Oncol 2008;26(Suppl 1):Abstract 10592.
Stacchiotti S, Tamborini E, Marrari A, et al. Response to sunitinib malate in advanced alveolar soft part sarcoma. Clin Cancer Res 2009;15:1096-1104.
Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 2008;358:140-151.
Chen F, Omasa M, Kondo N, et al. Sirolimus treatment for recurrent lymphangioleiomyomatosis after lung transplantation. Ann Thorac Surg 2009;87:6-7.
Davies DM, Johnson SR, Tattersfield AE, et al. Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med 2008;358:200-203.
Wagner AJ, Malinowska-Kolodziej I, Morgan JA, et al. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol 2010;28:835-840.
Taille C, Debray MP, Crestani B. Sirolimus treatment for pulmonary lymphangioleiomyomatosis. Ann Intern Med 2007;146:687-688.
Casali PG, Stacchiotti S, Grosso F, et al. Adding cisplatin (CDDP) to imatinib (IM) re-establishes tumor response following secondary resistance to IM in advanced chordoma [abstract]. J Clin Oncol 2007;25(Suppl 1):Abstract 10038.
Stacchiotti S, Marrari A, Tamborini E, et al. Response to imatinib plus sirolimus in advanced chordoma. Ann Oncol 2009;20:1886-1894.
Singhal N, Kotasek D, Parnis FX. Response to erlotinib in a patient with treatment refractory chordoma. Anticancer Drugs 2009;20:953-955.
Geoerger B, Morland B, Ndiaye A, et al. Target-driven exploratory study of imatinib mesylate in children with solid malignancies by the Innovative Therapies for Children with Cancer (ITCC) European Consortium. Eur J Cancer 2009;45:2342-2351.
Casali PG, Messina A, Stacchiotti S, et al. Imatinib mesylate in chordoma. Cancer 2004;101:2086-2097.
| Individual Disclosure for the NCCN Soft Tissue Sarcoma Panel | |||||
|---|---|---|---|---|---|
| Panel Member | Clinical Research Support | Advisory Boards, Speakers Bureau, Expert Witness, or Consultant | Patent, Equity, or Royalty | Other | Date Completed |
| Scott Antonia, MD, PhD | Novartis Pharmaceuticals Corporation; Ziopharm; and Pfizer Inc. | None | None | None | 12/17/09 |
| Robert S. Benjamin, MD | Novartis Pharmaceuticals Corporation | Novartis Pharmaceuticals Corporation | Johnson & Johnson; and Pfizer Inc. | None | 12/29/09 |
| Marilyn M. Bui, MD, PhD | None | None | None | None | 8/13/09 |
| Ephraim S. Casper, MD | None | Novartis Pharmaceuticals Corporation | None | None | 9/29/09 |
| Ernest U. Conrad III, MD | Stryker Corporation; and Zimmer Corporation | None | None | None | 10/29/09 |
| Thomas F. DeLaney, MD | None | None | None | None | 7/3/09 |
| George D. Demetri, MD | ARIAD Pharmaceuticals, Inc.; Daiichi-Sankyo Co.; Novartis Pharmaceuticals Corporation; Ziopharm; and Pfizer Inc. | ARIAD Pharmaceuticals, Inc.; Daiichi-Sankyo Co.; Johnson & Johnson; Novartis Pharmaceuticals Corporation; Koltan Pharmaceuticals; Ziopharm; and Pfizer Inc. | None | None | 12/22/09 |
| Kristen N. Ganjoo, MD | None | Genentech, Inc.; and GlaxoSmithKline | None | None | 1/5/10 |
| Martin J. Heslin, MD | None | None | None | None | 1/7/10 |
| Raymond J. Hutchinson, MD | None | None | None | None | 1/5/10 |
| John M. Kane III, MD | None | None | None | None | 9/8/09 |
| G. Douglas Letson, MD | None | Stryker Corporation | None | None | 12/22/09 |
| Sean V. McGarry, MD | None | None | None | Musculoskeletal Transplant Foundation | 10/29/09 |
| Richard J. O’Donnell, MD | None | None | None | None | 10/29/09 |
| I. Benjamin Paz, MD | Agendia BV | None | None | None | 1/8/10 |
| John D. Pfeifer, MD, PhD | None | None | None | None | 7/9/09 |
| Raphael E. Pollock, MD, PhD | None | None | None | None | 11/23/09 |
| R. Lor Randall, MD | None | Musculoskeletal Tumor Society | None | None | 11/4/09 |
| Richard F. Riedel, MD | None | ARIAD Pharmaceuticals, Inc.; Novartis Pharmaceuticals Corporation; and Pfizer Inc. | None | None | 12/8/09 |
| Karen D. Schupak, MD | None | None | None | None | 7/2/09 |
| Herbert S. Schwartz, MD | None | None | None | Musculoskeletal Transplant Foundation | 7/1/09 |
| Katherine Thornton, MD | Amgen Inc.; ARIAD Pharmaceuticals, Inc.; Celgene Corporation; and Genentech, Inc. | Pfizer Inc. | None | None | 12/9/09 |
| Margaret von Mehren, MD | Johnson & Johnson; Merck & Co., Inc.; Novartis Pharmaceuticals Corporation; and Pfizer Inc. | ARIAD Pharmaceuticals, Inc.; Johnson & Johnson; Novartis Pharmaceuticals Corporation; and Pfizer Inc. | None | None | 2/26/10 |
| Jeffrey Wayne, MD | None | Novartis Pharmaceuticals Corporation | None | None | 4/7/10 |
Footnotes
The NCCN guidelines staff have no conflicts to disclose.
Information from the FDA label. For more detailed information, review the full content at www.fda.gov.
See Principles of Ancillary Techniques Useful in the Diagnosis of Sarcomas (page 647 and 648).
Fletcher CD, Unni K, Mertens F, eds. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2002.
Molecular genetic analysis involves highly complex test methods. None of the methods are absolutely sensitive or provide results that are absolutely specific; test results must always be interpreted in the context of the clinical and pathologic features of the case. Testing should therefore be performed by a pathologist with expertise in sarcoma diagnosis and molecular diagnostic techniques.
This table is not exhaustive for either sarcomas with characteristic genetic changes or the genes involved. Consultation with a pathologist who has expertise in sarcoma diagnosis and molecular diagnostic techniques should be obtained before testing.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.D’Amato G, Steinert DM, McAuliffe JC, Trent JC. Update on the biology and therapy of gastrointestinal stromal tumors. Cancer Control. 2005;12:44–56. doi: 10.1177/107327480501200106. [DOI] [PubMed] [Google Scholar]
- 3.Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol. 2002;33:459–465. doi: 10.1053/hupa.2002.123545. [DOI] [PubMed] [Google Scholar]
- 4.Zahm S, Fraumeni JJ. The epidemiology of soft tissue sarcoma. Semin Oncol. 1997;24:504–514. [PubMed] [Google Scholar]
- 5.Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer. 2001;91:1914–1926. doi: 10.1002/1097-0142(20010515)91:10<1914::aid-cncr1214>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 6.Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin. 2004;54:94–109. doi: 10.3322/canjclin.54.2.94. [DOI] [PubMed] [Google Scholar]
- 7.Domanski HA. Fine-needle aspiration cytology of soft tissue lesions: diagnostic challenges. Diagn Cytopathol. 2007;35:768–773. doi: 10.1002/dc.20765. [DOI] [PubMed] [Google Scholar]
- 8.Antonescu CR. The role of genetic testing in soft tissue sarcoma. Histopathology. 2006;48:13–21. doi: 10.1111/j.1365-2559.2005.02285.x. [DOI] [PubMed] [Google Scholar]
- 9.Pfeifer JD, Hill DA, O’Sullivan MJ, Dehner LP. Diagnostic gold standard for soft tissue tumours: morphology or molecular genetics? Histopathology. 2000;37:485–500. doi: 10.1046/j.1365-2559.2000.01107.x. [DOI] [PubMed] [Google Scholar]
- 10.Hill DA, O’Sullivan MJ, Zhu X, et al. Practical application of molecular genetic testing as an aid to the surgical pathologic diagnosis of sarcomas: a prospective study. Am J Surg Pathol. 2002;26:965–977. doi: 10.1097/00000478-200208000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Sorensen PH, Lynch JC, Qualman SJ, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar thabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2002;20:2672–2679. doi: 10.1200/JCO.2002.03.137. [DOI] [PubMed] [Google Scholar]
- 12.Guillou L, Benhattar J, Bonichon F, et al. Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol. 2004;22:4040–4050. doi: 10.1200/JCO.2004.11.093. [DOI] [PubMed] [Google Scholar]
- 13.Ladanyi M, Antonescu CR, Leung DH, et al. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patients. Cancer Res. 2002;62:135–140. [PubMed] [Google Scholar]
- 14.Antonescu CR, Tschernyavsky SJ, Decuseara R, et al. Prognostic impact of P53 status, TLS-CHOP fusion transcript structure, and histological grade in myxoid liposarcoma: a molecular and clinicopathologic study of 82 cases. Clin Cancer Res. 2001;7:3977–3987. [PubMed] [Google Scholar]
- 15.Guillou L, Coindre J, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol. 1997;15:350–362. doi: 10.1200/JCO.1997.15.1.350. [DOI] [PubMed] [Google Scholar]
- 16.Dileo P, Demetri GD. Update on new diagnostic and therapeutic approaches for sarcomas. Clin Adv Hematol Oncol. 2005;3:781–791. [PubMed] [Google Scholar]
- 17.Mocellin S, Rossi C, Brandes A, Nitti D. Adult soft tissue sarcomas: conventional therapies and molecularly targeted approaches. Cancer Treat Rev. 2006;32:9–27. doi: 10.1016/j.ctrv.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Brennan MF, Casper ES, Harrison LB, et al. The role of multimodality therapy in soft-tissue sarcoma. Ann Surg. 1991;214:328–336. doi: 10.1097/00000658-199109000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim B, Chen YL, Kirsch DG, et al. An effective preoperative three-dimensional radiotherapy target volume for extremity soft tissue sarcoma and the effect of margin width on local control. Int J Radiat Oncol Biol Phys. 2010 doi: 10.1016/j.ijrobp.2009.06.086. in press. [DOI] [PubMed] [Google Scholar]
- 20.Fleming JB, Berman RS, Cheng SC, et al. Long-term outcome of patients with American Joint Committee on Cancer stage IIB extremity soft tissue sarcomas. J Clin Oncol. 1999;17:2772–2780. doi: 10.1200/JCO.1999.17.9.2772. [DOI] [PubMed] [Google Scholar]
- 21.Gerrand CH, Wunder JS, Kandel RA, et al. Classification of positive margins after resection of soft-tissue sarcoma of the limb predicts the risk of local recurrence. J Bone Joint Surg Br. 2001;83:1149–1155. doi: 10.1302/0301-620x.83b8.12028. [DOI] [PubMed] [Google Scholar]
- 22.Pisters PW, Leung DH, Woodruff J, et al. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol. 1996;14:1679–1689. doi: 10.1200/JCO.1996.14.5.1679. [DOI] [PubMed] [Google Scholar]
- 23.Lewis JJ, Leung D, Espat J, et al. Effect of reresection in extremity soft tissue sarcoma. Ann Surg. 2000;231:655–663. doi: 10.1097/00000658-200005000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer. 2003;97:2544–2553. doi: 10.1002/cncr.11367. [DOI] [PubMed] [Google Scholar]
- 25.Ghert MA, Abudu A, Driver N, et al. The indications for and the prognostic significance of amputation as the primary surgical procedure for localized soft tissue sarcoma of the extremity. Ann Surg Oncol. 2005;12:10–17. doi: 10.1007/s10434-004-1171-3. [DOI] [PubMed] [Google Scholar]
- 26.Stojadinovic A, Jaques DP, Leung DH, et al. Amputation for recurrent soft tissue sarcoma of the extremity: indications and outcome. Ann Surg Oncol. 2001;8:509–518. doi: 10.1007/s10434-001-0509-3. [DOI] [PubMed] [Google Scholar]
- 27.Williard WC, Hajdu SI, Casper ES, Brennan MF. Comparison of amputation with limb-sparing operations for adult soft tissue sarcoma of the extremity. Ann Surg. 1992;215:269–275. doi: 10.1097/00000658-199203000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeLaney TF, Trofimov AV, Engelsman M, Suit HD. Advanced-technology radiation therapy in the management of bone and soft tissue sarcomas. Cancer Control. 2005;12:27–35. doi: 10.1177/107327480501200104. [DOI] [PubMed] [Google Scholar]
- 29.Leibel SA, Fuks Z, Zelefsky MJ, et al. Intensity-modulated radiotherapy. Cancer J. 2002;8:164–176. doi: 10.1097/00130404-200203000-00010. [DOI] [PubMed] [Google Scholar]
- 30.Davis A, O’Sullivan B, Bell R, et al. Function and health status outcomes in a randomized trial comparing preoperative and postoperative radiotherapy in extremity soft tissue sarcoma. J Clin Oncol. 2002;20:4472–4477. doi: 10.1200/JCO.2002.03.084. [DOI] [PubMed] [Google Scholar]
- 31.Sadoski C, Suit H, Rosenberg A, et al. Preoperative radiation, surgical margins, and local control of extremity sarcomas of soft tissues. J Surg Oncol. 1993;52:223–230. doi: 10.1002/jso.2930520405. [DOI] [PubMed] [Google Scholar]
- 32.Alektiar KM, Velasco J, Zelefsky MJ, et al. Adjuvant radiotherapy for margin-positive high-grade soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys. 2000;48:1051–1058. doi: 10.1016/s0360-3016(00)00753-7. [DOI] [PubMed] [Google Scholar]
- 33.Cahlon O, Spierer M, Brennan MF, et al. Long-term outcomes in extremity soft tissue sarcoma after a pathologically negative re-resection and without radiotherapy. Cancer. 2008;112:2774–2779. doi: 10.1002/cncr.23493. [DOI] [PubMed] [Google Scholar]
- 34.Alektiar KM, Leung D, Zelefsky MJ, et al. Adjuvant brachytherapy for primary high-grade soft tissue sarcoma of the extremity. Ann Surg Oncol. 2002;9:48–56. doi: 10.1245/aso.2002.9.1.48. [DOI] [PubMed] [Google Scholar]
- 35.Tran QN, Kim AC, Gottschalk AR, et al. Clinical outcomes of intraoperative radiation therapy for extremity sarcomas. Sarcoma. 2006;2006:91671. doi: 10.1155/SRCM/2006/91671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark MA, Thomas JM. Amputation for soft-tissue sarcoma. Lancet Oncol. 2003;4:335–342. doi: 10.1016/s1470-2045(03)01113-6. [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg. 1982;196:305–315. doi: 10.1097/00000658-198209000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baldini EH, Goldberg J, Jenner C, et al. Long-term outcomes after function-sparing surgery without radiotherapy for soft tissue sarcoma of the extremities and trunk. J Clin Oncol. 1999;17:3252–3259. doi: 10.1200/JCO.1999.17.10.3252. [DOI] [PubMed] [Google Scholar]
- 39.Lin PP, Guzel VB, Pisters PW, et al. Surgical management of soft tissue sarcomas of the hand and foot. Cancer. 2002;95:852–861. doi: 10.1002/cncr.10750. [DOI] [PubMed] [Google Scholar]
- 40.O’Sullivan B, Davis A, Turcotte R, et al. Five-year results of a randomized phase III trial of pre-operative vs post-operative radiotherapy in extremity soft tissue sarcoma [abstract] J Clin Oncol. 2004;22(Suppl 1) Abstract 9007. [Google Scholar]
- 41.Yang JC, Chang AE, Baker AR, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol. 1998;16:197–203. doi: 10.1200/JCO.1998.16.1.197. [DOI] [PubMed] [Google Scholar]
- 42.Wilson AN, Davis A, Bell RS, et al. Local control of soft tissue sarcoma of the extremity: the experience of a multidisciplinary sarcoma group with definitive surgery and radiotherapy. Eur J Cancer. 1994;30A:746–751. doi: 10.1016/0959-8049(94)90286-0. [DOI] [PubMed] [Google Scholar]
- 43.Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol. 1996;14:859–868. doi: 10.1200/JCO.1996.14.3.859. [DOI] [PubMed] [Google Scholar]
- 44.Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol. 2008;26:3440–3444. doi: 10.1200/JCO.2008.16.6249. [DOI] [PubMed] [Google Scholar]
- 45.Kepka L, DeLaney TF, Suit HD, Goldberg SI. Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys. 2005;63:852–859. doi: 10.1016/j.ijrobp.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 46.Clasby R, Tilling K, Smith MA, Fletcher CD. Variable management of soft tissue sarcoma: regional audit with implications for specialist care. Br J Surg. 1997;84:1692–1696. [PubMed] [Google Scholar]
- 47.Demas BE, Heelan RT, Lane J, et al. Soft-tissue sarcomas of the extremities: comparison of MR and CT in determining the extent of disease. AJR Am J Roentgenol. 1988;150:615–620. doi: 10.2214/ajr.150.3.615. [DOI] [PubMed] [Google Scholar]
- 48.Hanna SL, Fletcher BD. MR imaging of malignant soft-tissue tumors. Magn Reson Imaging Clin N Am. 1995;3:629–650. [PubMed] [Google Scholar]
- 49.Heslin MJ, Smith JK. Imaging of soft tissue sarcomas. Surg Oncol Clin N Am. 1999;8:91–107. [PubMed] [Google Scholar]
- 50.Schwab JH, Boland P, Guo T, et al. Skeletal metastases in myxoid liposarcoma: an unusual pattern of distant spread. Ann Surg Oncol. 2007;14:1507–1514. doi: 10.1245/s10434-006-9306-3. [DOI] [PubMed] [Google Scholar]
- 51.Schwab JH, Boland PJ, Antonescu C, et al. Spinal metastases from myxoid liposarcoma warrant screening with magnetic resonance imaging. Cancer. 2007;110:1815–1822. doi: 10.1002/cncr.22992. [DOI] [PubMed] [Google Scholar]
- 52.Tateishi U, Hasegawa T, Beppu Y, et al. Prognostic significance of MRI findings in patients with myxoid-round cell liposarcoma. AJR Am J Roentgenol. 2004;182:725–731. doi: 10.2214/ajr.182.3.1820725. [DOI] [PubMed] [Google Scholar]
- 53.Portera CA, Jr, Ho V, Patel SR, et al. Alveolar soft part sarcoma: clinical course and patterns of metastasis in 70 patients treated at a single institution. Cancer. 2001;91:585–591. doi: 10.1002/1097-0142(20010201)91:3<585::aid-cncr1038>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 54.Schuetze SM. Utility of positron emission tomography in sarcomas. Curr Opin Oncol. 2006;18:369–373. doi: 10.1097/01.cco.0000228744.49294.12. [DOI] [PubMed] [Google Scholar]
- 55.Folpe AL, Lyles RH, Sprouse JT, et al. (F-18) fluorodeoxyglucose positron emission tomography as a predictor of pathologic grade and other prognostic variables in bone and soft tissue sarcoma. Clin Cancer Res. 2000;6:1279–1287. [PubMed] [Google Scholar]
- 56.Schuetze SM, Rubin BP, Vernon C, et al. Use of positron emission tomography in localized extremity soft tissue sarcoma treated with neoadjuvant chemotherapy. Cancer. 2005;103:339–348. doi: 10.1002/cncr.20769. [DOI] [PubMed] [Google Scholar]
- 57.Eary JF, O’Sullivan F, Powitan Y, et al. Sarcoma tumor FDG uptake measured by PET and patient outcome: a retrospective analysis. Eur J Nucl Med Mol Imaging. 2002;29:1149–1154. doi: 10.1007/s00259-002-0859-5. [DOI] [PubMed] [Google Scholar]
- 58.Geer RJ, Woodruff J, Casper ES, Brennan MF. Management of small soft-tissue sarcoma of the extremity in adults. Arch Surg. 1992;127:1285–1289. doi: 10.1001/archsurg.1992.01420110027007. [DOI] [PubMed] [Google Scholar]
- 59.Karakousis CP, Emrich LJ, Rao U, Khalil M. Limb salvage in soft tissue sarcomas with selective combination of modalities. Eur J Surg Oncol. 1991;17:71–80. [PubMed] [Google Scholar]
- 60.Pisters PW, O’Sullivan B, Maki RG. Evidence-based recommendations for local therapy for soft tissue sarcomas. J Clin Oncol. 2007;25:1003–1008. doi: 10.1200/JCO.2006.09.8525. [DOI] [PubMed] [Google Scholar]
- 61.Pisters PW, Pollock RE, Lewis VO, et al. Long-term results of prospective trial of surgery alone with selective use of radiation for patients with T1 extremity and trunk soft tissue sarcomas. Ann Surg. 2007;246:675–681. doi: 10.1097/SLA.0b013e318155a9ae. discussion 681–682. [DOI] [PubMed] [Google Scholar]
- 62.Kraybill WG, Harris J, Spiro IJ, et al. Phase II study of neoadjuvant chemotherapy and radiation therapy in the management of high-risk, high-grade, soft tissue sarcomas of the extremities and body wall: Radiation Therapy Oncology Group trial 9514. J Clin Oncol. 2006;24:619–625. doi: 10.1200/JCO.2005.02.5577. [DOI] [PubMed] [Google Scholar]
- 63.Grobmyer SR, Maki RG, Demetri GD, et al. Neo-adjuvant chemotherapy for primary high-grade extremity soft tissue sarcoma. Ann Oncol. 2004;15:1667–1672. doi: 10.1093/annonc/mdh431. [DOI] [PubMed] [Google Scholar]
- 64.DeLaney TFSI, Suit HD, Gebhardt MC, et al. Neoadjuvant chemotherapy and radiotherapy for large extremity soft-tissue sarcomas. Int J Radiat Oncol Biol Phys. 2003;56:1117–1127. doi: 10.1016/s0360-3016(03)00186-x. [DOI] [PubMed] [Google Scholar]
- 65.Pisters PW, Patel SR, Varma DG, et al. Preoperative chemotherapy for stage IIIB extremity soft tissue sarcoma: long-term results from a single institution. J Clin Oncol. 1997;15:3481–3487. doi: 10.1200/JCO.1997.15.12.3481. [DOI] [PubMed] [Google Scholar]
- 66.Pisters PW, Ballo MT, Patel SR. Preoperative chemoradiation treatment strategies for localized sarcoma. Ann Surg Oncol. 2002;9:535–542. doi: 10.1007/BF02573888. [DOI] [PubMed] [Google Scholar]
- 67.Maki RG. Role of chemotherapy in patients with soft tissue sarcomas. Expert Rev Anticancer Ther. 2004;4:229–236. doi: 10.1586/14737140.4.2.229. [DOI] [PubMed] [Google Scholar]
- 68.Tierney JF, Mosseri V, Stewart LA, et al. Adjuvant chemotherapy for soft-tissue sarcoma: review and meta-analysis of the published results of randomised clinical trials. Br J Cancer. 1995;72:469–475. doi: 10.1038/bjc.1995.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet. 1997;350:1647–1654. [PubMed] [Google Scholar]
- 70.Bramwell V, Rouesse J, Steward W, et al. Adjuvant CYVADIC chemotherapy for adult soft tissue sarcoma—reduced local recurrence but no improvement in survival: a study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol. 1994;12:1137–1149. doi: 10.1200/JCO.1994.12.6.1137. [DOI] [PubMed] [Google Scholar]
- 71.Cormier JN, Huang X, Xing Y, et al. Cohort analysis of patients with localized, high-risk, extremity soft tissue sarcoma treated at two cancer centers: chemotherapy-associated outcomes. J Clin Oncol. 2004;22:4567–4574. doi: 10.1200/JCO.2004.02.057. [DOI] [PubMed] [Google Scholar]
- 72.Frustaci S, De Paoli A, Bidoli E, et al. Ifosfamide in the adjuvant therapy of soft tissue sarcomas. Oncology. 2003;65:80–84. doi: 10.1159/000073366. [DOI] [PubMed] [Google Scholar]
- 73.Frustaci S, Gherlinzoni F, De Paoli A, et al. Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: results of the Italian randomized cooperative trial. J Clin Oncol. 2001;19:1238–1247. doi: 10.1200/JCO.2001.19.5.1238. [DOI] [PubMed] [Google Scholar]
- 74.Woll PJ, van Glabbeke M, Hohenberger P, et al. Adjuvant chemotherapy (CT) with doxorubicin and ifosfamide in resected soft tissue sarcoma (STS): interim analysis of a randomised phase III trial [abstract] J Clin Oncol. 2007;25(Suppl 1) Abstract 10008. [Google Scholar]
- 75.Alektiar KM, Leung D, Zelefsky MJ, Brennan MF. Adjuvant radiation for stage II-B soft tissue sarcoma of the extremity. J Clin Oncol. 2002;20:1643–1650. doi: 10.1200/JCO.2002.20.6.1643. [DOI] [PubMed] [Google Scholar]
- 76.Antman K, Crowley J, Balcerzak S, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol. 1993;11:1276–1285. doi: 10.1200/JCO.1993.11.7.1276. [DOI] [PubMed] [Google Scholar]
- 77.Antman KH, Judson EA. Dana-Farber Cancer Institute studies in advanced sarcoma. Semin Oncol. 1990;1:7–15. [PubMed] [Google Scholar]
- 78.Bramwell VH, Anderson D, Charette ML. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev. 2003:CD003293. doi: 10.1002/14651858.CD003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Edmonson J, Ryan L, Blum R, et al. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol. 1993;11:1269–1275. doi: 10.1200/JCO.1993.11.7.1269. [DOI] [PubMed] [Google Scholar]
- 80.Elias A, Ryan L, Sulkes A, et al. Response to mesna, doxorubicin, ifosfamide, and dacarbazine in 108 patients with metastatic or unresectable sarcoma and no prior chemotherapy. J Clin Oncol. 1989;7:1208–1216. doi: 10.1200/JCO.1989.7.9.1208. [DOI] [PubMed] [Google Scholar]
- 81.Lorigan P, Verweij J, Papai Z, et al. Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol. 2007;25:3144–3150. doi: 10.1200/JCO.2006.09.7717. [DOI] [PubMed] [Google Scholar]
- 82.Santoro A, Tursz T, Mouridsen H, et al. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first- line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol. 1995;13:1537–1545. doi: 10.1200/JCO.1995.13.7.1537. [DOI] [PubMed] [Google Scholar]
- 83.Zalupski M, Metch B, Balcerzak S, et al. Phase III comparison of doxorubicin and dacarbazine given by bolus versus infusion in patients with soft-tissue sarcomas: a Southwest Oncology Group study. J Natl Cancer Inst. 1991;83:926–932. doi: 10.1093/jnci/83.13.926. [DOI] [PubMed] [Google Scholar]
- 84.Siehl JM, Thiel E, Schmittel A, et al. Liposomal anthracyclines and ifosfamide in the first line treatment of advanced soft tissue sarcomas: a two cohort phase II study [abstract] J Clin Oncol. 2006;24(Suppl 1) Abstract 9563. [Google Scholar]
- 85.Siehl JM, Thiel E, Schmittel A, et al. Ifosfamide/liposomal daunorubicin is a well tolerated and active first-line chemotherapy regimen in advanced soft tissue sarcoma: results of a phase II study. Cancer. 2005;104:611–617. doi: 10.1002/cncr.21211. [DOI] [PubMed] [Google Scholar]
- 86.Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol. 2002;20:2824–2831. doi: 10.1200/JCO.2002.11.050. [DOI] [PubMed] [Google Scholar]
- 87.Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002. J Clin Oncol. 2007;25:2755–2763. doi: 10.1200/JCO.2006.10.4117. [DOI] [PubMed] [Google Scholar]
- 88.Leu KM, Ostruszka LJ, Shewach D, et al. Laboratory and clinical evidence of synergistic cytotoxicity of sequential treament with gemcitabine followed by docetaxel in the treatment of sarcoma. J Clin Oncol. 2004;22:1706–1712. doi: 10.1200/JCO.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 89.Bay JO, Ray-Coquard I, Fayette J, et al. Docetaxel and gemcitabine combination in 133 advanced soft-tissue sarcomas: a retrospective analysis. Int J Cancer. 2006;119:706–711. doi: 10.1002/ijc.21867. [DOI] [PubMed] [Google Scholar]
- 90.Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer. 2007;109:1863–1869. doi: 10.1002/cncr.22609. [DOI] [PubMed] [Google Scholar]
- 91.Garcia del Muro X, Lopez-Pousa A, Martin J, et al. A phase II trial of temozolomide as a 6-week, continuous, oral schedule in patients with advanced soft tissue sarcoma: a study by the Spanish Group for Research on Sarcomas. Cancer. 2005;104:1706–1712. doi: 10.1002/cncr.21384. [DOI] [PubMed] [Google Scholar]
- 92.Talbot SM, Keohan ML, Hesdorffer M, et al. A phase II trial of temozolomide in patients with unresectable or metastatic soft tissue sarcoma. Cancer. 2003;98:1942–1946. doi: 10.1002/cncr.11730. [DOI] [PubMed] [Google Scholar]
- 93.Le Cesne A, Blay JY, Judson I, et al. Phase II study of ET-743 in advanced soft tissue sarcomas: a European Organisation for the Research and Treatment of Cancer (EORTC) Soft Tissue and Bone Sarcoma Group trial. J Clin Oncol. 2005;23:576–584. doi: 10.1200/JCO.2005.01.180. [DOI] [PubMed] [Google Scholar]
- 94.Garcia-Carbonero R, Supko JG, Manola J, et al. Phase II and pharmacokinetic study of ecteinascidin 743 in patients with progressive sarcomas of soft tissues refractory to chemotherapy. J Clin Oncol. 2004;22:1480–1490. doi: 10.1200/JCO.2004.02.098. [DOI] [PubMed] [Google Scholar]
- 95.Garcia-Carbonero R, Supko JG, Maki RG, et al. Ecteinascidin-743 (ET-743) for chemotherapy-naive patients with advanced soft tissue sarcomas: multicenter phase II and pharmacokinetic study. J Clin Oncol. 2005;23:5484–5492. doi: 10.1200/JCO.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 96.Yovine A, Riofrio M, Blay JY, et al. Phase II study of ecteinascidin-743 in advanced pretreated soft tissue sarcoma patients. J Clin Oncol. 2004;22:890–899. doi: 10.1200/JCO.2004.05.210. [DOI] [PubMed] [Google Scholar]
- 97.Laverdiere C, Kolb EA, Supko JG, et al. Phase II study of ecteinascidin 743 in heavily pretreated patients with recurrent osteosarcoma. Cancer. 2003;98:832–840. doi: 10.1002/cncr.11563. [DOI] [PubMed] [Google Scholar]
- 98.Issels RD, Lindner LH, Wust P, et al. Regional hyperthermia (RHT) improves response and survival when combined with systemic chemotherapy in the management of locally advanced, high grade soft tissue sarcomas (STS) of the extremities, the body wall and the abdomen: a phase III randomised pros [abstract] J Clin Oncol. 2007;25(Suppl 1) Abstract 10009. [Google Scholar]
- 99.Issels R, Schlemmer M. Current trials and new aspects in soft tissue sarcoma of adults. Cancer Chemother Pharmacol. 2002;49(Suppl 1):S4–8. doi: 10.1007/s00280-002-0445-3. [DOI] [PubMed] [Google Scholar]
- 100.Grunhagen DJ, Brunstein F, ten Hagen TL, et al. TNF-based isolated limb perfusion: a decade of experience with antivascular therapy in the management of locally advanced extremity soft tissue sarcomas. Cancer Treat Res. 2004;120:65–79. doi: 10.1007/1-4020-7856-0_4. [DOI] [PubMed] [Google Scholar]
- 101.Grunhagen DJ, de Wilt JH, Graveland WJ, et al. Outcome and prognostic factor analysis of 217 consecutive isolated limb perfusions with tumor necrosis factor-alpha and melphalan for limb-threatening soft tissue sarcoma. Cancer. 2006;106:1776–1784. doi: 10.1002/cncr.21802. [DOI] [PubMed] [Google Scholar]
- 102.Whooley BP, Mooney MM, Gibbs JF, Kraybill WG. Effective follow-up strategies in soft tissue sarcoma. Semin Surg Oncol. 1999;17:83–87. doi: 10.1002/(sici)1098-2388(199907/08)17:1<83::aid-ssu11>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 103.Whooley BP, Gibbs JF, Mooney MM, et al. Primary extremity sarcoma: what is the appropriate follow-up? Ann Surg Oncol. 2000;7:9–14. doi: 10.1007/s10434-000-0009-x. [DOI] [PubMed] [Google Scholar]
- 104.Kane JM., III Surveillance strategies for patients following surgical resection of soft tissue sarcomas. Curr Opin Oncol. 2004;16:328–332. doi: 10.1097/01.cco.0000127879.62254.d3. [DOI] [PubMed] [Google Scholar]
- 105.Lewis JJ, Leung D, Casper ES, et al. Multifactorial analysis of long-term follow-up (more than 5 years) of primary extremity sarcoma. Arch Surg. 1999;134:190–194. doi: 10.1001/archsurg.134.2.190. [DOI] [PubMed] [Google Scholar]
- 106.Fleming JB, Cantor SB, Varma DG, et al. Utility of chest computed tomography for staging in patients with T1 extremity soft tissue sarcomas. Cancer. 2001;92:863–868. doi: 10.1002/1097-0142(20010815)92:4<863::aid-cncr1394>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 107.Singer S, Antman K, Corson JM, Eberlein TJ. Long-term salvageability for patients with locally recurrent soft-tissue sarcomas. Arch Surg. 1992;127:548–553. doi: 10.1001/archsurg.1992.01420050068009. discussion 553–544. References 108–127 are cited in the section on “Retroperitoneal/Intra-Abdominal Soft Tissue Sarcomas,” and are available in the version of these guidelines online at www.NCCN.org. [DOI] [PubMed] [Google Scholar]
- 128.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 129.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 130.Hirota S, Ohashi A, Nishida T, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125:660–667. doi: 10.1016/s0016-5085(03)01046-1. [DOI] [PubMed] [Google Scholar]
- 131.Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23:70–83. doi: 10.1053/j.semdp.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 132.Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130:1466–1478. doi: 10.5858/2006-130-1466-GSTROM. [DOI] [PubMed] [Google Scholar]
- 133.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 134.Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–625. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 135.Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–1134. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 136.Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626–632. doi: 10.1200/JCO.2007.13.4452. [DOI] [PubMed] [Google Scholar]
- 137.Dagher R, Cohen M, Williams G, et al. Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8:3034–3038. [PubMed] [Google Scholar]
- 138.Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology. 2008;53:245–266. doi: 10.1111/j.1365-2559.2008.02977.x. [DOI] [PubMed] [Google Scholar]
- 139.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 140.Debiec-Rychter M, Dumez H, Judson I, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40:689–695. doi: 10.1016/j.ejca.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 141.Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26:5360–5367. doi: 10.1200/JCO.2008.17.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zalcberg JR, Verweij J, Casali PG, et al. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer. 2005;41:1751–1757. doi: 10.1016/j.ejca.2005.04.034. [DOI] [PubMed] [Google Scholar]
- 143.Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–1103. doi: 10.1016/j.ejca.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 144.Gastrointestinal stromal tumor meta-analysis group comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 Patients. J Clin Oncol. 2010;28:1247–1253. doi: 10.1200/JCO.2009.24.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42–47. doi: 10.1002/jso.21160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McAuliffe JC, Hunt KK, Lazar AJ, et al. A randomized, phase II study of preoperative plus postoperative imatinib in GIST: evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Ann Surg Oncol. 2009;16:910–919. doi: 10.1245/s10434-008-0177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Eisenberg BL, Judson I. Surgery and imatinib in the management of GIST: emerging approaches to adjuvant and neoadjuvant therapy. Ann Surg Oncol. 2004;11:465–475. doi: 10.1245/ASO.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 148.Gold JS, Dematteo RP. Combined surgical and molecular therapy: the gastrointestinal stromal tumor model. Ann Surg. 2006;244:176–184. doi: 10.1097/01.sla.0000218080.94145.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.DeMatteo RP, Lewis JJ, Leung D, et al. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231:51–58. doi: 10.1097/00000658-200001000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dematteo RP, Antonescu CR, Chadaram V, et al. Adjuvant imatinib mesylate in patients with primary high risk gastrointestinal stromal tumor (GIST) following complete resection: safety results from the U.S. Intergroup phase II trial ACOSOG Z9000 [abstract] J Clin Oncol. 2005;23(Suppl 1) Abstract 9009. [Google Scholar]
- 151.Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373:1097–1104. doi: 10.1016/S0140-6736(09)60500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Guilhot F. Indications for imatinib mesylate therapy and clinical management. Oncologist. 2004;9:271–281. doi: 10.1634/theoncologist.9-3-271. [DOI] [PubMed] [Google Scholar]
- 153.Trent JC, Patel SS, Zhang J, et al. Rare incidence of congestive heart failure in gastrointestinal stromal tumor and other sarcoma patients receiving imatinib mesylate. Cancer. 2010;116:184–192. doi: 10.1002/cncr.24683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Benjamin RS, Debiec-Rychter M, Le Cesne A, et al. Gastrointestinal stromal tumors II: medical oncology and tumor response assessment. Semin Oncol. 2009;36:302–311. doi: 10.1053/j.seminoncol.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 155.Maleddu A, Pantaleo MA, Nannini M, et al. Mechanisms of secondary resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumours (review) Oncol Rep. 2009;21:1359–1366. doi: 10.3892/or_00000361. [DOI] [PubMed] [Google Scholar]
- 156.Heinrich MC, McArthur GA, Demetri GD, et al. Clinical and molecular studies of the effect of imatinib on advanced aggressive fibromatosis (desmoid tumor) J Clin Oncol. 2006;24:1195–1203. doi: 10.1200/JCO.2005.04.0717. [DOI] [PubMed] [Google Scholar]
- 157.Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 158.George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45:1959–1968. doi: 10.1016/j.ejca.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 159.Chu D, Lacouture ME, Weiner E, Wu S. Risk of hand-foot skin reaction with the multitargeted kinase inhibitor sunitinib in patients with renal cell and non-renal cell carcinoma: a meta-analysis. Clin Genitourin Cancer. 2009;7:11–19. doi: 10.3816/CGC.2009.n.002. [DOI] [PubMed] [Google Scholar]
- 160.Zhu X, Stergiopoulos K, Wu S. Risk of hypertension and renal dysfunction with an angiogenesis inhibitor sunitinib: systematic review and meta-analysis. Acta Oncol. 2009;48:9–17. doi: 10.1080/02841860802314720. [DOI] [PubMed] [Google Scholar]
- 161.Chu TF, Rupnick MA, Kerkela R, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007;370:2011–2019. doi: 10.1016/S0140-6736(07)61865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Torino F, Corsello SM, Longo R, et al. Hypothyroidism related to tyrosine kinase inhibitors: an emerging toxic effect of targeted therapy. Nat Rev Clin Oncol. 2009;6:219–228. doi: 10.1038/nrclinonc.2009.4. [DOI] [PubMed] [Google Scholar]
- 163.Sepe PS, Moparty B, Pitman MB, et al. EUS-guided FNA for the diagnosis of GI stromal cell tumors: sensitivity and cytologic yield. Gastrointest Endosc. 2009;70:254–261. doi: 10.1016/j.gie.2008.11.038. [DOI] [PubMed] [Google Scholar]
- 164.Van den Abbeele AD. The lessons of GIST—PET and PET/CT: a new paradigm for imaging. Oncologist. 2008;13(Suppl 2):8–13. doi: 10.1634/theoncologist.13-S2-8. [DOI] [PubMed] [Google Scholar]
- 165.Van Den Abbeele AD, Badawi RD, Manola J, et al. Effects of cessation of imatinib mesylate (IM) therapy in patients (pts) with IM-refractory gastrointestinal stromal tumors (GIST) as visualized by FDG-PET scanning [abstract] J Clin Oncol. 2004;22(Suppl 1) Abstract 3012. [Google Scholar]
- 166.Blay JY, Le Cesne A, Ray-Coquard I, et al. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol. 2007;25:1107–1113. doi: 10.1200/JCO.2006.09.0183. [DOI] [PubMed] [Google Scholar]
- 167.Dematteo RP, Gold JS, Saran L, et al. Tumor mitotic rate, size, and location independently predict recurrence after resection of primary gastrointestinal stromal tumor (GIST) Cancer. 2008;112:608–615. doi: 10.1002/cncr.23199. [DOI] [PubMed] [Google Scholar]
- 168.Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localised primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045–1052. doi: 10.1016/S1470-2045(09)70242-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Raut CP, Posner M, Desai J, et al. Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitors. J Clin Oncol. 2006;24:2325–2331. doi: 10.1200/JCO.2005.05.3439. [DOI] [PubMed] [Google Scholar]
- 170.Wiebe L, Kasza KE, Maki RG, et al. Activity of sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitinib (SU)-resistant (RES) gastrointestinal stromal tumors (GIST): a phase II trial of the University of Chicago Phase II Consortium [abstract] J Clin Oncol. 2008;26(Suppl 1) Abstract 10502. [Google Scholar]
- 171.Blay JY, Casali PG, Reichardt P, et al. A phase I study of nilotinib alone and in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors (GIST): study update [abstract] J Clin Oncol. 2008;26(Suppl 1) Abstract 10553. [Google Scholar]
- 172.Demetri GD, Lo Russo P, MacPherson IR, et al. Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin Cancer Res. 2009;15:6232–6240. doi: 10.1158/1078-0432.CCR-09-0224. [DOI] [PubMed] [Google Scholar]
- 173.Fumagalli E, Coco P, Morosi C, et al. Rechallenge with imatinib in GIST patients resistant to second or third line therapy. Presented at the 2009 Connective Tissue Oncology Society 15th Annual Meeting; November 5–7, 2009; Miami Beach, Florida. Abstract 39404. References 174–201 are cited in the section on “Desmoid Tumors (Fibromatoses),” and are available in the version of these guidelines online at www.NCCN.org. [Google Scholar]