

Abstract
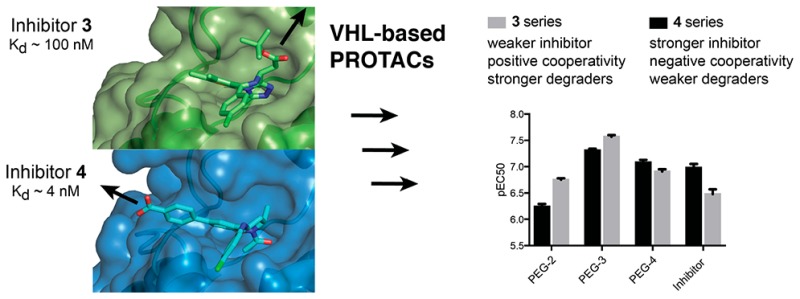
The design of proteolysis-targeting chimeras (PROTACs) is a powerful small-molecule approach for inducing protein degradation. PROTACs conjugate a target warhead to an E3 ubiquitin ligase ligand via a linker. Here we examined the impact of derivatizing two different BET bromodomain inhibitors, triazolodiazepine JQ1 and the more potent tetrahydroquinoline I-BET726, via distinct exit vectors, using different polyethylene glycol linkers to VHL ligand VH032. Triazolodiazepine PROTACs exhibited positive cooperativities of ternary complex formation and were more potent degraders than tetrahydroquinoline compounds, which showed negative cooperativities instead. Marked dependency on linker length was observed for BET-degrading and cMyc-driven antiproliferative activities in acute myeloid leukemia cell lines. This work exemplifies as a cautionary tale how a more potent inhibitor does not necessarily generate more potent PROTACs and underscores the key roles played by the conjugation. The provided insights and framework for structure–activity relationships of bivalent degraders are anticipated to have wide future applicability.
Introduction
Targeted protein degradation by exploiting the ubiquitin proteasome system has recently emerged as a new modality of intervention for medicinal chemistry.1−3 One approach to induce protein degradation is to design heterobifunctional molecules called proteolysis-targeting chimeras (also known as PROTACs) which comprise a ligand binding an E3 ubiquitin ligase conjugated to a ligand binding the target protein.4,5 First introduced by Crews and Deshaies in 2001 (ref (6)), developments of the technology over the following decade were in large part hampered by poor druglikeness of the early generation compounds that typically incorporated peptidic binders for E3 ligases.6,7 Recently discovered high-affinity small molecules for the Cullin RING E3 ubiquitin ligases (CRLs),8 in particular against von Hippel–Lindau (VHL, e.g., 1 (VH032), Chart 1)9−11 and cereblon (CRBN, e.g., 2 (pomalidomide), Chart 1)12−15 greatly contributed to full realization of the technology’s potential. As a result of these developments, we and others recently reported potent activities and specificity in cells and in vivo of both VHL-based5,16−20 and CRBN-based18,20−25 PROTACs against several targets, including the bromodomain and extra-terminal (BET) proteins Brd2, Brd3, and Brd4.16,19,21,22 BET proteins are particularly attractive targets, with a dozen of BET inhibitors from different scaffolds,26,27 that are in >20 clinical trials against a variety of diseases, mainly solid and hematological cancers including acute myeloid leukemia (AML) and mixed lineage leukemia (MLL)28,29 as well as NUT-midline carcinomas.30 BET-targeting PROTACs could provide advantageous therapeutic profiles over BET inhibitors.19 In addition to their therapeutic potential, BET-targeting PROTACs provide useful chemical tools for posttranslational protein knockdown. The acute, profound, and reversible effect of this class of compounds make it an alternative and advantageous approach to genetic knockdowns to study the function of BET proteins in physiological and disease cellular state.
Chart 1. Chemical Structures of Ligands for VHL (1)10 and CRBN (2) and BET Inhibitors 3 (JQ1)34 and 4 (I-BET726)36.

One potential advantage of transforming inhibitors into degraders using the PROTAC approach is that removal of the entire protein is expected to be mechanistically different from blockade of a single domain interaction with an inhibitor and to more closely phenocopy genetic downregulation. This limitation is exemplified by small-molecule inhibitors of the bromodomain of SMARCA2 and SMARCA4, which fail to display the antiproliferative phenotype expected based on genetic protein knockdown.31 A second advantage of ligand directed protein degradation is the potential to enhance selectivity of target modulation over and above the binary target engagement selectivity of the constitutive inhibitor.5,16 Selective targeting of a single BET protein while sparing its paralogs would allow to better decipher their individual physiological roles.32 This is particularly relevant given traditional genetic techniques have proven challenging, exemplified by the embryonic lethality of BET gene knockouts.30 While selective inhibition of BET bromodomains can be achieved using allele-selective bump-and-hole approaches,33 single-point mutations need to be introduced ideally using isogenic knock-ins to enable selective target inhibition.
We previously reported VHL-targeting PROTAC compounds 6 (MZ1) and analogue 7 (MZ2) (Chart 2, see ref (16)) that induced preferential depletion of a single BET member, Brd4, over Brd2 and Brd3, despite binding the different BET bromodomains with comparable affinities.16 Our recent work disclosing the crystal structure of VHL–6–Brd4 ternary complex, the first crystal structure of a PROTAC bound to both target protein and E3 ligase, showed how PROTAC 6 folds into itself to allow the two proteins to form productive interactions.5 Our discovery provided structural insights into ligand-induced protein–protein interactions driving cooperative and preferential formation of ternary complexes as a basis for effective target degradation.5 This realization has important implications for PROTACs, as it demonstrates an added layer of target depletion selectivity through PROTAC-induced interactions between the target and the ligase, and supports important roles for the derivatization mode of the two warhead ligands via the linker. All BET-degrading PROTACs reported so far by us and others16,19,21,22 are based on the pan-selective triazolodiazepine-based BET inhibitor 3 (Chart 1).34 However, while this manuscript was under review, a study has reported active CRBN-based BET degraders based on an azacarbazole containing BET inhibitor.35 To interrogate the impact of using a different, more potent BET inhibitor than 3, and of exploring a different vector out of the warhead, on the activity and intra-BET selectivity profile of BET-targeting PROTACs, we here report novel VHL-recruiting PROTACs derived from a high-affinity BET ligand, the tetrahydroquinoline-based BET inhibitor 4 (Chart 1).36
Chart 2. Chemical Structures of VHL-Targeting PROTACs Based on 4 and 3 Used in This Study and Chemical Structure of CRBN-Targeting PROTAC 11 (ARV-825).

Results and Discussion
Crystal structures of 4 (Kd for Brd4 tandem bromodomain is 4 nM;36 compare to Kd of 100 nM for 3, ref (34)) bound to BET bromodomains show that the free carboxylic acid of the BET inhibitor is solvent exposed and is not involved in direct interactions with the protein (Figure 1b).36,37 We therefore hypothesized that the carboxylate group could be exploited to readily conjugate a linker, e.g., via amide bond formation, without impairing binding to BET bromodomains. Superposition of the cocrystal structures of 3 and 4 each bound to the N-terminal bromodomain of Brd4 (Figure 1) additionally showed that the benzoic acid group of 4 extends in a different direction from the tert-butyl ester group of 3. We therefore became interested in exploring the tolerance of the PROTAC approach to different exit vectors from BET inhibitor scaffolds. On the basis of this design strategy, 4 was connected to the terminal acetamide group of VHL ligand 1 (ref (10)) to obtain PROTACs 8 (MZP-61), 9 (MZP-54), and 10 (MZP-55) which bear a 2-, 3-, and 4-unit PEG linker, respectively, consistent with 5 (MZ4), 6, and 7 (Chart 2). Cereblon-based compound 11 (Chart 2, ref (22)) was also included to provide a first direct comparison with VHL-based PROTACs.
Figure 1.
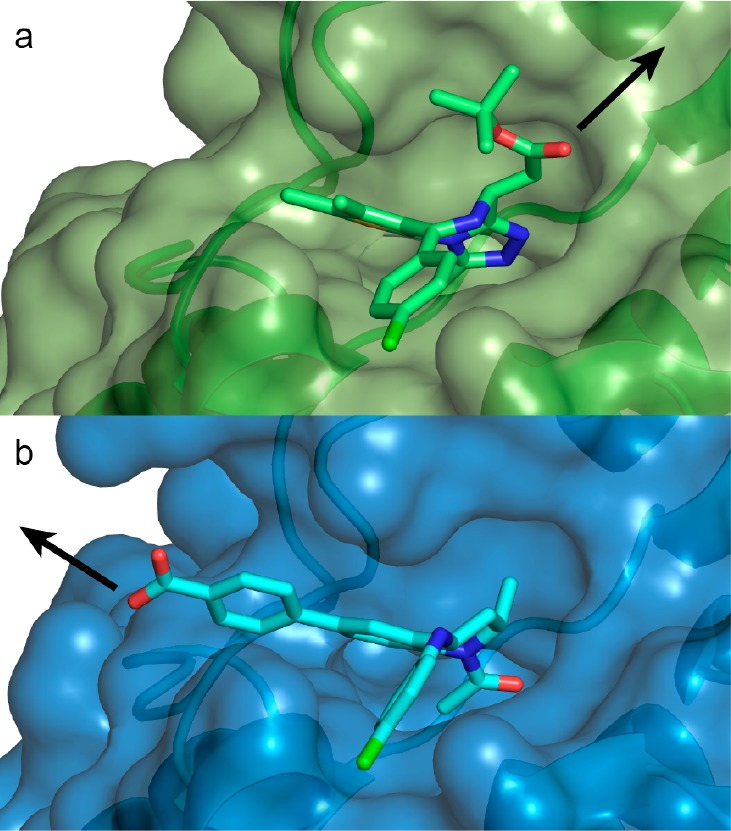
Co-crystal structures to guide PROTAC linking design. First bromodomain of Brd4 with bound (a) 3 (green carbons, PDB code 3MXF(34)) and (b) 4 (cyan carbons, PDB code 4BJX(37)). Arrows highlight exit vectors for linking.
To assess BET degradation activities, compounds were first profiled in HeLa cancer cells because these cells are less susceptible to the cytotoxic effects of BET knockdown or inhibition (Figure 2 and Figure S1; see full blots in Figure S5). Representative PROTACs 10 and 7 (each containing a PEG-4 linker unit) induced marked concentration-dependent knockdown of BET proteins (Figure 2).
Figure 2.

Protein degradation profile of VHL-based BET degraders. HeLa cells were treated for 24 h. Protein levels are shown from one representative of two biological replicates, visualized by immunoblot (a, c) and quantified relative to DMSO control (b, d). Intensity values were quantified as described in the Experimental Section.
Interestingly, tetrahydroquinoline-based compound 10 showed depletion selectivity for Brd4 and Brd3 over Brd2, in contrast to 7 that is a Brd4-selective degrader (Figure 2).16 A similar pattern of BET proteins degradation was observed with PEG-3 linked compounds 9 and 6 (Figure S1c,d,g,h). In contrast, PEG-2 linked PROTACs 5 (Figure S1e,f), and 8 (Figure S1a,b) showed lower activity over all BET proteins. Similar to tetrahydroquinoline-based PROTACs, 11 showed some preference for degrading Brd3/4 over Brd2, although all BET proteins were potently depleted at 100 nM (Figure S1i,j). Interestingly, treatments with tetrahydroquinoline-based PROTACs 9 and 10 revealed increased levels of BET proteins at the higher concentration (1–10 μM, Figure S1c,d and Figure 2a,b), thought to be due to the “hook effect”.4 Brd2 levels even increased beyond vehicle control level (Figure S1c,d and Figure 2a,b). These effects were largely recapitulated when the degradation assays were repeated with shorter treatments of 6 h (Figure S2), suggesting that the observed increase in protein levels is not due to secondary effects at the longer time point. Control treatments with the parent BET inhibitors 3 and 4 also led to increased levels of BET proteins (Figure S1k–n). Marked up-regulation was seen for Brd2 with inhibitor 4 treatment (Figure S1k,l) and for Brd4 long isoform with inhibitor 3 (Figure S1m,n). Similar up-regulation of Brd4 with 3 were observed in Burkitt’s lymphoma cell lines.22 Together, the data suggest that tetrahydroquinoline based PROTACs function more as inhibitors than as degraders at the higher concentrations. These results underscore the importance to identify suitable concentrations to dissect effects due to PROTAC-induced degradation activity from those due to inhibitory activity and potential cellular feedback mechanisms, which could compensate pharmacological activity. Nonetheless, compounds 9 and 10 act as selective degraders of BRD3/4 within appropriate window of concentration (30–100 nM).
The distinctive activity profile of tetrahydroquinoline-based PROTACs prompted us to compare and contrast thermodynamics of ternary complex formation equilibria for this class of PROTACs relative to the triazolodiazepine-based series. We applied an isothermal titration calorimetry (ITC) based assay setup that we recently developed to circumvent potential hook-effects in ternary complex formation and that we used to characterize thermodynamics and cooperativities for binding of 6 to VHL and different BET bromodomains.5 In our previous work, we showed how 6 forms highly cooperative and stable complexes between VHL and BET bromodomains and preferentially with the second bromodomain of Brd4 (Brd4BD2).5 We therefore set out to measure dissociation constants Kd of binary and ternary complexes formed between compounds 5–10, the VHL-EloC-EloB protein (VCB), and Brd4BD2, and the resulting cooperativities (Table 1, see also Figure S3). At the binary level, the bromodomain warheads of the PROTACs 8–10 bound the BET bromodomain consistently with higher potency that the corresponding bromodomain ligand warheads within 5–7, while the VHL ligand warhead bound VCB with comparable affinities across all PROTACs (Table 1). However, strikingly, all tetrahydroquinoline based PROTACs exhibited negative cooperativities of ternary complex formation, meaning that they bound the first protein more tightly on their own than in the presence of the second protein (α < 1, where α values are defined as ratio between binary and ternary Kd values,5Table 1; see Figure 3 for representative binary and ternary titrations of VCB into 10 in the absence and presence of bromodomain).
Table 1. ITC Results of Binary and Ternary Complex Formation for PROTACs 5–10 and Brd4BD2 and VCBa.
| protein in syringe | species in cell | Kd (nM) | ΔG (kcal·mol–1) | ΔH (kcal·mol–1) | –TΔS (kcal·mol–1) | stoichiometry N | α | ΔpKd ± uncertainty | no. of replicates |
|---|---|---|---|---|---|---|---|---|---|
| Brd4BD2 | 8b | 3 ± 2 | –11.7 ± 0.4 | –10.0 ± 0.1 | –1.71 ± 0.43 | 0.804 ± 0.003 | 1 | ||
| Brd4BD2 | 9b | 4 ± 2 | –11.5 ± 0.3 | –9.74 ± 0.10 | –1.73 ± 0.30 | 1.15 ± 0.01 | 1 | ||
| Brd4BD2 | 10 | 8 ± 4 | –11.1 ± 0.3 | –10.8 ± 0.05 | –0.33 ± 0.38 | 0.86 ± 0.06 | 2 | ||
| Brd4BD2 | 5 | 17 ± 2 | –10.6 ± 0.06 | –11.2 ± 0.04 | –0.65 ± 0.47 | 0.81 ± 0.04 | 4 | ||
| Brd4BD2 | 6 | 26 ± 2 | –10.3 ± 0.04 | –11.1 ± 0.8 | 0.77 ± 0.80 | 0.91 ± 0.06 | 3 | ||
| Brd4BD2 | 7 | 27 ± 2 | –10.3 ± 0.04 | –10.6 ± 0.5 | 0.31 ± 0.53 | 0.79 ± 0.03 | 2 | ||
| VCB | 8b | 116 ± 24 | –9.46 ± 0.13 | –4.07 ± 0.07 | –5.39 ± 0.14 | 1.12 ± 0.01 | 1 | ||
| 9b | 105 ± 24 | –9.52 ± 0.13 | –6.18 ± 0.12 | –3.34 ± 0.18 | 0.96 ± 0.01 | 1 | |||
| 10 | 109 ± 8 | –9.50 ± 0.04 | –8.01 ± 0.25 | –1.50 ± 0.21 | 0.92 ± 0.15 | 2 | |||
| VCB | 5 | 147 ± 24 | –9.34 ± 0.10 | –5.72 ± 0.47 | –3.61 ± 0.37 | 0.8 ± 0.05 | 3 | ||
| 6 | 69 ± 8 | –9.77 ± 0.07 | –7.76 ± 0.92 | –2.02 ± 0.9 | 0.81 ± 0.07 | 3 | |||
| 7 | 73 ± 15 | –9.75 ± 0.13 | –8.79 ± 0.42 | –0.96 ± 0.29 | 0.76 ± 0.03 | 2 | |||
| VCB | Brd4BD2−8b | 781 ± 60 | –8.33 ± 0.05 | –7.02 ± 0.11 | –1.31 ± 0.12 | 1.07 ± 0.01 | 0.2 | –0.83 ± 0.10 | 1 |
| Brd4BD2–9b | 228 ± 33 | –9.06 ± 0.09 | –6.90 ± 0.11 | –2.16 ± 0.14 | 1.44 ± 0.02 | 0.5 | –0.34 ± 0.12 | 1 | |
| Brd4BD2–10 | 183 ± 29 | –9.20 ± 0.10 | –7.58 ± 0.05 | –1.62 ± 0.15 | 0.87 ± 0.02 | 0.6 | –0.22 ± 0.08 | 2 | |
| VCB | Brd4BD2–5 | 26 ± 7 | –10.4 ± 0.2 | –5.36 ± 0.77 | –5.05 ± 0.62 | 0.76 ± 0.06 | 5.7 | 0.78 ± 0.16 | 3 |
| Brd4BD2–6 | 9 ± 5 | –11.1 ± 0.3 | –8.47 ± 2.83 | –2.59 ± 0.69 | 0.83 ± 0.04 | 7.4 | 0.95 ± 0.29 | 2 | |
| Brd4BD2–7 | 15 ± 1 | –10.7 ± 0.05 | –10.6 ± 1.3 | –0.07 ± 1.3 | 0.76 ± 0.07 | 4.7 | 0.66 ± 0.10 | 2 |
Values reported are the mean ± SEM, unless specified otherwise.
Errors are generated by the Origin program and reflect the quality of the fit between the nonlinear least-squares curve and the experimental data.
Figure 3.

Measuring cooperativities of ternary complex formation by ITC: (a) VCB titrated into 10 alone; (b) VCB titrated into Brd4BD2–10 binary complex; (c) VCB titrated into Brd2BD1–10. VCB binds more strongly to 10 alone (Kd = 110 nM) than to Brd4BD2–10 (Kd = 180 nM) or Brd2BD1–10 (Kd = 330 nM), highlighting negative cooperativity.
Negative cooperativities were confirmed against all six BET bromodomains, as shown for representative compound 10, with the Brd2 bromodomains showing the lowest α values (Table S1). This feature was in stark contrast to the triazolodiazepine-based series 5–7, which all showed positive cooperativities (α values of >1, Table 1). The thermodynamic data highlight an important feature; that is, cooperativities of PROTACs ternary complex formation do not follow the binding affinities of the target warheads. Our data exemplify how PROTACs made from more potent target warhead ligands can form ternary complexes less productively. It is interesting that despite being negatively cooperative, compounds 8–10 can still act as effective degraders at low concentration, underscoring the power of the substoichiometric catalytic activity of PROTACs. The observation of stronger hook effects for 8–10 compared to 5–7 in the degradation assays is however consistent with their negative cooperativity, i.e., with them behaving more like inhibitors than degraders at higher concentration. We previously demonstrated the importance of the ligand-induced protein–protein contacts in dictating the large positive cooperativity of the VHL–6–Brd4 system.5 It is therefore likely that the different exit vector from the tetrahydroquinoline warhead forces an unfavorable relative orientation between the E3 ligase and the bromodomain. Comparing the different linker lengths within a given series, it was found that PEG-3 linked 6 showed the highest cooperativity among the triazolodiazepine-based series, whereas PEG-2 linked 8 showed the lowest cooperativity among the tetrahydroquinoline-based series (Table 1). In both series overall, short linker proved to be less efficient in forming ternary complex and inducing protein degradation.
To provide a functional downstream readout of the cellular activity of BET degraders, we assessed antiproliferative effects of PROTACs in AML MV4;11 (Figure 4a,b) and HL60 (Figure S4a,b), as these are well characterized BET-sensitive cell lines (see full blots in Figure S6). All compounds showed marked antiproliferative activity in both cell lines. Although some PROTAC compounds exhibited comparable nanomolar half-maximal antiproliferative concentrations (pEC50 values) relative to the constitutive inhibitors alone, the maximal response to baseline level at the higher concentrations (Emax) of all VHL-based PROTACs presented here exceeded that of the BET inhibitors (see Figure 4a,b and Figure S4a,b and values tabulated in Table 2). This activity is likely owing to the more profound effect associated with removing the entire protein compared to blocking an individual binding site, which leaves other parts and domains of the proteins (e.g., the extra-terminal ET domain) still functional. PEG-3 and PEG-4 based PROTACs proved overall more potent than PEG-2, consistent with the trends in degradation activities in HeLa and cooperativities (Figure 4c and Table 2).
Figure 4.

Antiproliferative and Myc-suppression activity of BET degraders and inhibitors: (a, b) MV4;11 cells treated with PROTACs and their corresponding BET targeting ligands for 48 h prior to quantitation of cell viability; (c) half-effective concentrations of BET degraders and corresponding inhibitors; (d) MV4;11 cells treated for 4 h with BET PROTACs or inhibitors (50 nM) or DMSO control. Protein levels are shown from one representative of two biological replicates.
Table 2. BET Reduction by PROTACs in HeLa Cells, Antiproliferative Activity, and Brd4/cMyc Reduction in AML Cellsa.
| pDC50/Dmax (%) in HeLa cells |
pEC50 |
Emax (%) |
Brd4/cMyc depletion (%) |
cooperativity (α) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Brd4 short | Brd4 long | Brd3 | Brd2 | MV4;11 | HL60 | MV4;11 | HL60 | MV4;11 | HL60 | Brd4–BD2 | |
| 8 | 6.9/94 | 6.7/78 | 6.8/74 | –/37 | 6.24 ± 0.05 | 6.17 ± 0.03 | 88.1 ± 1.0 | 96.6 ± 0.1 | 11/11 | –156/14 | 0.15 |
| 9 | 8.1/98 | 7.6/95 | 7.3/91 | –/43 | 7.31 ± 0.03 | 6.57 ± 0.02 | 94.2 ± 0.2 | 98.3 ± 0.1 | 87/73 | 28/50 | 0.46 |
| 10 | 8.1/95 | 7.5/93 | 7.7/92 | –/26 | 7.08 ± 0.05 | 6.37 ± 0.03 | 96.4 ± 0.2 | 98.3 ± 0.1 | 81/60 | 22/47 | 0.59 |
| 5 | 7.0/96 | 7.0/97 | 6.5/97 | 6.2/93 | 6.75 ± 0.03 | 5.84 ± 0.06 | 91.4 ± 0.4 | 91.4 ± 0.3 | 51/27 | –172/4 | 5.7 |
| 6 | 8.1/98 | 8.6/100 | 7.0/100 | 7.4/98 | 7.57 ± 0.03 | 6.66 ± 0.05 | 96.1 ± 0.3 | 92.0 ± 0.4 | 96/84 | 82/68 | 7.4 |
| 7 | 8.4/99 | 8.0/100 | 6.5/99 | 6.7/97 | 6.91 ± 0.04 | 5.90 ± 0.05 | 95.2 ± 0.1 | 91.7 ± 0.1 | 93/66 | 20/23 | 4.7 |
| 11 | 9.2/97 | 9.0/100 | 9.1/98 | 8.2/83 | 7.77 ± 0.06 | 7.46 ± 0.03 | 83.5 ± 2.3 | 88.0 ± 0.1 | 64/70 | 32/57 | nd |
| 4 | 6.98 ± 0.07 | 6.69 ± 0.06 | 73.3 ± 0.6 | 82.4 ± 4.3 | 29/67 | –157/42 | |||||
| 3 | 6.48 ± 0.09 | 6.13 ± 0.09 | 79.1 ± 2.5 | 73.7 ± 2.5 | –29/49 | –243/20 | |||||
DC50: concentration in molar causing 50% reduction of protein level relative to vehicle control treatment in 24 h. Dmax: maximum reduction of protein level relative to vehicle control treatment. pEC50 was measured after 48 h treatment. Errors on pEC50 values reflect the quality of the curve fitting. Protein depletion % are for 50 nM treatments (4 h) in MV4;11/HL60. nd: not determined.
To confirm Brd4 degradation and downstream impact on cMyc levels, we examined protein levels in the same cellular context following acute pharmacological intervention (4 h treatments). In each of the two series, PEG-3 and PEG-4 based PROTACs (6 and 7, 9 and 10) induced superior depletion of both Brd4 and cMyc over their respective PEG-2 analogues 5 and 8 in both cell lines, at two different concentrations (Figure 4d and Figure S4c-e). PROTACs 6 and 7 and 9 also showed higher depletion of cMyc levels compared to their inhibitor counterparts, indicating a greater downstream response with more efficient chemical degraders, while 5 and 8 induce lower cMyc depletion than the corresponding inhibitors 3 and 4, respectively (Figure 4d). Together, the results confirmed the PEG-2 linker length to be too short for optimal PROTAC activity.
Structure–activity relationschips (SARs) are typically quantified by measuring binding or inhibition constants (Kd, Ki) or inhibitory dose response curves (IC50). Because induced protein degradation features catalytic depletion of protein levels over time, different parameters are needed to quantify compounds potency and efficacy. To evaluate SAR in a quantitative fashion, we evaluted the following: pDC50 (concentration causing 50% reduction of protein level relative to vehicle) and Dmax (maximum reduction of protein level relative to vehicle) for HeLa protein degradation responses; pEC50 (half-maximal effective concentrations) and Emax (maximal response to baseline level at the highest concentrations) from cell viability assays; % reduction of Brd4 and cMyc levels in AML cell lines; cooperativity (α) of ternary complex formation with VCB and Brd4BD2 (values reported in Table 2). To evaluate the main drivers of the observed antiproliferative effects, we plotted PROTACs pEC50 values from AML cell viability assays relative to other parameters (Figure 5). Strong correlation was found between pEC50 in MV4;11 and pDC50 on the long isoform of Brd4 in HeLa (r2 = 0.84, Figure 5a). The antiproliferative activities against AML of BET PROTACs and their parent inhibitors correlated well with depletion of cellular levels of cMyc in both MV4;11 (r2 = 0.69, Figure 5b) and HL60 (r2 = 0.62), consistent with AML cells proliferation being cMyc-driven.38 Overall, PEG-3 linked 6 and 9 were confirmed to be the most effective among the VHL-based PROTACs, with 6 performing comparably to CRBN-based PROTAC 11. Importantly, for a given linker length, the trends confirmed tetrahydroquinoline-based PROTACs to be less effective degraders than the triazolodiazepine based, despite the constitutive ligand 4 confirming to be a more potent BET inhibitor than 3 in these cell lines (Figure 5c).
Figure 5.
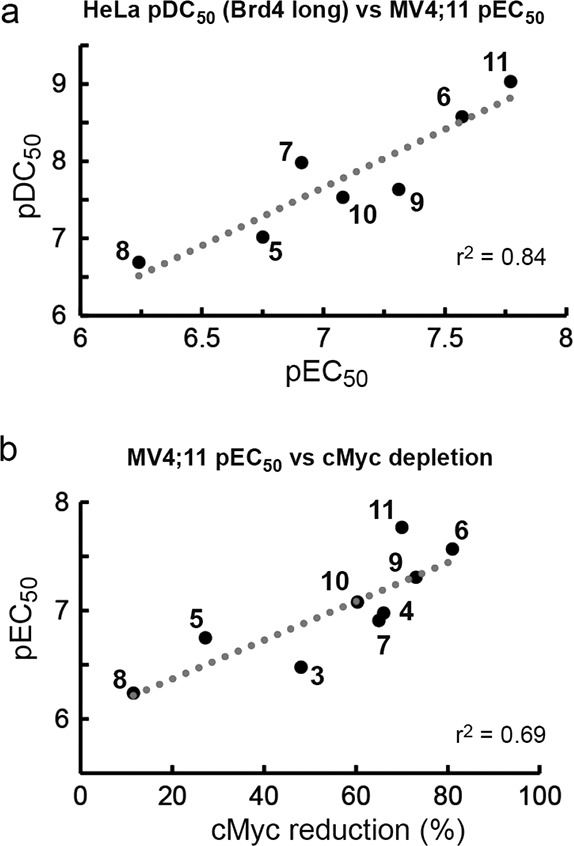
PROTACs’ SAR correlation plots. Anti-AML activities (48 h treatments) are plotted against (a) HeLa degradation of Brd4 long isoform (24 h) and (b) reduction in cMyc levels in MV4;11 (4 h).
Conclusions
We describe novel VHL-targeting BET degraders designed based on a high-affinity tetrahydroquinoline inhibitor and explore the impact of varying the BET-recruiting scaffold and the linkage vector on PROTAC ternary complex recognition and cellular activity. Despite being derivatized from a more potent BET inhibitor, the tetrahydroquinoline based series showed negative cooperativities of ternary complex formation and proved to be less effective degraders than the positively cooperative triazolodiazepine series. These results exemplify how more potent inhibitors do not necessarily generate more potent PROTACs and underscore how the ability to strongly form the ternary complex is critical to the mechanism of action of bivalent degraders. Side-by-side comparisons demonstrated remarkable dependency of cellular activity on the linker length, with a trend of PEG-3 > PEG-4 ≫ PEG-2 observed for both chemical series, potentially suggesting a “sweet-spot” for optimal linking within a given E3 ligase–target pair. We also show how by changing the BET-recruiting warhead and linkage vector, the intra-BET degradation selectivity profile could be tuned from Brd4-selective for one series to Brd3/4 selective for another. Further SAR on either the linker or warhead ligand and exit vector could optimize potency and selectivity of degrading the different BET proteins. Future work assessing the impact of varying other parameters, such as the nature of the E3 ligase recruited and the E3 warhead used, is also warranted. More generally, we provide a framework for establishing future structure–activity relationships of chemical degraders based on measurable in vitro parameters that we anticipate will prove useful to the burgeoning new field of inducing protein degradation with small molecules.
Experimental Section
A. Chemistry
All chemicals, unless otherwise stated, were commercially available and used without further purification. Enantiopure (+)-3 and 4 were purchased from Medchemexpress LLC, Princeton, NJ, USA. (+)-3 was deprotected to the carboxylic acid form 6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepine-6-acetic acid, as previously described.1611 was synthesized as described previously.226 and 7 were synthesized as described.16 Reactions were magnetically stirred; commercially available anhydrous solvents were used. NMR spectra were recorded on a Bruker Ascend 400. Chemical shifts are quoted in ppm and referenced to the residual solvent signals: 1H δ = 7.26 (CDCl3), 13C δ = 77.16; signal splitting patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad (br). Coupling constants (JH–H) are measured in Hz. High resolution mass spectra (HRMS) were recorded on a Bruker microTOF. Low resolution MS and analytical HPLC traces were recorded on an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole LC–MS, connected to an Agilent diode array detector. Preparative HPLC was performed on a Gilson preparative HPLC system with a Waters X-Bridge C18 column (100 mm × 19 mm; 5 μm particle size) and a gradient of 5–95% acetonitrile in water over 10 min, flow 25 mL/min, with 0.1% ammonia in the aqueous phase. The purity of all compounds was analyzed by HPLC–MS (ESI) and was >95%.
General Procedure for Synthesis of VHL Ligand–Linker Conjugates
The azide-(PEG)n derivatives of compound 1 were synthesized as previously described.16 Analytical data for azide-(PEG)3 and azide-(PEG)4 were reported previously.16
(2S,4R)-1-((S)-2-(2-(2-(2-Azidoethoxy)ethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide
The azide-(PEG)2 derivative was prepared accordingly to the general procedure for synthesis of VHL ligand–linker conjugates. Obtained 411 mg, colorless oil, 68% yield. 1H NMR (CDCl3, 400 MHz) δ ppm: 8.68 (s, 1H), 7.38–7.33 (m, 5H), 7.24 (d, J = 8.5 Hz, 1H), 4.75 (t, J = 7.9 Hz, 1H), 4.59–4.54 (m, 2H), 4.48 (d, J = 8.6 Hz, 1H), 4.33 (dd, J = 14.9 Hz, J = 5.2 Hz, 1H), 4.12–4.09 (m, 1H), 4.06–3.96 (m, 2H), 3.70–3.66 (m, 6H), 3.61 (dd, J = 11.4 Hz, J = 3.7 Hz, 1H), 3.41–3.38 (m, 2H), 2.89 (br s, 1H), 2.63–2.58 (m, 1H), 2.52 (s, 3H), 2.13–2.08 (m, 1H), 0.95 (s, 9H). 13C NMR (CDCl3, 101 MHz) δ ppm: 171.6, 170.7, 170.6, 150.7, 138.4, 130.4, 129.7, 128.6, 128.4, 127.6, 71.2, 70.5, 70.3, 70.2, 67.2, 58.5, 57.3, 56.8, 50.7, 43.4, 35.8, 34.9, 26.5, 16.0. MS calcd for C28H39N7O6S 601.3, found 602.3 [M + H+].
General Procedure for Synthesis of Final PROTAC Molecules
The azide-(PEG)n derivative of compound 1 (40 μmol) was dissolved in methanol (5 mL). Catalytic amount of Pd on charcoal (10%, dry) was added and the reaction mixture stirred under an atmosphere of hydrogen for 3 h at 25 °C. The reaction mixture was filtered through a plug of Celite and the resulting solution evaporated to dryness to obtain the desired amine. The resulting amines (35 μmol, 1.4 equiv) and 4 or the carboxylic acid form of (+)-3 (25 μmol, 1 equiv) were dissolved in DCM (2 mL). HATU (14.3 mg, 37.5 μmol, 1.5 equiv) was added and the pH adjusted to >9 by adding DIPEA (17.5 μL, 100 μmol, 4 equiv). After stirring the reaction mixture at 25 °C for 18 h the solvent was removed in vacuum. The crude was purified by preparative HPLC as described above.
(2S,4R)-1-((S)-2-(tert-Butyl)-14-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-4,13-dioxo-6,9-dioxa-3,12-diazatetradecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (5)
White amorphous powder. Yield: 22.2 mg (66%). 1H NMR (CDCl3, 400 MHz) δ ppm: 8.65 (s, 1H), 8.30–8.25 (m, 2H), 7.63 (d, J = 10.0 Hz, 1H), 7.36 (d, J = 8.7 Hz, 2H), 7.24 (d, J = 8.3 Hz, 2H), 7.11 (d, J = 8.0 Hz, 2H), 7.02 (d, J = 8.0 Hz, 2H), 4.94–4.86 (m, 2H), 4.63–4.57 (m, 2H), 4.21 (d, J = 5.8 Hz, 2H), 4.07–3.81 (m, 5H), 3.72–3.52 (m, 7H), 3.44 (dd, J = 15.9 Hz, J = 3.0 Hz, 1H), 3.13–3.08 (m, 1H), 2.55 (s, 3H), 2.45 (s, 3H), 2.39–2.35 (m, 4H), 2.27–2.20 (m, 1H), 1.66 (s, 3H), 1.09 (s, 9 H). 13C NMR (CDCl3, 101 MHz) δ ppm: 172.1, 171.1, 170.6, 170.4, 163.3, 156.2, 150.2, 149.9, 148.4, 138.5, 136.9, 136.7, 131.9, 131.6, 131.4, 131.3, 131.2, 130.2, 130.1, 129, 128.8, 127.7, 71.5, 70.4, 70.3, 69.8, 59.3, 57.5, 56.5, 53.9, 42.7, 39.7, 38.3, 37.2, 36.3, 26.6, 16.2, 14.5, 13.3, 11.8. HRMS m/z calcd for C46H57N9ClO7S2 expected 957.3505, found 958.3498 [M + H+].
(2S,4R)-1-((S)-1-(4-((2S,4R)-1-Acetyl-4-((4-chlorophenyl)amino)-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl)phenyl)-12-(tert-butyl)-1,10-dioxo-5,8-dioxa-2,11-diazatridecan-13-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (8)
White amorphous powder. Yield: 14.5 mg (42%). 1H NMR (CDCl3, 400 MHz) δ ppm: 8.66 (s, 1H), 7.91 (d, J = 8.2 Hz, 2H), 7.79–7.77 (m, 1H), 7.60–7.50 (m, 5H), 7.32 (d, J = 7.9 Hz, 2H), 7.22 (d, J = 8.1 Hz, 2H), 7.13 (d, J = 9.0 Hz, 2H), 6.63–6.57 (m, 3H), 4.89 (br s, 1H), 4.54–4.37 (m, 4H), 4.22–4.15 (m, 2H), 4.07–3.90 (m, 3H), 3.81–3.47 (m, 10H), 3.20 (br s, 1H), 2.71–2.64 (m, 1H), 2.50 (s, 3H), 2.38–2.32 (m, 1H), 2.24 (s, 3H), 1.99–1.93 (m, 1H), 1.36–1.25 (m, 1H), 1.18 (d, J = 6.4 Hz, 3H), 0.85 (s, 9H). 13C NMR (CDCl3, 101 MHz) δ ppm: 171.1, 170.7, 170.6, 169.4, 167.5, 150.3, 148.5, 145.8, 143.4, 137.8, 136.3, 133.4, 131.5, 131.0, 129.5, 129.4, 128.3, 128.1, 126.9, 126.6, 126.0, 122.9, 122.4, 114.5, 77.3, 77.0, 76.7, 71.7, 70.9, 70.7, 70.5, 70.0, 58.8, 57.0, 56.9, 50.5, 43.3, 41.1, 39.8, 36.2, 36.0, 26.4, 23.1, 21.3, 16.1. HRMS m/z calcd for C53H63ClN7O8S [M + H+] 992.4142, found 992.4091.
(2S,4R)-1-((S)-1-(4-((2S,4R)-1-Acetyl-4-((4-chlorophenyl)amino)-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl)phenyl)-15-(tert-butyl)-1,13-dioxo-5,8,11-trioxa-2,14-diazahexadecan-16-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (9)
White amorphous powder. Yield: 17.1 mg (71%). 1H NMR (CDCl3, 400 MHz) δ ppm: 8.66 (s, 1H), 7.80 (d, J = 8.5 Hz, 2H), 7.53–7.48 (m, 4H), 7.36–7.30 (m, 6H), 7.22–7.19 (m, 2H), 7.14 (d, J = 8.7 Hz, 2H), 6.64 (d, J = 8.6 Hz, 2H), 4.88 (br s, 1H), 4.60 (t, J = 7.7 Hz, 1H), 4.52 (d, J = 19.7 Hz, 2H), 4.42 (br s, 1H), 4.34–4.18 (m, 3H), 3.94–3.72 (m, 3H), 3.68–3.51 (m, 14H), 3.21 (br s, 1H), 2.70–2.64 (m, 1H), 2.49 (s, 3H), 2.44–2.36 (m, 1H), 2.22 (s, 3H), 2.05–2.01 (m, 1H), 1.34–1.25 (m, 1H), 1.18 (d, J = 6.3 Hz, 3H), 0.94 (s, 9H). 13C NMR (CDCl3, 101 MHz) δ ppm: 171.1, 170.9, 170.2, 169.5, 167.4, 150.3, 145.8, 143.2, 138.2, 137.7, 136.3, 133.4, 130.9, 129.5, 129.3, 128.1, 127.8, 126.9, 126.5, 125.8, 122.7, 122.6, 114.6, 77.4, 77.0, 76.7, 71.0, 70.4, 70.3, 70.1, 70.0, 69.6, 58.5, 56.9, 56.8, 50.2, 47.6, 43.2, 41.0, 40.0, 36.2, 35.4, 26.4, 23.1, 21.3, 16.1. HRMS m/z calcd for C55H67ClN7O9S [M + H+] 1036.4404, found 1036.4356.
(2S,4R)-1-((S)-1-(4-((2S,4R)-1-Acetyl-4-((4-chlorophenyl)amino)-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl)phenyl)-18-(tert-butyl)-1,16-dioxo-5,8,11,14-tetraoxa-2,17-diazanonadecan-19-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (10)
White amorphous powder. Yield: 14.6 mg (58%). 1H NMR (CDCl3, 400 MHz) δ ppm: 8.66 (s, 1H), 7.84 (d, J = 8.1 Hz, 2H), 7.54–7.49 (m, 4H), 7.36–7.30 (m, 5H), 7.26–7.22 (m, 2H), 7.15–7.12 (m, 3H), 6.62 (d, J = 8.6 Hz, 2H), 4.89 (br s, 1H), 4.69 (t, J = 7.9 Hz, 1H), 4.57–4.46 (m, 3H), 4.33 (dd, J = 5.6, 14.7 Hz, 1H), 4.24–4.15 (m, 2H), 4.00–3.80 (m, 3H), 3.66–3.55 (m, 18H), 3.46 (br s, 1H), 2.69–2.63 (m, 1H), 2.50–2.44 (m, 4H), 2.22 (s, 3H), 2.10–2.06 (m, 1H), 1.36–1.25 (m, 1H), 1.18 (d, J = 6.2 Hz, 3H), 0.93 (s, 9H). 13C NMR (CDCl3, 101 MHz) δ ppm: 170.2, 169.5, 167.3, 150.3, 145.8, 143.2, 138.2, 137.8, 136.3, 133.4, 129.5, 129.3, 128.1, 127.8, 126.9, 126.5, 125.9, 122.6, 114.5, 71.0, 70.6, 70.2, 69.8, 58.6, 57.0, 56.8, 50.3, 47.5, 43.2, 41.1, 39.9, 36.1, 35.3, 26.4, 23.1, 21.3, 16.1. HRMS m/z calcd for C57H71ClN7O10S [M + H+] 1080.4666, found 1080.4623.
B. Biology
HeLa cells were kept in DMEM medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), l-glutamine (Gibco), penicillin, streptomycin. MV4;11 and HL60 cells were kept in RPMI medium (Gibco) supplemented with 10% FBS, l-glutamine, penicillin, and streptomycin. Cells were kept at 37 °C, 5% CO2.
Testing Compounds in Cells
HeLa cells were seeded at 3 × 105 per well on a standard 6-well plate. After a day, cells were treated with compounds for the desired time. Cells were washed with PBS twice and lysed with RIPA buffer (Sigma), supplemented with protease inhibitor cocktail (Roche), Benzonase (Merck), and 0.5 mM MgCl2. Lysate was briefly sonicated and centrifuged at 20 000g for 10 min at 4 °C. Supernatant was collected and protein concentration measured by BCA assay. For MV4;11 and HL60, 1.2 × 107 cells in 15 mL of medium were treated with compound for the desired time. Cells were washed with PBS twice and lysed with hypotonic buffer (10 mM HEPES, 10 mM KCl, protease inhibitor cocktail, Benzonase, and 0.5 mM MgCl2) for 30 min by vortexing twice between the incubation period to disrupt cell outer membrane and release nuclei. Nuclei were pelleted by centrifugation at 2000g for 15 min. The pellet was resuspended in RIPA buffer, supplemented with protease inhibitor cocktail, Benzonase, and 0.5 mM MgCl2. The suspension was briefly sonicated and centrifuged at 20 000g for 10 min at 4 °C. Supernatant was collected and protein concentration measured by BCA assay.
Immunoblotting
Protein on gel was transferred to nitrocellulose membrane using iBlot2 (Life Technology) according to manufacturer guidelines. Blots were probed with anti-Brd4 (AbCam, ab128874), anti-Brd3 (AbCam, ab50818), anti-Brd2 (AbCam, ab139690), anti-β-actin (Cell Signaling, no. 4970), anti-cMyc (AbCam, ab32072), anti-lamin B1 (AbCam, ab133741) antibodies. Blots were developed with secondary anti-mouse IgG (Licor, 926-32210) or anti-rabbit IgG (Licor, 926-32213) antibodies from Licor and bands visualized using Licor Odessey Sa imaging system.
Western Blot Quantification
Image processing and band intensity quantification were performed using Licor Image Studio software version 5.2.5. Reported band intensities are normalized to loading control, i.e., β-actin for total lysates and lamin B1 for nuclear extracts. DC50 values were determined by assuming a linear model between the two data points across the 50% protein level mark. Dmax was determined as the highest protein depletion across the concentrations tested.
Cell Viability Assay
MV4;11 or HL60 cells were incubated with compounds at the desired concentration for 48 h on a clear-bottom 384-well plate. Cells were kept in RPMI medium supplemented with 10% FBS, l-glutamine, penicillin, and streptomycin. Initial cell density was 3 × 105 per mL. Cells were treated with various concentrations of compound or 0.05% DMSO. After treatment, cell viability was measured with Promega CellTiter-Glo luminescent cell viability assay kit according to the manufacturer instructions. Signal was recorded on a BMG Labtech Pherastar luminescence plate reader with recommended settings. Data were analyzed with Graphpad Prism software to obtain EC50 values of each test compound.
Protein Expression and Purification
Bromodomain and VCB complex constructs and protein preparation were described in a previous publication.5 Wild-type version of human proteins VHL (UniProt accession number P40337), ElonginC (Q15369), ElonginB (Q15370), and the bromodomains of Brd2 (P25440), Brd3 (Q15059), and Brd4 (O60885) were used for all protein expression. In brief, the His6-tagged constructs were transformed into E. coli BL21(DE3) and induced with IPTG to produce the desired proteins. E. coli cells were homogenized at 4 °C, and His6-tagged proteins were purified from the soluble lysate by passing through a Ni affinity column. After cleaving the His-tag by TEV protease, a second Ni affinity column purification was performed to obtain tag-free protein in the flow-through. VCB was then additionally purified by anion exchange using MonoQ (GE Healthcare). For all proteins, purity was further polished by gel filtration chromatography.
Isothermal Titration Calorimetry (ITC)
Titrations were performed on an ITC200 microcalorimeter (GE Healthcare) as previously reported.5 The titrations were in ITC buffer (20 mM Bis-Tris propane, 150 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), pH 7.4), supplemented with either 0.2% or 3% DMSO and consisted of 19 injections of 2 μL of protein solution at a rate of 0.5 μL/s at 120 s time intervals. An initial injection of protein (0.4 μL) was made and discarded during data analysis. All experiments were performed at 25 °C, while stirring at 600 rpm. PROTACs were diluted from a 10 mM DMSO stock solution to 20 μM in ITC buffer with the final concentration of DMSO to be 0.2% for triazolodiazepine-based PROTAC or 3% for tetrahydroquinoline-based PROTAC. Bromodomain protein in the same buffer was titrated into the PROTAC in the cell. At the end of the titration, the excess of solution was removed from the cell, the syringe was washed and dried, and VCB complex (168 μM, in the same buffer) was loaded in the syringe and titrated into the complex of PROTAC–bromodomain. The concentration of the complex in the cell (C) after the first titration (16.8 μM) was calculated as follows:
where C0 is the initial concentration of the PROTAC in the cell (20 μM), Vcell is the volume of the sample cell (200.12 μL), and Vinj is the volume of titrant injected during the first titration (38.4 μL). Titrations for the binary complex PROTAC–VCB were performed in the same manner with VCB titrated into 16.8 μM PROTAC in the cell. For titration with 8 and 9, concentrations of PROTAC and proteins were halved due to compound solubility. The data were fitted to a single-binding-site model to obtain the stoichiometry n, the dissociation constant Kd, and the enthalpy of binding ΔH using the Microcal LLC ITC200 Origin software provided by the manufacturer.
Acknowledgments
We are thankful to L. Finn for support with tissue culture (MRC-PPU) and to the Ferguson lab for access to LI-COR equipment. This work was supported by the European Research Council (Starting Grant ERC-2012-StG-311460 DrugE3CRLs to A.C.); the UK Biotechnology and Biological Sciences Research Council (BBSRC Grant BB/J001201/2 to A.C.); the European Commission (Marie Skłodowska-Curie Actions Individual Fellowship H2020-MSCA-IF-2014-655516 to K.-H.C.); and the Wellcome Trust (Strategic Award 100476/Z/12/Z for biophysics and drug discovery to the Division of Biological Chemistry and Drug Discovery).
Glossary
Abbreviations Used
- BET
bromodomain and extra-terminal
- Brd2/3/4
bromodomain-containing protein 2/3/4
- CRBN
cereblon
- DIPEA
N,N-diisopropylethylamine
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- PROTAC
proteolysis-targeting chimera
- VHL
von Hippel–Lindau
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01912.
Dose-dependent Western blots and quantification of protein levels in HeLa cells; ITC titration curves and results; antiproliferative activities in HL60; Brd4 and cMyc-suppression in MV4,11 and HL60; original blot images; NMR spectra of tetrahydroquinoline-based PROTACs (PDF)
Molecular formula strings (CSV)
Author Contributions
All authors have given approval to final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Huang X.; Dixit V. M. Drugging the Undruggables: Exploring the Ubiquitin System for Drug Development. Cell Res. 2016, 26 (4), 484–498. 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Crews C. M. Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discovery 2017, 16 (2), 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins I.; Wang H.; Caldwell J. J.; Chopra R. Chemical Approaches to Targeted Protein Degradation Through Modulation of the Ubiquitin-Proteasome Pathway. Biochem. J. 2017, 474 (7), 1127–1147. 10.1042/BCJ20160762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottis P.; Crews C. M. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 2017, 12 (4), 892–898. 10.1021/acschembio.6b01068. [DOI] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K.-H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13 (5), 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (15), 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth J. S.; Fonseca F. N.; Koldobskiy M.; Mandal A.; Deshaies R.; Sakamoto K.; Crews C. M. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J. Am. Chem. Soc. 2004, 126 (12), 3748–3754. 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- Bulatov E.; Ciulli A. Targeting Cullin-RING E3 Ubiquitin Ligases for Drug Discovery: Structure, Assembly and Small-Molecule Modulation. Biochem. J. 2015, 467 (3), 365–386. 10.1042/BJ20141450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Van Molle I.; Gareiss P. C.; Tae H. S.; Michel J.; Noblin D. J.; Jorgensen W. L.; Ciulli A.; Crews C. M. Targeting the Von Hippel-Lindau E3 Ubiquitin Ligase Using Small Molecules to Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 2012, 134 (10), 4465–4468. 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein-Protein Interaction Between the Von Hippel-Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57 (20), 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost J.; Galdeano C.; Soares P.; Gadd M. S.; Grzes K. M.; Ellis L.; Epemolu O.; Shimamura S.; Bantscheff M.; Grandi P.; Read K. D.; Cantrell D. A.; Rocha S.; Ciulli A. Potent and Selective Chemical Probe of Hypoxic Signalling Downstream of HIF-A Hydroxylation via VHL Inhibition. Nat. Commun. 2016, 7, 13312. 10.1038/ncomms13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E. S.; Böhm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E. J.; Harper J. W.; Jenkins J. L.; Thomä N. H. Structure of the DDB1-CRBN E3 Ubiquitin Ligase in Complex with Thalidomide. Nature 2014, 512 (7512), 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain P. P.; Lopez-Girona A.; Miller K.; Carmel G.; Pagarigan B.; Chie-Leon B.; Rychak E.; Corral L. G.; Ren Y. J.; Wang M.; Riley M.; Delker S. L.; Ito T.; Ando H.; Mori T.; Hirano Y.; Handa H.; Hakoshima T.; Daniel T. O.; Cathers B. E. Structure of the Human Cereblon-DDB1-Lenalidomide Complex Reveals Basis for Responsiveness to Thalidomide Analogs. Nat. Struct. Mol. Biol. 2014, 21 (9), 803–809. 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- Ito T.; Ando H.; Suzuki T.; Ogura T.; Hotta K.; Imamura Y.; Yamaguchi Y.; Handa H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327 (5971), 1345–1350. 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- Lopez-Girona A.; Mendy D.; Ito T.; Miller K.; Gandhi A. K.; Kang J.; Karasawa S.; Carmel G.; Jackson P.; Abbasian M.; Mahmoudi A.; Cathers B.; Rychak E.; Gaidarova S.; Chen R.; Schafer P. H.; Handa H.; Daniel T. O.; Evans J. F.; Chopra R. Cereblon Is a Direct Protein Target for Immunomodulatory and Antiproliferative Activities of Lenalidomide and Pomalidomide. Leukemia 2012, 26 (11), 2326–2335. 10.1038/leu.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K.-H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10 (8), 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55 (2), 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K.; Lu J.; Qian Y.; Altieri M.; Gordon D.; Rossi A. M. K.; Wang J.; Chen X.; Dong H.; Siu K.; Winkler J. D.; Crew A. P.; Crews C. M.; Coleman K. G. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (26), 7124–7129. 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurz R. P.; Dellamaggiore K.; Dou H.; Javier N.; Lo M.-C.; McCarter J. D.; Mohl D.; Sastri C.; Lipford J. R.; Cee V. J.. A “Click Chemistry Platform” for the Rapid Synthesis of Bispecific Molecules for Inducing Protein Degradation. J. Med. Chem. [Online early access]. DOI: 10.1021/acs.jmedchem.6b01781. Published Online: Apr 5, 2017. [DOI] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348 (6241), 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22 (6), 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebraud H.; Wright D. J.; Johnson C. N.; Heightman T. D. Protein Degradation by in-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2 (12), 927–934. 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remillard D.; Buckley D. L.; Paulk J.; Brien G. L.; Sonnett M.; Seo H.-S.; Dastjerdi S.; Wühr M.; Dhe-Paganon S.; Armstrong S. A.; Bradner J. E. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew. Chem., Int. Ed. 2017, 56, 5738–5743. 10.1002/anie.201611281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiedel M.; Herp D.; Hammelmann S.; Swyter S.; Lehotzky A.; Robaa D.; Oláh J.; Ovádi J.; Sippl W.; Jung M.. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. [Online early access]. DOI: 10.1021/acs.jmedchem.6b01872. Published Online: Apr 5, 2017. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Smith S. G.; Zhou M.-M. Discovery of Chemical Inhibitors of Human Bromodomains. Chem. Rev. 2015, 115 (21), 11625–11668. 10.1021/acs.chemrev.5b00205. [DOI] [PubMed] [Google Scholar]
- Galdeano C.; Ciulli A. Selectivity on-Target of Bromodomain Chemical Probes by Structure-Guided Medicinal Chemistry and Chemical Biology. Future Med. Chem. 2016, 8 (13), 1655–1680. 10.4155/fmc-2016-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A.; Prinjha R. K.; Dittmann A.; Giotopoulos G.; Bantscheff M.; Chan W.-I.; Robson S. C.; Chung C.-W.; Hopf C.; Savitski M. M.; Huthmacher C.; Gudgin E.; Lugo D.; Beinke S.; Chapman T. D.; Roberts E. J.; Soden P. E.; Auger K. R.; Mirguet O.; Doehner K.; Delwel R.; Burnett A. K.; Jeffrey P.; Drewes G.; Lee K.; Huntly B. J. P.; Kouzarides T. Inhibition of BET Recruitment to Chromatin as an Effective Treatment for MLL-Fusion Leukaemia. Nature 2011, 478 (7370), 529–533. 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature 2011, 478 (7370), 524–528. 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrieu G.; Belkina A. C.; Denis G. V. Clinical Trials for BET Inhibitors Run Ahead of the Science. Drug Discovery Today: Technol. 2016, 19, 45–50. 10.1016/j.ddtec.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangamudi B.; Paul T. A.; Shah P. K.; Kost-Alimova M.; Nottebaum L.; Shi X.; Zhan Y.; Leo E.; Mahadeshwar H. S.; Protopopov A.; Futreal A.; Tieu T. N.; Peoples M.; Heffernan T. P.; Marszalek J. R.; Toniatti C.; Petrocchi A.; Verhelle D.; Owen D. R.; Draetta G.; Jones P.; Palmer W. S.; Sharma S.; Andersen J. N. The SMARCA2/4 ATPase Domain Surpasses the Bromodomain as a Drug Target in SWI/SNF-Mutant Cancers: Insights From cDNA Rescue and PFI-3 Inhibitor Studies. Cancer Res. 2015, 75 (18), 3865–3878. 10.1158/0008-5472.CAN-14-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Vakoc C. R. The Mechanisms Behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54 (5), 728–736. 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud M. G. J.; Lin-Shiao E.; Cardote T.; Tallant C.; Pschibul A.; Chan K.-H.; Zengerle M.; Garcia J. R.; Kwan T. T. L.; Ferguson F. M.; Ciulli A. A Bump-and-Hole Approach to Engineer Controlled Selectivity of BET Bromodomain Chemical Probes. Science 2014, 346 (6209), 638–641. 10.1126/science.1249830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective Inhibition of BET Bromodomains. Nature 2010, 468 (7327), 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Hu J.; Xu F.; Chen Z.; Bai L.; Fernandez-Salas E.; Lin M.; Liu L.; Yang C.-Y.; Zhao Y.; McEachern D.; Przybranowski S.; Wen B.; Sun D.; Wang S.. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem. [Online early access]. DOI: 10.1021/acs.jmedchem.6b01816. Published Online: Mar 24, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosmini R.; Nguyen V. L.; Toum J.; Simon C.; Brusq J.-M. G.; Krysa G.; Mirguet O.; Riou-Eymard A. M.; Boursier E. V.; Trottet L.; Bamborough P.; Clark H.; Chung C.-W.; Cutler L.; Demont E. H.; Kaur R.; Lewis A. J.; Schilling M. B.; Soden P. E.; Taylor S.; Walker A. L.; Walker M. D.; Prinjha R. K.; Nicodeme E. The Discovery of I-BET726 (GSK1324726A), a Potent Tetrahydroquinoline ApoA1 Up-Regulator and Selective BET Bromodomain Inhibitor. J. Med. Chem. 2014, 57 (19), 8111–8131. 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- Wyce A.; Ganji G.; Smitheman K. N.; Chung C.-W.; Korenchuk S.; Bai Y.; Barbash O.; Le B.; Craggs P. D.; McCabe M. T.; Kennedy-Wilson K. M.; Sanchez L. V.; Gosmini R. L.; Parr N.; McHugh C. F.; Dhanak D.; Prinjha R. K.; Auger K. R.; Tummino P. J. BET Inhibition Silences Expression of MYCN and BCL2 and Induces Cytotoxicity in Neuroblastoma Tumor Models. PLoS One 2013, 8 (8), e72967. 10.1371/journal.pone.0072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado M. D.; León J. Myc Roles in Hematopoiesis and Leukemia. Genes Cancer 2010, 1 (6), 605–616. 10.1177/1947601910377495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.