

Abstract
Despite considerable progress in identifying causal genes for lipodystrophy syndromes, the molecular basis of some peculiar adipose tissue disorders remains obscure. In an Israeli–Arab pedigree with a novel autosomal recessive, multiple symmetric lipomatosis (MSL), partial lipodystrophy and myopathy, we conducted exome sequencing of two affected siblings to identify the disease-causingmutation. The 41-year-old female proband and her 36-year-old brother reported marked accumulation of subcutaneous fat in the face, neck, axillae, and trunk but loss of subcutaneous fat from the lower extremities and progressive distal symmetric myopathy during adulthood. They had increased serum creatine kinase levels, hypertriglyceridemia and low levels of high-density lipoprotein cholesterol. Exome sequencing identified a novel homozygous NC_000019.9:g.42906092C>A variant on chromosome 19, leading to a NM_005357.3:c.3103G>T nucleotide change in coding DNA and corresponding p.(Glu1035*) protein change in hormone sensitive lipase (LIPE) gene as the disease-causing variant. Sanger sequencing further confirmed the segregation of the mutation in the family. Hormone sensitive lipase is the predominant regulator of lipolysis from adipocytes, releasing free fatty acids from stored triglycerides. The homozygous null LIPE mutation could result in marked inhibition of lipolysis from some adipose tissue depots and thus may induce an extremely rare phenotype of MSL and partial lipodystrophy in adulthood associated with complications of insulin resistance, such as diabetes, hypertriglyceridemia and hepatic steatosis.
Keywords: multiple symmetric lipomatosis, lipodystrophy, hormone sensitive lipase, myopathy, insulin resistance
INTRODUCTION
Familial partial lipodystrophies (FPLD) are characterized by selective loss of body fat from the extremities and predisposition to complications of insulin resistance [Garg, 2011]. In the last two decades, considerable progress has been made in identifying several causal genes, such as LMNA, PPARG, PLIN1, AKT2, and CIDEC; and more recently, LIPE, for FPLD [Patni and Garg, 2015]. Despite this progress, the molecular genetic basis of some patients with peculiar body fat distribution still remains obscure. Therefore, in this study, we investigated the underlying basis of excess fat accumulation in the cervical and truncal region suggestive of multiple symmetric lipomatosis (MSL), partial lipodystrophy of the legs, and myopathy in a consanguineous Israeli–Arab pedigree using exome sequencing approach.
MATERIALS AND METHODS
The protocol was approved by the Institutional Review Board of UT Southwestern; and all the patients and their family members gave the written informed consent. Clinical features, metabolic parameters and anthropometric variables of the three affected adults and their parents and siblings, from MSL 36 pedigree, are described below and summarized in Table I.
TABLE I.
Metabolic Parameters and Anthropometric Variables of the Affected and Unaffected Subjects From the Family
| Affected
|
Unaffected
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Reference | |||||||||
| ID | MSL36.3 | MSL36.4b | MSL36.9c | MSL36.1 | MSL 36.2a | MSL36.5 | MSL36.6 | MSL36.7 | MSL36.8 | |
| Genotype | Mut/Mut | Mut/Mut | Mut/Mut | Mut/WT | Mut/WT | WT/WT | Mut/WT | Mut/WT | Mut/WT | |
| Age (y)/sex | 41/F | 36/M | 23/M | 70/M | 62/F | 45/F | 43/M | 38/M | 29/F | |
| Height (m) (%ile) | 1.59 (55) | 1.70 (45) | 1.68 (35) | 1.65 (21) | 1.65 (83) | 1.68 (92) | 1.73 (61) | 1.83 (95) | 1.64 (79) | |
| Weight (kg) | 56 | 102 | 66 | 92 | 84 | 75 | 71 | 85 | 59 | |
| BMI (kg/m2) | 22.1 | 35.3 | 23.4 | 33.8 | 30.9 | 26.6 | 23.7 | 25.4 | 21.9 | |
| Total-C | 179 | 220 | 124 | 167 | 142 | 140 | 131 | 145 | 132 | 125–200 mg/dL |
| HDL-C | 33 | 56 | 36 | 28 | 72 | 39 | 36 | 52 | 57 | 40 mg/dL |
| Triglycerides | 531d | 240e | 380 | 390 | 149 | 198 | 105 | 95 | 80 | <150 mg/dL |
| Glucose | 144 | 76 | 80 | 242 | 96 | 75 | 83 | 63 | 107 | 65–99 mg/dL |
| HbA1c | 6.2 | 5.3 | 5.7 | 7.1 | 6.2 | 5 | 5.3 | 5.4 | 5 | <5.7% |
| AST | 40 | 38 | 45 | 15 | 36 | 37 | 27 | 25 | 25 | 10–40 U/L |
| ALT | 32 | 50 | 66 | 18 | 38 | 46 | 23 | 33 | 25 | 9–46 U/L |
| GGT | 34 | 97 | 32 | 96 | 72 | 28 | 30 | 18 | 24 | 3–70 U/L |
| Uric acid | 7.9 | 7.4 | 8.7 | 9.9 | 6.7 | 6.2 | 3.8 | 5.8 | 5 | 4.0–8.0 mg/dL |
| Creatine kinase | 685 | 771 | 446 | 40 | 40 | 49 | 71 | 75 | 165 | 44–196 U/L |
Mut, mutation; WT, wild-type allele; AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, gamma-glutamyl transferase; M, male; F, female; C, cholesterol; HDL-C, high density lipoprotein-cholesterol; HbA1c, hemoglobin A1c; BMI, body mass index.
Nonfasting values.
On simvastatin 20 mg daily, enalapril 10 mg daily, and omeprazole 20 mg daily.
On simvastatin 40 mg daily.
Non-fasting level 1895 mg/dL.
Non fasting level 894 mg/dL.
Height centiles calculated from https://tall.life/height-percentile-calculator-age-country/ for Jordan to match for Arabic ancestry.
CLINICAL REPORTS
MSL 36.3
This 41-year-old female proband noted pain and weakness in the hands at age 30, followed by leg weakness, progressing gradually from the feet to thighs. Serum creatine kinase level was 631 U/L. She also had hypertension and bilateral pes cavus. She had excessive fat accumulation in the face and neck and in the armpits and groins but loss of subcutaneous fat and muscles from the extremities (Fig. 1A and B). In addition, hypopigmented lesions were noticed on the fingers and ankles and psoriatic lesions on elbows (Supplementary Fig. S1A and B). At age 32, a right deltoid muscle biopsy revealed muscle atrophy (Fig. 1C and Supplementary Fig. S1C). Magnetic resonance imaging (MRI) of the neck at the age 36 showed symmetric accumulation of fat at the posterior neck and back (Fig. 1D–F). A dual energy X-ray absorptiometry (DEXA) scan revealed total fat of 22.9% with increased upper extremities fat (24.6%) and truncal fat (24.1%) but reduced lower extremities fat (17.1%) compared to normal values (Supplementary data).
FIG. 1.
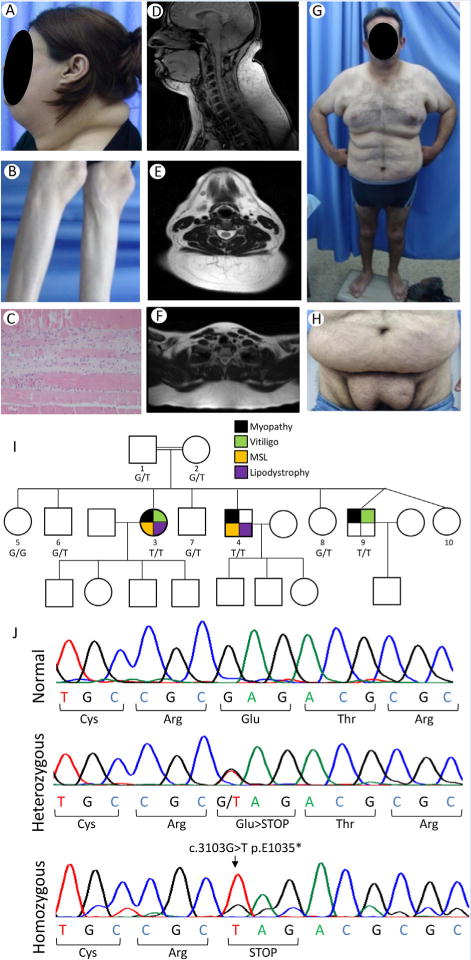
Phenotypic manifestations of our patients, pedigree and electropherograms showing LIPE mutations. A: Lateral view of the head and neck of the patient MSL 36.3 showing marked increase in facial, submental and dorsocervical fat. B: Lateral view of the lower extremities of the patient MSL 36.3 showing loss of subcutaneous fat from the calves and loss of muscle mass. C: Photomicrograph of the deltoid muscle biopsy of MSL 36.3 showing atrophic muscle fibers and regenerating basophilic skeletal muscle cells surrounded by fibrosis. D: T-1 weighted midline sagittal magnetic resonance image of the head and neck of MSL 36.3 showing excess fat accumulation in the anterior and posterior neck region, which measured 6 cm at the paravertebral muscle at the level of cervical five vertebra. E: T-1 weighted axial magnetic resonance image of the neck of MSL 36.3 showing excess fat accumulation in the posterior neck region as well as in the anterior neck. F: T-1 weighted midline axial magnetic resonance image of the upper chest of MSL 36.3 showing excess fat accumulation in the posterior region. G: Anterior view of the patient MSL 36.4 showing marked excess fat accumulation in the anterior neck, upper arms, chest and abdomen. H: Anterior view of the patient MSL 36.4 showing excess fat accumulation in the lower abdomen, pubic and inguinal region. I: MSL 36 Pedigree with consanguinity among parents. Filled symbols indicate affected subjects and unfilled symbols unaffected subjects. The genotypes are indicated under the symbols: G/G wild-type; G/T heterozygotes; T/T homozygotes. Circles denote females and squares denote males. Black color in symbols indicates presence of myopathy; green color, vitiligo; yellow color multiple symmetric lipomatosis (MSL) phenotype; and purple, lipodystrophy of the lower extremities. J: Electropherograms of the part of exon 10 of LIPE showing normal sequence, heterozygous and homozygous c.3013G>T mutation resulting in stop codon at position 1,035 instead of glutamic acid residue. [Color figure can be viewed at wileyonlinelibrary.com].
MSL 36.4
This 36-year-old male was obese since age 15. He is a chronic smoker since age 23 and developed hypertension and hyperlipidemia at age 20. He had fat accumulation in the upper body and symmetrical lipomatous deposits in the armpits and pubic area (Fig. 1G and H). Marked loss of subcutaneous fat, and weakness and muscular atrophy were noticed in the lower limbs, especially in the calves. The facial and anterior cervical fat was normal. He had bilateral pes cavus and acanthosis nigricans. Despite repeated resection of fat tissue from the axillae and pubis, it re-accumulated. Adipose tissue was normal on histology. An echocardiogram at age 33 demonstrated mild mitral regurgitation and diastolic dysfunction. An abdominal ultrasound revealed a normal size liver with mild steatosis. Electromyography at age 35 revealed myopathy affecting mainly distal legs. A DEXA scan at the age 33 revealed total fat of 35.4% with increased upper extremities (45.2%) and truncal fat (37.2%) but reduced lower extremities fat (27.2%) compared to normal values (Supplementary data).
MSL 36.9
This 23-year-old male reported muscle wasting of the lower extremities and difficulty in climbing stairs recently. He had mild lordosis of the lumbar spine, bilateral thinning of the calves and thighs, bilateral pes cavus, and plantar flexion of all the toes. His body fat distribution appeared to be normal. He had diffuse hypopigmented lesions on the dorsum of all the fingers (Supplementary Fig. S1D) and psoriatic lesions on the elbows.
Sanger Sequencing
DNA of the proband, MSL 36.3, was Sanger sequenced for LMNA. Exon 10 of LIPE containing the candidate variant was Sanger sequenced using ABI Prism 3100.
Exome Sequencing (ES)
ES was performed on DNA of MSL 36.3 and 36.4 using the SureSelect Human All Exon V4 kit on the Illumina platform. Given the parental consanguinity (Fig. 1I), we hypothesized that a homozygous mutation shared by the two affected subjects was most likely and therefore filtered for rare missense, nonsense, splicing or frameshift variants with minor allele frequency (MAF) <0.1% in the 1000 Genomes Project and Exome Aggregation Consortium (ExAC) database and GERP++ score >2.0.
RESULTS
The two older affected patients had a peculiar body fat distribution that was most consistent with MSL, as well as atypical FPLD. Sanger and ES in the two affected subjects did not reveal any disease-causing variants in LMNA, PPARG, PLIN1, AKT2, CIDEC, WRN, PCYT1A, mtDNA, and MFN2. ES revealed five potentially pathogenic homozygous mutations using the filters (Supplementary Table SI), including a novel NC_000019.9:g.42906092C>A variant in hormone sensitive lipase (HSL)—encoding gene, LIPE, on chromosome 19; NM_005357.3:c.3103G>T; p.(Glu1035*) (Fig. 1J) which was considered as the disease-causing. Sanger sequencing confirmed its segregation in the family (Fig. 1I).
The three affected subjects had mild to severe hypertriglyceridemia with mildly elevated aminotransferase and uric acid levels and consistently high creatine kinase levels as compared to the other unaffected siblings (Table I). One of them had diabetes and another one had hepatic steatosis. The father (36.1) had diabetes, hyperlipidemia, coronary heart disease, chronic renal insufficiency and was on hemodialysis. Mother (36.2) had diabetes, hypertension and psoriasis. One sister (36.5) had psoriasis, Crohn’s disease, hypertension and nephrolithiasis. Other unaffected siblings (36.6, 36.7, and 36.8) were healthy.
DISCUSSION
Our report extends the clinical manifestations of HSL deficiency (Supplementary Table SII). Albert et al. [2014] reported three Old Older Amish females and one male, age 42–56, harboring a homozygous c.2300_2318del; p.(Val767Glyfs* 102) LIPE mutation. All four had diabetes, three had mild hypertriglyceridemia and two had hepatic steatosis. Despite normal total body fat in the affected females, increased android and arm fat but reduced gynoid and leg fat was reported. One of them had mildly increased submental fat; another had vitiligo, with little or no fat on the lower extremities. All four patients had normal neurological examination.
Carboni et al. [2014] reported a homozygous variant, c.ins1519CG; p.(Ser508Profs*56) in a 50-year-old female and a 49-year-old male from a consanguineous Italian pedigree. The female had abnormal accumulation of subcutaneous fat in the neck, back, abdomen, clavicular regions, axillae, below the triceps and labia majora, consistent with MSL. However, she also had reduced fat in the legs since age 33 suggestive of FPLD. She had mild proximal muscle weakness. The affected male also had increased fat on the abdomen and axillae and reduced fat in the legs. Both developed diabetes after age 40 and one of them had hypertriglyceridemia. Both of them had high serum creatine kinase levels but the male had normal muscle strength. Muscle biopsy from the affected female showed dystrophic changes.
Our report confirms association of HSL deficiency with progressive adult-onset myopathy and suggests association with an MSL and partial lipodystrophy phenotype and likely with vitiligo. However, why patients with homozygous p.(Val767Glyfs*102) mutation had no myopathy remains unclear. Previously, Carboni et al. [2014] reported late-onset of lipodystrophy and therefore, not surprisingly, the 23-year-old affected male in our study had not developed the lipodystrophy so far. We also see variable association of HSL deficiency with diabetes, hypertriglyceridemia, low HDL cholesterol and hepatic steatosis. The phenotype of heterozygous mutation carriers, however, was not detectable in our pedigree.
HSL is the predominant mediator of the hydrolysis of diglycerides, cholesterol esters, and retinyl esters in human white adipose tissue. LIPE mRNA is widely expressed with high expression in the adipose tissue, breast, adrenal, ovary, testis, heart and skeletal muscle (www.genecards.org). Thus many different organ systems may be affected in HSL deficiency and there may be marked heterogeneity in clinical features.
Albert et al. [2014] reported >80% reduction of triglyceride lipase and cholesterol esterase activities in the subcutaneous abdominal adipose tissue of two HSL deficient subjects with no detectable mutant protein. They also reported increased macrophage infiltration in adipose tissue. In contrast, our patient’s adipose tissue was normal on histology and whether p.(Glu1035*) mutant is expressed in adipocytes remains unclear for lack of availability of adipose tissue from our patients. Interestingly, similar to the HSL deficient mouse phenotype of reduced abdominal fat mass but increased interscapular brown fat [Osuga et al., 2000], we see MSL phenotype overlapping with partial lipodystrophy in HSL-deficient humans. Whether adipocyte hypertrophy in some regions but atrophy in others is related to differential expression, promiscuity or different functions of HSL [Giuseppe et al., 2012], remains unclear. However, in contrast to normoglycemia, reduced hepatic triglyceride levels and azoospermia in HSL-deficient mice [Osuga et al., 2000], humans with HSL-deficiency develop diabetes, hypertriglyceridemia and hepatic steatosis, and have normal fertility [Albert et al., 2014; Carboni et al., 2014; Farhan et al., 2014].
Finally, we conclude that HSL deficiency in humans is associated with adult-onset, progressive, MSL, lower extremity lipodystrophy, and myopathy along with metabolic abnormalities such as diabetes, hypertriglyceridemia and hepatic steatosis.
Supplementary Material
Acknowledgments
We thank Frank M. Vuitch, M.D., for reviewing the muscle biopsy photomicrographs, Pei-Yun Tseng, B.S., for illustrations and mutational screening; and the McDermott Center Sequencing and Bioinformatics Cores for sequencing and analysis.
Grant sponsor: National Institutes of Health; Grant numbers: R01-DK105448, UL1TR001105; Grant sponsor: Southwest Medical Foundation.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- Albert JS, Yerges-Armstrong LM, Horenstein RB, Pollin TI, Sreenivasan UT, Chai S, Blaner WS, Snitker S, O’Connell JR, Gong DW, Breyer RJ, 3rd, Ryan AS, McLenithan JC, Shuldiner AR, Sztalryd C, Damcott CM. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N Engl J Med. 2014;370:2307–2315. doi: 10.1056/NEJMoa1315496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboni N, Brancati F, Cocco E, Solla E, D’Apice MR, Mateddu A, McIntyre A, Fadda E, Mura M, Lattanzi G, Piras R, Maioli MA, Marrosu G, Novelli G, Marrosu MG, Hegele RA. Partial lipodystrophy associated with muscular dystrophy of unknown genetic origin. Muscle Nerve. 2014;49:928–930. doi: 10.1002/mus.24157. [DOI] [PubMed] [Google Scholar]
- Farhan SM, Robinson JF, McIntyre AD, Marrosu MG, Ticca AF, Loddo S, Carboni N, Brancati F, Hegele RA. A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can J Cardiol. 2014;30:1649–1654. doi: 10.1016/j.cjca.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Garg A. Clinical review#: Lipodystrophies: Genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96:3313–3325. doi: 10.1210/jc.2011-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuseppe M, Luigia M, Elena P, Yan F, Luigi M. Enzyme promiscuity in the hormone-sensitive lipase family of proteins. Protein Pept Lett. 2012;19:144–154. doi: 10.2174/092986612799080400. [DOI] [PubMed] [Google Scholar]
- Osuga J, Ishibashi S, Oka T, Yagyu H, Tozawa R, Fujimoto A, Shionoiri F, Yahagi N, Kraemer FB, Tsutsumi O, Yamada N. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc Natl Acad Sci USA. 2000;97:787–792. doi: 10.1073/pnas.97.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patni N, Garg A. Congenital generalized lipodystrophies-new insights into metabolic dysfunction. Nat Rev Endocrinol. 2015;11:522–534. doi: 10.1038/nrendo.2015.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.