

Abstract
The most common solid tumors show intrinsic multidrug resistance (MDR) or inevitably acquire suchwhen treated with anticancer drugs. In this work, we describe the discovery of a peripherally restricted, potent, competitive NMDA receptor antagonist 1l by a structure-activity-study of the broad-acting ionotropic glutamate receptor antagonist 1a. Subsequently, we demonstrate that 1l augments the cytotoxic action of sorafenib in murine hepatocellular carcinoma (HCC) cells. The underlying biological mechanism was shown to be interference with the lipid signaling pathway, leading to reduced expression of MDR transporters and therebyan increased accumulation of sorafenib in the cancer cells. Interference with lipid signaling pathwaysby NMDA receptor inhibition is a novel and promising strategy for reversing transporter-mediated chemoresistance in cancer cells.
Graphical abstract
Introduction
Over time, solid tumors inevitably acquire resistance against anticancer therapy - a phenomenon known as multi-drug resistance (MDR).1 But more devastatingare cancers, such as hepatocellular carcinoma (HCC), which exertsintrinsic drug-resistance.2 A key mechanism underlyingMDR is increased expression of ATP-binding cassette (ABC) transporters, whichexpel a broad range of chemotherapeutic agents from the cancer cells.3 According to current knowledge, the principal ABC transporters responsible for chemoresistance in humans areATP-binding cassette subfamily B member 1 (ABCB1, Pgp), ATP-binding cassette subfamily G member 2 (ABCG2, BCRP) and ATP-binding cassette subfamily C (ABCC, MRP).4–7 In regard to HCC, these transporters are responsible for expelling the first line HCC drug sorafenib from the cancer cells.8–10 The general strategy to reverseMDR has been to co-administer ABC transporter inhibitors with anticancer drugs.11 However, several complications limit the use of ABC transporter inhibitors, amongst them the fact that ABC transporters are also present in normal cells, leading to undesired drug accumulation.11 Thus, a change of strategy is needed to overcome ABC transporter-mediated chemoresistance.
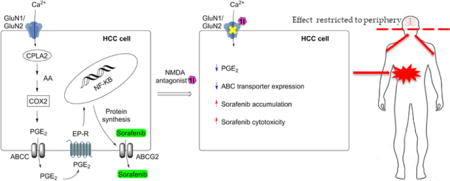
One such strategy is to down-regulate efflux transporter expression by alternating the cell lipid signaling pathwayby blocking theN-methyl-D-aspartate (NMDA) receptors.12 NMDA receptors are a subclass of ionotropic glutamate receptors (iGluRs), which also comprises the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and the kainic acid (KA) receptors.13 NMDA receptor activation increases calcium entry, whichactivates cytoplasmic phospholipase A2 (cPLA2), leading to increased production of arachidonic acid (Figure 1).14,15 This fatty acid is further converted to the proinflammatory lipid, prostaglandin-E2 (PGE2) and actively transported into the extracellular space by ABCC transporters, where it binds to the prostaglandin E receptors (EP-Rs), leading to NF-κB activation and creating a positive feedback loop creating an inflammation microenvironment.12,14 It has been shown that NF-κB activation results in increased cancercell survival and proliferationas well aschemoresistance due to elevated ABC transporterand CYP enzyme expression.12,16,17
Figure 1.
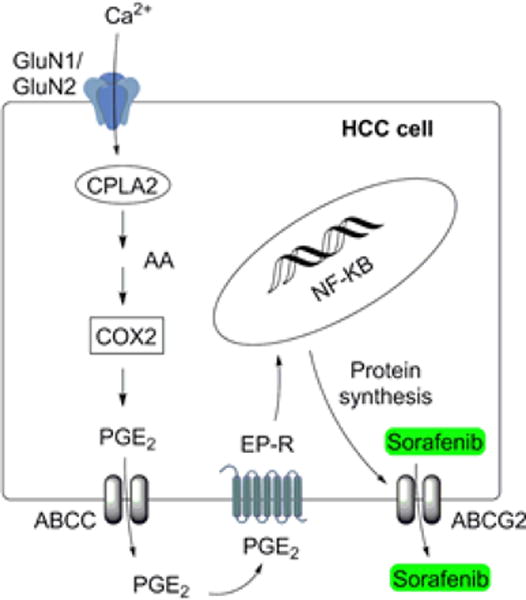
Activation of the lipid signaling pathway by GluN1/GluN2A receptor and the creation of transporter mediated drug resistance. NMDA binding to the GluN1/GluN2A receptor allows the influx of Ca2+ into the cell, which leads to cPLA2 activation and the release of arachidonic acid (AA) from phospholipids. Arachidonic acid is metabolized to PGE2, which is transported by ABCC transporters out of the cell allowing the PGE2 binding to EP-R. The EP-R activation leads to nuclear translocation of NF-κB followed by ABCG2 protein transcription and MDR.12,14–17
NMDA receptors are over-expressed on the cell membrane of many types of cancer cells, including HCC.18 The non-competitive NMDA receptor antagonist MK-801 (Figure 2) has previously been shown to augment the antiproliferative efficacy of antiestrogens in melanoma cells19 and suppress the growthof HCC cells.20 The antiproliferative effect of MK-801 in HCC cells was shown to be mediated through the FOXO/XTNIP pathway, which is not connected to pro-inflammatory lipid signaling. Moreover, the impact of NMDA receptor antagonists on transporter expression, anticancer drug accumulation, and efficacy in cancers has not been studied. Most importantly, a competitiveNMDA receptor antagonist for use as an augmentative drug for the treatment of peripheral solid tumors must notbe capable ofcrossing the blood-brain barrier (BBB), as this would otherwise lead tosevere adverse effects, including psychosis.21
Figure 2.
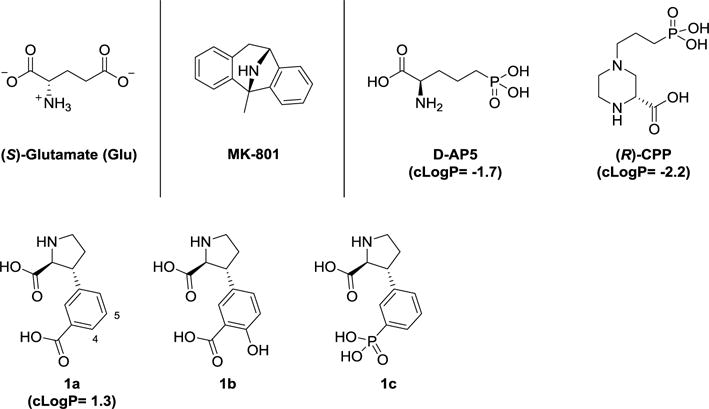
Chemical structures of Glu, selective un-competitive NMDA receptor antagonist MK-801, selective competitive NMDA receptor antagonists D-AP5, (R)-CPP, and competitive iGluR antagonist 1a, including published analogs 1b,c with relevanceto SAR study reported herein.
In the present study, we report the design and synthesis of the novel peripherally acting potent NMDA receptor antagonist 1l. We describe the ability of 1l to modulate the cPLA2 activation-dependent lipid signaling pathway, downregulate ABC transporter expression andthereby augmentthe cytotoxic efficacy of the anticancer drug sorafenib in murine HCC cells.
Results and Discussion
The two commonly studied competitive NMDA receptor antagonists which do not penetrate the BBB are D-AP5 and (R)-CPP (Figure 2). Both are amino acid analogs and highly polar with calculated partition coefficient values in octanol:water (cLogP(o/w)) well below zero (−1.7 and −2.2, respectively). Incomparisonwith recommended cLogP(o/w) values of −0.4 to 5.622 for oral bioavailability, we believed that these NMDA receptor antagonists were not attractive candidates for this study.
We therefore turned to the previously reported non-selective iGluR antagonist 1a (Figure 2 and Table 1).23 The fact that 1a does not penetrate the BBB in mouse in situ brain perfusion and that its cLogP(o/w) value is calculated at 1.3, makes it an attractive starting point for the purpose of developing orally available, peripherally restricted, competitive NMDA receptor antagonist.22
Table 1.
Binding affinities of 1a-n at native AMPA, KA and NMDA receptors (rat synaptosomes), and cloned homomeric receptors GluK1-3.
| Cmpd No |
|
AMPA IC50 (μM) |
KA IC50 (μM) |
NMDA Ki (μM) |
GluK1 Ki (μM) |
GluK2 Ki (μM) |
GluK3 Ki (μM) |
|---|---|---|---|---|---|---|---|
| 1a23 |
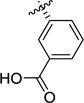
|
51 | 22 | 6.0 | 4.3 | >100 | 8.1 |
| 1b24 |
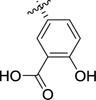
|
2.0 | 1.4 | 1.0 | 4.8 | 10–100 | 0.87 |
| 1c24 |
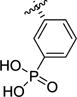
|
>100 | >100 | >100 | 126 | >1000 | 78 |
| 1d |
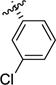
|
>100 | >100 | >100 | >100 | >100 | >100 |
| 1e |
|
>100 | >100 | >100 | >100 | >100 | >100 |
| 1f |
|
>100 | >100 | >100 | – | – | – |
| 1g |
|
>100 | >100 | >100 | >1000 | >1000 | >1000 |
| 1h |
|
>100 | >100 | >100 | >100 | >100 | >100 |
| 1i |
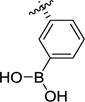
|
>100 | >100 | >100 | >1000 | >1000 | >100 |
| 1j |
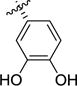
|
>100 | >100 | 35 [ 4.46 ± 0.04] |
– | – | – |
| 1k |
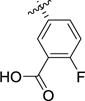
|
> 100 | 59 [4.23 ± 0.05] |
4.6 [5.34 ± 0.04] |
12 ± 0.5 |
>100 | 11 ± 0.97 |
| 1l |
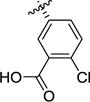
|
>100 | >100 | 0.63 [6.22 ± 0.10] |
154 ± 13 |
>100 | 131 ± 13 |
| 1m |
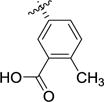
|
>100 | >100 | 17 [4.78 ± 0.04] |
>100 | >100 | >100 |
| 1n |
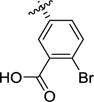
|
>100 | >100 | 0.62 [6.22 ± 0.07] |
> 100 | > 100 | > 100 |
–: not tested. Radioligands: AMPA, [3H]AMPA; KA, [3H]KA; NMDA, [3H]CGP-39653; GluK1, [3H]SYM2081; GluK2 and GluK3, [3H]KA. Data are mean values of three to six individual experiments performed in triplicate. For AMPA and KA: pIC50 values with SEM in brackets. For NMDA: pKi values with SEM in brackets.
We have previously reported a first structure-activity-relationship (SAR) study on 1a, which disclosed the 4-position on the aryl ring as a hotspot for induction of KA receptor subtype selectivity (compound 1b, Figure 2 and Table 1). Also, it was seen that simple lipophilic substituents in the 5-position did not lead to any significant improvement in receptor selectivity or higher affinity (structures not shown).24 The 3-carboxylic acid functionality was displaced with a phosphonic acid group, compound 1c (Figure 2), which led to a significant reduction in binding affinity at the iGluRs (Table 1).24
Withthis SAR information in hand, we set out to investigate two strategies with the aim of improving iGluR class selectivity. Firstly, we wanted to substitute the 3-carboxylic acid functionality with different functional groups (analogs 1d-j). While it is well-accepted that the γ-carboxylate functionality in Glu is mandatory for agonist activity at the iGluRs,13 it remains an open question if non-ionizable distal functional groups can stabilize the NMDA receptor in itsopen antagonist state. Based on synthetic tractability, the following seven analogs were thus designed: 3-chloro (1d), 3-trifluoromethyl (1e), 3-amino (1f), 3-cyano (1g), 3-carbamido (1h), 3-boronic acid (1i), and 3,4-dihydroxy (1j). The synthesis of 1d-j was carried out by a stereoselective rhodium(I)-catalyzed addition of an arylboronic acid to protected enone 224 as the key step (Scheme 1).25 Hereby, the 2,3-trans stereochemistry on the proline ring was set, and subsequent functional group transformations in accordance with earlier reported strategies (1d-f; Scheme 2), (1g,h; Scheme 3), (1i;Scheme 4) and (1j; Scheme 5), gave the free amino acids 1d-j ready for pharmacological evaluation.23,24
Scheme 1.
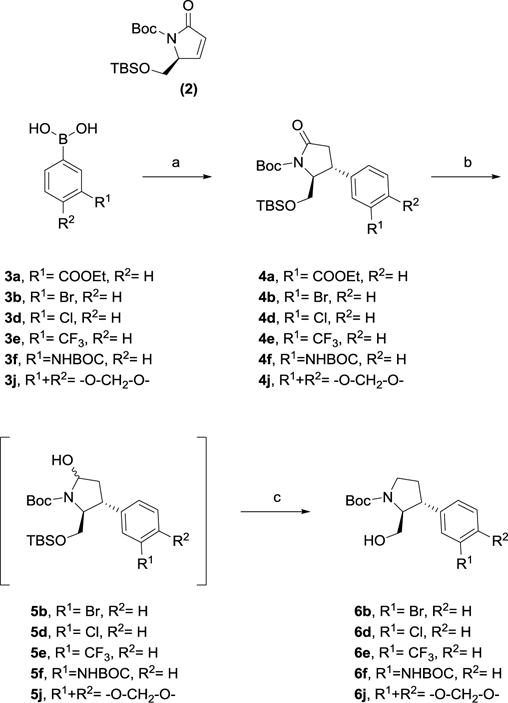
Synthesis of key alcohol intermediates 6b,d-f,j via rhodium(I)-catalyzed 1–4 addition of respective boronic acids 3a,b,d-f,j to enone 2.
Reagents and conditions. a) [Rh(cod)Cl]2, H2O, Cs2CO3, enone 2, THF or dioxane, rt (52–67%). b) LiBEt3H, THF, −78°C. c) HSiEt3, BF3•Et2O, DCM, −78°C (31–78%).
Scheme 2.
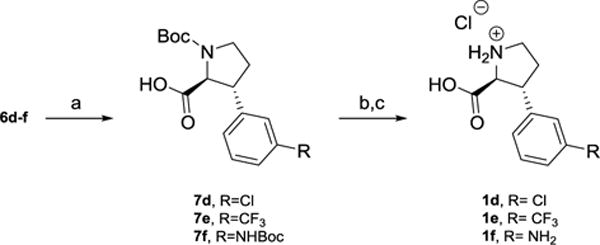
Synthesis of1d-f from alcohols6d-f, respectively
Reagents and conditions. a) RuCl3•xH2O, NaIO4, EtOAc:MeCN:H2O (49–86%). b) TFA, DCM, rt. c) 1 M HCl (12–78%).
Scheme 3.
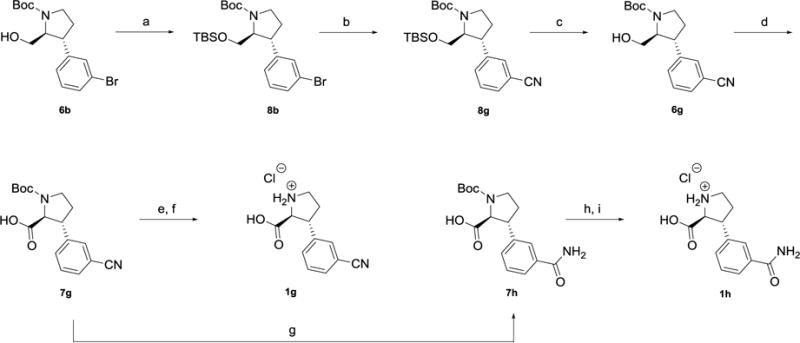
Synthesis of 3-cyano analog 1gfrom alcohol 6b, and3-carbamido analog 1h from7g.
Reagents and conditions. a) TBSCl, imidazole, DMF, rt (73%, three steps from 4b). b) Zn(CN)2, Pd2(dba)3, dppf, DMA, 120°C. c) TBAF, THF, rt (71%, two steps). d) RuCl3•xH2O, NaIO4, EtOAc:MeCN:H2O (76%). e) TFA, DCM, rt. f) 1 M HCl (81%, two steps). g) 30% H2O2, K2CO3, EtOH/H2O, rt (66%). h) TFA, DCM, rt. i) 1 M HCl (59% after three steps).
Scheme 4.
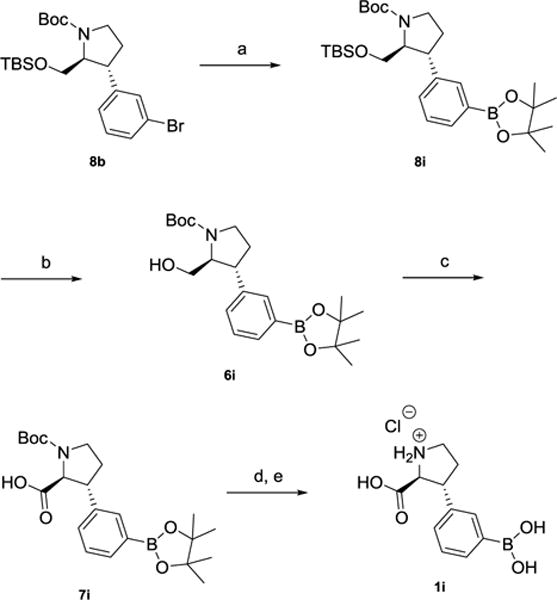
Synthesis of 3-boronic acid analog 1i, starting from bromine8b
Reagents and conditions. a) KOAc, Pd2(dba)3, dppf, DMF (61%). b) TBAF, THF, rt (60%). c) RuCl3•xH2O, NaIO4, EtOAc:MeCN:H2O (65%). d) TFA, DCM, rt. e) 1 M HCl (40%, two steps).
Scheme 5.
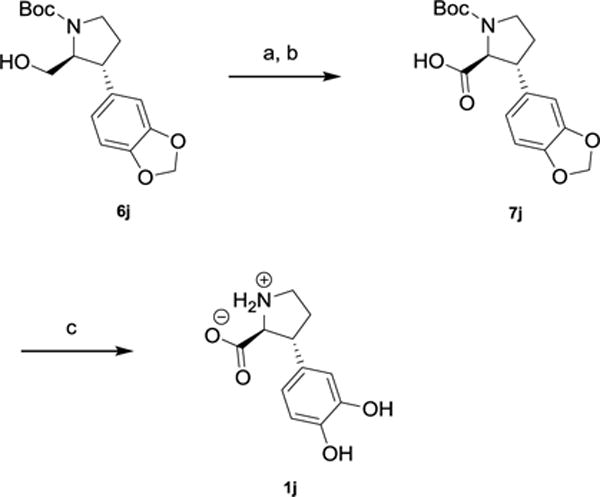
Synthesis of 1j from alcohol 6j.
Reagents and conditions. a) IBX, DMSO, rt. b) NaClO2, NaH2PO4, 2-methyl-2-butene, tert-BuOH/H2O, rt (66%, two steps). c) BBr3, DCM (8% after recrystallization from MeOH).
The second series of analogs, compounds 1k-n, aimed to further explore the impact of substituents in the4-position of the aryl ring. We have previously shown that introduction of a 4-hydroxy group (analog 1b) resulted in generally enhanced affinity for iGluRs with a 10-fold improvement for the GluK3 subunit (Table 1).24 Given the hydrogen bond donor and acceptor abilities of the 4-hydroxyl group, three new analogs were designed to address the influence of these features: 4-fluoro (1k), 4-chloro (1l) and 4-methyl (1m). For 1k and 1l, the syntheses were based on the stereoselective conjugate addition of an in situ formed aryl cuprateto enone 2,24,26 whereas for 1m, the afore applied protocol of rhodium(I)-catalyzed addition of a boronic ester to enone 2 was used (Scheme 6). Subsequent functional group transformations to obtain the free amino acids 1k-m followed the strategy for comparable analogs previously reported by us.23,24
Scheme 6.
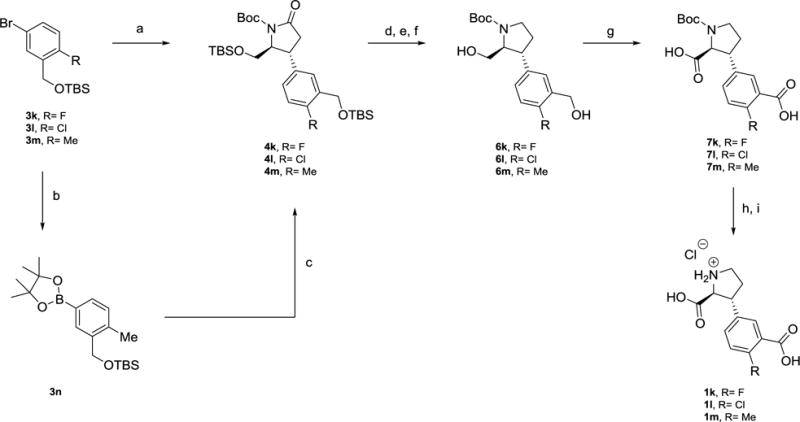
Synthesis of 1k-lvia copper(I) catalyzed addition to enone 2, and 1m via rhodium(I) catalyzed addition to enone 2.
Reagents and conditions. a) for 3k,l: n-BuLi, CuCN, then enone2, Et2O, −78 to −42°C (46% and 73%). b) for 3m: (Bpin)2, KOAc, (PPh3)2PdCl2 (quant). c) [Rh(cod)Cl]2, H2O, Cs2CO3, enone 2, rt (41%). d) for 4k,l: LiBEt3H, THF, −78°C, then HSiEt3, BF3•Et2O, DCM, −78°C. e) for 4m: BH3•SMe2, THF, reflux. f) TBAF, THF, rt. (20% and 49%). g) RuCl3•xH2O, NaIO4, EtOAc:MeCN:H2O (20–97%). h) TFA, DCM, rt. i) 1 M HCl (56–60%).
Binding affinities of the synthesized amino acids 1d-n were determined at native AMPA, KA and NMDA receptors (rat synaptosomes) and cloned rat homomeric subtypes GluK1-3 and results summarized in Table 1. The 3-chloro, 3-trifluoromethyl, 3-amino and 3-cyano analogs, 1d-g respectively, all showed insignificant binding affinity for any of the iGluRs (IC50 or Ki>100 μM). Furthermore, 3-carbamido analog 1h and 3-boronic acid analog 1i did not display notable binding affinities for native iGluRs nor for homomeric GluK1-3 receptors (IC50 or Ki>100 μM). Finally, the 3,4-dihydroxy analog 1j was a weak binder at the NMDA receptors (Ki = 35 μM). While these results were all together disappointing, the affinity profiles for the 4-substituted analogs 1k-m were in contrast exciting. In comparison with the 4-hydroxy analog 1b, the 4-fluoro analog 1k displayed first steps towards selectivity for the NMDA receptors by showing a lower affinity for native AMPA and KA receptors as well as for homomeric GluK1-3 receptors. This trend was boosted significantly for 4-chloro analog 1l, which proved to be selectivefor native NMDA receptors with submicromolarbinding affinity (Ki = 0.63 μM). Also for 4-bromo analog 1n,27 full selectivity for NMDA receptors was observed with binding affinity similar to 1l (Ki = 0.62 μM). In contrast to this important finding, the binding affinity for native NMDA receptors dropped 30-fold for the 4-methyl analog 1m.
The cLogP(o/w) of 1l and 1n were calculated at 1.9 and 2.1, respectively, placing both analogs in the recommended cLogP intervalof −0.4 to 5.6 for good bioavailability.22 Given that an aryl bromide is a less attractive functional group in compounds for biological administration compared to an aryl chloride, compound 1l was selected for further functional studies at NMDA receptors.
Functional characterization of 1l at NMDA receptor subtypes
Compound 1l was next evaluated in a functional assay at the four NMDA receptor subtypes, GluN1/GluN2A-D (Figure 3 and Table 2). At all four subtypes, 1l was found to be a competitive antagonist with Kivalues of 4.7, 10, 24 and 41 μM, respectively, and the selectivity profile of 1l was similar to D-AP5 (Ki values of 0.39, 2.8, 5.9 and 21, respectively28).
Figure 3.
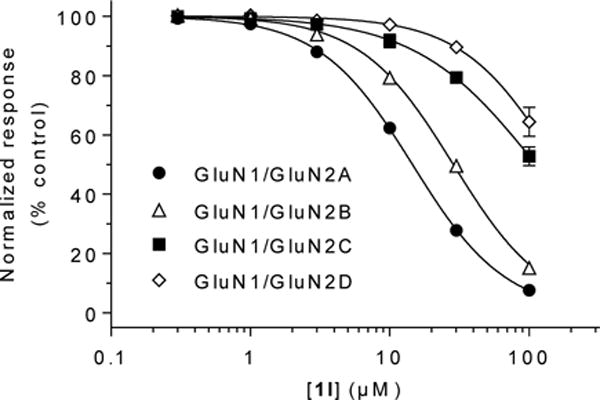
Concentration-inhibition data for1lat recombinant NMDA receptor subtypes GluN1/GluN2A-D. Responses were measured using two-electrode voltage-clamp electrophysiology and were activated by co-application of 100 μM glycine and GlutoXenopusoocytes expressing recombinant NMDA receptors subtypes. 10 μM Glu was used for GluN1/GluN2A, 3 μM Glu for GluN1/GluN2B and GluN1/GluN2C, and 1 μM Glu for GluN1/GluN2D receptors. Data are mean ± SD (error bars are mostly contained within the symbols). See Table 2 for IC50 and estimated Ki values.
Table 2.
Inhibition of recombinant NMDA receptor subtypes by 1l. IC50 values for inhibition of current responses activated by co-application of 100 μM glycine and GlutoXenopusoocytes expressing recombinant rat GluN1/GluN2A-D NMDA receptors. Responses were activated by Glu concentrations 2- to 3-fold higher than the EC50 at the respective NMDA receptor subtypes; 10 μM Glu was used for GluN1/GluN2A, 3 μMGlu for GluN1/GluN2B and GluN1/GluN2C, and 1 μM Glu for GluN1/GluN2D receptors. Ki values were estimated using the Cheng-Prusoff relationship29 andpreviouslydetermined Glu EC50 values30. IC50 and Ki values are mean ± SEM, nH is the Hillslope, and N is the number of oocytes.
| IC50(μM) | nH | Estimated Ki (μM) | N | |
|---|---|---|---|---|
| GluN1/GluN2A | 15 ± 1 | 1.3 | 4.7 ± 0.1 | 7 |
| GluN1/GluN2B | 29 ± 1 | 1.3 | 10 ± 1 | 5 |
| GluN1/GluN2C | 110 ± 8 | 1.0 | 24 ± 2 | 4 |
| GluN1/GluN2D | 170 ± 10 | 1.3 | 41 ± 3 | 7 |
Pharmacokinetics of Compound 1l in Mice
With the attractive pharmacological profileof 1l in hand, we turned to determine its pharmacokinetics and brain permeation. The pharmacokinetic analysis was performed by 10 mg/kg i.p. injection at five time points between 10 and 240 min. The apparent pharmacokinetic parameters, area under the concentration-time curve from time zero to 240 min (AUC0-240 min), the maximum concentration after dosing (Cmax), time to reach Cmax (tmax) and elimination half-life (t½β) in plasma, liver and brain, calculated from the in vivo data are presented in Table 3. The compound was absorbed from the injection site and concentrations above the Ki value were detected from both plasma and liver. The Kpliver/plasma value was 0.19. Importantly, compound 1l did not exhibit BBBpermeation and was detected only at 30 and 60 min time points with concentrations of 0.3 and 0.1 nmol/g, respectively. We confirmed the poor BBBpermeationusing an in situ mouse brain perfusion technique. The brain concentration of compound 1l followingperfusion with 100 μM of the compound was below the detection limit of our analytical method (5 pmol/g). Moreover, there were no observable changes in the behavior of the mice after 1l injection. This supports our findings from the in vivo pharmacokinetic experiments that 1l is a fullyperipherally restricted NMDA receptor antagonistsuitable for affecting peripheral tumors. In addition to hepatocellular carcinoma, NMDA receptor expression has been reported to be elevated in colon, prostate and breast cancers compared to healthy tissues.18 Moreover, the lack of pharmacological effect in the CNS provides the possibility to utilize the compound as a research tool for investigating the role of peripheral NMDA receptors in other diseasesand/or conditions such as chronic pain.
Table 3.
Pharmacokinetic parameters of compound 1l in plasma, liver and brain calculated from in vivo data after a single dose of 10 mg/kg i.p. in mice.
| Plasma | Liver | Brain | |
|---|---|---|---|
| AUC(0–240min) (nmol/g×min) | 3020 | 575 | 13 |
| Cmax (nmol/g) | 43.4 | 8.7 | 0.3 |
| tmax (min) | 15 | 30 | 30 |
| t½β (min) | 30 | 36 | ND |
The ability of compound 1l to inhibit NMDA receptor expressed in murine HCC cells
With compound 1l in hand, we proceeded to studies ofits in vitro efficacy in mice. First, we confirmed the expression of the GluN1 NMDA receptor subunit in the murine HCC cell line by Western immunoblotting (data not shown). In order to assess the function of NMDA receptors in the murine HCC cell culture we used calcium imaging (Figure 4). To compare the abilities of 1l and D-AP5 to inhibit NMDA to activation, we measured the intracellular Ca2+ transients induced by 150 μM NMDA alone and in presence of the compound 1l (25 and 41 μM) or D-AP5 (20 μM). Both compounds were able to inhibit NMDA activation significantly in the murine HCC cells suggesting their ability to antagonise the lipid signaling pathway in cancer cells.
Figure 4.
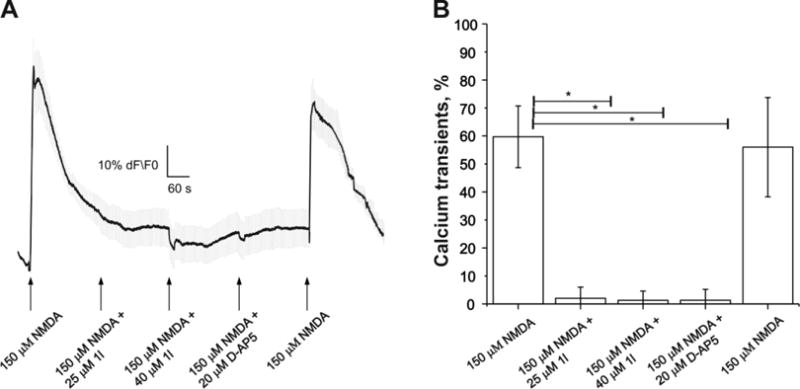
Ca2+ transients via NMDA receptors in murine HCC cells. A) Ca2+ transients in HCC cells activated by NMDA applications (15 s) with and without NMDA receptor antagonists (1l and D-AP5). B) Histogram showing percentage of induced Ca2+ transients in a response to NMDA application and absence of the activation while applied together with 1l at 25 μM 41 μM concentration and D-AP5 at 20 μM concentration. Quantitative data were expressed as mean ± SEM (n=5). The statistical significance was assessed with the Student-paired t test or Mann-Whitney t test for non-parametric data. Statistically significant differences were set at *P < 0.05.
Ability of compound 1l to decrease PGE2 concentration in murine HCC cells
The ability of compound 1l to decreasethe extracellular PGE2 concentration in murine HCC cells was investigated by incubating the cells in 24-well plates for 24 h with 100 μM 1l. In addition, the PGE2 concentration wasmeasured from HCC cells with lipopolysaccharide (LPS) induced inflammation using the same compound 1l concentration and incubation time (Figure 5). Compound 1l significantly decreased the PGE2 concentration in both the presence and absence of LPS, confirming that the compound is able to interfere with the synthesis of PGE2. This inhibition of PGE2 production will reducethe proinflammatory lipid signaling pathway in HCC cells, which has been suggested to down-regulateABC transporters in cancer cells.12,14–16
Figure 5.
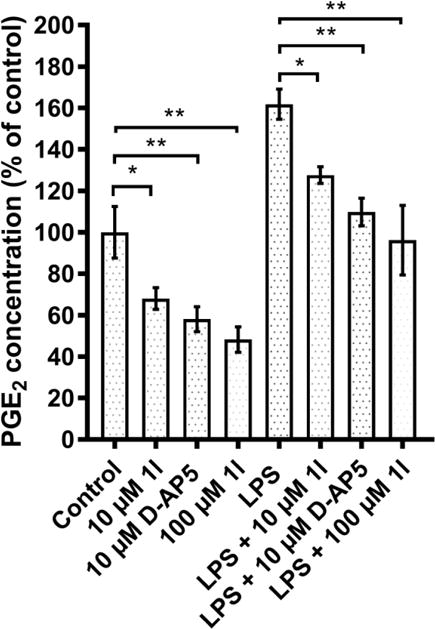
The effect of compound 1l and D-AP5 on PGE2 levels in HCC cells. Compounds 1l and D-AP5 at 10 μM and compound 1l at 100 μM reduced the concentration of PGE2 after 24 h incubation to 68± 5 %, 58 ± 6 % and 48 ± 6%, respectively. The addition of LPS (2.5 μg/mL) increased the PGE2 concentration to 162 ± 7 % %, and the addition of compounds 1l and D-AP5 at 10 μM as well as compound 1l at 100 μM prevented the effect of LPS, reducing the PGE2 concentrations to 127 ± 4 %, 110 ± 7 and 96 ± 17, respectively. The data is presented as mean ± SEM, n=3. The statistical significance of differences in PGE2 concentrations between treatments was determined using One-way ANOVA and Tukey`s test. (*P < 0.05, **P< 0.01).
Effect of the compound 1l on transporter expression in mouse HCC cells
Expression levels of Abcb1, Abcg2, Abcc2 and Abcc4 as well as Slc7a5 and Slc2a1 in HCC cells was determined by selected/multiple reaction monitoring (SRM/MRM) analysis in liquid chromatography-tandem mass spectrometry (LCMS-MS),31 following administration of 10 μM 1l and compared to control. Compound 1l reduced the expression levels of Abcb1 and Abcg2 transporters in the crude membrane fraction by 39% and34%, respectively (Table 4). Abcc4 protein expression was reduced by 14%, but the reduction was not statistically significant and, Abcc2 protein expression was below the lower limit of quantification. Interestingly, the expression of Slc2a1 and Slc7a5 transporter proteins was decreased by 42% and 24%, respectively. However, the difference in the case of Slc7a5 was not statistically significant. In all, these findings evidence that NMDA receptor antagonism results in downregulation of ABC transporter expression in HCC cells. In addition, the expression of the two investigated nutrient transporters was reduced significantly. In order to further study the significance of the transporter downregulation, we investigated the cell accumulation of transporter probes and sorafenib.
Table 4.
TheABC transporter expression levels in mouse HCC cell crude membrane fraction measured by SMR/MRM analysis.
| Protein expression level (fmol/μg of total protein)
|
||||||
|---|---|---|---|---|---|---|
| Abcb1 (Pgp) | Abcg2 (Bcrp) | Abcc2 (Mrp2) | Abcc4 (Mrp4) | Slc2a1 (Glut1) | Slc7a5 (Lat1) | |
| Control | 0.64 ± 0.08 | 1.88 ± 0.04 | a | 0.31 ± 0.04 | 22.7 ± 0.84 | 7.55 ± 0.28 |
| 10 μM 1l | 0.39± 0.06* | 1.24± 0.13** | a | 0.27 ± 0.04 | 13.2± 0.53*** | 5.72± 1.61 |
below the lower limit of quantification; The statistical difference between compound 1l-treated cells and the control cells was determined using unpaired two-tailed t-test (*P < 0.05, **P< 0.01, ***P< 0.001). Data are presented as mean ± SEM (n=4).
Ability of NMDA receptor antagonist 1l to alterthe cell accumulation of transporter probes and sorafenib
To investigatethe ability of compound 1l to reverse ABC transporter mediated MDR and decrease the uptake of essential nutrients in murine HCC cells, we determined the intracellularaccumulation of known ABC transporter substrates:Abcb1 probe [3H]-digoxin,32 Abcc1-5 probe fluorescein,33 Abcb1 and Abcg2 substrate sorafenib (Figure 6) as well as Slc7a5 substrate [14C]-L-leucine and Slc2a1 substrate [14C]-D-glucose (Figure 7).8–10 The cells were incubated with or without 10 μM of compound 1l in the growth medium for 24 h. Accumulation of theefflux probes in the cells was significantly increased after incubating the cells with compound 1l. In addition, the cell uptake of [14C]-L-leucine was reduced significantly. Therefore, the data provides evidence that the reduced transporter expression levels are significant enough to lead to reduced transporter activity. The riskof compound 1l to interfere with transporter function was minimized by washing the cells before uptake experiments. Thus, the increased cell accumulation of transporter probes is not likely due to compound 1l acting as a transporter inhibitor, but as a modulator of transporter expression.
Figure 6.
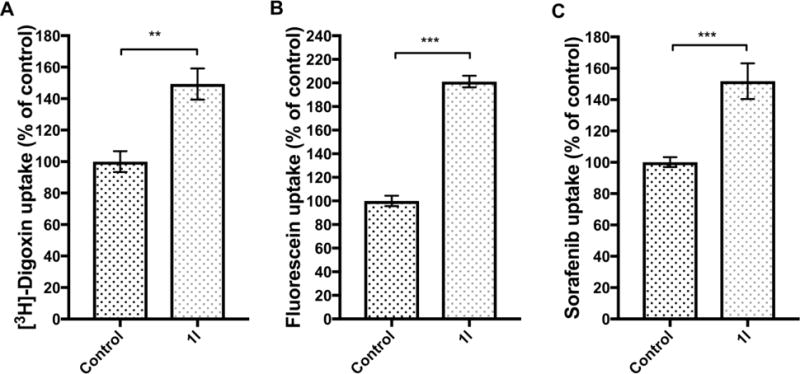
The effect of compound 1l on cell accumulation of efflux probes [3H]-digoxin, fluorescein and sorafenib. The incubation of the HCC cells with 10 μM compound 1l increased the cell accumulation of (A) [3H]-digoxin, (B) fluorescein and (C) sorafenib to 149 % ±10 %, 201 % ±9 % and 152 % ± 11 %, respectively. The analyte concentrations are normalized by the amount of protein in each sample. The statistical difference between compound 1l-treated cell and the control cells was determined using unpaired two-tailed t-test (**P< 0.01, ***P < 0.001). Data are presented as mean ± SEM (n=3).
Figure 7.
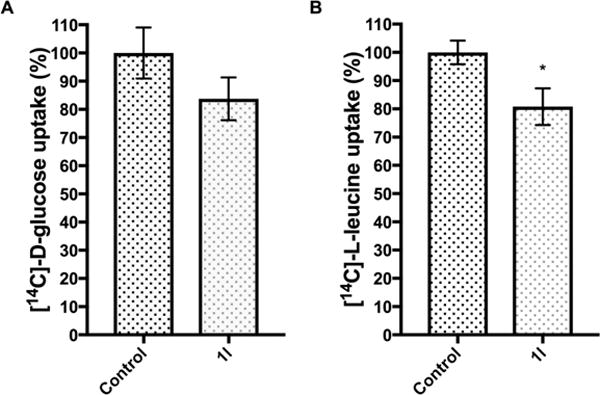
The effect of compound 1l on cell accumulation of Slc2a1 and Slc7a5 transportersubstrates [14C]-D-glucose and [14C]-L-leucine. The incubation of the HCC cells with 10 μM compound 1l reduced the cell accumulation of (A)[14C]-D-glucose and (B)[14C]-L-leucineto 84 % ±8 % and 81 % ±6 %, respectively. The analyte concentrations are normalized by the amount of protein in each sample. The statistical difference between compound 1l-treated cell and the control cells was determined using unpaired two-tailed t-test (*P< 0.05). Data are presented as mean ± SEM (n=4).
Effect of compound 1l on sorafenib cytotoxicity in mouse HCC cells
Thehalf maximal inhibitory concentration (IC50) value of sorafenib on HCC cell viability was investigated at 72 h using concentration range from 0.1 to 200 μM (Figure 8A) The effect of 1l on HCC cell proliferation and the possible potentiating effect on sorafenib cytotoxicity were evaluated by incubating the HCC cells for 72 h atdifferent sorafenib concentrations with and without a 100 μM concentration of 1l (Figure 8B). Interestingly, 1l significantlyaugmented the efficacy of sorafenib at 1, 2.5 and 5 μM concentrations. At 10 μM sorafenib concentration, NMDA receptor antagonist 1l also augmentedsorafenib efficacy, although the effect was less pronounced. On its own the NMDA antagonist 1l was only able to reduce HCC cell proliferation slightly. In addition, D-AP5 had potentiating effect on sorafenib cytotoxicity at all sorafenib concentrations investigated (Figure 8C). Previously it was shown that at above 100 μM concentrations a non-competitive NMDA receptor antagonist has antiproliferative effects on HCC cells affecting the FOXO/TXNIPpathway. Therefore, the antiproliferative effect of 1l alone is likely mediated via the same pathway. However, as sorafenib exerts its effect through the RAF/MEK/ERK pathway,34 the potentiating effect of 1l is more likely mediated through the lipid signaling pathway and downregulation of efflux transporter expression.
Figure 8.
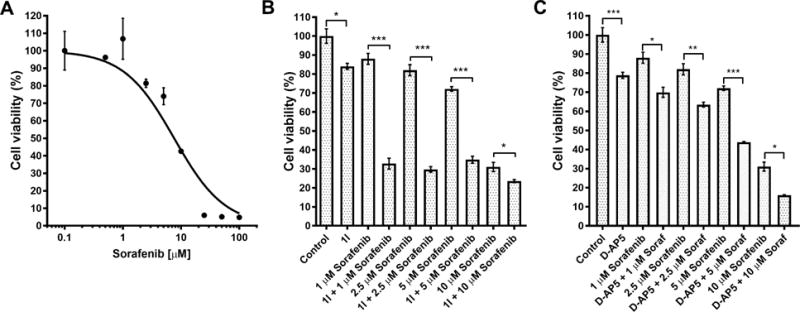
A) Concentration-dependent antiproliferative efficacy of sorafenib in HCC cells after 72 h. IC50 value was 7.4 ± 1.7 μM. Ability of compound 1l to potentiate the cytotoxic efficacy of sorafenib in mouse HCC cells. B) The potentiating effect of compound 1l on sorafenib antiproliferative efficacy in murine HCC cells. Compared to sorafenib effect alone, the cell viability was further reduced in the presence of 100 μM compound 1lfrom 88 ± 3 % to 33 ± 3 %,82 ± 3 % to 30 ±2 %, 72 ± 1 % to 35 ±2 % and from 31 ± 2 % to 24 ± 1 %, at 1 μM, 2.5 μM, 5 μM and 10 μM sorafenib, respectively. Compound 1l at 100 μM without sorafenib was able to reduce the cell viability to 84 ± 2 %. C) The potentiating effect of D-AP5 on sorafenib antiproliferative efficacy in murine HCC cells. The cell viability wasreduced to 70 ± 3 %, 63 ± 1 %, 44 ± 1 % and 16 ± 1 %, in combination of 100 μM D-AP5and1 μM, 2.5 μM, 5 μM and 10 μM sorafenib, respectively. D-AP5 at 100 μM without sorafenib was able to reduce the cell viability to 79 ± 2 %. The remaining viability is presented as mean ± SEM (n = 3˗6). The statistical difference between groups were determined by one-way ANOVA followed by Tukey`s multiple comparison test (*P< 0.05,**P < 0.01,***P < 0.001).
Conclusion
Starting from the broad-acting iGluR antagonist 1a, we have developed apotent and selective competitive NMDA receptor antagonist, compound 1l, which action is restricted to peripheral tissues and which displays good drug-like properties. Application of 1l to murine HCC cells reduced the intracellular concentration of PGE2 and thereby interferedwith the proinflammatory lipid signaling pathway resulting in downregulation of MDR transporters. It was subsequentlyshown that 1l augmentscell accumulation of MDR transporter substrates and that cell accumulation of the HCC anti-cancer agentand ABC transporter substrate sorafenib was significantly increased leading to augmentedcytotoxic efficacy. Another important finding was the significant downregulation of nutrient transporters, which are essential for the rapid growth of cancer cells. In summary, this proof-of-conceptstudy demonstrates that competitive NMDA receptor antagonistsrepresenta novel and promisingstrategy to reverseMDRinperipheral solid tumors, such as HCC.
Experimental Section
Chemistry
All reagents were obtained from commercial suppliers and used without further purification. Dry solvents were obtained differently. THF was distilled over sodium/benzophenone. Et2O was dried over neatly cut sodium. All solvents were tested for water content using a Carl Fisher apparatus. Water - or air sensitive reactions were conducted in flame dried glassware under nitrogen with syringe-septum cap technique. Purification by DCVC (dry columnvacuum chromatography) was performed with silica gel size 25–40μm (Merck, Silica gel 60). For TLC, Merck TLC Silica gel F254 plateswere used with appropriatespray reagents: KMnO4 or Molybdenum blue. 1H NMR and 13C NMR spectrawere obtained on a Varian Mercury Plus (300 MHz) and a Varian Gemini 2000 instrument (75 MHz), respectively, unless otherwise noted. Dioxane was used as internal reference for NMR spectra run in D2O. Preparative HPLC was performed using either a Spectraseries UV100 detector with a JASCO 880-PU HPLC pump and an XTerra®Prep MS C18 (10μm, 10×300 mm) column or an Agilent Prep HPLC system, equipped with a 1100 series pump, a 1200 seriesmultiplewavelength detector, and aZorbax 300 SB-C18 (21.2 × 250 mm,7 μm) column. LC-MS was performed using an Agilent 1200 HPLCsystem coupled to an Agilent 6400 triple quadrupole mass spectrometer equipped with an electrospray ionizationsource. A Zorbax Eclipse XDB-C18 (4.6 × 50 mm) column and gradients of 10% aqueous acetonitrile + 0.05% formic acid (buffer A) and 90% aqueous acetonitrile + 0.046% formic acid (buffer B) were employed. Optical rotation was measured using a Perkin-Elmer 241 spectrometer, with a Na lamp at 589 nm. Melting points were measured using anautomated melting point apparatus, MPA100 OptiMelt (SRS) and are uncorrected. Compounds were dry either under high vacuum or freeze dried usinga Holm & Halby, Heto LyoPro 6000 freeze drier. Compounds for pharmacological characterization were all with a purity of >95%, determined by HPLC (254 nM).
(2S,3R)-2-Carboxy-3-(3-chlorophenyl)pyrrolidin-1-ium chloride (1d)
Acid 7d (120 mg, 0.37 mmol, 1.0 equiv) was dissolved in a 1:1 mixture of TFA:DCM (7.2 mL). The reaction mixture was allowed to stir for 1 hour at rt, then evaporated to dryness under reduced pressure. The solid was dissolved in 1M HCl (2 mL) and evaporated to dryness (3×) to give the crude product, which was recrystallized from MeOH/CHCl3 to afford the title compoundas white needles (17 mg, 18%).1H NMR (CDCl3) δ (two rotamers) 9.64 (br s, 1H), 7.27–7.22 (m, 3H), 7.15–7.13 (m, 1H), 4.40 (d, J = 5.5 Hz, 0.4H), 4.25 (d, J = 6.5 Hz, 0.6H), 3.79–3.44 (m, 3H), 2.38–2.28 (m, 1H), 2.06–1.96 (m, 1H), 1.49 (s, 4H), 1.42 (s, 5H).13C NMR (CDCl3) δ (two rotamers) 177.6, 175.7, 155.2, 153.7, 142.9, 142.4, 134.6, 130.1, 127.5, 127.4, 127.1, 125.2, 81.2, 80.1, 65.4, 64.9, 49.4, 47.5, 46.1, 45.9, 32.7, 32.2, 28.3, 28.2. MS (m/z) calcd. for C11H13ClNO2 [M+H]+ 226.1, found 226.1..Mp: decomposition.
(2S,3R)-2-Carboxy-3-(3-(trifluoromethyl)phenyl)pyrrolidin-1-ium chloride (1e)
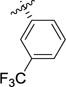
Compound 7e (135 mg) was dissolved in a 1:1 mixture TFA:DCM (7.0 mL). The reaction mixture was allowed to stir for 1h at rt, then the solvent was evaporated under reduced pressure. The crude was dissolved in HCl 1M (30 mL) and the solvent was evaporated to afford the corresponding HCl salt (63 mg) as a white solid. The compound was recrystallized from MeOH/CHCl3 to afford the title compound as a white powder (51 mg, 46%). 1H NMR (CDCl3) δ (two rotamers) 9.64 (br s, 1H), 7.27–7.22 (m, 3H), 7.15–7.13 (m, 1H), 4.40 (d, J = 5.5 Hz, 0.4H), 4.25 (d, J = 6.5 Hz, 0.6H), 3.79–3.44 (m, 3H), 2.38–2.28 (m, 1H), 2.06–1.96 (m, 1H), 1.49 (s, 4H), 1.42 (s, 5H). 13C NMR (CDCl3) δ (two rotamers) 177.6, 175.7, 155.2, 153.7, 142.9, 142.4, 134.6, 130.1, 127.5, 127.4, 127.1, 125.2, 81.2, 80.1, 65.4, 64.9, 49.4, 47.5, 46.1, 45.9, 32.7, 32.2, 28.3, 28.2. MS (m/z) calcd. forC12H13F3NO2 [M+H]+295.06, found 295.1. [α]25D +58.9 (c= 0.38, MeOH). Mp: 180.7–182.5 °C.
(2S,3R)-3-(3-Aminophenyl)pyrrolidine-2-carboxylic acid dihydrochloride (1f)
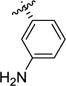
Acid 7f (41 mg, 0.101 mmol, 1 equiv) was dissolved in a 1:1 mixture TFA:DCM (2.0 mL). The reaction mixture was allowed to stir for 1.5h at rt, then the solvent was evaporated under reduced pressure. The oily residue was dissolved in 1M HCl (10 mL) and the solvent was evaporated (3×) to give the crude product, which was recrystallized from MeOH/CHCl3 to afford the title compound as a white solid (22 mg, 78%).1H NMR (MeOD) δ 7.58 – 7.31 (m, 3H), 7.31 (d, J = 7.2 Hz, 1H), 4.45 (d, J = 9.1 Hz, 1H), 3.77 – 3.58 (m, 2H), 3.57 – 3.42 (m, 1H), 2.66 – 2.49 (m, 1H), 2.36 – 2.17 (m, 1H).13C NMR (CDCl3) δ 168.5, 144.4, 140.6, 131.18, 131.17, 124.8, 118.6, 66.7, 49.6, 46.8, 34.4. MS (m/z) calcd. for C11H15N2O2 [M+H]+ 207.1, found 207.1.[α]25D +62.7° (c= 0.22, MeOH). Mp: decomposition.
(2S,3R)-3-(3-Cyanophenyl)pyrrolidine-2-carboxylic acid hydrochloride (1g)
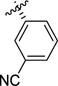
Acid 7g (65 mg, 0.205 mmol, 1 equiv) was dissolved in a 1:1 mixture of TFA:DCM (2.0 mL). The reaction mixture was allowed to stir for 3 hours at rt, then the solvent was evaporated under reduced pressure. The oily residue was dissolved in HCl 1M (10 mL) and evaporated to dryness (3×) to afford the title compound as a white solid (42 mg, 81%). 1H NMR (D2O) δ 7.82 (t, J = 1.6 Hz, 1H), 7.78 – 7.70 (m, 2H), 7.59 (t, J = 7.8 Hz, 1H), 4.44 (d, J = 9.9 Hz, 1H), 3.80 – 3.68 (m, 2H), 3.57 (ddd, J = 11.8, 10.3, 6.9 Hz, 1H), 2.61 (dtd, J = 10.5, 7.1, 3.3 Hz, 1H), 2.35 – 2.22 (m, 1H).13C NMR (D2O+dioxane) δ 171.4, 140.3, 133.4, 132.5, 132.1, 130.5, 120.0, 112.3, 65.41, 48.0, 46.4, 33.4. LC-MS (m/z) calcd. for C12H13N2O2 [M+H]+ 217.10, found 217.0. Mp: 210.8–212.9 °C.
(2S,3R)-3-(3-Carbamoylphenyl)-2-carboxypyrrolidin-1-ium chloride (1h)
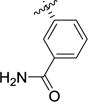
H2O2 (4.23 mmol, 0.43 mL of 30% solution in H2O, 6 equiv.) was added dropwise to a solution of 7g (223 mg, 0.71 mmol, 1.0 equiv.) and K2CO3 (390 mg, 2.82 mmol, 4 equiv.) in EtOH:H2O (1:1,4.86 mL). The reaction mixture was stirred at rt for 70 min, then slowly acidified with 1M HCl. The aqueous phase was extracted with EtOAc (3 × 10 mL) and the combined organic layers washed with brine (10 mL), dried over MgSO4 and concentrated to give a pale yellow solid. The crude was dissolved in DCM (5.8 mL) and TFA (5.8 mL) was added. The reaction mixture was stirred for 2 hours at rt. The solvent was evaporated and the oily residue was dissolved in 1M HCl and evaporated to dryness (3 × 10 mL). The crude pink solid was recrystallized from H2O/Acetone to afford the title compound as a white solid (95 mg, 59% over two steps).1H NMR (D2O) δ 7.85 (d, J = 1.6 Hz, 1H), 7.83 – 7.77 (m, 1H), 7.70 – 7.65 (m, 1H), 7.59 (t, J = 7.7 Hz, 1H), 4.38 (d, J = 9.7 Hz, 1H), 3.77 – 3.67 (m, 2H), 3.64 – 3.52 (m, 1H), 2.61 (dtd, J = 13.9, 7.1, 3.4 Hz, 1H), 2.39 – 2.25 (m, 1H).13C NMR (D2O) δ 173.4, 172.3, 139.9, 134.1, 132.2, 130.1, 127.4, 127.2, 66.1, 48.7, 46.3, 33.7. LC-MS (m/z) calcd. for C12H15N2O3 [M+H]+ 235.1, found 235.1. Mp: decomposition.
(2S,3R)-3-(3-Boronophenyl)pyrrolidine-2-carboxylic acid hydrochloride (1i)
To a solution of 7i (50 mg, 0.120 mmol, 1.0 equiv.) in acetone (9.4 mL) was added 0.1M NH4OAc (27.7 mg, 0.36 mmol, 3.0 equiv, in H2O) and NaIO4 (76.8 mg, 0.36 mmol, 3 equiv). The reaction mixture was stirred for 48 hours at rt. Acetone was removed under reduced pressure and the aqueous layer diluted with 2M NaOH (4 mL) and washed with EtOAC, The aqueous phase was then acidified with 1M HCl until pH~3 and extracted with EtOAc (4 × 5 mL). The combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated to dryness. The crude product was dissolved in DCM (1.0 mL) and TFA (1.0 mL) and the reaction mixture stirred for 2.5h at rt. The solvents were evaporated and the oily residue was dissolved in 1M HCl and evaporated to dryness (3 × 10 mL) to give an off-white solid, which was recrystallized from H2O/acetone to give the title compound as an off-white solid (13 mg, 43% over two steps).1H NMR (D2O) δ 7.80 – 7.72 (m, 2H), 7.58 – 7.46 (m, 2H), 4.41 (d, J = 9.8 Hz, 1H), 3.75 – 3.65 (m, 2H), 3.56 (ddd, J = 11.7, 10.3, 6.9 Hz, 1H), 2.57 (dtd, J = 13.9, 7.1, 3.4 Hz, 1H), 2.38 – 2.20 (m, 1H).13C NMR (D2O+dioxane) δ 171.4, 137.9, 133.1, 132.6, 130.1, 128.7, 65.2, 48.2, 45.8, 33.1. LC-MS (m/z) calcd. for C11H15BNO4 [M+H]+ 236.1, found 236.1. Mp: decomposition.
(2S,3R)-3-(3,4-Dihydroxyphenyl)pyrrolidine-2-carboxylic acid (1j)
In a flame-dried flask a solution of 7j (70 mg, 0.21 mmol, 1.0 equiv) in dry DCM (3 mL) was cooled to 0°C. A 1M solution of BBr3 (in DCM) (0.53 mL, 0.522mmol, 2.5 equiv) was added dropwise and the reaction mixture was allowed to stir 3hat rt. The reaction mixture was quenched with H2O (1 mL) and the solvent evaporated. The crude product was purified by prep. HPLC and then recrystallized from MeOH to afford the title compound as a white solid (3.8 mg, 8% yield).1H NMR (CDCl3) δ 6.89 - 6.94 (m, 2H), 6.79 - 6.86 (m, 1H), 4.02 (d, J = 9.03 Hz, 1H), 3.54 - 3.62 (m, 1H), 3.36 - 3.51 (m, 2H), 2.43 (dtd, J = 3.64, 6.73, 13.52 Hz, 1H), 2.09 - 2.23 (m, 1H). MS (m/z) calcd. for C11H13NO4 [M+H]+ 224.09, found 224.1.
(2S,3R)-2-Carboxy-3-(3-carboxy-4-fluorophenyl)pyrrolidin-1-ium chloride (1k)
Acid 7k (85 mg, 0.24 mmol, 1 equiv) was dissolved in a 1:1 mixture TFA:DCM (4.66 mL). The reaction mixture was allowed to stir for 2h at rt, then the solvent was evaporated under reduced pressure. The oily residue was dissolved in HCl 1M (10 mL) and the solvent was evaporated (3×) to afford the title compound as a white solid (42 mg, 60%). 1H NMR (D2O, 400 MHz) δ 7.97 (dd, J = 6.9, 2.6 Hz, 1H), 7.69 (ddd, J = 8.6, 4.6, 2.5 Hz, 1H), 7.31 (dd, J = 11.0, 8.6 Hz, 1H), 4.36 (d, J = 9.7 Hz, 1H), 3.77 – 3.66 (m, 2H), 3.57 (ddd, J = 11.8, 10.2, 6.9 Hz, 1H), 2.59 (dtd, J = 13.9, 7.0, 3.3 Hz, 1H), 2.29 (dtd, J = 13.4, 10.4, 8.2 Hz, 1H).13C NMR (D2O) δ 172.0, 168.4, 162.5, 160.8, 135.34, 135.31, 135.2, 135.1, 131.5, 119.2, 119.1, 118.2, 118.1, 67.2, 65.9, 47.8, 46.3, 33.5. MS (m/z) calcd. for C12H13FNO4 [M+H]+ 254.1, found 254.1. Mp: 196.8→dec.
(2S,3R)-3-(3-Carboxy-4-chlorophenyl)pyrrolidine-2-carboxylic acid hydrochloride (1l)
Diacid 7l (190 mg, 0.514 mmol, 1 equiv) was dissolved in a 1:1 mixture TFA:DCM (10 mL). The reaction mixture was allowed to stir for 2 hours at rt, then the solvent was evaporated under reduced pressure. The oily residue was dissolved in 1M HCl (10 mL) and the solvent was evaporated (3 times) to afford the corresponding HCl salt (135 mg). The crude product was recrystallized from H2O/acetone to afford the title compound as an off-white solid (89 mg, 56%). 1H NMR (D2O) δ 7.85 (d, J = 1.8 Hz, 1H), 7.68 – 7.49 (m, 2H), 4.37 (d, J = 9.7 Hz, 1H), 3.71 (ddd, J = 11.8, 9.2, 5.1 Hz, 2H), 3.62 – 3.52 (m, 1H), 2.60 (dtd, J = 13.9, 7.1, 3.4 Hz, 1H), 2.35 – 2.23 (m, 1H). 13C NMR (D2O) δ 171.9, 170.7, 138.3, 132.6, 131.9, 131.8, 131.7, 130.3, 65.7, 47.8, 46.2, 33.4. MS (m/z) calcd. for C12H13ClNO4 [M+H]+ 270.0, found 270.0. Mp 239.4–241.3 °C.
(2S,3R)-3-(3-Carboxy-4-methylphenyl)pyrrolidine-2-carboxylic acid hydrochloride (1m)
A solution of NaIO4 (638 mg, 2.98 mmol, 8.2 equiv) and RuCl3·xH2O (4.5 mg, 0.022 mmol, 0.06 equiv) in H2O (4.5 mL) was added to a solution of 6m (117 mg, 0.36 mmol) in MeCN:EtOAc (1:1, 5.2 mL) cooled to 0 °C. The reaction mixture was stirred for 30 min and then filtered through celite and the filter cake washed with EtOAc. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried over MgSO4 and concentrated to give the corresponding di-acid, which was used without further purification. The diacid 7m was dissolved in a 1:1 mixture of TFA:DCM (6.0 mL), and the reaction mixture allowed to stir for 2.5h at rt, then the solvent was evaporated under reduced pressure. The oily residue was dissolved in 1M HCl (10 mL) and evaporated to dryness (3×). The crude product was purified by preparative HPLC (0 to 40% B in A) and then evaporated with 1M HCl (3 × 1 mL) to afford the title compound, as a white solid (21 mg, 20% over two steps). 1H NMR (600 MHz, D2O) δ 7.78 (d, J = 2.0 Hz, 1H), 7.45 (dd, J = 7.9, 2.1 Hz, 1H), 7.29 (d, J = 7.9 Hz, 1H), 4.39 (d, J = 9.8 Hz, 1H), 3.69 – 3.61 (m, 2H), 3.51 (ddd, J = 11.7, 10.2, 6.9 Hz, 1H), 2.50 (dtd, J = 10.5, 7.1, 3.2 Hz, 1H), 2.44 (s, 3H), 2.25 – 2.17 (m, 1H). 13C NMR (151 MHz, D2O) δ 171.54, 170.63, 139.15, 135.72, 132.31, 131.28, 130.16, 129.04, 64.56, 47.25, 45.81, 32.82, 20.04. MS (m/z) calcd. for C13H16NO4 [M+H]+ 250.3, found 250.3. Mp: decomposition.
(2S,3R)-3-(3-Carboxy-4-bromophenyl)pyrrolidine-2-carboxylic acid hydrochloride (1n)
1H NMR (600 MHz, D2O) δ 7.67 (d, J = 8.3 Hz, 1H), 7.63 (d, J = 2.3 Hz1H), 7.36 (dd, J = 8.3, 2.4 Hz, 1H), 4.18 (d, J = 9.4 Hz, 1H), 3.61–3.54 (m, 1H), 3.48–3.43 (m, 1H), 2.51–2.46 (m, 1H), 2.21–2.14 (m, 1H). 13C NMR (600 MHz, D2O) δ 171.9, 171.6, 138.6, 134.8, 134.2, 131.4, 128.8, 118.4, 65.6, 47.5, 45.6, 32.8, MS (m/z) calcd. forC12H12BrNO4 [M+H]+312.9 & 314.9, found 313.9 & 315.9. Mp:250.1–257.3 °C.
5-Bromo-2-methylbenzyl)oxy)(tert-butyl)dimethylsilane (3m)
To a flame-dried round bottom flask was charged with5-bromo-2-methylphenyl)methanol35 (4.34 g, 21.6 mmol), anhydrous DMF (50 mL), imidazole (4.41 g, 64.8 mmol), TBSCl (6.51 g, 43.2 mmol) and DMAP (264 mg, 2.16 mmol). The reaction mixture was stirred for 16h at rt and subsequently diluted with EtOAc. The resulting mixture was washed twice with 1M HCl, twice with brine, dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by consecutive flash chromatography (heptane:EtOAc, 2:1 and 5:1), to provide title compound as a clear oil (4.63 g, 68%). 1H NMR (600 MHz, CDCl3) δ 7.56 (d, J = 2.0 Hz, 1H), 7.27 (dd, J = 8.0, 2.2 Hz, 1H), 6.98 (d, J = 8.0 Hz, 1H), 4.65 (s, 2H), 2.19 (s, 3H), 0.95 (s, 9H), 0.11 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 141.60, 133.72, 131.50, 129.77, 129.30, 119.78, 62.76, 26.09, 18.56, 18.16, −5.17. Rf 0.84 (heptane:EtOAc, 1:1).
tert-Butyldimethyl((2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)oxy)silane (3n)
A dry, Ar-filled 20 mL vial was charged with bromine 3m (419 mg, 1.33 mmol), (Bpin)2 (354 mg, 1.40 mmol), KOAc (394 mg, 4.06 mmol) and (PPh3)2PdCl2 (46.9 mg, 66.5 μmol). The vialwas then evacuated and backfilled with Ar, followed by addition of degassed dioxane (13.2 mL). The vial was capped with a screw cap and stirred for 16h at 100 °C. The reaction mixture was then diluted with EtOAc and washed three times with H2O. The organic partition was washed with brine, dried over MgSO4, filtered through celite and evaporated. The crude product was purified by DCVC (heptane:EtOAc, 10:1) to yield the title compound (407 mg, contains 5 mol% PPh3) which was used without further purification.1H NMR (600 MHz, CDCl3) δ 7.76 (s, 1H), 7.62 (dd, J = 7.4, 1.0 Hz, 1H), 7.15 (d, J = 7.5 Hz, 1H), 4.70 (s, 2H), 2.34 (s, 3H), 1.33 (s, 12H), 0.93 (s, 9H), 0.09 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 139.94, 138.40, 134.16, 134.03, 129.75, 83.71, 64.14, 26.11, 25.01, 19.19, 18.57, −5.11. Rf 0.54 (heptane:EtOAc, 10:1).
(2S,3R)-tert-Butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-3-(3-(ethoxycarbonyl)phenyl)-5-oxopyrrolidine-1-carboxylate (4a)
[Rh(cod)Cl]2 (37.6 mg, 0.076 mmol, 0.05 equiv), (S)-tert-butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (2) (500 mg, 1.53 mmol, 1.0 equiv) and (3-(ethoxycarbonyl)phenyl)boronic acid (3a, 474 mg, 2.44 mmol, 1.6 equiv) were placed in a 50 mL flask which was evacuated and backfilled with N2. Degassed dioxane (13.0 mL) was added and to the stirred clear solution, 1M NaOH (2.44 mL, 1.6 equiv, degassed water) was added dropwise. The reaction mixture was stirred at rt for 4.5 hours. The reaction mixture was diluted with H2O and the aqueous phase was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine (1 × 20 mL), dried over MgSO4 and concentrated to give 743 mg. The crude product was purified by flash chromatography (heptanes:EtOAc, 9:1) to afford the title compound as an off-white solid (382 mg, 52%). 1H NMR (CDCl3) δ 7.95 (dt, J = 7.4, 1.6 Hz, 1H), 7.87 (s, 1H), 7.39 (tt, J = 4.6, 3.3 Hz, 2H), 4.38 (q, J = 7.1 Hz, 2H), 4.09 (dt, J = 4.0, 2.1 Hz, 1H), 4.01 (dd, J = 10.6, 3.9 Hz, 1H), 3.82 (dd, J = 10.5, 2.2 Hz, 1H), 3.54 (dt, J = 9.6, 2.4 Hz, 1H), 3.17 (dd, J = 17.9, 9.6 Hz, 1H), 2.53 (dd, J = 17.9, 2.8 Hz, 1H), 1.53 (s, 9H), 1.39 (t, J = 7.1 Hz, 3H), 0.91 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H). 13C NMR (CDCl3) δ 173.7, 166.4, 150.0, 144.6, 131.4, 130.6, 129.4, 128.5, 128.0, 83.3, 66.6, 63.7, 61.3, 40.1, 38.8, 28.2, 26.0, 18.3, 14.5, -5.3. LC-MS (m/z) calcd. for C25H39NO6Si [M+H]+ 478.2, found 378.2 [(M+H)-Boc]+. Rf 0.25 (heptanes:EtOAc, 8:2).
(2S,3R)-tert-Butyl 3-(3-bromophenyl)-2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxopyrrolidine-1-carboxylate (4b)
To a solution at rt of (S)-tert-butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (2) (1.0 g, 3.05 mmol, 1.0 equiv) and (3-bromophenyl)boronic acid (3b, 981 mg, 4.89 mmol, 1.6 equiv) in degassed THF (26.3 mL) under nitrogen was added [Rh(cod)Cl]2 (75.3 mg, 0.153 mmol, 0.05 equiv). A 1M NaOH (4.9 mL, 1.6 equiv) solution was then added dropwise and the reaction mixture stirred at rt for 4 hours. The reaction mixture was diluted with H2O and the aqueous phase was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine (1 × 20 mL), dried over MgSO4 and concentrated to give 1.56 g. The crude product was purified by flash chromatography (heptanes:EtOAc, 9:1) to afford the title compound as a white solid (803 mg, 54%). 1H NMR (CDCl3,400 MHz) δ 7.40 (ddd, J = 7.9, 1.9, 1.0 Hz, 1H), 7.34 (t, J = 1.8 Hz, 1H), 7.21 (t, J = 7.8 Hz, 1H), 7.14 – 7.09 (m, 1H), 4.06 (dt, J = 3.9, 2.0 Hz, 1H), 3.99 (dd, J = 10.5, 3.9 Hz, 1H), 3.80 (dd, J = 10.5, 2.2 Hz, 1H), 3.42 (dt, J = 9.6, 2.2 Hz, 1H), 3.14 (dd, J = 17.9, 9.6 Hz, 1H), 2.50 (dd, J = 17.9, 2.6 Hz, 1H), 1.53 (s, 9H), 0.91 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H). 13C NMR (CDCl3, 400 MHz) δ 173.6, 150.0, 146.6, 130.8, 130.5, 129.9, 125.0, 123.2, 83.4, 66.6, 63.8, 39.94, 38.7, 28.2, 26.0, 18.3, −5.3, −5.4. Rf 0.21 (heptanes:EtOAc, 9:1).
(2S,3R)-tert-Butyl-2-(((tert-butyldimethylsilyl)oxy)methyl)-3-(3-chlorophenyl)-5-oxopyrrolidine-1-carboxylate (4d)
To a solution of (S)-tert-butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (2) (500 mg, 1.53 mmol, 1.0 equiv) and (3-chlorophenyl)boronic acid (3d, 382 mg, 2.44 mmol, 1.6 equiv) in THF:H2O (9:1, 15 mL) under nitrogen at rt, was added [Rh(cod)Cl]2 (37.6 mg, 0.076 mmol, 0.05 equiv). Then, NaOH 1M (2.5 mL, 1.6 equiv) was added dropwise and the reaction mixture was stirred at rt for 4.5h. The reaction mixture was diluted with H2O and the aqueous phase was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine (1 × 20 mL), dried over MgSO4 and concentrated to give 690 mg. The crude product was purified by flash chromatography (heptanes:EtOAc, 4:1) to afford the title compound as a white solid (387 mg, 58%). 1H NMR (CDCl3) δ 7.55–7.38 (m, 4H, Ar-H), 4.09 (m, 1H, CH-N), 4.01 (m, 1H), 3.83 (dd, J = 10.5, 2.3 Hz, 1H), 3.54 (dt, J = 9.5, 2.3 Hz, 1H), 3.20 (dd, J = 17.9, 9.7 Hz, 1H), 2.53 (dd, J = 17.8, 2.5 Hz, 1H), 1.54 (s, 9H), 0.92 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H). 13C NMR (CDCl3) δ 173.3, 149.8, 145.1, 131.3 (q, J =32 Hz, C-CF3), 129.7, 129.4, 124.0 (q, J =3.7 Hz, C-Ar), 123.9 (q, J =270 Hz, CF3), 123.6 (q, J = 3.7, C-Ar), 83.3, 66.3, 63.3, 39.8, 38.7, 28.0, 25.8, 18.1, −5.5, −5.6. LC-MS (m/z) calcd. for C22H35ClNO4Si [M+H]+ 440.2, found 340.1 [(M+H)-Boc]+. [α]25D −26.5 (c= 0.27, MeOH). Mp: 71.4–73.9 °C. Rf 0.29 (heptanes:EtOAc, 9:1).
(2S,3R)-tert-Butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxo-3-(3-(trifluoromethyl)phenyl)pyrrolidine-1-carboxylate (4e)
To a solution of (S)-tert-butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (2) (1.0 g, 3.053 mmol, 1 equiv) and commercially available (3-(trifluormomethyl)phenyl)boronic acid (3e, 928 mg, 4.89 mmol, 1.6 equiv) in THF:H2O (9:1, 30.0 mL) under nitrogen at rt, was added [Rh(cod)Cl]2 (75.3 mg, 0.153 mmol, 0.05 equiv). Then, NaOH 1M (5.0 mL, 1.6 equiv) was added dropwise and the reaction mixture was stirred at rt for 4 hours. The reaction mixture was diluted with H2O and the aqueous phase was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine (1 × 30 mL), dried over MgSO4 and concentrated. The crude product was purified by flash chromatography (heptanes:EtOAc, 9:1) to afford the title compound as a white solid (727 mg, 56%). 1H NMR (CDCl3) δ 7.55–7.38 (m, 4H), 4.09 (m, 1H), 4.01 (m, 1H), 3.83 (dd, J = 10.5, 2.3 Hz, 1H), 3.54 (dt, J = 9.5, 2.3 Hz, 1H), 3.20 (dd, J = 17.9, 9.7 Hz, 1H), 2.53 (dd, J = 17.8, 2.5 Hz, 1H), 1.54 (s, 9H), 0.92 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H).13C NMR (CDCl3) δ 173.3, 149.8, 145.1, 131.3 (q, J =32 Hz), 129.7, 129.4, 124.0 (q, J =3.7 Hz), 123.9 (q, J =270 Hz), 123.6 (q, J = 3.7 Hz), 83.3, 66.3, 63.3, 39.8, 38.7, 28.0, 25.8, 18.1, −5.5, −5.6. LC-MS (m/z) calcd. for C23H35F3NO4Si [M+H]+ 474.2, found 374.1 [(M+H)-Boc]+. [α]25D -26.4 (c= 0.32, MeOH). Mp: 95.3–96.9 °C. Rf 0.29 (heptanes:EtOAc, 9:1).
(2S,3R)-tert-Butyl 3-(3-((tert-butoxycarbonyl)amino)phenyl)-2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxopyrrolidine-1-carboxylate (4f)
[Rh(cod)Cl]2 (37.6 mg, 0.076 mmol, 0.05 equiv), (S)-tert-butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (500 mg, 1.53 mmol, 1.0 equiv) and 3-(tert-butoxycarbonylamino)boronic acid (3f, 579 mg, 2.44 mmol, 1.6 equiv) were placed in a 50 mL flask which was evacuated and backfilled with N2. Degassed dioxane (13.0 mL) was added followed by dropwise addition of 1M NaOH (2.44 mL, 1.6 equiv, degassed water). The reaction mixture was stirred at rt for 3.5h. The reaction mixture was diluted with H2O and the aqueous phase was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine (1 × 20 mL), dried over MgSO4 and concentrated to give 743 mg. The crude product was purified by flash chromatography (heptanes:EtOAc, 9:1) to afford the title compound as a pale yellow oil (471 mg, 59% yield). 1H NMR (CDCl3) δ 7.19 - 7.30 (m, 3H), 6.82 - 6.90 (m, 1H), 6.49 (s, 1H), 4.07 - 4.12 (m, 1H), 4.02 (dd, J = 3.51, 10.54 Hz, 1H), 3.80 (dd, J = 1.88, 10.67 Hz, 1H), 3.43 (td, J = 1.88, 9.54 Hz, 1H), 3.15 (dd, J = 9.54, 17.82 Hz, 1H), 2.50 (dd, J = 2.26, 17.82 Hz, 1H), 1.52 (s, 9H), 0.91 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H).13C NMR (CDCl3) δ 174.1, 152.6, 149.9, 145.4, 138.9, 129.7, 120.7, 117.2, 116.6, 83.0, 80.7, 66.6, 63.8, 40.2, 38.9, 28.3, 28.1, 25.8, 18.2, −5.5. LC-MS (m/z) calcd. for C27H45N2O6Si [M+H]+ 521.3, found 365.2 [(M+H)-Boc -t-Bu]. [α]25D −19.8 (c= 0.55, MeOH). Rf 0.28 (heptanes:EtOAc, 8:2).
(2S,3R)-tert-Butyl 3-(benzo[d][1,3]dioxol-5-yl)-2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxopyrrolidine-1-carboxylate (4j)
[Rh(cod)Cl]2 (112.9 mg, 0.229 mmol, 0.05 equiv) was added to a solution of (S)-tert-butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (2) (1.50 g, 4.58 mmol, 1.0 equiv) and benzo[d][1,3]dioxol-5-ylboronic acid (1.22 g, 7.33 mmol, 1.6 equiv) in THF (39.0 mL) under nitrogen at rt. Aqueous 1M NaOH (7.5 mL, 1.6 equiv) was then added dropwise and the reaction mixture was stirred at rt for 4h. The reaction mixture was diluted with H2O and the aqueous phase was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine (1 × 50 mL), dried over MgSO4 and concentrated. The crude product was purified by flash chromatography (heptanes:EtOAc, 9:1) to afford the title compound as a white solid (1.38 g, 67%). 1H NMR (CDCl3) δ 6.75 (d, J = 8.0 Hz, 1H), 6.68 (d, J 1.8 Hz, 1H), 6.64 (dd, J = 8.0, 1.8 Hz, 1H), 5.95 (s, 2H), 4.03 (m, 1H), 3.98 (m, 1H), 3.78 (dd, J = 10.4, 2.1 Hz, 1H), 3.37 (dt, J = 9.5, 2.3 Hz, 1H), 3.12 (dd, J = 17.8, 9.5 Hz, 1H), 2.47 (dd, J = 17.8, 2.8 Hz, 1H), 1.53 (s, 9H), 0.91 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H). 13C NMR (CDCl3) δ 173.9, 149.8, 148.2, 146.6, 138.1, 119.3, 108.5, 106.7, 101.1, 83.0, 66.9, 63.5, 40.2, 38.5, 28.1, 25.8, 18.2, −5.5. LC-MS (m/z) calcd. for C23H36NO6Si [M+H]+ 450.2, found 350.1 [(M+H)-Boc]+. [α]25D -25.6 (c= 0.42, MeOH). Mp: 106.6–107.6 °C. Rf 0.36 (heptanes:EtOAc, 8:2).
(2S,3R)-tert-Butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-3-(3-(((tert-butyldimethylsilyl)oxy)methyl)-4-fluorophenyl)-5-oxopyrrolidine-1-carboxylate (4k)
A flamed-dry round-bottomed flask was charged with a solution of tert-BuLi in pentane (8.98 mL, 15.265 mmol, 5.0 equiv.) and cooled to −78 °C. A solution of bromine 3k (2.44 g, 7.634 mmol, 2.5 equiv.) in dry Et2O (25 mL) was added dropwise and the clear, yellow solution was stirred at −78 °C for 10 minutes. A suspension of CuCN in dry Et2O (2.5 mL) was added portion wise at −78 °C. The resulting suspension was stirred at −78 °C for 5 minutes and then at −42 °C for 10 min (clear solution), after which it was re-cooled to −78 °C. Enone 2 was dissolved in dry Et2O (3.0 mL) and added dropwise to the cuprate mixture at −78 °C, which resulted in a color change to bright dark red. The temperature was raised to −42 °C and the reaction mixture was stirred at this temperature for 1 hour. The brown solution was quenched by addition of a freshly prepared sat. NaHCO3 (5 mL), allowed to warm up to rt and then transferred to a separating funnel with water (15 mL) and EtOAc (15 mL). The organic layer was separated and the aqueous layer was extracted with EtOAc (2 × 15 mL). The combined organic layers were washed with brine (1 × 15 mL), dried over anhydrous MgSO4, filtered and evaporated in vacuo to dryness. The crude product was purified by column chromatography (heptanes:EtOAc, 9:1) to afford the title compound as a pale yellow oil (793 mg, 1.40 mmol, 46% yield). 1H NMR (CDCl3) δ 7.30 (dd, J = 6.8, 2.3 Hz, 1H), 7.04 (ddd, J = 7.5, 4.9, 2.5 Hz, 1H), 6.95 (dd, J = 9.6, 8.5 Hz, 1H), 4.77 (s, 2H), 4.04 (dd, J = 3.9, 2.0 Hz, 1H), 3.99 (dd, J = 10.4, 4.0 Hz, 1H), 3.79 (dd, J = 10.4, 2.1 Hz, 1H), 3.45 (dt, J = 9.6, 2.3 Hz, 1H), 3.13 (dd, J = 17.9, 9.6 Hz, 1H), 2.49 (dd, J = 17.9, 2.7 Hz, 1H), 1.52 (s, 9H), 0.94 (s, 9H), 0.91 (s, 9H), 0.11 (s, 3H), 0.11 (s, 3H), 0.08 (s, 3H), 0.07 (s, 3H). 13C NMR (CDCl3) δ 173.9, 159.8, 158.1, 150.0, 140.12, 140.10, 129.2, 129.1, 126.64, 126.61, 126.25, 126.19, 115.6, 115.4, 83.2, 66.9, 63.7, 59.1, 59.0, 40.3, 38.3, 28.2, 26.1, 26.0, 18.6, 18.3, −5.17, −5.20, −5.3. Rf 0.32 (heptanes:EtOAc, 9:1).
(2S,3R)-tert-Butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-3-(3-(((tert-butyldimethylsilyl)oxy)methyl)-4-chlorophenyl)-5-oxopyrrolidine-1-carboxylate (4l)
A flamed-dry round-bottomed flask was charged with a solution of tert-BuLi in pentane (8.98 mL, 15.265 mmol, 5.0 equiv) and cooled to −78 °C. A solution of 3l (2.56g, 7.63 mmol, 2.5 equiv) in dry Et2O (25 mL) was added dropwise and the clear, yellow/orange solution was stirred at −78 °C for 10 minutes. A suspension of CuCN in dry Et2O (2.4 mL) was added portion wise at −78 °C. The resulting suspension was stirred at −78 °C for 5 minutes and then at −42 °C for 10 min (clear solution), after which it was re-cooled to −78 °C. Enone 2 was dissolved in dry Et2O (3.0 mL) and added dropwise to the cuprate mixture at −78 °C, which resulted in a color change to bright dark red. The temperature was raised to −42 °C and the reaction mixture was stirred at this temperature for 1 hour. The brown solution was quenched by addition of a freshly prepared sat. NaHCO3 (5 mL), allowed to warm up to rt and then transferred to a separating funnel with water (15 mL) and EtOAc (15 mL). The organic layer was separated and the aqueous layer was extracted with EtOAc (2 × 15 mL). The combined organic layers were washed with brine (1 × 15 mL), dried over anhydrous MgSO4, filtered and evaporated in vacuo to dryness. The crude product was purified by column chromatography (heptanes:EtOAc, 9:1) to afford the title compound as a low-melting white solid (1.31 g, 73%). 1H NMR (CDCl3) δ 7.39 (d, J = 2.3 Hz, 1H), 7.26 (d, J = 8.2 Hz, 1H), 7.01 (dd, J = 8.2, 2.3 Hz, 1H), 4.77 (s, 2H), 4.04 (dt, J = 3.9, 2.0 Hz, 1H), 4.00 (dd, J = 10.4, 3.9 Hz, 1H), 3.78 (dd, J = 10.4, 2.0 Hz, 1H), 3.45 (dt, J = 9.6, 2.3 Hz, 1H), 3.14 (dd, J = 17.9, 9.6 Hz, 1H), 2.50 (dd, J = 17.9, 2.8 Hz, 1H), 1.52 (s, 9H), 0.96 (s, 9H), 0.90 (s, 9H), 0.13 (s, 3H), 0.13 (s, 3H), 0.07 (s, 3H), 0.06 (s, 3H). 13C NMR (CDCl3) δ 173.7, 150.0, 143.1, 139.6, 130.2, 129.6, 125.9, 125.8, 83.2, 66.7, 63.7, 62.4, 40.1, 38. 6, 28.2, 26.1, 26.0, 18.6, 18.4, −5.1, −5.2, −5.34, −5.35. Rf 0.47 (heptanes:EtOAc, 8:2).
tert-Butyl (2S,3R)-2-(((tert-butyldimethylsilyl)oxy)methyl)-3-(3-(((tert-butyldimethylsilyl)oxy)methyl)-4-methylphenyl)-5-oxopyrrolidine-1-carboxylate (4m)
[Rh(cod)Cl]2 (23.2 mg, 0.047 mmol, 0.05 equiv), (S)-tert-butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (2) (300 mg, 0.92 mmol), tert-butyldimethyl((2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)oxy) silane (3n) (532 mg, 1.47 mmol) and Cs2CO3 (477 mg, 1.46 mmol) were placed in a 25 mL flask which was evacuated and backfilled with Ar. Degassed, anhydrous THF (8.9 mL) was added, followed by addition of degassed H2O (21 μL, 1.1 mmol). The reaction mixture was stirred at rt for 24 hours. The reaction mixture was diluted with H2O and the aqueous phase was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine, dried over MgSO4 and concentrated. The crude product was purified by flash chromatography (heptane:EtOAc, 10:1 to 5:2) to afford the title compound as a pale yellow oil (211 mg, 41% yield). 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 1.4 Hz, 1H), 7.09 (d, J = 7.8 Hz, 1H), 7.00 (dd, J = 7.7, 2.0 Hz, 1H), 4.68 (s, 2H), 4.11 – 4.05 (m, 1H), 4.01 (dd, J = 10.5, 3.8 Hz, 1H), 3.79 (dd, J = 10.5, 2.0 Hz, 1H), 3.46 – 3.41 (m, 1H), 3.13 (dd, J = 17.9, 9.6 Hz, 1H), 2.53 (dd, J = 17.8, 2.8 Hz, 1H), 2.25 (s, 3H), 1.52 (s, 9H), 0.94 (s, 9H), 0.91 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H), 0.08 (s, 3H), 0.07 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.29, 150.02, 141.83, 139.94, 134.14, 130.65, 124.93, 124.83, 82.97, 67.01, 63.70, 63.41, 40.18, 38.57, 28.19, 26.08, 25.97, 18.52, 18.32, 18.26, −5.14, −5.15, −5.38, −5.40. Rf 0.43 (heptane:EtOAc, 5:1).
(2S,3R)-tert-Butyl 3-(3-chlorophenyl)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (6d)
In a flame dried flask a solution of 4d (368 mg, 0.84 mmol, 1.0 equiv) in dry THF (2.2 mL) was cooled to −78 °C. LiBEtH3 1 M (1.0 mL, 1.00 mmol, 1.2 equiv) was added via syringe dropwise and the reaction mixture was stirred for 1h, then quenched with NaHCO3 sat. sol. (3 mL) and warmed to rt. The aqueous phase was extracted with EtOAc (3 × 5 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated to give the corresponding hemiaminal 5d as a colorless oil was which was used in the next step without further purification.
In a flame dried flask a solution of the crude hemiaminal 5d (0.84 mmol, 1.0 equiv) in dry DCM (2.6 mL) was cooled to −78 °C. HSiEt3 (0.27 mL, 1.670 mmol, 2 equiv) and BF3·Et2O (0.23 mL, 1.837 mmol, 2.2 equiv) were added sequentially via syringe and the reaction mixture stirred for 6 hours. The reaction mixture was quenched with sat. NaHCO3 (4 mL) and warmed to rt. The mixture was diluted with DCM and the organic phase was separated, washed with sat. NH4Cl (5 mL), dried over MgSO4 and concentrated. The crude product was purified by flash chromatography (heptanes:EtOAc, 2:1) to afford the title compounds as a colorless gummy oil (154 mg, 59% over two steps). 1H NMR (CDCl3) δ 7.31–7.26 (m, 3H, Ar-H), 7.16–7.14 (m, 1H, Ar-H), 3.94 (m, 1H, CH-N), 3.78 (m, 1H), 3.67 (m, 1H), 3.40 (m, 1H), 3.05 (broad s, 1H), 2.20 (m, 1H), 1.97 (m, 1H), 1.54 (s, 9H). 13C NMR (CDCl3) δ 156.5, 143.0, 134.5, 130.0, 127.6, 127.2, 125.7, 80.5, 66.7, 65.5, 47.2, 46.8, 32.6, 28.4. MS (m/z) calcd. for C16H23ClNO3 [M-H]+ 312.14, found 212.1 [(M+H)-Boc]+. [α]25D −13.1 (c= 0.92, MeOH). Rf 0.49 (heptanes:EtOAc, 1:1 + 1% AcOH).
(2S,3R)-tert-Butyl 2-(hydroxymethyl)-5-oxo-3-(3-(trifluoromethyl)phenyl)pyrrolidine-1-carboxylate (6e)
In a flame dried flask a solution of 4e (500 mg, 1.06 mmol, 1.0 equiv) in dry THF (2.8 mL) was cooled to −78 °C. 1M LiBEtH3 (solution in THF) (1.27 mL, 1.27 mmol, 1.2 equiv) was added via syringe dropwise and the reaction mixture was stirred for 40 minutes, then was quenched with NaHCO3 sat. sol. (3 mL) and warmed to rt. The aqueous phase was extracted with EtOAc (3 × 10 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated to give the corresponding hemiaminal 5e as a colorless oil which was used in the next step without further purification.
In a flame dried flask a solution of the crude hemiaminal 5e (1.06 mmol, 1 equiv) in dry DCM (3.3 mL) was cooled to −78 °C. HSiEt3 (0.34 mL, 2.11 mmol, 2 equiv) and BF3·Et2O (0.29 mL, 2.315 mmol, 2.2 equiv) were sequentially added via syringe and the reaction mixture was stirred for 6h. After the complete consumption of the substrate the reaction was quenched with sat. NaHCO3 (4 mL) and warmed to rt. The mixture was diluted with DCM and the organic phase was separated, washed with sat. NH4Cl (5 mL), dried over MgSO4 and concentrated to give 371 mg (crude). The crude was purified by flash chromatography (heptanes:EtOAc, 2:1) to afford the title compound as a colorless gummy oil (286 mg, 78% yield over two steps). 1H NMR (CDCl3) 7.49–7.39 (m, 4H), 4.94 (br s, 1H, OH), 3.92 (br s, 1H), 3.75–3.58 (m, 3H), 3.35 (ddd, J = 11.2, 8.9, 6.8, 1H), 3.04 (m, 1H), 2.18 (br s, 1H), 1.99–1.89 (m, 1H), 1.46 (s, 9H). 13C NMR (CDCl3) δ (two rotamers) 156.3, (154.3), (143.9), 142.1, 130.9 (q, J = 31.5 Hz, C-CF3), 130.8, 129.1, 124.1, 123.9 (q, J = 272.2 Hz, CF3), 123.8, 80.4, (80.1), 66.6, (65.6), 65.0, (62.5), 47.1, 46.7, (46.2), (32.6), 31.7. MS (m/z) calcd. for C17H23F3NO3 [M+H]+ 346.2, found 246.1 [(M+H)-Boc]+. [α]25D −10.3 (c= 0.43, MeOH). Rf 0.23 (heptanes:EtOAc, 2:1).
(2S,3R)-tert-Butyl 3-(3-((tert-butoxycarbonyl)amino)phenyl)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (6f)
In a flame dried flask a solution of 4f (371 mg, 0.71 mmol, 1.0 equiv) in dry THF (1.86 mL) was cooled to −78 °C. 1M LiBEtH3 (solution in THF) (1.7 mL, 1.72 mmol, 2.4 equiv) was added via syringe dropwise and the reaction mixture was stirred for 1h, then was quenched with sat. NaHCO3 (3 mL) and warmed to rt. The aqueous phase was extracted with EtOAc (3 × 5 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated to give the corresponding hemiaminal 4f as a colorless oil which was used in the next step without further purification.
In a flame dried flask a solution of the crude hemiaminal 4f (0.714 mmol, 1 equiv) in dry DCM (2.2 mL) was cooled to −78 °C. HSiEt3 (0.23 mL, 1.43 mmol, 2.0 equiv) and BF3·Et2O (0.27 mL, 2.14 mmol, 3.0 equiv) were sequentially added via syringe and the reaction mixture was stirred for 5h. After the complete consumption of the substrate the reaction was quenched with NaHCO3 sat. sol. (4 mL) and warmed to rt. The mixture was diluted with DCM and the organic phase was separated, washed with sat. NH4Cl (5 mL), dried over MgSO4 and concentrated. The crude product was purified by flash chromatography (heptanes:EtOAc, 2:1) to afford the title compound as a colorless oil (110 mg, 31% over two steps).1H NMR (CDCl3) δ 7.39 (s, 1H), 7.21 (t, J = 7.8 Hz, 1H), 7.12 (d, J = 8.1 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 6.57 (s, 1H), 3.90 (d, J = 6.8 Hz, 1H), 3.79 – 3.68 (m, 2H), 3.60 (dd, J = 11.5, 6.7 Hz, 1H), 3.39 – 3.27 (m, 1H), 2.92 (s, 1H), 2.14 (dtd, J = 9.1, 6.5, 2.8 Hz, 1H), 1.96 (dtd, J = 12.4, 10.1, 8.1 Hz, 1H), 1.51 (s, 9H), 1.49 (s, 9H).13C NMR (CDCl3) δ 152.8, 141.1 (b), 139.0, 129.4, 122.3, 117.7, 117.4, 80.7, 80.6, 67.0, 48.0, 47.2, 32.9, 28.6, 28.5. MS (m/z) calcd. for C21H32N2O5 [M+H]+ 392.23, found 293.1 [(M-Boc) +H]+. [α]25D −8.6 (c= 0.31, MeOH). Rf 0.16 (heptanes:EtOAc, 2:1).
(2S,3R)-tert-Butyl 3-(3-cyanophenyl)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (6g)
Under nitrogen atmosphere, TBAF (1.74 mmol, 3 equiv,1.74 mL of 1M solution in THF) was added to a solution of 8g (242 mg, 0.581 mmol, 1 equiv) in dry THF (5.6 mL). The reaction mixture was stirred at rt for 2h, then diluted with water (10 mL) and sat. NaHCO3 (10 mL). The aqueous layer was extracted with EtOAc (2 × 10 mL) and the combined organic layers were washed with brine, dried over MgSO4 and concentrated. The crude product was purified by column chromatography (heptanes:EtOAc, 1:1) to give the title compound as a pale yellow oil (202 mg, 63% yield over two steps). 1H NMR (CDCl3) δ 7.51 – 7.34 (m, 4H), 4.78 (br s, 1H), 3.86 (br s, 1H), 3.73 – 3.54 (m, 3H), 3.33 (ddd, J = 11.1, 8.8, 6.8 Hz, 1H), 3.01 (bs, 1H), 2.17 (m, 1H), 1.95 – 1.81 (m, 1H), 1.43 (s, 9H). 13C NMR (CDCl3) δ 156.2, 154.2, 144.6, 142.9, 132.0, 131.1, 130.7, 129.6, 118.6, 112.7, 80.6, 80.3, 66.5, 65.5, 64.9, 62.4, 46.9, 46.8, 46.3, 32.5, 31.7, 28.4. Rf 0.21 (heptane:EtOAc, 1:1).
(2S,3R)-tert-Butyl 2-(hydroxymethyl)-3-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)pyrrolidine-1-carboxylate (6i)
Under nitrogen atmosphere, TBAF (1.311 mmol, 3 equiv, 1.31 mL of 1M solution in THF), was added to a solution of 9 (226 mg, 0.437 mmol, 1 equiv) in dry THF (5.3 mL). The reaction mixture was stirred at rt for 2.5h, then was diluted with sat. NaHCO3 (10 mL). The aqueous layer was extracted with EtOAc (3 × 10 mL) and the combined organic layers were washed with brine, dried over MgSO4 and concentrated. The crude product was purified by column chromatography (heptanes:EtOAc, 2:1) to give the title compound as a colorless oil (106 mg, 63%). 1H NMR (CDCl3) δ 7.72 – 7.63 (m, 2H), 7.32 – 7.29 (m, 2H), 3.96 (bs, 1H), 3.72 (m, 2H), 3.59 (dd, J = 11.5, 6.7 Hz, 1H), 3.33 (td, J = 10.5, 6.5 Hz, 1H), 2.89 (bs, 1H), 2.10 (bs, 1H), 2.04 – 1.89 (m, 1H), 1.48 (s, 9H), 1.33 (s, 12H). 13C NMR (CDCl3) δ 156.9, 139.9, 133.9, 133.7, 130.7, 129.6, 128.2, 83.9, 80.5, 67.1, 66.0, 48.0, 47.2, 33.1, 28.5, 24.9. LC-MS (m/z) calcd. for C17H23N2O3 [M+H]+ 302.17, found 203.1 [(M+H)-Boc]+. Rf 0.17 (heptanes:EtOAc, 2:1).
(2S,3R)-tert-Butyl 3-(benzo[d][1,3]dioxol-5-yl)-2-(hydroxymethyl)-5-oxopyrrolidine-1-carboxylate (6j)
In a flame dried flask a solution of 4j (687 mg, 1.53 mmol, 1.0 equiv) in dry THF (4 mL) was cooled to −78 °C. 1M LiBEtH3 (THF solution) (1.83 mL, 1.834 mmol, 1.2 equiv) was added via syringe dropwise and the reaction mixture was stirred for 45 minutes, then was quenched with NaHCO3 sat. sol. (4 mL) and warmed to rt. The aqueous phase was extracted with EtOAc (3 × 10 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated to give the corresponding hemiaminal 5j as a colorless oil, which was used in the next step without further purification.
In a flame dried flask a solution of the crude hemiaminal (1.53 mmol, 1 equiv) in dry DCM (4.8 mL) was cooled to −78 °C. HSiEt3 (0.49 mL, 3.06 mmol, 2 equiv) and BF3·Et2O (0.42 mL, 3.37 mmol, 2.2 equiv) were added sequentially via syringe and the reaction mixture was stirred for 6h. The reaction was quenched with sat. NaHCO3 (4 mL) and warmed to rt. The mixture was diluted with DCM and the organic phase was separated, washed with sat. NH4Cl (5 mL), dried over MgSO4 and concentrated. The crude product was purified by flash chromatography (heptanes:EtOAc, 2:1) to afford the title compound as a pale yellow oil (334 mg, 69% yield over two steps).1H NMR (CDCl3) δ 6.69 – 6.64 (m, J = 4.7, 3.2 Hz, 2H), 6.61 (dd, J = 8.0, 1.6 Hz, 1H), 5.85 (s, 2H), 3.82 – 3.58 (m, 3H), 3.53 (dd, J = 11.4, 6.0 Hz, 1H), 3.26 (ddd, J = 11.0, 9.4, 6.7 Hz, 1H), 2.88 – 2.73 (m, 1H), 2.05 (br s, 1H), 1.89 – 1.72 (m, 1H), 1.42 (s, 9H).13C NMR (CDCl3) δ (two rotamers) 156.4, 154.4, 147.9, 146.4, 136.7, 134.7, 120.7, 120.4, 108.2, 107.5, 100.9, 80.3, 80.0, 66.9, 66.0, 65.2, 62.4, 47.3, 46.8, 46.4, 32.8, 32.0, 28.3. MS (m/z) calcd. for C17H24NO5 [M+H]+ 322.16, found 222.1 [(M+H)-Boc]+. [α]25D −18.3 (c= 1.2, MeOH). Rf 0.38 (heptanes:EtOAc, 2:1).
(2S,3R)-tert-Butyl 3-(4-fluoro-3-(hydroxymethyl)phenyl)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (6k)
In a flame dried flask a solution of 4k (793 mg, 1.40 mmol, 1.0 equiv) in dry THF (4.3 mL) was cooled to −78 °C. LiBEtH3 1 M (1.68 mL, 1.675 mmol, 1.2 equiv) was added via syringe dropwise and the reaction mixture was stirred for 1h, then quenched with sat. NaHCO3 (5 mL). The aqueous phase was extracted with EtOAc (3 × 10 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated to give the corresponding hemiaminal 5k as a colorless oil, which was used in the next step without further purification.
In a flame dried flask a solution of the crude hemiaminal (1.40 mmol, 1.0 equiv) in dry DCM (4.43 mL) was cooled to −78 °C. HSiEt3 (0.44 mL, 2.79 mmol, 2 equiv) and BF3·Et2O (0.38 mL, 3.07 mmol, 2.2 equiv) were added sequentially via syringe and the reaction mixture stirred for 5h at −78 °C. The reaction mixture was quenched with sat. NaHCO3 (5 mL), warmed to rt and diluted with DCM. The organic phase was separated, washed with sat. NH4Cl (5 mL), dried over MgSO4 and concentrated. The crude was dissolved in dry THF under N2 atmosphere at rt and 1M TBAF (THF solution) (1.02 mL, 1.02 mmol, 3 equiv) was added dropwise. The reaction mixture was allowed to stir overnight, then quenched with sat. NaHCO3 (5 mL) and portioned between EtOAc (10 mL) and H2O (10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with brine (1 × 10 mL), dried over MgSO4 and concentrated. The crude product was purified by flash chromatography (heptanes:EtOAc, 2:3) to afford the title compound as a colorless oil (92 mg, 20% over three steps). 1H NMR (CDCl3,400 MHz) δ 7.35 – 7.26 (m, 1H), 7.08 (ddd, J = 7.7, 5.0, 2.4 Hz, 1H), 6.95 (dd, J = 9.7, 8.4 Hz, 1H), 5.13 (br s, 1H), 4.68 (s, 2H), 3.95 – 3.53 (m, 4H), 3.31 (td, J = 10.4, 6.4 Hz, 1H), 2.90 (br s, 1H), 2.22 – 2.05 (m, 1H), 1.89 (ddd, J = 12.6, 10.5, 8.2 Hz, 1H), 1.47 (s, 9H). 13C NMR (CDCl3, 600 MHz) δ 160.3, 158.6, 156.8, 154.7, 138.3, 136.6, 128.6, 128.5, 128.13, 128.10, 115.5, 115.4, 80.7, 67.1, 66.1, 65.6, 62.8, 58.94, 58.91, 47.2, 47.1, 46.7, 46.5, 33.0, 32.3, 28.5. MS (m/z) calcd. for C17H25FNO4 [M+H]+ 326.2, found 226.1 [(M-Boc) +H]+. Rf 0.18 (heptanes:EtOAc, 1:1).
(2S,3R)-tert-Butyl 3-(4-chloro-3-(hydroxymethyl)phenyl)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (6l)
In a flame dried flask a solution of 4l (1.31 g, 2.24 mmol, 1 equiv) in dry THF (6.85 mL) was cooled to −78 °C. LiBEtH3 1 M (2.69 mL, 2.69 mmol, 1.2 equiv) was added via syringe dropwise and the reaction mixture was stirred for 1h, then quenched with sat. NaHCO3 (6 mL) and warmed to rt. The aqueous phase was extracted with EtOAc (3 × 10 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated to give the corresponding hemiaminal 5l as a colorless oil was used in the next step without further purification.
In a flame dried flask a solution of the crude hemiaminal 5l (2.24 mmol, 1 equiv) in dry DCM (7.1 mL) was cooled to −78 °C. HSiEt3 (0.71 mL, 4.476 mmol, 2 equiv) and BF3·Et2O (0.62 mL, 4.94 mmol, 2.2 equiv) were sequentially added via syringe and the reaction mixture was stirred for 5h at −78 °C. After the complete consumption of the substrate the reaction was quenched with sat. NaHCO3 (4 mL) and warmed to rt. The mixture was diluted with DCM and the organic phase was separated, washed with sat. NH4Cl (5 mL), dried over MgSO4 and concentrated to give 1.12 g. The crude product was dissolved in dry THF under N2 atmosphere at rt and 1M TBAF-THF solution (6.7 mL, 6.71 mmol, 3 equiv) was added dropwise. After 2h, the reaction mixture was quenched with sat. NaHCO3 (5 mL), EtOAc (10 mL) and H2O (10 mL). The layers were separated and the aqueous layer extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with brine (1 × 10 mL), dried over MgSO4 and concentrated to dryness. The crude product was purified by flash chromatography (heptanes:EtOAc, 2:1 → 3:2) to afford the title compounds as a colorless oil (370 mg, 49% over three steps).1H NMR (CDCl3) δ 7.37 (d, J = 1.9 Hz, 1H), 7.25 (d, J = 8.2 Hz, 1H), 7.04 (dd, J = 8.2, 2.2 Hz, 1H), 4.69 (s, 2H), 4.00 – 3.48 (m, 6H), 3.41 – 3.24 (m, 1H), 2.92 (s, 1H), 2.12 (dtd, J = 9.4, 6.4, 2.9 Hz, 1H), 1.89 (dq, J = 12.3, 9.7 Hz, 1H), 1.47 (s, 9H).13C NMR (CDCl3) δ 156.7, 139.9, 139.0, 130.9, 129.5, 127.5, 127.5, 80.7, 66.8, 65.5, 62.3, 47.2, 47.0, 32.8, 28.5. MS (m/z) calcd. for C17H25ClNO4 [M+H]+ 342.1, found 242.1 [(M-Boc) +H]+. Rf 0.25 (heptanes:EtOAc, 6:4).
tert-Butyl (2S,3R)-2-(hydroxymethyl)-3-(3-(hydroxymethyl)-4-methylphenyl)pyrrolidine-1-carboxylate (6m)
Compound 4m (211 mg, 0.037 mmol) was dissolved in dry THF (3.1 mL) and dimethylsulfide borane complex (0.11 mL, 10.5 M in THF) was added dropwise. The reaction mixture was refluxed for 2.5h, after which it was allowed to cool down to rt and diluted with Et2O, quenched with sat. NH4Cl and the organic portion separated. The organic partition was washed with brine, dried over MgSO4, filtered through celite and concentrated in vacuo. The crude product was dissolved in THF (4.7 mL) and treated with TBAF (1M, 2.24 mL) at rt for 23h. The mixture was diluted with EtOAc and washed consecutively with NH4Cl (1M), H2O, brine and dried over MgSO4. The solution was concentrated under reduced pressure and purified by flash column chromatography (heptane:EtOAc, 10:1 −> EtOAc) to provide the title compound as a colorless syrup (117 mg, 97%). Two rotamers.1H NMR (400 MHz, CDCl3) δ 7.25 (s, 1H), 7.11 – 7.07 (m, 1H), 7.06 – 7.01 (m, 1H), 4.68 – 4.50 (m, 2H), 3.99 – 3.84 (m, 1H), 3.72 (m, 2H), 3.65 – 3.55 (m, 1H), 3.38 – 3.26 (m, 1H), 2.87 (m, 1H), 2.30 – 2.25 (m, 3H), 2.15 – 2.05 (m, 1H), 2.00 – 1.86 (m, 1H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 156.93, 139.42, 130.66, 128.87, 126.69, 126.45, 114.21, 80.66, 67.02, 65.78, 63.11, 47.56, 47.22, 33.00, 28.57, 18.27. Rf 0.05 (heptane:EtOAc, 10:1).
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-chlorophenyl)pyrrolidine-2-carboxylic acid (7d)
A solution of NaIO4 (434 mg, 2.03 mmol, 4.1 equiv) and RuCl3·xH2O (3.1 mg, 0.015 mmol, 0.03 equiv) in H2O (6.2 mL) was added to a solution of 6d (154 mg, 0.494 mmol, 1 equiv) in MeCN:EtOAc (1:1, 7.0 mL) cooled to 0 °C. The reaction mixture was stirred for 30 min then filtered through celite and the filter cake washed with EtOAc. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried over MgSO4 and concentrated. The crude product was purified by flash chromatography (heptanes:EtOAc, 1:1 +1% AcOH) to afford the title compound as an off-white solid (138 mg, 86%). 1H NMR (CDCl3) δ (two rotamers) 9.64 (br s, 1H), 7.27–7.22 (m, 3H), 7.15–7.13 (m, 1H), 4.40 (d, J = 5.5 Hz, 0.4H), 4.25 (d, J = 6.5 Hz, 0.6H), 3.79–3.44 (m, 3H), 2.38–2.28 (m, 1H), 2.06–1.96 (m, 1H), 1.49 (s, 4H), 1.42 (s, 5H).13C NMR (CDCl3) δ (two rotamers) 177.6, 175.7, 155.2, 153.7, 142.9, 142.4, 134.6, 130.1, 127.5, 127.4, 127.1, 125.2, 81.2, 80.1, 65.4, 64.9, 49.4, 47.5, 46.1, 45.9, 32.7, 32.2, 28.3, 28.2. LC-MS (m/z) calcd. for C16H21ClNO4 [M+H]+ 326.12, found 226.0 [(M+H)-Boc]+. [α]25D +40.5 (c= 0.55, MeOH). Rf 0.27 (heptanes:EtOAc, 1:1 +1% AcOH). Mp: decomposition.
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-(trifluoromethyl)phenyl)pyrrolidine-2-carboxylic acid (7e)
A solution of NaIO4 (380.6 mg, 1.78 mmol, 4.1 equiv) and RuCl3·xH2O (2.7 mg, 0.013 mmol, 0.03 equiv) in H2O (5.45 mL) was added to a solution of 6e in MeCN:EtOAc (1:1, 6.2 mL) cooled to 0 °C. The reaction mixture was stirred for 30 min then was filtered through Celite and the filter cake was washed with EtOAc. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried over MgSO4 and concentrated to give the title compound as a colorless sticky oil (95 mg, 61%). 1H NMR (CDCl3) δ (two rotamers) 9.26 (br s, 1H), 7.55–7.44 (m, 4H), 4.43 (d, J = 5.8 Hz, 0.4H), 4.27 (d, J = 6.5 Hz, 0.6H), 3.81–3.52 (m, 3H), 2.42–2.31 (m, 1H), 2.10–1.99 (m, 1H), 1.50 (s, 4H), 1.42 (s, 5H).13C NMR (CDCl3) δ (two rotamers) 177.4, 175.4, 155.3 153.7, 141.8, 141.3, 131.1 (q, J = 33.0 Hz, C-CF3), 130.4, 130.3, 129.4, 129.3, 124.2, 124.1, 123.9 (q, J = 272.2 Hz, CF3), 123.8, 123.7, 81.3, 81.1, 65.4, 65.0, 49.5, 47.6, 46.1, 45.9, 32.8, 32.3, 28.3, 28.1.[α]25D +27.9 (c= 0.64, MeOH). Rf 0.26 (heptanes:EtOAc, 1:1 + 1% AcOH).
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-((tert-butoxycarbonyl)amino)phenyl)pyrrolidine-2-carboxylic acid (7f)
A solution of NaIO4 (179 mg, 0.836 mmol, 4.1 equiv) and RuCl3·xH2O (1.27 mg, 0.006 mmol, 0.03 equiv) in H2O (2.55 mL) was added to a solution of 6f (80 mg, 0.204 mmol, 1 equiv) in MeCN:EtOAc (1:1, 2.9 mL) cooled to 0 °C. The reaction mixture was stirred for 30 min then was filtered through filter paper and the filter cake was washed with EtOAc. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried over MgSO4 and concentrated to give 128 mg. The crude product was purified by flash chromatography (heptanes:EtOAc, 1:1 + 1% AcOH) to afford the title compound as a colorless oil (41 mg, 49% yield). 1H NMR (CDCl3) δ (two rotamers) 10.51 (s, 1H), 7.30 (bs, 1H), 7.25 – 7.16 (m, 2H), 6.94 – 6.89 (m, 1H), 6.73 (bs, 1H), 4.41 (d, J = 5.4 Hz, 0.4H), 4.25 (d, J = 6.5 Hz, 0.6H), 3.85 – 3.35 (m, 3H), 2.42 – 2.18 (m, 1H), 2.07 – 1.96 (m, 1H), 1.51 (s, 9H), 1.49 (s, 4H), 1.41 (s, 5H). 13C NMR (CDCl3) δ (two rotamers) 178.1, 177.1, 175.8, 155.6, 153.9, 142.0, 141.5, 139.0, 138.9, 129.5, 121.7, 117.7, 117.4, 81.3, 80.9, 65.7, 65.3, 60.6, 50.0, 47.8, 46.4, 46.1, 32.9, 32.5, 28.5, 28.5, 28.4. [α]25D +48.4 (c= 0.28, MeOH). Rf 0.21 (heptanes:EtOAc, 1:1 + 1% AcOH).
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-cyanophenyl)pyrrolidine-2-carboxylic acid (7g)
A solution of NaIO4 (435 mg, 2.03 mmol, 4.1 equiv) and RuCl3·xH2O (3.09 mg, 0.015 mmol, 0.03 equiv) in H2O (6.19 mL) was added to a solution of 6g (150 mg, 0.496 mmol, 1 equiv) in MeCN:EtOAc (1:1, 7.04 mL) cooled to 0 °C. The reaction mixture was stirred for 1h, then filtered through filter paper and the filter cake was washed with EtOAc. The aqueous layer was extracted with EtOAc (3 × 5 mL) and the combined organic layers were washed with brine, dried over MgSO4 and concentrated. The crude product was purified by flash chromatography (heptanes:EtOAc, 1:2 +1% AcOH) to afford the title compound as a white solid (119 mg, 76%). 1H NMR (CDCl3) δ (two rotamers) 10.56 (bs, 1H), 7.66 – 7.37 (m, 4H), 4.37 (d, J = 5.6 Hz, 0.5H), 4.23 (d, J = 6.6 Hz, 0.5H), 3.83 – 3.40 (m, 3H), 2.36 (dt, J = 11.9, 6.5 Hz, 1H), 2.01 (dq, J = 12.7, 7.9 Hz, 1H), 1.48 (s, 4H), 1.41 (s, 5H). 13C NMR (CDCl3) δ (two rotamers) 177.3, 175.3, 155.4, 153.7, 142.5, 142.1, 131.8, 131. 7, 131.2, 131.1, 130.8, 130.7, 129.9, 118.6, 113.1, 81.7, 81.3, 65.4, 65.1, 49.3, 47.4, 46.2, 46.0, 32.7, 32.3, 28.5, 28.3. MS (m/z) calcd. for C17H21N2O4 [M+H]+ 317.15, found 217.1 [(M+H)-Boc]+. Mp: 141.4–143.1 °C. Rf 0.26 (heptanes:EtOAc, 1:2 + 1% AcOH).
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carbamoylphenyl)pyrrolidine-2-carboxylic acid (7h)
H2O2 30% (w/w) in H2O (0.089 mL, 0.870 mmol, 6 equiv) was added dropwise to a solution of 7g (46 mg, 0.145 mmol, 1 equiv) and K2CO3 (80 mg, 0.58 mmol, 4 equiv) in EtOH:H2O (1:1, 1 mL). The reaction mixture was stirred for 3 hours at rt, then cautiously acidified with HCl 1M (until pH~3) and extracted with EtOAc (3 × 5 mL). The combined organic layers were washed with brine (1 × 15 mL), dried over MgSO4 and concentrated to give the title compound (32 mg, 66%).
1H NMR (CDCl3) δ (two rotamers) 8.61 (br s, 1H), 7.87 – 7.61 (m, 2H), 7.48 – 7.30 (m, 2H), 7.12 – 6.72 (m, 2H), 4.50 (d, J = 6.6 Hz, 0.5H), 4.21 (d, J = 7.3 Hz, 0.5H), 3.83 – 3.40 (m, 3H), 2.44 – 2.17 (m, 1H), 2.12 – 1.94 (m, 1H), 1.47 (s, 5H), 1.40 (s, 4H). 13C NMR (CDCl3) δ (two rotamers) 176.4, 175.0, 171.0, 170.9, 155.4, 154.0, 141.2, 141.1, 133.6, 133.3, 131.2, 130.7, 129.3, 129.2, 127.3, 126.8, 126.6, 126.5, 81.3, 81.0, 66.1, 65.3, 50.1, 48.5, 46.7, 46.2, 33.4, 32.4, 28.6, 28.4. LC-MS (m/z) calcd. for C17H23N2O5 [M+H]+ 335.16, found 235.1 [(M+H)-Boc]+. Mp: 136.9–139.4 °C Rf 0.13 (heptanes:EtOAc, 1:3 + 1% AcOH).
(2S,3R)-1-(tert-butoxycarbonyl)-3-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)pyrrolidine-2-carboxylic acid (7i)
A solution of NaIO4 (231 mg, 1.08 mmol, 4.1 equiv) and RuCl3·xH2O (1.63 mg, 0.008 mmol, 0.03 equiv) in H2O (3.27 mL) was added to a solution of 6i (106 mg, 0.263 mmol, 1.0 equiv) in MeCN:EtOAc (1:1, 3.7 mL) cooled to 0 °C. The reaction mixture was stirred for 30 minutes then filtered through filter paper and the filter cake was washed with EtOAc. The aqueous layer was extracted with EtOAc (3 × 5 mL) and the combined organic layers were washed with brine, dried over MgSO4 and concentrated to afford the title compound as an off-white solid (72 mg, 66%). 1H NMR (CDCl3) δ (two rotamers) 7.77 – 7.65 (m, 2H), 7.35 – 7.32 (m, 2H), 4.46 (d, J = 5.6 Hz, 0.4H), 4.30 (d, J = 6.7 Hz, 0.6H), 3.85 – 3.42 (m, 3H), 2.43 – 2.23 (m, 1H), 2.07 (dt, J = 16.9, 8.1 Hz, 1H), 1.50 (s, 4H), 1.42 (s, 5H), 1.34 (s, 12H). 13C NMR (CDCl3) δ (two rotamers) 178.1, 175.5, 155.7, 153.8, 140.2, 139.76, 134.0, 133.8, 133.3, 133.3, 130.2, 129.6, 128.3, 84.0, 81.3, 80.8, 65.8, 65.3, 50.1, 47.8, 46.4, 46.2, 33.3, 32.8, 28.5, 28.4, 25.0. LC-MS (m/z) calcd. for C22H33BNO6 [M+H]+ 418.2, found 318.1 [(M+H)-Boc]+. Mp: decomposition. Rf 0.40 (heptanes:EtOAc, 1:2 + 1% AcOH).
(2S,3R)-3-(Benzo[d][1,3]dioxol-5-yl)-1-(tert-butoxycarbonyl)-5-oxopyrrolidine-2-carboxylic acid (7j)
IBX (340 mg, 1.21 mmol, 2 equiv) was added to a solution of 6j (195.0 mg, 0.607 mmol, 1 equiv) in DMSO (2.4 mL) at rt. The reaction mixture was allowed to stir until the total consumption of the starting material (3.5h) then was quenched with sat. NaHCO3 (3 mL). The aqueous phase was extracted with EtOAc (3 × 5 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated. The crude aldehyde (colorless oil) was used in the next step without further purification.
The crude aldehyde (0.607 mmol, 1 equiv), NaH2PO4·2H2O (284 mg, 1.82 mmol, 3 equiv) and 2-methyl-2-butene (0.32 mL, 3.04 mmol, 5 equiv) were dissolved in tert-BuOH:H2O (3:1, 3 mL). NaClO2 was then added and the reaction mixture was stirred for 1.5h at rt. After the complete consumption of the starting material, the reaction was quenched with pH 7 phosphate buffer (3 mL) and the aqueous phase was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated. The crude product was purified by flash chromatography (heptanes: EtOAc, 2:1 + 1% AcOH) to afford the title compound as an off-white solid (135 mg, 66% over two steps). 1H NMR (CDCl3) δ (two rotamers) 7.93 (br s, 1H), 6.72 (m, 3H), 5.94 (s, 2H), 4.33 (d, J = 5.3 Hz, 0.4H), 4.18 (d, J = 6.6 Hz, 0.6H), 3.80 – 3.36 (m, 3H), 2.33 – 2.22 (m, 1H), 2.07 – 1.88 (m, 1H), 1.50 (s, 4H), 1.42 (s, 5H). 13C NMR (CDCl3) δ (two rotamers) 178.2, 175.1, 155.9, 153.8, 148.2, 146.9, 146.80, 134.8, 134.3, 120.4, 120.3, 108.5, 107.4, 101.2, 81.6, 80.9, 66.0, 65.6, 49.9, 47.4, 46.4, 46.1, 33.1, 32.7, 28.5, 28.4. LC-MS (m/z) calcd. for C17H22NO6 [M+H]+ 336.1, found 236.1 [(M+H)-Boc]+. [α]25D +43.8 (c= 1.0, MeOH). Mp: 129.5–131.7. Rf 0.32 (heptanes:EtOAc, 1:1 + 1% AcOH).
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-4-fluorophenyl)pyrrolidine-2-carboxylic acid (7k)
A solution of NaIO4 (496 mg, 2.32 mmol, 8.2 equiv) and RuCl3·xH2O (3.5 mg, 0.017 mmol, 0.06 equiv) in H2O (3.51 mL) was added to a solution of 6k (92 mg, 0.283 mmol, 1 equiv) in MeCN:EtOAc (1:1, 3.98 mL) cooled to 0 °C. The reaction mixture was stirred for 2 hours then was filtered through filter paper and the filter cake was washed with EtOAc. The aqueous layer was extracted with EtOAc (3 × 10 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated to give 93 mg. The crude was purified by column chromatography (eluent: hept/EtOAc 1/3 + 1% AcOH) to afford the title compound as white needles (89 mg, 89% yield). 1H NMR (CDCl3,400 MHz) δ (two rotamers) 7.91 (dd, J = 6.8, 2.5 Hz, 1H), 7.49 (ddd, J = 8.6, 4.4, 2.5 Hz, 1H), 7.15 (dt, J = 14.4, 7.2 Hz, 1H), 4.39 (d, J = 6.4 Hz, 0.5H), 4.22 (d, J = 7.3 Hz, 0.5H), 3.88 – 3.45 (m, 3H), 2.33 (dt, J = 12.4, 6.1 Hz, 1H), 2.10 – 1.99 (m, 1H), 1.51 (s, 5H), 1.43 (s, 4H). 13C NMR (MeOD) δ (two rotamers) 177.9, 176.4, 174.4, 168.4, 168.3, 162.7, 162.6, 161.0, 160.9, 156.2, 153.7, 136.7, 136.2, 134.53, 134.47, 134.0, 133.9, 131.8, 131.4, 118.0, 117.90, 117.86, 117.7, 82.2, 81.3, 65.8, 65. 6, 49.3, 46.8, 46.6, 46.2, 33.0, 32.7, 28.5, 28.4, 20.7. LC-MS (m/z) calcd. for C17H21FNO6 [M+H]+ 354.1, found 254.1 [(M+H)-Boc]+. Mp: 168.5 – 170.6 °C. Rf 0.16 (heptanes:EtOAc, 3:1 + 1% AcOH).
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-4-chlorophenyl)pyrrolidine-2-carboxylic acid (7l)
A solution of NaIO4 (164 mg, 7.68 mmol, 8.2 equiv) and RuCl3·xH2O (11.6 mg, 0.056 mmol, 0.06 equiv) in H2O (11.6 mL) was added to a solution of 6l (320 mg, 0.936 mmol, 1 equiv) in MeCN:EtOAc (1:1, 13.2 mL) cooled to 0 °C. The reaction mixture was stirred for 2 hours then was filtered through filter paper and the filter cake was washed with EtOAc. The aqueous layer was extracted with EtOAc (3 × 10 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated. The crude product was purified by column chromatography (heptanes:EtOAc, 1:3 + 1% AcOH) to afford the title compound as a white solid (205 mg, 51% yield). 1H NMR (CDCl3) δ (two rotamers) 7.88 (d, J = 2.1 Hz, 1H), 7.46 (dd, J = 8.1, 4.4 Hz, 1H), 7.39 (d, J = 8.3 Hz, 1H), 6.91 (br s, 2H), 4.40 (d, J = 6.2 Hz, 0.5H), 4.24 (d, J = 7.2 Hz, 0.5H), 3.85 – 3.48 (m, 3H), 2.41 – 2.30 (m, 1H), 2.03 (dt, J = 9.1, 8.6 Hz, 1H), 1.51 (s, 5H), 1.44 (s, 4H). 13C NMR (MeOD) δ (two rotamers) 175.7, 175.3, 169.0, 168.9, 156.0, 155.6, 141.7, 141.2, 132.93, 132.87, 132.80, 132.76, 132.7, 132.31, 132.27, 132.2, 131.1, 130.9, 82.0, 81.6, 67.2, 66.6, 50.5, 47.3, 47.1, 33.8, 33.2, 28.7, 28.5. LC-MS (m/z) calcd. for C17H21ClNO6 [M+H]+ 370.1, found 270.0 [(M+H)-Boc]+. Mp: 184.2–185.6 °C. Rf 0.26 (heptanes:EtOAc, 3:1 + 1% AcOH).
(2S,3R)-tert-Butyl 3-(3-bromophenyl)-2-(((tert-butyldimethylsilyl)oxy)methyl)pyrrolidine-1-carboxylate (8b)
In a flame dried flask a solution of 4b (803 mg, 1.66 mmol, 1 equiv) in dry THF (5.1 mL) was cooled to −78 °C. 1M LiBEtH3 (THF solution) (1.99 mL, 1.99 mmol, 1.2 equiv) was added via syringe dropwise and the reaction mixture was stirred for 1 hour, then was quenched with sat. NaHCO3 (3 mL) and warmed to rt. The aqueous phase was extracted with EtOAc (3 × 5 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4 and concentrated to give the corresponding hemiaminal 5b, which was used in the next step without further purification.
In a flame dried flask a solution of the crude hemiaminal 5b (1.66 mmol, 1 equiv) in dry DCM (5.3 mL) was cooled to −78 °C. HSiEt3 (0.53 mL, 3.31 mmol, 2 equiv) and BF3·Et2O (0.46 mL, 3.65 mmol, 2.2 equiv) were added sequentially via syringe and the reaction mixture stirred for 4.5 hours. The reaction was quenched with sat. NaHCO3 (4 mL) and warmed to rt. The mixture was diluted with DCM and the organic phase was separated, washed with sat. NH4Cl (5 mL), dried over MgSO4 and concentrated to dryness to give crude alcohol 6b.
TBSCl (300 mg, 1.99 mmol, 1.2 equiv) was added to a solution of the crude alcohol 6b (1.66 mmol, 1 equiv) and imidazole (282 mg, 4.14 mmol, 2.5 equiv) in dry DMF (11.7 mL) under nitrogen at rt. The reaction mixture was stirred overnight, then poured into H2O. The aqueous layer was extracted with Et2O (3 × 10 mL) and the collective organic layers were washed with 1M HCl (10 mL) and brine (10 mL), dried over MgSO4 and concentrated to give 839 mg (crude). The crude product was purified by flash chromatography (heptanes:EtOAc, 9:1) to afford the title compound as a colorless oil (572 mg, 73% over three steps). 1H NMR (CDCl3,400MHz) δ 7.39 – 7.32 (m, 2H), 7.16 (dd, J = 16.8, 9.0 Hz, 2H), 4.03 – 3.27 (m, 6H), 2.35 – 2.19 (m, 1H), 1.96 – 1.80 (m, 1H), 1.48 (s, 9H), 0.89 (s, 9H), 0.04 (s, 3H), 0.04 (s, 3H). 13C NMR (CDCl3) δ 154.3, 146.6, 146.2, 130.7, 130.6, 130.6, 130.3, 129.7, 126.1, 126.0, 122.8, 79.8, 79.4, 65.5, 65.4, 63.2, 61.8, 47.0, 46.6, 46.4, 45.5, 32.8, 31.7, 28.7, 26.0, 18.3, -5.3. Rf 0.27 (heptanes:EtOAc, 9:1).
(2S,3R)-tert-Butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-3-(3-cyanophenyl)pyrrolidine-1-carboxylate (8g)
Aryl bromide 8b (500 mg, 1.06 mmol, 1 equiv), Zn(CN)2 (75 mg, 0.64 mmol, 0.6 equiv), Zn powder (8.3 mg, 0.127 mmol, 0.12 equiv), Pd2(dba)3 (48.6 mg, 0.053 mmol, 0.05 equiv) and dppf (58.9 mg, 0.106 mmol, 0.1 equiv) were placed in a 10 mL oven-dried flask which was evacuated and backfilled with N2. Dry dimethylacetamide (2.1 mL) was added and the reaction mixture was stirred at 120 °C for 75 minutes, then cooled to rt and diluted with EtOAc (5 mL). The organic phase was washed with sat. NaHCO3, brine, dried over MgSO4 and concentrated. The crude product was purified by column chromatography (heptanes:EtOAc, 95:5) to give the title compound a colorless oil.1H NMR (CDCl3) δ 7.53 – 7.35 (m, 4H), 3.99 – 3.58 (m, 4H), 3.57 – 3.49 (m, 1H), 3.42 – 3.29 (m, 1H), 2.30 (dtd, J = 12.8, 7.3, 5.6 Hz, 1H), 1.87 (dq, J = 14.3, 7.2 Hz, 1H), 1.47 (s, 9H), 0.86 (s, 9H), 0.03 (s, 3H), 0.02 (s, 3H).13C NMR (CDCl3) δ 154.1, 145.5, 145.3, 131.9, 131.7, 131.0, 130.2, 129.5, 118.8, 112.7, 79.9, 79.5, 65.2, 63.2, 61.8, 46.7, 46.6, 46.1, 45.4, 32.5, 31.6, 28.6, 25.8, 18.2, -5.4. LC-MS (m/z) calcd. for C18H29N2OSi [(M+H)-Boc]+ 317.2, found 317.2. Rf 0.59 (heptanes:EtOAc, 2:1)
(2S,3R)-tert-Butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-3-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)pyrrolidine-1-carboxylate (8i)
(BPin)2 (380 mg, 1.50 mmol, 1.1 equiv), PdCl2(dppf) (49.7 mg, 0.068 mmol, 0.05 equiv) and KOAc (400 mg, 4.08 mmol, 3.0 equiv) were placed in an oven-dried flask which was evacuated and backfilled with nitrogen. A solution of aryl bromide 8 (640 mg, 1.36 mmol, 1.0 equiv) in dry DMF (8.2 mL) was added via syringe and the reaction mixture was stirred at 80 °C for 5 hours. The reaction mixture was cooled to rt and diluted with H2O (10 mL). The aqueous layer was extracted with Et2O (3 × 10 mL) and the combined organic layers were washed with HCl 1M (1 × 10 mL), brine (1 × 10 mL), dried over MgSO4 and concentrated. The crude product was purified by column chromatography (heptanes:EtOAc, 95:5) to give the title compound as a colorless oil (431 mg, 61%). 1H NMR (CDCl3) δ 7.71 – 7.64 (m, 2H), 7.36 – 7.27 (m, 2H), 4.10 – 3.49 (m, 5H), 3.43 – 3.23 (m, 1H), 2.31 – 2.19 (m, 1H), 1.99 – 1.88 (m, 1H), 1.49 (s, 9H), 1.34 (s, 12H), 0.89 (s, 9H), 0.04 (s, 6H). 13C NMR (CDCl3) δ (two rotamers) 154.4, 154.3, 143.2, 142.7, 133.9, 133.7, 133.0, 130.5, 130.3, 129.4 (bs), 128.2, 83.9, 79.5, 79.1, 65.5, 65.4, 63.0, 61.5, 47.2, 46.7, 46.5, 45.7, 33.0, 31.8, 28.7, 26.0, 25.0, 25.0, 18.3, -5.3. LC-MS (m/z) calcd. for C28H49BNO5Si [M+H]+ 518.3, found 336.2 [(M+H)-Boc-Pin]+. Rf 0.16 (heptanes:EtOAc, 95:5).
Pharmacological studies
All reagents and solvents were commercial and high purity of analytical grade or ultra-gradient HPLC-grade purchased from Sigma, (St. Louis, MO, USA), J.T. Baker (Denventer, The Netherlands), Merck (Darmstadt, Germany) or Riedel-de Haën (Seelze, Germany). Water was purified using a Milli-Q Gradient system (Millipore, Milford, MA, USA). LPS from Escherichia coli 055:B5 and fluorescein were purchased from Sigma (St. Louis, MO, USA). [3H]-digoxin, 250μCi (9.25MBq) and Emulsifier safe liquid scintillation cocktail were purchased from PerkinElmer (Boston, MA, USA). Prostaglandin E2 was purchased from Bio-techne Ltd (Abingdon, UK) and sorafenib from Cayman Chemical (Ann Arbor, MI, USA).
Liquid Chromatographic and Mass Spectrometric (LC-MS/MS) Analyses
An Agilent 1200 Series Rapid Resolution LC System was used together with a Poroshell 120 EC-C-18column (50 mm × 2.1 mm, 2.7 μm) for liquid chromatography prior to MS analysis of compound 1l, sorafenib and PGE2 with Agilent 6410 triple quadrupole mass spectrometer equipped with an electrospray ionization source (Agilent Technologies Inc., Wilmington, DE). The high-performance liquid chromatography eluents were water (A) containing 0.1% (v/v) formic acid and acetonitrile (B). For the compound 1l a gradient elution with 5–60% B was applied over 1–3 minutes, followed by 1 minute of isocratic elution with 60% B and 3 minutes column equilibration, giving a total time of 7 minutes/injection. For sorafenib, a gradient elution with 20–90% B was applied over 1–4 minutes, followed by 1 minute of isocratic elution with 90% B and 3 minutes column equilibration, giving a total time of 8 minutes/injection. For PGE2 an isocratic method with 10 % B was used. For all compounds the eluent flow rate was 0.2 mL/min, the column temperature was 40°C and injection volume 5 μL. The following mass spectrometry conditions were used for the compound 1l, sorafenib and PGE2. Electrospray ionization, positive ion mode for compound 1l, sorafenib and negative ion mode for PGE2; drying gas (nitrogen) temperature, 300°C; drying gas flow rate, 8 L/min; nebulizer pressure, 20 psi; and capillary voltage, 3500 V. Analyte detection was performed using multiple reaction monitoring, the transitions being 270.1 → 114.5 and 236.1 → 190 for compound 1l and 1a (internal standard), respectively. The transitions for sorafenib and the used internal standard, diclofenac were 465.1 → 252.2, 296.1 → 250, respectively. The transitions for PGE2 was 315.4 → 314.5. Fragmentor voltages used for compound 1l, 1a, sorafenib, diclofenac and PGE2 were 60V, 140V, 140V 100V and 180V, and the collision energies were 40V, 16V, 30V, 10V and 6V, respectively. Agilent MassHunter Workstation Acquisition software (Data Acquisition for Triple Quadrupole Mass Spectometer, version B.03.01) was used for data acquisition, and Quantitative Analysis (B.04.00) software was used for the data processing and analysis. The compound 1l lower limit of quantification for the brain, liver and plasma samples were 0.02 nmol/g, 0.02 nmol/g and 0.5 μM respectively. The lower limit of quantification for sorafenib and PGE2 in cell samples was 1 nM. Linearity of the calibration curves was evaluated by a quadratic regression analysis. The method was also selective, accurate, and precise over the calibration range. Within-run accuracy and precision were calculated from the results of the quality control samples at the three concentrations. The accuracies and precisions for quality control concentrations of 20% were considered to be acceptable.
Preparation of crude membrane fractions of mouse HCC cells
Mouse Nras driven p53−/− HCC cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with L-glutamine (2 mM), heat-inactivated fetal bovine serum (10%), penicillin (50 U/mL) and streptomycin (50 μg/mL). The cells were incubated for 48 h with or without 10 μM of compound 1l, washed three times with HBSS, followed by centrigfugation of the cells into a pllet and rapid freezing in liquid nitrogen. ProteoExtract® Subcellular Proteome Extraction Kit (Merck KGaA, Darmstadt, Germany) was used for the isolation of crude membrane fraction from the cell pellets following the manufacturer`s instructions. The protein concentrations in the crude membrane fractions were determined as mean of three samples by Bio-Rad Protein Assay (EnVision, Perkin Elmer, Inc., Waltham, MA, USA) and aliquots containing 50 μg of total protein were taken for further sample preparation.
Protein quantification by Multiplexed Selected/Multiple Reaction Monitoring by LC−MS/MS
The protein expressionsof the investigated transporters were simultaneously determined by means of multiplexed multiplereaction monitoring analysis. The aliquots containing 50 μg of protein from the crude membrane fractions were solubilized in 500 mM Tris–HCl (pH 8.5), 7 Mguanidine hydrochloride, 10 mM EDTA, and the proteins were S-carbamoylmethylated with iodoacetamide following dithiothreitol treatment. The alkylated proteins were precipitated using methanol andchloroform. The precipitates were dissolved in 6 M urea in 100 mMTris–HCl (pH 8.5), diluted fivefold with 100 mM Tris–HCl (pH 8.5), spiked with internal standard peptides and treated with Protease-Max surfactant (Promega, Madison, WI, USA). The dilutions were treated with lysyl endopeptidase (Lys-C:Wako Pure Chemical Industries, Osaka, Japan) at rtfor 3 h. After which tosylphenylalanyl chloromethyl ketone-treatedtrypsin (Promega, Madison, WI, USA) at an enzyme/substrate ratio of 1:100 was added into the samples and incubated at 37°C for 16 h. The tryptic digests were mixed formic acid, and then centrifuged at 4°C and 14 000 g for 5 min. The supernatant was mixed with water prior to LC-MS/MS analysis.
LC−MS/MS analysis was performed by coupling anAgilent 1290Infinity LC (Agilent Technologies, Waldbronn, Germany) instrumentationto a Agilent 6495 Triple Quadrupole Mass Spectrometer with an electrospray ionization (ESI) source (Agilent Technologies, Palo Alto, CA, USA) using multiple reaction monitoring (MRM). The following conditions were applied: positive ionization mode, the drying gas (nitrogen) was maintained at 210 °C, drying gas flow rate was 16 L/min, nebulizer gas pressure was 45 psi, sheath gas temperature 300 °C, sheath gas flow 11 L/min, fragmentor voltage was 380 V and the mass-spectrometer (MS) capillary voltage was 3.0 kV. The following ion funnel parameters were used for both positive and negative ionization: high- pressure ion funnel RF voltage 200 V and low-pressure ion funnel RF voltage 100 V. An injection volume of 15 μL was used and analytes were separated by AdvanceBio Peptide Map 2.1 × 250 mm, 2.7 μm. Mobile phases A and B respectively consisted of 0.1% formicacid in milli-Q water and acetonitrile. Thegradient sequence was as follows: flow rate of 0.30 mL/min, 50 min of total run time, 97:3 to 60:40 (A:B) during 40 min after injection, 5:95at 44 min, 97:3 at 45 min and constant 97:3 for 5 min. The eluted peptides were selectively and simultaneouslyanalyzed by SRM/MRM mode with LC−MS/MS. For each target protein, oneunique peptide was chosen according to previously published work.36 These peptides were monitored with two orthree different SRM/MRM transition sets (Table 5) derived from one set of stable isotope-labeled peptides purchased from JPT Peptide Technologies GmbH (Berlin, Germany) and unlabeled peptides.
Table 5.
Probe peptides and MRM transitions for the LC-MS/MS analysis of investigated transporters
| Transporter | St/Is | Probe peptide sequence | MRM transitions (m/z)
|
|||
|---|---|---|---|---|---|---|
| Q1 | Q3.1 | Q3.2 | Q3.3 | |||
| Abcb1 | St | NTTGALTTR | 467.8 | 618.4 | 719.4 | 516.3 |
| Is | NTTGALTTR* | 472.6 | 628.4 | 729.4 | 571.3 | |
| Abcg2 | St | SSLLDVLAAR | 522.8 | 757.5 | 644.3 | 529.3 |
| Is | SSLLDVLAAR* | 527.8 | 767.5 | 654.4 | 539.4 | |
| Abcc2 | St | LTIIPQDPILFSGNLR | 899.0 | 1356.7 | 678.9 | - |
| Is | LTIIPQDPILFSGNLR* | 904.0 | 1366.7 | 683.9 | - | |
| Abcc4 | St | APVLFFDR | 482.8 | 796.4 | 697.4 | 584.3 |
| Is | APVLFFDR* | 487.8 | 806.4 | 707.4 | 594.3 | |
| Slc7a5 | St | VQDAFAAAK | 460.8 | 693.4 | 578.3 | - |
| Is | VQDAFAAAK* | 464.8 | 701.4 | 586.3 | - | |
| Slc2a1 | St | TFDEIASGFR | 571.8 | 894.4 | 537.3 | - |
| Is | TFDEIASGFR* | 576.8 | 904.4 | 547.3 | - | |
denotes 13C labeled arginine and lysine
The ability of compound 1l to alter the cell accumulation of transporter probes and sorafenib
The ability of compound 1l to increase the cell accumulation of Abcb1 probe [3H]-digoxin, Abcc1-5 probe fluorescein, Abcb1 and Abcg2 substrate sorafenib as well as to decrease cell accumulation of Slc7a5 probe [14C]-L-leucine and Slc2a1 probe [14C]-D-glucose was determined in mouse Nras driven p53−/− HCC cells. The cells were cultured in DMEM supplemented with L-glutamine (2 mM), heat-inactivated fetal bovine serum (10%), penicillin (50 U/mL) and streptomycin (50 μg/mL). HCC cells were seeded at the density of 1 × 105 cells/well onto 24-well plates. After seeding the cells were incubated for 24 h with or without 10 μM of compound 1l. In order to ensure that compound 1l does not inhibit the function of the efflux transporters, the incubation medium was removed and the cells were carefully washed three times with pre-warmed Hank’s balance salt solution (HBSS) containing 125 mM choline chloride, 4.8 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1.3 mM CaCl2, 5.6 mM glucose, 100 μM leucine and 25 mM HEPES (pH 7.4). After which transporter probes, [3H]-digoxin,[14C]-L-leucine, [14C]-D-glucose, 20 μM fluorescein or 10 μM sorafenib was added in pre-warmed HBSS buffer (250 μL) on the top of the cell layer and incubating at 37 °C for 30 min for [3H]-digoxin, fluorescein and sorafenib or 5 min for[14C]-L-leucine and [14C]-D-glucose. Subsequently, the cells were washed three times with ice-cold HBSS and lysed with 500 μL of 0.1 M NaOH. The protein concentrations in each well were determined by Bio-Rad Protein Assay (EnVision, Perkin Elmer, Inc., Waltham, MA, USA). The lysate from the [3H]-digoxin, [14C]-L-leucine and [14C]-D-glucose samples was mixed with 3.5 mL of Emulsifier safe liquid scintillation cocktail. The radioactivity was measured by liquid scintillation counting (Wallac 1450 MicroBeta; Wallac Oy, Finland). The fluorescein samples were measured using fluorescein detector (EnVision, Perkin Elmer, Inc., Waltham, MA, USA) and the sorafenib concentrations were determined by the LC-MS/MS.
Live imaging of murine HCC cell culture
The cells were loaded with the Ca2+ sensitive fluorescent dye Fluo-4-AM (5 μM) for 45 min followed by a 20 min washout in the BSS containing the following (in millimolars): 2.5 KCl, 152 NaCl, 10 glucose, 2 CaCl2, and 10 HEPES at pH 7.4. Fluorescence was visualized using the imaging setup (TILL Photonics GmbH) consisting of a monochromatic light source and a CCD camera (SensiCam). Cells Loaded with Fluo-4 were imaged with an excitation light of 495 nm (exposure time 100 ms, binning 2). Chemicals were applied using a fast perfusion system (Rapid Solution Changer RSC-200, BioLogic Science). Data were analysed offline using the TILL Photonics and Origin 8 software.
Ability of 1l to decrease PGE2 concentration in the cells
The ability of compound 1l to decrease PGE2 synthesis was studied in HCC cells with and without LPS-induced inflammation. The LPS concentration in the growth medium was 2.5 μg/mL. The cells were incubated 24 h with or without 100 μM compound 1l. Subsequently, the cells were washed three times with ice-cold HBSS and then lysed with 500 μL of acetonitrile on ice. The PGE2 concentrations from the cell lysates were determined by the LC-MS/MS.
Anti-proliferative activity in vitro
The mouse HCC cells were seeded at the density of 2 × 104 cells/well onto collagen-coated 96-well plates and the cells were used for the experiments one day after seeding. A concentration of 1 μM, 2.5 μM, 5 μM and 10 μM of sorafenib, 100 μM of compound 1l, as well as, the combination of all the mentioned sorafenib concentrations with 100 μM compound 1l were added into the growth medium and incubated for 3 days. Each day the medium was changed and after the 72 h incubation the cell viability was determined by resazurin cell proliferation kit (Sigma, St. Louis, MO, USA), which is directly proportional to aerobic respiration and cellular metabolism of cells. The samples were measured fluorometrically by monitoring the increase in fluorescence at a wavelength of 590 nm using an excitation wavelength of 560 nm (EnVision, Perkin Elmer, Inc., Waltham, MA, USA). The cell death was confirmed in the decrease of cell amount by visualizing the wells with microscopy.
Animals
Adult male mice weighing 25 ± 5 g were supplied by the National Laboratory Animal Centre (Kuopio, Finland). Mice were housed in stainless steel cages on a 12 h light (07:00–19:00) and 12 h dark (19:00–07:00) cycle at an ambient temperature of 22 ± 1 °C with a relative humidity of 50–60%. All experiments were carried out during the light phase. Tap water and food pellets (Lactamin R36; Lactamin AB, Södertälje, Sweden) were available ad libitum. All procedures with the animals were performed according to European Community Guidelines and Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication no. 85–23, revised in 1985). The procedures were reviewed and approved by the Finnish National Animal Experiment Board.
In Situ mouse brain perfusion
Mice were anesthetized with intraperitoneal (i.p.) injections of ketamine (120 mg/kg) and xylazine (8 mg/mL), and their right carotid artery system was exposed. The right external carotid artery was ligated, and the right common carotid artery was cannulated with catheter filled with 100 IU/mL heparin. The perfusions were performed at 37 °C with a flow rate of 2.5 mL/min, for 60 s followed by the washing of the capillaries for 2 s with 4 °C drug free perfusion buffer. The method is described in more detail by Gynther et al. 2016.37
In vivo pharmacokinetics of compound 1l
A concentration of 5.0 mM concentration of compound 1l was dissolved in a vehicle containing 0.9 % (w/v) NaCl in water. A dose of 10 mg/kg of compound 1l was administered as a bolus injection (i.p.) to mice. The mice were sacrificed by decapitation at selected time points between (10–240 min) and plasma, brain and liver were collected for analysis.
Plasma and tissue sample preparation
Plasma samples were prepared by precipitating of 100 μL of plasma with 200 μL of acetonitrile containing the internal standard (1a). Samples were vortexed and centrifuged for 10 min at 14,000 g at 4 °C. Then 100 μL of supernatant was mixed with 100 μL of ultrapure water prior to LC-MS/MS analysis. Tissue samples were weighed and homogenized with ultrapure water (1:3). An aliquot of 100 μL was taken, and the analyte was isolated from the samples by protein precipitation with 300 μL of acetonitrile containing the internal standard. Samples were vortexed and centrifuged for 10 min at 14,000 g at 4 °C. Then 200 μL of supernatant was mixed with 100 μL of ultrapure water prior LC-MS/MS analysis.
Two-Electrode Voltage-Clamp electrophysiology
Rat cDNAs encoding GluN1-1a (Genbank accession number U11418 and U08261; hereafter GluN1), GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), and GluN2D (L31611) were generously provided by Dr. S. Heinemann (Salk Institute, La Jolla, CA), Dr. S. Nakanishi (Osaka Bioscience Institute, Osaka, Japan), and Dr. P. Seeburg (University of Heidelberg). The cDNA encoding rat GluN2B was modified without changing the amino acid sequence to remove a T7 RNA polymerase termination site located in the C-terminal domain.38 For expression in Xenopus laevis oocytes, cDNAs were linearized using restriction enzymes and used as templates to synthesize cRNA using the mMessage mMachine kit (Ambion, Life Technologies, Paisley, UK). Xenopus oocytes were obtained from Rob Weymouth (Xenopus 1, Dexter, MI) and prepared as previously described.39 The oocytes were injected with cRNAs encoding GluN1 and GluN2 in a 1:2 ratio, and maintained at 18°C in Barth’s solution containing 88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 0.82 mM MgSO4, 0.33 mM Ca(NO3)2, 0.91 mM CaCl2, 10 mM HEPES (pH 7.5 with NaOH) supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin and 50μg/ml gentamycin (Invitrogen, Life Technologies, Paisley, UK). Two-electrode voltage-clamp recordings were performed on Xenopus oocytes at room temperature 2–6 days post-injection with the extracellular recording solution comprised of 90 mM NaCl, 1 mM KCl, 10 mM HEPES, 0.5 mM BaCl2 and 0.01 mM EDTA (pH 7.4 with NaOH) at a holding potential of −40 mV essentially as previously described.39 NMDA receptor ligands were dissolved in extracellular recording solution and applied to the oocyte by gravity-driven perfusion.
Data analysis
All statistical analyses were performed using GraphPad Prism v. 5.03 software (GraphPad Software, San Diego, CA, USA). Statistical differences between groups were tested using two-tailed t-test (Figure 1 and Figure 2). Concentration-inhibition data measured using two-electrode voltage-clamp electrophysiology were fitted to the Hill equation to obtain IC50 values for individual oocytes as previously described.39 The sorafenib IC50 value for cytotoxicity in murine HCC cells was calculated by nonlinear regression analysis and presented as the mean ± SEM. The pharmacokinetic parameters, AUC0–240 min, Cmax, tmax and t½β in plasma, brain and liver, were obtained from the pharmacokinetic data.
Radioligand Binding
Ligand affinities at native AMPA, KA, and NMDA receptors (rat brain synaptosomes) were determined using [3H]AMPA, [3H]KA, and [3H]CGP-39653, respectively as previously described.40 Ligand affinities at recombinant homomeric rat GluK1-3 were determined using [3H]KA as the radioligand as previously detailed (Sagot et al, 2008; Alcaide et al, 2016).41,42 Data were analyzed using GraphPad Prism 7 (GraphPad Software, San Diego, CA) do determine ligand IC50 and Ki values.
In silico studies
Calculation of LogP(o/w) and total polar surface area (TPSA) was done in MOE version 2016.08.02 released by Chemcomp corporation.
Acknowledgments
We would like to thank the Lundbeck Foundation and the Danish Medical Research Council for financial support of the medicinal chemistry work reported herein. Emil Aaltonen Foundation and The Finnish Cultural Foundation is acknowledged for the funding regarding the pharmacokinetic analysis, transporter expression modulation and cytotoxicity studies. Finally, the National Institutes ofHealth (P20GM103546 and R01NS097536) is acknowledged for financial support in regard to the functional characterization of 1l at NMDA receptors. Mrs. Sari Ukkonen is acknowledged for technical assistance.
List of abbreviations
- ABC
ATP-binding cassette
- Abcb1
ATP-binding cassette subfamily B member 1
- Abcc
ATP-binding cassette subfamily C
- Abcg2
ATP-binding cassette subfamily G member 2
- AUC
area under the concentration curve
- BBB
blood-brain barrier
- Cmax
maximum concentration
- cPLA2
cytoplasmic phospholipase A2
- DCM
dichloromethane
- DCVC
dry collum vacuum chromatography
- DMAP
N,N-dimethyl-4-aminopyridine
- DMEM
Dulbecco’s Modified Eagle Medium
- DMF
N,N-dimethylformamide
- DMSO
N,N-dimethylsulfonamide
- HBSS
Hank’s balance salt solution
- HCC
hepatocellular carcinoma
- IBX
2-Iodoxybenzoic acid
- LC-MS/MS
liquid chromatography-mass spectrometry
- LPS
lipopolysaccharide
- MDR
multi drug resistance
- MRM
multiple reaction monitoring
- PGE2
prostaglandin E2
- SRM
selected reaction monitoring
- TBAF
tetrabutylammonium flouride
- TBSCl
tert-butyldimethylsilyl chloride
- TFA
trifluoroacetic acid
- Tmax
time to reach the maximum concentration
- T½β
elimination half-life
References
- 1.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer Drug Resistance: An Evolving Paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 2.Rudalska R, Dauch D, Longerich T, McJunkin K, Wuestefeld T, Kang T-WT-WT-W, Hohmeyer A, Pesic M, Leibold J, von Thun A, Schirmacher P, Zuber J, Weiss K-HK-H, Powers S, Malek NPNP, Eilers M, Sipos B, Lowe SWSWSW, Geffers R, Laufer S, Zender L, Von A, Wuestefeld T, Kang T-WT-WT-W, Hohmeyer A, Pesic M, Leibold J, von Thun A, Schirmacher P, Zuber J, Weiss K-HK-H, Powers S, Malek NPNP, Eilers M, Sipos B, Lowe SWSWSW, Geffers R, Laufer S, Zender L, et al. In Vivo RNAi Screening Identifies a Mechanism of Sorafenib Resistance in Liver Cancer. Nat Med. 2014;20:1138–1146. doi: 10.1038/nm.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting Multidrug Resistance in Cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y-K, Wang Y-J, Gupta P, Chen Z-S. Multidrug Resistance Proteins (MRPs) and Cancer Therapy. AAPS J. 2015;17:802–812. doi: 10.1208/s12248-015-9757-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stacy AE, Jansson PJ, Richardson DR. Molecular Pharmacology of ABCG2 and Its Role in Chemoresistance. Mol Pharmacol. 2013;84:655–669. doi: 10.1124/mol.113.088609. [DOI] [PubMed] [Google Scholar]
- 6.Gillet JP, Efferth T, Remacle J. Chemotherapy-Induced Resistance by ATP-Binding Cassette Transporter Genes. Biochim Biophys Acta. 2007;1775:237–262. doi: 10.1016/j.bbcan.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Wu CP, Hsieh CH, Wu YS. The Emergence of Drug Transporter-Mediated Multidrug Resistance to Cancer Chemotherapy. Mol Pharm. 2011;8:1996–2011. doi: 10.1021/mp200261n. [DOI] [PubMed] [Google Scholar]
- 8.Rigalli JP, Ciriaci N, Arias A, Ceballos MP, Villanueva SSM, Luquita MG, Mottino AD, Ghanem CI, Catania VA, Ruiz ML. Regulation of Multidrug Resistance Proteins by Genistein in a Hepatocarcinoma Cell Line: Impact on Sorafenib Cytotoxicity. PLoS One. 2015;10:e0119502. doi: 10.1371/journal.pone.0119502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang WC, Hsieh YL, Hung CM, Chien PH, Chien YF, Chen LC, Tu CY, Chen CH, Hsu SC, Lin YM, Chen YJ. BCRP/ABCG2 Inhibition Sensitizes Hepatocellular Carcinoma Cells to Sorafenib. PLoS One. 2013;8:e83627. doi: 10.1371/journal.pone.0083627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gnoth MJ, Sandmann S, Engel K, Radtke M. In Vitro to in Vivo Comparison of the Substrate Characteristics of Sorafenib Tosylate toward P-Glycoprotein. Drug Metab Dispos. 2010;38:1341–1346. doi: 10.1124/dmd.110.032052. [DOI] [PubMed] [Google Scholar]
- 11.Callaghan R, Luk F, Bebawy M. Inhibition of the Multidrug Resistance P-Glycoprotein: Time for a Change of Strategy? Drug Metab Dispos. 2014;42:623–631. doi: 10.1124/dmd.113.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC Transporters in Cancer: More than Just Drug Efflux Pumps. Nat Rev Cancer. 2010;10:147–156. doi: 10.1038/nrc2789. [DOI] [PubMed] [Google Scholar]
- 13.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qosa H, Miller DS, Pasinelli P, Trotti D. Regulation of ABC Efflux Transporters at Blood-Brain Barrier in Health and Neurological Disorders. Brain Res. 2015;1628:298–316. doi: 10.1016/j.brainres.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leslie CC. Properties and Regulation of Cytosolic Phospholipase A2. J Biol Chem. 1997;272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- 16.Maeda S, Omata M. Inflammation and Cancer: Role of Nuclear Factor-kappaB Activation. Cancer Sci. 2008;99:836–842. doi: 10.1111/j.1349-7006.2008.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi Y, Wang S-Y, Yao M, Sai W-L, Wu W, Yang J-L, Cai Y, Zheng W-J, Yao D-F. Chemosensitization of HepG2 Cells by Suppression of NF-kappaB/p65 Gene Transcription with Specific-siRNA. World J Gastroenterol. 2015;21:12814–12821. doi: 10.3748/wjg.v21.i45.12814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deutsch SI, Tang AH, Burket JA, Benson AD. NMDA Receptors on the Surface of Cancer Cells: Target for Chemotherapy? Biomed Pharmacother. 2014;68:493–496. doi: 10.1016/j.biopha.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ribeiro MPC, Nunes-Correia I, Santos AE, Custódio JBA. The Combination of Glutamate Receptor Antagonist MK-801 with Tamoxifen and Its Active Metabolites Potentiates Their Antiproliferative Activity in Mouse Melanoma K1735-M2 Cells. Exp Cell Res. 2014;321:288–296. doi: 10.1016/j.yexcr.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi F, Hirata Y, Akram H, Kamitori K, Dong Y, Sui L, Tokuda M. FOXO/TXNIP Pathway Is Involved in the Suppression of Hepatocellular Carcinoma Growth by Glutamate Antagonist MK-801. BMC Cancer. 2013;(13):468. doi: 10.1186/1471-2407-13-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergeron R, Coyle JT. NAAG, NMDA Receptor and Psychosis. Curr Med Chem. 2012;19:1360–1364. doi: 10.2174/092986712799462685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghose AK, Viswanadhan VN, Wendoloski JJ. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J Comb Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 23.Larsen AM, Venskutonytė R, Valadés EA, Nielsen B, Pickering DS, Bunch L. Discovery of a New Class of Ionotropic Glutamate Receptor Antagonists by the Rational Design of (2S,3R)-3-(3-Carboxyphenyl)-Pyrrolidine-2-Carboxylic Acid. ACS Chem Neurosci. 2011;2:107–114. doi: 10.1021/cn100093f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krogsgaard-Larsen N, Storgaard M, Møller C, Demmer CS, Hansen J, Han L, Monrad RN, Nielsen B, Tapken D, Pickering DS, Kastrup JS, Frydenvang K, Bunch L. Structure-Activity Relationship Study of Ionotropic Glutamate Receptor Antagonist (2S,3R)-3-(3-Carboxyphenyl)pyrrolidine-2-Carboxylic Acid. J Med Chem. 2015;58:6131–6150. doi: 10.1021/acs.jmedchem.5b00750. [DOI] [PubMed] [Google Scholar]
- 25.Ogura A, Yamada K, Yokoshima S, Fukuyama T. Total Synthesis of (−)-Anisatin. Org Lett. 2012;14:1632–1635. doi: 10.1021/ol300390k. [DOI] [PubMed] [Google Scholar]
- 26.Bunch L, Krogsgaard-Larsen P, Madsen U. Mixed Cyano-Gilman Cuprates-Advances in Conjugate Addition to Alpha, Beta-Unsaturated Pyroglutaminol. Synthesis (Stuttg) 2002;2002:31–33. [Google Scholar]
- 27.Synthesis Will Be Reported Elsewhere.
- 28.Hansen KB, Bräuner-Osborne H, Egebjerg J. Pharmacological Characterization of Ligands at Recombinant NMDA Receptor Subtypes by Electrophysiological Recordings and Intracellular Calcium Measurements. Comb Chem High Throughput Screen. 2008;11:304–315. doi: 10.2174/138620708784246040. [DOI] [PubMed] [Google Scholar]
- 29.Yung-Chi C, Prusoff WH. Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 30.Hedegaard M, Hansen KB, Andersen KT, Bräuner-Osborne H, Traynelis SF. Molecular Pharmacology of Human NMDA Receptors. Neurochem Int. 2012;61:601–609. doi: 10.1016/j.neuint.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uchida Y, Tachikawa M, Obuchi W, Hoshi Y, Tomioka Y, Ohtsuki S, Terasaki TA. Study Protocol for Quantitative Targeted Absolute Proteomics (QTAP) by LC-MS/MS: Application for Inter-Strain Differences in Protein Expression Levels of Transporters, Receptors, Claudin-5, and Marker Proteins at the Blood– Brain Barrier in ddY, FVB, an. Fluids Barriers CNS. 2013;(10):21. doi: 10.1186/2045-8118-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rautio J, Humphreys JE, Webster LO, Balakrishnan A, Keogh JP, Kunta JR, Serabjit-Singh CJ, Polli JW. In Vitro P-Glycoprotein Inhibition Assays for Assessment of Clinical Drug Interaction Potential of New Drug Candidates: A Recommendation for Probe Substrates. Drug Metab Dispos. 2006;34:786–792. doi: 10.1124/dmd.105.008615. [DOI] [PubMed] [Google Scholar]
- 33.Löscher W, Potschka H. Blood-Brain Barrier Active Efflux Transporters: ATP-Binding Cassette Gene Family. NeuroRX. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib Blocks the RAF/MEK/ERK Pathway, Inhibits Tumor Angiogenesis, and Induces Tumor Cell Apoptosis in Hepatocellular Carcinoma Model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 35.Si C, Myers AG. A Versatile Synthesis of Substituted Isoquinolines. Angew Chemie - Int Ed. 2011;50:10409–10413. doi: 10.1002/anie.201104769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akazawa T, Uchida Y, Tachikawa M, Ohtsuki S, Terasaki T. Quantitative Targeted Absolute Proteomics of Transporters and Pharmacoproteomics-Based Reconstruction of P-Glycoprotein Function in Mouse Small Intestine. Mol Pharm. 2016;13:2443–2456. doi: 10.1021/acs.molpharmaceut.6b00196. [DOI] [PubMed] [Google Scholar]
- 37.Gynther M, Pickering DS, Spicer JA, Denny WA, Huttunen KM. Systemic and Brain Pharmacokinetics of Perforin Inhibitor Prodrugs. Mol Pharm. 2016;13:2484–2491. doi: 10.1021/acs.molpharmaceut.6b00217. [DOI] [PubMed] [Google Scholar]
- 38.Hansen KB, Ogden KK, Yuan H, Traynelis SF. Distinct Functional and Pharmacological Properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA Receptors. Neuron. 2014;81:1084–1096. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansen KB, Tajima N, Risgaard R, Perszyk RE, Jørgensen L, Vance KM, Ogden KK, Clausen RP, Furukawa H, Traynelis SF. Structural Determinants of Agonist Efficacy at the Glutamate Binding Site of N-Methyl-D-Aspartate Receptors. Mol Pharmacol. 2013;84:114–127. doi: 10.1124/mol.113.085803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Assaf Z, Larsen AP, Venskutonytė R, Han L, Abrahamsen B, Nielsen B, Gajhede M, Kastrup JS, Jensen Aa, Pickering DS, Frydenvang K, Gefflaut T, Bunch L. Chemoenzymatic Synthesis of New 2,4-Syn-Functionalized (S)-Glutamate Analogues and Structure-Activity Relationship Studies at Ionotropic Glutamate Receptors and Excitatory Amino Acid Transporters. J Med Chem. 2013;56:1614–1628. doi: 10.1021/jm301433m. [DOI] [PubMed] [Google Scholar]
- 41.Sagot E, Pickering DS, Pu X, Umberti M, Stensbøl TB, Nielsen B, Chapelet M, Bolte J, Gefflaut T, Bunch L, Stensboel TB, Nielsen B, Chapelet M, Bolte J, Gefflaut T, Bunch L. Chemo-Enzymatic Synthesis of a Series of 2,4-Syn-Functionalized (S)-Glutamate Analogues: New Insight into the Structure-Activity Relation of Ionotropic Glutamate Receptor Subtypes 5, 6, and 7. J Med Chem. 2008;51:4093–4103. doi: 10.1021/jm800092x. [DOI] [PubMed] [Google Scholar]
- 42.Alcaide A, Marconi L, Marek A, Haym I, Nielsen B, Møllerud S, Jensen M, Conti P, Pickering DS, Bunch L. Synthesis and Pharmacological Characterization of the Selective GluK1 Radioligand (S)-2-Amino-3-(6-[ 3 H]-2,4-Dioxo-3,4-dihydrothieno[3,2-D]pyrimidin-1(2H)-Yl)propanoic Acid ([3H]-NF608) Med Chem Commun. 2016;7:2136–2144. [Google Scholar]