

ABSTRACT
Recurrent mutations at key lysine residues in the histone variant H3.3 are thought to play an etiologic role in the development of distinct subsets of pediatric gliomas and bone and cartilage cancers. H3.3K36M is one such mutation that was originally identified in chondroblastomas, and its expression in these tumors contributes to oncogenic reprogramming by triggering global depletion of dimethylation and trimethylation at H3K36 with a concomitant increase in the levels of H3K27 trimethylation. H3.3K36M expression can also cause epigenomic changes in cell types beyond chondrocytic cells. Here we show that expression of H3.3K36M in HT1080 fibrosarcoma cancer cells severely impairs cellular proliferation, which contrasts its role in promoting transformation of chondrocytic cells. H3.3K36M-associated cellular toxicity phenocopies the specific depletion of H3K36me2, but not loss of H3K36me3. We further find that the H3K36me2-associated toxicity is largely independent of changes in H3K27me3. Together, our findings lend support to the argument that H3K36me2 has distinct roles in cancer cells independent of H3K36me3 and H3K27me3, and highlight the use of H3.3K36M as an epigenetic tool to study H3K36 and H3K27 methylation dynamics in diverse cell types.
KEYWORDS: chromatin, H3K36, lysine methylation, oncohistone, NSD2, SETD2
Introduction
Recent studies have revealed recurrent mutations in the histone variant H3.3 encoded by H3F3A and H3F3B, including p.Lys36Met (K36M) and p.Gly34Trp/Leu (G34W/L) in bone and cartilage cancers, and p.Lys27Met (K27M) and p.Gly34Arg/Val (G34R/V) in pediatric glioblastomas [1]. Notably, these mutations occur at, or near, two key lysine residues (K27 and K36) involved in chromatin regulation and collectively, these and other histone mutations found in cancer are referred to as “oncohistones.” [2] The K27M mutation on H3.3 induces a dramatic loss of trimethylation at Lys27 (H3K27me3) and global reprogramming of regulatory histone modifications in pediatric brain cancers [3]. Interestingly, this study showed that although the H3.3K27M oncohistone comprised only a small fraction of the total H3 in cells, it mediated loss of global H3K27me3 in a dominant negative manner, depleting this mark from the majority of endogenous H3. Assays testing the interaction of the H3K27 lysine methyltransferase (KMT) complex PRC2 with H3 tail peptides carrying K27M suggested that the catalytic SET domain is sequestered by the mutation and thus not available to methylate wild-type H3 protein [3-6]. A subsequent study demonstrated the use of the H3.3K9M mutation as a tool to deplete H3K9 methylation in Drosophila and study inhibition of H3K9 methyl-associated heterochromatin formation in vivo [7].
The H3.3K36M oncohistone has also been shown to exert a dominant negative effect on H3K36 methylation, depleting both dimethyl (me2) and trimethyl (me3) modifications [3]. To date, this mutation has been largely investigated in the context of acting as a driver of chondroblastomas. Recent work showed that H3.3K36M mediates oncogenesis in mesenchymal progenitor cells via global alteration of the repressive H3K27me3 mark, exhaustion of PRC1, and misregulated cell differentiation pathways [8]. Another study in cultured chondrocytes attributed the oncogenic effect of the K36M mutation to redistribution of H3K36 methylation resulting in upregulation of cancer pathways [9]. Together, these studies suggest that the high tumor-type specificity of these oncohistones indicates distinct functions in different cell types.
[1,8] In this regard, many studies in other cell types have demonstrated distinct biological roles for H3K36 me2 and me3, where upregulation of H3K36me2 is a driver of oncogenesis [10,11] but H3K36me3 is associated with tumor suppressive functions [12-14]. Thus, while H3.3K36M and loss of H3K36 methylation leads to disease states in certain cell types, the effect of this histone mutation and depletion of both H3K36 me2 and me3 simultaneously in other cell types remains to be characterized.
Here, we show that H3.3K36M expression in a commonly cultured cell line leads to global reduction in both H3K36me2 and H3K36me3, concomitant with gain in H3K27me3. Cells expressing H3.3K36M show significantly impaired growth that phenocopies loss of H3K36me2, but not H3K36me3, with respect to cell proliferation. Using the H3.3K36M mutant as a tool to study H3K36 methylation biology, we interrogate the functional effect of the crosstalk between H3K36 and H3K27 methylation. We find the H3K36me2-associated growth impairment is largely immune to co-treatment with EZH2 inhibitor to reverse the increase in H3K27me3. Our findings highlight the impact of the H3.3K36M mutation on H3K36me2 levels in cells, and the need to understand at the molecular level the distinct roles of dimethyl vs. trimethyl modifications at H3K36 in different tissue types.
Results
H3.3 K36M in non-chondrocyte cell line depletes global H3K36 me2 and me3
To study the effects of the K36M mutation on histone H3.3 in commonly cultured non-chondrocyte cells, we chose as our cell system the HT1080 fibrosarcoma cell line. As shown in Figure 1A, stable expression of FLAG-H3.3K36M in HT1080 cells resulted in a global decrease in H3K36 di- and tri-methylation compared to cells expressing the H3.3 wild-type control, consistent with previous studies [3,8,9]. In contrast to H3K36me2/3, significant changes in H3K36me1 levels were not observed. In addition, no significant changes were observed for H3K4me3, H3K9me3, or H3K79me3. However, we did observe an increase in H3K27me3, which is consistent with previous data [15,16] and is further elaborated on below.
Figure 1.
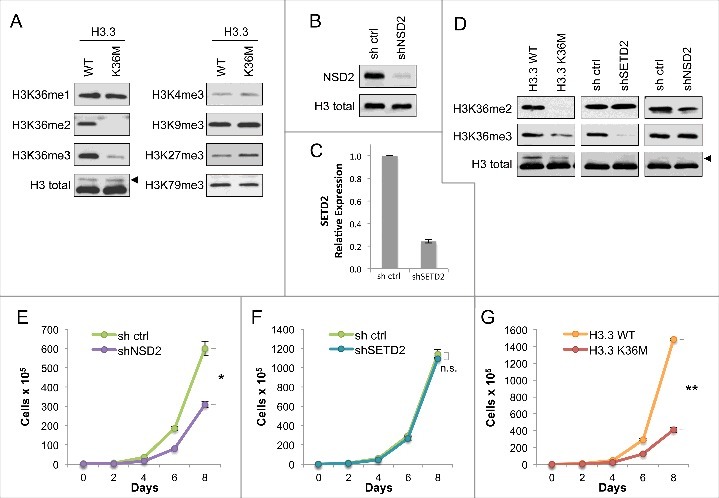
Expression of H3.3K36M depletes global di- and tri- methylation at H3K36 but recapitulates loss of H3K36me2 in cell proliferation (A) Western blot analysis lysates from HT1080 cells stably expressing H3.3 wild-type or K36M, using the indicated antibodies. Arrowhead, FLAG-H3.3 fusion construct appears as a higher molecular weight band than endogenous H3. (B) Western blot analysis of lysates from HT1080 cells stably expressing shNSD2 or control (sh ctrl) probed with anti-NSD2 or anti-H3 as a loading control. (C) Real-time PCR of SETD2 mRNA expression from RNA extracted from HT1080 cells stably expressing shSETD2 or sh ctrl. (D) Western blot analysis of H3K36 methylation levels in lysates from HT1080 cells stably expressing FLAG-H3.3 K36M mutant or KMT shRNA knockdown, using the indicated antibodies. Arrowhead, as in (A). (E-G) Proliferation assays of cells from (D), showing growth of cells expressing shNSD2 compared to control shRNA in (E), shSETD2 compared to control shRNA in (F), and H3.3K36M compared to H3.3WT control in (G). Cells were maintained in selection media and counted every two days for the duration of the assay. Error bars indicate s.e.m. from three experiments. p-values were calculated using a two-tailed Student's t test. *, p < 0.05. **, p < 0.01. n.s., not significant.
Expression of H3.3K36M in HT1080 cells recapitulates the phenotype associated with H3K36me2 depletion with respect to cell proliferation
Dimethylation and trimethylation at H3K36 are associated with opposing biological outcomes, as H3K36me2 is linked to oncogenic potential [10,17] whereas H3K36me3 is associated with tumor suppressor functions [12-14,18]. Since expression of H3.3K36M depletes both modifications, we asked which of the two states of methylation at H3K36 (me2 and me3) it would phenotypically recapitulate, using cell proliferation as a readout in which the phenotypes for depletion of each of the two methyl states can be distinguished. First, we established HT1080 cell lines depleted of either NSD2 (shNSD2) (Figure 1B), which produces the bulk of dimethylation at this residue [10], or SETD2 (shSETD2), which is responsible for all trimethylation at H3K36 independent of the presence of H3K36me210,19 (Figure 1C; note we have been unable to date to find a suitable antibody to reliably detect endogenous SETD2 and thus assayed mRNA). As shown in Figure 1D, a decrease in H3K36me2 was found in shNSD2 cells but not shSETD2 cells, while a decrease in H3K36me3 was observed in shSETD2 cells but not in shNSD2 cells. As described above, H3.3K36M expression resulted in depletion of both H3K36me2 and H3K36me3 (Figure 1, A and D).
We next investigated the role of H3K36 methylation on cellular proliferation. The shNSD2 cells showed significantly slower proliferation (Figure 1E), consistent with previous reports that depletion of NSD2 and H3K36me2 loss leads to impaired cell growth, with the only cells that grow out likely being those that have escaped NSD2 silencing [10,17,20,21]. In contrast to H3K36me2 depletion, the loss of only H3K36me3 did not impact cell proliferation, as the shSETD2 cells showed growth comparable to control cells (Figure 1F). Notably, the cells expressing H3.3K36M also showed significantly retarded proliferation compared to the corresponding H3.3WT control cells (Figure 1G), phenocopying the effect of NSD2 depletion rather than SETD2 depletion. We note that levels of the FLAG-tagged H3.3 wild-type and K36M are initially comparable (see Figure 1A) but over time levels of H3.3K36M decrease compared to the H3.3 wild-type control as cells are passaged in proliferation assays (see Figure 1D). In light of their slow proliferation, we postulate that cells expressing higher levels of the H3.3K36M mutant may be toxic to these cells. Together, these results indicate that in HT1080 cells, with respect to cellular proliferation, the expression of H3.3K36M is similar to loss of H3K36me2 due to NSD2 depletion and not H3K36me3 due to SETD2 depletion.
Cells with loss of H3K36me2 have increased H3K27me3
Chondrocytic precursor cells expressing H3.3K36M showed diminished H3K36 methylation and a concomitant increase in H3K27me3 levels [8,9]. Methylation at H3K36 is known to antagonize EZH2-mediated deposition of the silencing histone modification H3K27me3 [15,16] and upregulation of either mark is associated with downregulation of the other [15,17]. We next probed this crosstalk in the HT1080 cell line described in Figure 1D. Expression of H3.3K36M and NSD2 depletion both resulted in modest increases in H3K27me3, whereas an increase in H3K27me3 was not observed in shSETD2 cells (Figure 2A). This difference is likely a result of the far greater abundance of H3K36me2 relative to H3K36me3 [10,22]. Thus, we conclude that the two cell lines that showed depletion of global H3K36me2 and impaired proliferation also had increased H3K27me3 levels.
Figure 2.
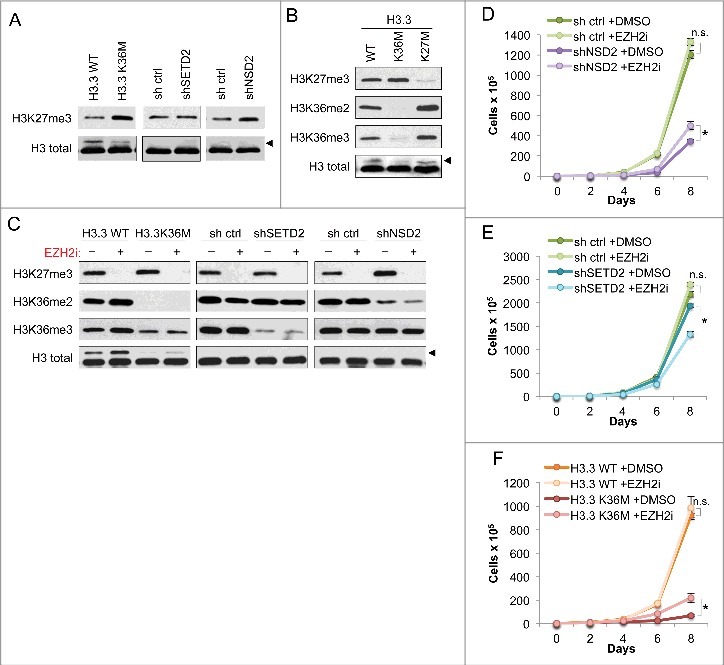
Growth impairment of H3K36me2-depleted cells is independent of antagonistic crosstalk between H3K36 and H3K27 methylation (A) Western blot analysis of H3K27me3 levels in lysates from the HT1080 cells from Figure 1B. Arrowhead, FLAG-H3.3 fusion construct appears as a higher molecular weight band than endogenous H3. (B) Western blot analysis of lysates from HT1080 cells stably expressing FLAG-H3.3 wild-type or mutant constructs, using the indicated antibodies. Arrowhead, as in (A). (C) Western blot analysis of lysates from HT1080 cells from Figure 1B under EZH2i or DMSO vehicle treatment, using the indicated antibodies. EZH2i, GSK126 small molecule inhibitor against EZH2. Arrowhead, as in (A). (D-F) Proliferation assays of the indicated cell lines treated with EZH2i or DMSO vehicle, showing growth of cells expressing shNSD2 with control shRNA in (D), shSETD2 and control shRNA in (E), and H3.3 K36M and H3.3 WT control in (F). Cells were maintained in selection media with drug or vehicle as indicated and counted every two days for the duration of the assay. Error bars indicate s.e.m. from three experiments. p-values were calculated using a two-tailed Student's t test. *, p < 0.05. n.s., not significant.
We used a similar system to test whether decrease in H3K27me3 leads to increase in H3K36me2/me3. To accomplish this, we compared the global levels of these modifications in HT1080s expressing H3.3 K36M or K27M (Figure 2B). The H3.3K36M cells consistently showed reduction in both H3K36me2 and me3, and a slight increase in H3K27me3, compared to the WT control cells. Cells expressing H3.3K27M showed a marked loss of H3K27me3 as well as increases in both H3K36me2 and me3. These results indicate that using H3.3 K36M or K27M to deplete lysine methyl modifications in HT1080 cells faithfully reflects the antagonistic crosstalk between H3K27 and H3K36 methylation.
H3K36me2-associated cellular proliferation is independent of H3K27me3
We considered that the observed decrease in proliferation rate of HT1080 cells expressing shNSD2 and H3.3K36M (see Figure 1) could be attributed to unrestricted spreading and re-distribution of H3K27me3 in the absence of H3K36me2, leading to aberrant gene silencing as previously reported in chondrocytic precursor cells [8]. To test this possibility, we treated H3K36me2/me3-depleted cells with an inhibitor of the H3K27 methyltransferase EZH2 (referred to as EZH2i) [23] to co-deplete H3K27me3 (Figure 2C). Western blot analysis demonstrated that EZH2i treatment effectively abolished generation of H3K27me3. We also note that both H3.3K36M and shNSD2 cells – which have reduced H3K36me2 – showed a slight increase in H3K27me3 modification under DMSO control treatment, consistent with our findings in Figure 2A. Reduction of H3K36 methylation in H3.3K36M or the NSD2 and SETD2 knockdown cells was unaffected by EZH2i. While in the H3.3WT cells, an increase in H3K36me2 and H3K36me3 was observed in response to EZH2 inhibition, this phenotype was not robust as it was not seen in the shRNA control cell lines (Figure 2C).
EZH2i treatment was next used to test whether the increase in H3K27me3 is required for the proliferation defect caused by H3.3K36M–and shNSD2–expression in HT1080 cells (Figure 2D-F). Loss of H3K27me3 was well tolerated by shRNA control and H3.3WT control cells, which grew comparably in response to EZH2i and DMSO treatment. Treatment of cells with EZH2i had little impact on proliferation of cells expressing H3.3K36M, showing only a slight increase in proliferation relative to DMSO treatment (Figure 2F). EZH2i treatment also largely failed to rescue proliferation of cells expressing shNSD2 (Figure 2D). shSETD2-expressing cells showed a modest proliferative decrease when treated with EZH2i in comparison to DMSO treatement (Figure 2E). Together, our data show that the EZH2i treatment does not rescue the proliferative defect caused by H3K36me2 depletion in HT1080 cancer cells, suggesting a specific molecular role for H3K36me2 that is independent of H3K27me3.
Discussion
Here we characterize the histone variant H3.3K36M mutant and its effects on H3K36 methylation in HT1080 cells, a commonly cultured, non-chondrocytic cancer cell line. We find that expression of H3.3K36M in HT1080s depletes both the H3K36 me2 and me3 modifications in this cell line (Figure 1A), consistent with previous reports in other cell types [3,8,9]. Notably, H3.3K36M expression results in slowed proliferation of HT1080s, mimicking loss of H3K36me2 but not H3K36me3 (Figure 1, E-G). In contrast, studies in mesenchymal progenitor cells showed that H3.3K36M mediates depletion of H3K36 methylation but shows proliferation rates comparable to, or greater than, H3.3WT [8,9]. Our results underline the difference in regulatory function of H3K36 methyl marks in distinct cell types.
Previous studies of misregulated H3K36 methylation have indicated changes in the crosstalk between this mark and the repressive H3K27me3 mark as an underlying disease mechanism [8,17,24] This same principle was applied in a proposed mechanism of oncogenicity for H3.3K36M in chondrocyte precursor cells [8], where depletion of H3K36 methylation resulted in increased global H3K27 methylation and led to redistribution of repressive complexes. Using the lysine-to-methionine mutation at H3K36 as an orthogonal tool to deplete H3K36 methylation in vivo, we interrogated the crosstalk between H3K36 and H3K27 methylation. By depleting H3K36me2 (shNSD2) or both H3K36 me2 and me3 (H3.3 K36M) in HT1080 cells, we observe a proliferation defect and increased levels of H3K27me3, consistent with previous studies. However, we find that the increase in this repressive histone modification does not account for the proliferation defect produced by H3K36me2 depletion, as abolishment of H3K27 methylation by EZH2i treatment has only a minor effect and does not restore normal proliferation (Figure 2, D and F).
Taken together, these data argue that in HT1080 cells, H3K36me2's role in regulating cell proliferation is largely independent of its function in blocking the spread of H3K27me3. The molecular mechanisms underlying the biological readout of this dimethyl modification remain to be elucidated. This work demonstrates the use of the H3.3K36M mutant as a tool to probe H3K36 methyl biology in diverse cell systems, and highlights the importance of understanding how lysine methylation signaling and crosstalk at the molecular level differs between cell types.
Methods
Plasmids
The wild-type human H3.3-FLAG sequence was subcloned from pEPI-H3.3 (EpiCypher) into pMSCVpuro (Clontech) for stable expression. Point mutations were introduced by site-directed mutagenesis. Plasmids were propagated in Stbl3 Chemically Competent E.coli (Invitrogen).
The shRNA oligos targetting NSD2 in ORF (5′ GGAAACTACTCTCGATTTATG 3′) or SETD2 3′UTR (5′ GTGCTATGTTGATAAGATT 3′) were designed using PSICOLIGOMAKER 1.5, [25] and synthesized oligos were cloned into pSicoR (Addgene, Plasmid ID 12084) as previously described [25]. Plasmids were propagated in Stbl3 Chemically Competent E.coli (Invitrogen).
Cell culture & proliferation assays
HEK 293T and HT1080 (ATCC CCL-121) cells were cultured in DMEM (Life Technologies) supplemented with 10% fetal bovine serum (Gibco/Life Technologies), penicillin-streptomycin (Life Technologies), L-glutamine (Life Technologies), sodium pyruvate (Life Technologies), and MEM non-essential amino acids (Life Technologies). Viral particles were produced in HEK 293T, and transductions of HT1080 were performed, as previously described [10] to generate stable knockdown or overexpression cell lines. Cells were selected under puromycin (Sigma) at 2 μg/mL. 2 μM EZH2i inhibitor GSK-12623 (Sigma), or equivalent volume of DMSO vehicle (Sigma), was added where indicated.
For proliferation assays, fully selected cells were seeded at 2 × 105 cells/well in triplicate in 6-well plates in selection media. Cells were trypsinized, counted, and re-seeded at a 1:5 dilution every two days for the duration of the assay. Graphs reflect dilution-corrected cell counts.
Cell lysates
To prepare whole cell extracts, 1 × 107 cells were collected, washed in PBS, and lysed in a RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.1% SDS, 1 mM DTT, cOmplete protease inhibitor tablet (Roche)) for 10 min on ice followed by sonication for 10 min in a Bioruptor. Samples were clarified by centrifugation at 21,000g for 10 min at 4°C. For normalization, protein contentrations were measured using the DC Protein Assay kit (BioRad).
Antibodies
The antibodies used were: anti-H3K36me1 (Cell Signaling Technology, Cat.# 14111S), anti-H3K36me2 (Cell Signaling Technology, Cat.# 2901S), anti-H3K36me3 (Cell Signaling Technology, Cat.# 4909S), H3 (generated at Covance), anti-H3K4me3 (EpiCypher, Cat.# 13–0004), anti-H3K9me3 (Abcam, Cat.# ab8898), anti-H3K27me3 (Cell Signaling Technology, Cat.# 9733S), anti-H3K79me3 (Cell Signaling Technology, Cat.# 4260), and anti-NSD2 (EpiCypher).
Real-time quantitiative PCR
RNA was extracted from cells using TRIzol (Invitrogen), and reverse transcribed using the SuperScript III First Strand Synthesis kit (Invitrogen) to make cDNA. Real-time quantitative PCR analysis was performed using Universal ProbeLibrary probes (Roche) on a Roche LightCycler 480. SETD2 mRNA levels (Universal ProbeLibrary probe #49) were normalized to GAPDH mRNA levels (Universal ProbeLibrary probe #60). qPCR primer sequences are listed below.
SETD2: 5′-AACAGCCAGATAAAACAGTGGAT-3′
5′-TCTTTGGAATTCGATATACCTCCT-3′
GAPDH: 5′-AGCCACATCGCTCAGACAC-3′
5′-GCCCAATACGACCAAATCC-3′
Funding Statement
This work was supported in part by grants from the NIH to O.G. (R01 GM079641).
Disclosure statement
O.G. is a co-founder of Epicypher, Inc. and Athelas Therapeutics, Inc.
Author contributions
O.G. and S.M.S. conceived of and designed the experiments and wrote the manuscript. S.M.S. generated reagents and performed the experiments.
References
- [1].Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et al.. Behjati et al. 2013 Nat Gen – distinct H3.3 driver mutations in cancers. Nature Publishing Group. 2013;45:1479-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Weinberg DN, Allis CD, Lu C. Oncogenic Mechanisms of Histone H3 Mutations. Cold Spring Harb Perspect Med. 2017;7:a026443. doi: 10.1101/cshperspect.a026443. PMID:27864305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013;340:857-61. doi: 10.1126/science.1232245. PMID:23539183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brown ZZ, Müller MM, Jain SU, Allis CD, Lewis PW, Muir TW. Strategy for “Detoxification” of a Cancer-Derived Histone Mutant Based on Mapping Its Interaction with the Methyltransferase PRC2. J Am Chem Soc. 2014;136:13498-501. doi: 10.1021/ja5060934. PMID:25180930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, et al.. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016;7:1-11. doi: 10.1038/ncomms11316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science. 2015;350:aac4383−3. doi: 10.1126/science.aac4383. PMID:26472914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Herz HM, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, Florens L, Washburn MP, Eissenberg JC, Shilatifard A. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science. 2014;345:1065-70. doi: 10.1126/science.1255104. PMID:25170156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, et al.. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 2016;352:844-9. doi: 10.1126/science.aac7272. PMID:27174990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fang D, Gan H, Lee J-H, Han J, Wang Z, Riester SM, Jin L, Chen J, Zhou H, Wang J, et al.. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science. 2016;352:1344-8. doi: 10.1126/science.aae0065. PMID:27229140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, et al.. NSD2 Links Dimethylation of Histone H3 at Lysine 36 to Oncogenic Programming. Mol Cell. 2011;44:609-20. doi: 10.1016/j.molcel.2011.08.042. PMID:22099308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wagner EJ, Carpenter PB. Wagner Carpenter 2012 Nat Rev Mol Cell Bio – language of H3K36me. Nature Publishing Group. 2012;13:115-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Duns G, van den Berg E, van Duivenbode I, Osinga J, Hollema H, Hofstra RMW, Kok K. Histone Methyltransferase Gene SETD2 Is a Novel Tumor Suppressor Gene in Clear Cell Renal Cell Carcinoma. Cancer Research. 2010;70:4287-91. doi: 10.1158/0008-5472.CAN-10-0120. PMID:20501857 [DOI] [PubMed] [Google Scholar]
- [13].Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, et al.. Wen et al 2014 Nature – ZMYND11 binds K36me3 and regulates PolII elongation. Nature. 2014;508:263-8. doi: 10.1038/nature13045. PMID:24590075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, Xu W, Mu S, Wen H, Qiu J, et al.. BS69/ZMYND11 Reads and Connects Histone H3.3 Lysine 36 Trimethylation-Decorated Chromatin to Regulated Pre-mRNA Processing. Mol Cell. 2014;56:298-310. doi: 10.1016/j.molcel.2014.08.022. PMID:25263594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. H3K36 Methylation Antagonizes PRC2-mediated H3K27 Methylation. J Biol Chem. 2011;286:7983-9. doi: 10.1074/jbc.M110.194027. PMID:21239496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schmitges FW, Prusty AB, Faty M, Stützer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, et al.. Histone Methylation by PRC2 Is Inhibited by Active Chromatin Marks. Mol Cell. 2011;42:330-41. doi: 10.1016/j.molcel.2011.03.025. PMID:21549310 [DOI] [PubMed] [Google Scholar]
- [17].Martinez-Garcia E, Popovic R, Min DJ, Sweet SMM, Thomas PM, Zamdborg L, Heffner A, Will C, Lamy L, Staudt LM, et al.. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211-20. doi: 10.1182/blood-2010-07-298349. PMID:20974671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Grosso AR, Leite AP, Carvalho S, Matos MR, Martins FB, Vítor AC, Desterro JM, Carmo-Fonseca M, de Almeida SF. Pervasive transcription read-through promotes aberrant expression of oncogenes and RNA chimeras in renal carcinoma. eLife. 2015;4:1-16. doi: 10.7554/eLife.09214. PMID:26575290.18046453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008;27:1-15. doi: 10.1038/sj.emboj.7601967. PMID:18046453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, Kaneda Y, Nimura, et al.. 2009 Nature – NSD2-mediated H3K36me3 links regulate Nkx2-5 to Wolf-Hirschorn syndrome. Nature. 2009;460:287-91. doi: 10.1038/nature08086. PMID:19483677 [DOI] [PubMed] [Google Scholar]
- [21].Sankaran SM, Wilkinson AW, Elias JE, Gozani O. A PWWP Domain of Histone-Lysine N-Methyltransferase NSD2 Binds to Dimethylated Lys-36 of Histone H3 and Regulates NSD2 Function at Chromatin. J Biol Chem. 2016;291:8465-74. doi: 10.1074/jbc.M116.720748. PMID:26912663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA. High throughput characterization of combinatorial histone codes. Mol Cell Proteomics. 2009;8:2266-84. doi: 10.1074/mcp.M900238-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, E Diaz, et al.. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108-12. doi: 10.1038/nature11606. PMID:23051747 [DOI] [PubMed] [Google Scholar]
- [24].Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, Shah MY, Zheng Y, Will CM, Small EC, et al.. Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation. PLoS Genet. 2014;10:e1004566. doi: 10.1371/journal.pgen.1004566. PMID:25188243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, Jacks T. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci USA. 2004;101:10380-5. doi: 10.1073/pnas.0403954101. PMID:15240889 [DOI] [PMC free article] [PubMed] [Google Scholar]