

ABSTRACT
Bromodomain and extra-terminal (BET) proteins are frequently overexpressed in various human cancers, therefore have been clinically pursed as attractive therapeutic anti-cancer targets. However, relatively little is known about the mechanism(s) underlying aberrant BET overexpression in human cancers. Recently, we reported that prostate cancer-derived SPOP mutants fail to interact with and promote BRD4 degradation, leading to accumulation of BRD4 in prostate cancer cells. As a result, prostate cancer cells harboring SPOP mutations are more resistant to BET inhibitors. Therefore, our results help to elucidate the tumor suppressor role of SPOP in the prostate cancer setting by negatively controlling BET proteins stability. More importantly, our results also provide a molecular basis for using combination with BET inhibitors and other inhibitors to treat prostate cancer patients with SPOP mutations.
KEYWORDS: BET, Cell proliferation, Resistance, SPOP, Ubiquitin
Acetylation of Lysine is one of key regulatory post-translational modification (PTM) of proteins, which is critical for regulation of transcription and cell signaling in a plethora of cellular processes.1 For example, acetylation of histones in chromatin is important for euchromatin establishment, resulting in transcription activations and subsequent gene expressions. Mechanistically, histone acetyltransferases (HATs) can add acetyl groups to histone Lys residues as writers, while histone deacetylases (HDACs) and Sirtuins (SIRTs) can remove acetyl groups as erasers.2 On the other hand, Bromodomain (BRD) proteins bind to acetylated lysine (Kac) and function as readers of lysine acetylation state to direct the downstream biological outputs.3 As such, the bromodomain and extra-terminal domain-containing (BET) proteins, which belongs to BRD protein family, have been characterized to play a pivotal role in regulation of cell cycle, apoptosis, cell motility, and metastasis in a broad range of human cancers.4,5
BET proteins share common structural features with two conserved acetyl-histone reading bromodomains (BD1 and BD2) in the N-terminus and an extra-terminal domain (ET domain) in the C-terminus.6 It is known that BET proteins include four mammalian members, namely the ubiquitously expressed BRD containing 2 (BRD2), BRD3, BRD4, and the germ-cell specific BRDT.7–9 The bromodomains as an epigenetic reader interact with not only lysine-acetylated histones, but also other non-histone proteins, whereas the ET domain largely mediates the interactions with various transcription regulators.8,9 Therefore, BET proteins largely function as transcriptional co-activators to recruit transcription modulators to specific acetylated chromatin site(s), such as the super-enhancer region enriched with H3K18ac and H3K27ac markers.10,11 Biologically, BET proteins exhibit the oncogenic function in part via upregulation of c-Myc or enhancing transcriptional activities of androgen receptor (AR) and ERG in cancer.12,13 BRD4, the most studied protein of the BET family, is associated with the P-TEFb (Positive transcription elongation factor) and involved in regulation of transcription by the RNA polymerase II.14,15
Deregulation of BET proteins is frequently observed and clinically associated with various types of human cancers, including NUT midline carcinoma, breast cancer and prostate cancer.5,16,17 Higher expression of BRD2 and BRD4 was also observed in human primary and metastatic melanoma. Moreover, inhibition of BRD2 and BRD4 led to downregulation of cytokines such as IL-6 and IL-8.18 BRD2 and BRD4 mRNA levels were significantly overexpressed in glioblastoma,19 whereas inhibition of BRD4 in glioblastoma cells reduced cell cycle progression and cell proliferation.19 These reports indicate that targeting BET proteins could be a potential therapeutic approach for treating human cancers with relatively high level of BET proteins. To this end, although BRD4 functions have been identified, the mechanism of how BRD4 was physiologically regulated is largely unclear. One study showed that BRD4 could be phosphorylated by casein kinase II (CK2) in its N-terminal phosphorylation sites, leading to its binding to acetylated chromatin and several transcription factors including p53, p50 and p65.20 This BRD4 phosphorylation event appears to be critical for tumor progression in triple-negative breast cancer, a process that could be antagonized via dephosphorylation by the protein phosphatase 2A (PP2A).21 Without a doubt, understanding modulation mechanism of the switch between dephosphorylation and phosphorylation of BRD4 is important to assess how BRD4 exerts their functions in cells to influence tumorigenesis.
In keeping with BETs as attractive therapeutic targets, two bromodomain inhibitors, JQ1 (a thieno-triazolo-diazepine) and iBET (a benzodiazepine), which compete with acetylated histones for binding to the bromodomain of BET proteins have been recently shown to antagonize tumor growth in part through selective repression of MYC and its downstream transcriptional targets in various types of cancers.12,22,3 It is noted that both JQ1 and iBET are pan-BET bromodomain inhibitors. Notably, JQ1 possesses cell growth inhibition and induces apoptosis in leukemia, medulloblastoma cells, and AR-positive prostate cancer cells.13,22,23 Similarly, JQ1 suppressed the estrogen receptor-α signaling pathway, leading to growth inhibition of tamoxifen resistant breast cancer cells.21 To this end, a recent study showed that JQ1 induced growth inhibition and resulted in G1 arrest and apoptosis in TNBC (triple-negative breast cancer) cells in both in vitro cell culture and in vivo murine TNBC xenograft models.24
However, there are emerging evidences of observed resistance to BET inhibitors that are partially attributed to hyper-phosphorylation status of BRD4 that affects its association with MED1 to dictates the downstream transcription events in TNBC setting.24 Furthermore, JQ1 has been reported to downregulate anti-apoptotic genes and JAK/STAT signaling pathways. JQ1 efficiently displaced BRD4 from MED1 in sensitive cells, but not in resistant cells, suggesting that increased recruitment of BRD4 to chromatin by MED1 underlies cellular resistance to JQ1.24 In another elegant study, Polycomb repressive complex 2 (PRC2) loss was found to amplify Ras-driven transcription and confer cellular sensitivity to BRD4 inhibitor-based combination therapies in high-grade gliomas and melanomas.25 On the other hand, in other cellular context such as acute myeloid leukaemia (AML), suppression of the PRC2 complex was found to promote JQ1 resistance.26 In this circumstance, PRC2 suppression failed to directly regulate Brd4-dependent transcript, but remodel regulatory pathways and restore Myc transcription that recruits WNT machinery to confer resistance to BET inhibition.26 In keeping with this finding, another independent study demonstrated that resistance to BET inhibitors such as JQ1 in human leukaemia cells is partly due to increased Wnt/beta-catenin signaling.27 Consistently, inhibition of this pathway leads to restoration of sensitivity to JQ1 in vitro and in vivo.27 Interestingly, in castration-resistant prostate cancer (CRPC), BET inhibitors exert anti-tumor function largely through attenuating the BRD4/AR/ERG signaling pathway. Although BET inhibitors have been shown promising outcomes in early clinical trials, recent studies indicate that cells may develop resistance to BET inhibitors during the treatment course.
However, detailed molecular mechanisms underlying the acquired BET inhibitor resistance are largely unknown. To this end, previous studies demonstrated that in most cases, accumulation of the target protein mediates the observed resistance to targeted therapies. Therefore, it will benefit clinic application of BET inhibitors for us to determine how BET protein stability is governed and whether deregulation of BET protein leads to resistance to BET inhibitors currently in clinical trials. To this end, our group and others have recently demonstrated that BRD4 is a target of Speckle-type POZ protein (SPOP) and deregulation of BRD4 contributes to resistance to BET inhibitors.28,29 SPOP is an E3 ubiquitin ligase adaptor protein, including an N-terminal MATH domain and a C-terminal BTB domain, which is often mutated in prostate and endometrial cancers. The MATH domain is involved in substrate recognition and interaction, while the BTB domain is essential for binding Cullin 3.30 SPOP regulates cellular function via targeting its substrates including death-associated protein 6, Marco H2A, AR, steroid receptor coactivator-3 (SRC-3), DEK, ERG, and SENP7.31–36 SPOP mutations were identified in about 10-15% prostate tumor samples and were early events in prostate tumorigenesis. Multiple studies indicate that SPOP plays a tumor suppressive role in several cancers including prostate cancer, endometrial cancer and breast cancer.37,38 In contrast, SPOP was identified to play a tumor promoting role in kidney cancer. Higher expression of SPOP was occurred in 99% of clear cell renal cell carcinoma and it controlled the ubiquitination and degradation of PTEN, ERK phosphatases, Daxx, and Gli2.39,40 In addition, one study demonstrated that SPOP mediated ubiquitination and destabilization of breast cancer metastasis suppressor 1 (BRMS1) in breast cancer, leading to de-repressing metastasis-associated genes.41 Therefore, SPOP likely elicits its biological functions in a tissue or cellular context-dependent manner.
The study from our group and others identified that prostate cancer-derived SPOP mutants fail to interact with and promote BRD4 degradation, leading to accumulation of BRD4 and subsequent resistance to BET bromodomain inhibitors in prostate cancer cells.28,29 Firstly, we found that the Cul3-SPOP E3 ubiquitin ligase negatively regulates the stability of BET protein. Depletion of endogenous Cul-3 increased BRD4 protein level, whereas ectopic expression of Cul-3 decreased the abundance of BRD4. Consistently, SPOP knockdown led to an increase in BRD4 expression, while overexpression of SPOP promoted BRD4 degradation. Secondly, Prostate cancer-associated SPOP-mutants enhanced prostate tumorigenesis through elevated BET proteins. Depletion of the MATH or BTB domains failed to degrade BRD4. Prostate cancer-associated SPOP mutations also affected BRD4 stability. Functionally, cells with these mutants exhibited enhanced growth and colony-formation ability. Clinically, SPOP mutations were closely associated with high expression of BRD4 in prostate cancer tumor tissues. Thirdly, wild-type SPOP promotes ubiquitination and destruction of BET proteins in a degron-dependent manner. Depletion of the identified degron disrupted the interaction between BRD4 and SPOP in cells and subsequent abolished SPOP-mediated degradation of BRD4, leading to promotion of cell growth and migration. Fourthly, BRD4 protein abundance triggers BET inhibitors resistance in SPOP-mutant prostate cancer cells.
In summary, SPOP mutations prevent SPOP-mediated BRD4 degradation in part due to disruption of the SPOP/BRD4 interaction, resulting in an elevated level of BRD4 and cooperation with other oncogenic proteins, most of which are transcription factors, such as AR and ERG to promote prostate cancer progression. This study therefore reveals a potential molecular basis into BET inhibitor resistance in prostate cancer cells largely through stabilizing BRD4 oncoprotein by escaping SPOP-mediated BRD4 degradation pathway. Given the critical oncogenic role of BRD4 and high frequency of SPOP mutation in prostate cancer, this study provides the molecular mechanism for further clinical investigation of a novel strategy to combat prostate cancer based on SPOP genetic status, which guides the future usage of BET inhibitors to treat SPOP-deficient cancer patients.
A study conducted by Zhang et al. has also shown that wild-type SPOP, but not mutant SPOP, binds to and induces ubiquitination and degradation of BET proteins.29 SPOP mutants lead to elevated BET expression and subsequent resistance to BET inhibitors. Moreover, this group found that GTPase RAC1 and cholesterol synthesis pathways are essential for SPOP-mutation-induced Akt-mTORC1 activation and BET inhibitor resistance.29 This finding suggests that Akt inhibitors combination with BET inhibitors could offer benefit for patients with SPOP-mutant prostate cancer. Interestingly, another independent study published in the same issue of Nature Medicine revealed that in endometrial cancer setting, the degradation of BRD2, BRD3 and BRD4 proteins was promoted by endometrial cancer-associated SPOP mutants, leading to sensitization to BET inhibitors.42 This concept is different from the conclusion of SPOP mutant-mediated BET degradation in prostate cancer cells, suggesting that SPOP mutants could have oppose drug susceptibilities in various types of human cancers (Fig. 1).
Figure 1.
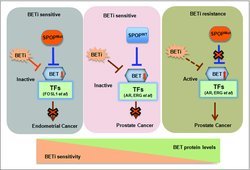
A schematic model illustrating how SPOP governs the BET protein stability and controls resistance to BET inhibitors. Prostate cancer cells with SPOP mutation exhibited increased resistance to BET inhibitor, where endometrial cancer-associated SPOP mutants lead to sensitization to BET inhibitors in endometrial cancer.
It is important to mention that there is no specific inhibitor for various BET family members. It could have various adverse effects due to that these BET inhibitors are not specific for each of the BET protein. Another challenge is to develop BET inhibitors that specifically target the cancer cells, but not normal cell types with basal expression of BET proteins. It is also important to identify biomarkers that could predict hypersensitivity to BET inhibitors. We believe that it is required to develop novel BET inhibitors with less toxicity and more sensitivity. JQ1-resistant cells retain sensitivity to other compounds such as CXCR2 and JAK2 inhibitors.24 Moreover, a significant synergy between JQ1 and CK2 inhibitor CX-4945, or BCL-xL inhibitor (ABT737) or the PP2A activator perphenazine (PPZ) has been observed, that offers potential therapeutic combination options to combat emerging JQ1 resistance.24 Along the same line, BET inhibitors in combination with other targeted inhibitors such as Akt inhibitor may also offer clinic benefits by overcoming resistance to BET inhibitors.29 However, further in-depth studies are required to assess their in vivo pre-clinical or clinical outputs.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
This work is support in part by the NIH grants to W.W. (GM094777 and CA177910). This work was also supported by grant from National Natural Science Foundation of China (NSFC number 81572936 and 81773186) and the priority academic program development of Jiangsu higher education institutions.
References
- [1].Fujisawa T, Filippakopoulos P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol. 2017;18:246–62. doi: 10.1038/nrm.2016.143. PMID:28053347 [DOI] [PubMed] [Google Scholar]
- [2].Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, et al.. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154:569–82. doi: 10.1016/j.cell.2013.07.013. PMID:23911322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fu LL, Tian M, Li X, Li JJ, Huang J, Ouyang L, Zhang Y, Liu B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget. 2015;6:5501–16. doi: 10.18632/oncotarget.3551. PMID:25849938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–56. doi: 10.1038/nrd4286. PMID:24751816 [DOI] [PubMed] [Google Scholar]
- [5].Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12:465–77. doi: 10.1038/nrc3256. PMID:22722403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–36. doi: 10.1016/j.molcel.2014.05.016. PMID:24905006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002;513:124–8. doi: 10.1016/S0014-5793(01)03309-9. PMID:11911891 [DOI] [PubMed] [Google Scholar]
- [8].Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282:13141–5. doi: 10.1074/jbc.R700001200. PMID:17329240 [DOI] [PubMed] [Google Scholar]
- [9].Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586:2692–704. doi: 10.1016/j.febslet.2012.04.045. PMID:22710155 [DOI] [PubMed] [Google Scholar]
- [10].Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al.. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–90. doi: 10.1016/j.ccr.2013.11.003. PMID:24332044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pott S, Lieb JD. What are super-enhancers? Nat Genet. 2015;47:8–12. doi: 10.1038/ng.3167. PMID:2554760321889194 [DOI] [PubMed] [Google Scholar]
- [12].Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al.. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. PMID:21889194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, et al.. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–82. doi: 10.1038/nature13229. PMID:24759320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–34. doi: 10.1016/j.molcel.2005.06.027. PMID:16109376 [DOI] [PubMed] [Google Scholar]
- [15].Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–45. doi: 10.1016/j.molcel.2005.06.029. PMID:16109377 [DOI] [PubMed] [Google Scholar]
- [16].French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 2003;63:304–7. PMID:12543779 [PubMed] [Google Scholar]
- [17].Crawford NP, Alsarraj J, Lukes L, Walker RC, Officewala JS, Yang HH, Lee MP, Ozato K, Hunter KW. Bromodomain 4 activation predicts breast cancer survival. Proc Natl Acad Sci U S A. 2008;105:6380–5. doi: 10.1073/pnas.0710331105. PMID:18427120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Klein K, Kabala PA, Grabiec AM, Gay RE, Kolling C, Lin LL, Gay S, Tak PP, Prinjha RK, Ospelt C, et al.. The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis. 2016;75:422–9. doi: 10.1136/annrheumdis-2014-205809. PMID:25467295 [DOI] [PubMed] [Google Scholar]
- [19].Pastori C, Daniel M, Penas C, Volmar CH, Johnstone AL, Brothers SP, Graham RM, Allen B, Sarkaria JN, Komotar RJ, et al.. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics. 2014;9:611–20. doi: 10.4161/epi.27906. PMID:24496381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell. 2013;49:843–57. doi: 10.1016/j.molcel.2012.12.006. PMID:23317504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, Lanz R, He B, Wang L, Fu X, et al.. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014;24:809–19. doi: 10.1038/cr.2014.71. PMID:24874954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al.. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–8. doi: 10.1038/nature10334. PMID:21814200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, et al.. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res. 2014;20:912–25. doi: 10.1158/1078-0432.CCR-13-2281. PMID:24297863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, et al.. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413–7. doi: 10.1038/nature16508. PMID:26735014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].De Raedt T Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H, et al.. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–51. PMID:25119042 [DOI] [PubMed] [Google Scholar]
- [26].Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, et al.. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525:543–7. doi: 10.1038/nature14898. PMID:26367798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, et al.. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525:538–42. doi: 10.1038/nature14888. PMID:26367796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, Liu S, Zhong Q, Guo J, Zhang J, et al.. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med. 2017;23:1063–71. doi: 10.1038/nm.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z, Wang S, Pan CW, Zhu Y, Yan Y, et al.. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med. 2017;23:1055–62. doi: 10.1038/nm.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mani RS. The emerging role of speckle-type POZ protein (SPOP) in cancer development. Drug Discov Today. 2014;19:1498–502. doi: 10.1016/j.drudis.2014.07.009. PMID:25058385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P, Varmeh S, Zhang J, Cheng L, Sun Y, et al.. SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol Cell. 2015;59:917–30. doi: 10.1016/j.molcel.2015.07.026. PMID:26344095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].An J, Ren S, Murphy SJ, Dalangood S, Chang C, Pang X, Cui Y, Wang L, Pan Y, Zhang X, et al.. Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Mol Cell. 2015;59:904–16. doi: 10.1016/j.molcel.2015.07.025. PMID:26344096 [DOI] [PubMed] [Google Scholar]
- [33].An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014;6:657–69. doi: 10.1016/j.celrep.2014.01.013. PMID:24508459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li C, Ao J, Fu J, Lee DF, Xu J, Lonard D, O'Malley BW. Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene. 2011;30:4350–64. doi: 10.1038/onc.2011.151. PMID:21577200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Theurillat JP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M, Wild PJ, Blattner M, Groner AC, Rubin MA, et al.. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science. 2014;346:85–9. doi: 10.1126/science.1250255. PMID:25278611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhu H, Ren S, Bitler BG, Aird KM, Tu Z, Skordalakes E, Zhu Y, Yan J, Sun Y, Zhang R. SPOP E3 Ubiquitin Ligase Adaptor Promotes Cellular Senescence by Degrading the SENP7 deSUMOylase. Cell Rep. 2015;13:1183–93. doi: 10.1016/j.celrep.2015.09.083. PMID:26527005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Le Gallo M O'Hara AJ, Rudd ML, Urick ME, Hansen NF, O'Neil NJ, Price JC, Zhang S, England BM, Godwin AK, et al.. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44:1310–5. doi: 10.1038/ng.2455. PMID:23104009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang P, Gao K, Jin X, Ma J, Peng J, Wumaier R, Tang Y, Zhang Y, An J, Yan Q, et al.. Endometrial cancer-associated mutants of SPOP are defective in regulating estrogen receptor-alpha protein turnover. Cell Death Dis. 2015;6:e1687. doi: 10.1038/cddis.2015.47. PMID:25766326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Guo ZQ, Zheng T, Chen B, Luo C, Ouyang S, Gong S, Li J, Mao LL, Lian F, Yang Y, et al.. Small-Molecule Targeting of E3 Ligase Adaptor SPOP in Kidney Cancer. Cancer Cell. 2016;30:474–84. doi: 10.1016/j.ccell.2016.08.003. PMID:27622336 [DOI] [PubMed] [Google Scholar]
- [40].Li G, Ci W, Karmakar S, Chen K, Dhar R, Fan Z, Guo Z, Zhang J, Ke Y, Wang L, et al.. SPOP promotes tumorigenesis by acting as a key regulatory hub in kidney cancer. Cancer Cell. 2014;25:455–68. doi: 10.1016/j.ccr.2014.02.007. PMID:24656772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim B, Nam HJ, Pyo KE, Jang MJ, Kim IS, Kim D, Boo K, Lee SH, Yoon JB, Baek SH, et al.. Breast cancer metastasis suppressor 1 (BRMS1) is destabilized by the Cul3-SPOP E3 ubiquitin ligase complex. Biochem Biophys Res Commun. 2011;415:720–6. doi: 10.1016/j.bbrc.2011.10.154. PMID:22085717 [DOI] [PubMed] [Google Scholar]
- [42].Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A, Bernasocchi T, Civenni G, Losa M, Svinkina T, et al.. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med. 2017;23:1046–54. doi: 10.1038/nm.4372. PMID:28805821 [DOI] [PMC free article] [PubMed] [Google Scholar]