

ABSTRACT
Hypertension-associated end-organ damage commonly leads to cardiac and renal fibrosis. As no effective anti-fibrotic therapy currently exists, the unchecked progression of fibrogenesis manifests as cardio-renal failure and early death. We have previously shown that FATp300—p300 with intrinsic factor acetyltransferase activity—is an essential epigenetic regulator of fibrogenesis, and is elevated in several fibrotic tissues. In this report, we investigate the therapeutic efficacy of a novel FATp300 inhibitor, L002, in a murine model of hypertensive cardio-renal fibrosis. Additionally, we examine the effects of L002 on cellular pro-fibrogenic processes and provide mechanistic insights into its antifibrogenic action. Utilizing cardiac fibroblasts, podocytes, and mesangial cells, we demonstrate that L002 blunts FATp300-mediated acetylation of specific histones. Further, incubating cells with L002 suppresses several pro-fibrogenic processes including cellular proliferation, migration, myofibroblast differentiation and collagen synthesis. Importantly, systemic administration of L002 in mice reduces hypertension-associated pathological hypertrophy, cardiac fibrosis and renal fibrosis. The anti-hypertrophic and anti-fibrotic effects of L002 were independent of blood pressure regulation. Our work solidifies the role of epigenetic regulator FATp300 in fibrogenesis and establishes it as a pharmacological target for reducing pathological matrix remodeling and associated pathologies. Additionally, we discover a new therapeutic role of L002, as it ameliorates hypertension-induced cardio-renal fibrosis and antagonizes pro-fibrogenic responses in fibroblasts, podocytes and mesangial cells.
KEYWORDS: Acetyltransferase p300, Angiotensin II, Cardiac Fibrosis, Epigenetics, Fibroblasts, Hypertension, Podocytes, Renal Fibrosis, Small molecule inhibitors, TGF-β
Introduction
Deposition of extracellular matrix (ECM) is necessary for physiological wound healing and tissue regeneration. However, when unchecked, excessive ECM protein deposition impairs tissue homeostasis, causes pathological matrix remodeling and leads to organ fibrosis. While fibrosis-associated organ dysfunction and failure contribute heavily to global morbidity and mortality, no effective anti-fibrotic therapy currently exists.1,2 Further, fibrogenesis is a complex physiopathological process characterized by simultaneous alterations in immunological, biochemical, cellular and molecular events.1,2 Transforming growth factor-β (TGF-β)—produced by immune and circulatory cells—plays a key role in the onset and progression of fibrogenesis, as it promotes cellular migration, proliferation, and myofibroblastic differentiation. Since the efforts to mitigate global TGF-β signaling are limited by severe side-effects,2 we propose that targeting TGF-β downstream effectors could provide a viable approach to combat fibrogenesis.
Epigenetic changes regulate numerous physiological processes and synchronize embryogenesis and mammalian development. In contrast, epigenetic dysregulation contributes to the onset and progression of numerous human diseases including organ fibrosis.3 We previously documented that epigenetic regulator FATp300—p300 with intrinsic factor acetyltransferase activity—is essential for profibrogenic-signal-induced Type I collagen synthesis and its level is significantly elevated in fibrotic tissues.4,5 As FATp300 levels are elevated in fibrotic tissues and hypertension induces fibrogenesis in heart and kidney, we hypothesize that pharmacological inhibition of FATp300 is an ideal approach to ameliorate hypertension-associated cardio-renal fibrosis.
Recently, utilizing a high-throughput screen, Yang et al 6 identified L002 as a novel, cell permeable inhibitor of p300 FAT activity. In addition to being structurally distinct from prior known inhibitors of FATp300, L002 binds the acetyl-CoA pocket and competitively inhibits the FATp300 catalytic domain. Here, we investigate the therapeutic efficacy of L002 in a murine model of hypertension-associated end-organ fibrosis. Additionally, we identify L002 as a novel suppressor of fibrogenic processes in diverse ECM producing cell types and provide mechanistic insights into its therapeutic action.
Results
FATp300 inhibitor L002 reduces differentiation, migration, and proliferation of cardiac fibroblasts
FATp300 plays a pivotal role in cellular fibrogenesis as it enhances synthesis of ECM proteins including collagen.5 We hypothesized that suppression of FATp300 activity could be an ideal approach for reducing cellular profibrogenic responses and organ fibrogenesis. Hence, we investigated the effects of cell permeable, FATp300 inhibitor small molecule L002 on TGF-β-induced fibrogenic responses in vitro. Cultures of human and rat cardiac fibroblasts (HCFs and RCFs) were treated with TGF-β to mimic a pro-fibrogenic environment. In parallel, cells were co-cultured with either L002 or DMSO (vehicle). We observed that pre-treatment with L002 suppresses TGF-β-induced Type I collagen synthesis in both HCFs (Fig. 1A, 1B) and RCFs (Fig. 1C). Further, L002 also blocks TGF-β-induced fibroblast-to-myofibroblast differentiation as suggested by a reduction in α-SMA mRNA and protein levels (Fig. 1A–C). Confirming our initial observations, inhibition of FATp300 by another inhibitor C646 also blunts TGF-β-mediated collagen upregulation (Fig. 1D, E), without altering α-SMA levels (Fig. 1D). Next, we tested the effect of L002 on overexpressed exogenous FATp300-induced collagen synthesis in HCFs. Our results showed that overexpressed wildtype FATp300, but not FAT deleted mutant p300, induces Type I collagen synthesis as expected.4 However, overexpressed wildtype FATp300 fails to stimulate collagen synthesis in the presence of L002, confirming target specificity and efficacy of L002 in inhibiting FATp300 activity (Online Figure 1A).
Figure 1.
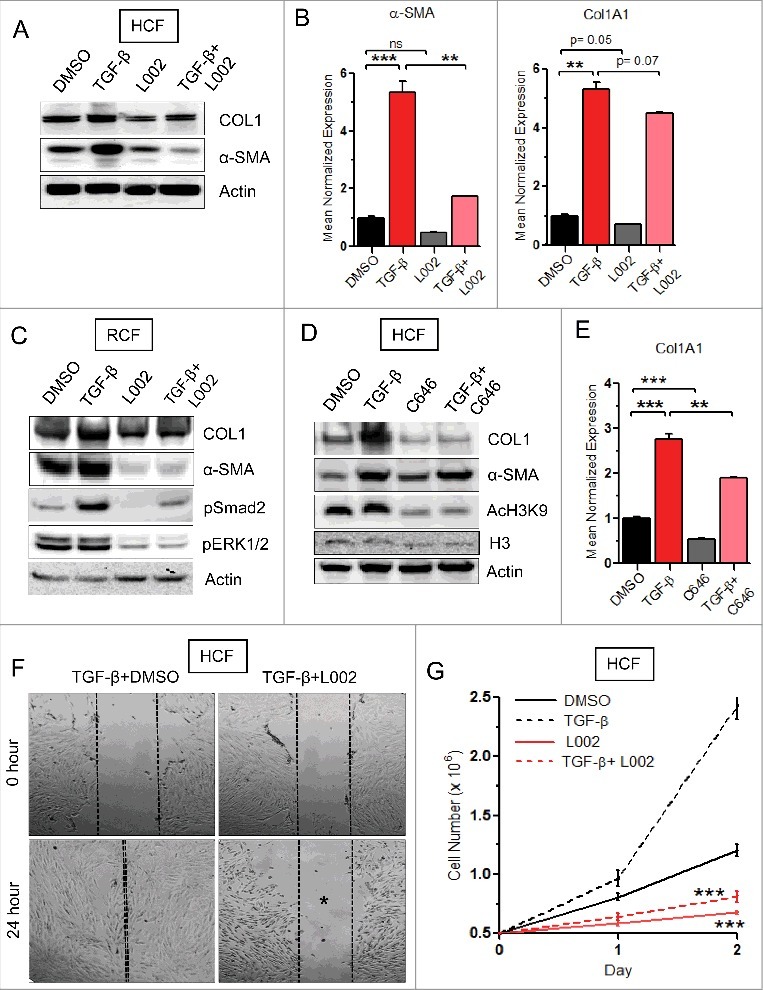
FATp300 inhibitor L002 reduces differentiation, migration and proliferation of cardiac fibroblasts. (A to E) Human cardiac fibroblasts (HCFs) or rat cardiac fibroblasts (RCFs) were cultured and treated in triplicate with L002 or C646 or DMSO in the presence or absence of TGF-β. Total protein were isolated, pooled and processed for Western blot (A, C, D) using indicated antibodies. Experiments were repeated two times. Total RNA were isolated and processed for qPCR in triplicate (B, E) using gene specific primers. (F) For migration study, the scratch wounds were made in monolayer cultures of HCFs. Cells were then pretreated in triplicate with L002 or DMSO for 1 hour followed by TGF-β treatment for 24 h. Photographs were taken at 0 hour and 24-hour post-treatment. (G) For proliferation study, HCFs were pretreated in triplicate with L002 or DMSO for 1 h followed by treatment with TGF-β. Cell numbers were counted at 24 h and 48 h. Data represented as mean ± SEM. **p < 0.01, ***p < 0.005.
Cellular migration and proliferation play a key role in physiological wound healing, and if unchecked leads to excessive scarring and fibrosis. Therefore, we next investigated the effects of L002 on cellular migration and proliferation. Using a scratch assay, we observed that L002 successfully blocks migration of HCFs in the absence (Online Fig. 1B) and presence of TGF-β (Fig. 1F). Additionally, L002 also reduces cellular proliferation as fewer cells were counted after incubating HCFs with L002 for 48 hours (Online Fig. 1C, 1D). This anti-proliferative effect of L002 was observed even after stimulating HCFs with proliferation-promoting TGF-β (Fig. 1G). Our results highlight the multi-faceted effect of L002 on cellular fibrogenic processes, as it reduces basal and TGF-β-mediated synthesis of collagen and α-SMA, cellular migration, and proliferation.
L002 reduces hypertension (HTN)-associated murine cardiac fibrosis and hypertrophy
HTN is a global health problem as chronically elevated high blood pressures (BP) lead to end-organ damage and fibrosis—especially within the heart and the kidneys. As L002 effectively antagonizes cellular fibrogenesis, and elevated levels of FATp300 are reported in fibrotic heart and kidneys,5 we next investigated the in vivo efficacy of L002 in a murine model of HTN. Murine BPs were raised by implanting Angiotensin II (Ang II) or saline (vehicle) containing mini-osmotic pumps (two weeks), as previously described.7 Additionally, saline- and Ang II-infused mice were also injected (intraperitoneally) with either L002 or DMSO. After completion of the study, M-Mode echocardiography analysis was undertaken, murine hearts were excised and processed for Masson's trichrome staining.
We observed that Ang II significantly increases perivascular and interstitial cardiac fibrosis as suggested by increased collagen accumulation (Fig. 2A, B). Most importantly, similar to our in vitro observations, we noticed that co-treatment with L002 significantly reduces Ang II-induced perivascular and interstitial collagen deposition (Fig. 2A, B). Interestingly, echocardiography data also reveals that L002 reduces thickness of the left ventricular wall (Fig. 2C, D, and Online Fig. 2A), while the post-mortem analysis of heart weight confirms anti-hypertrophic effect of L002 (Fig. 2E). Hence, our results identify L002 as an attractive candidate for treating hypertensive cardiac fibrosis as it reduces cardiac collagen deposition and protects against pathological cardiac hypertrophy. To address whether the reduction in cardiac hypertrophy and fibrosis is due to an unknown vasodilatory effect of L002, we measured murine blood pressures (BPs) by tail-cuff method. BPs were monitored and recorded basally (day 0), day 7 and day 14. We observed that despite its anti-fibrotic and anti-hypertrophic effects, L002 does not impart any vasodilatory benefits (Fig. 2F and Online Fig. 2B).
Figure 2.
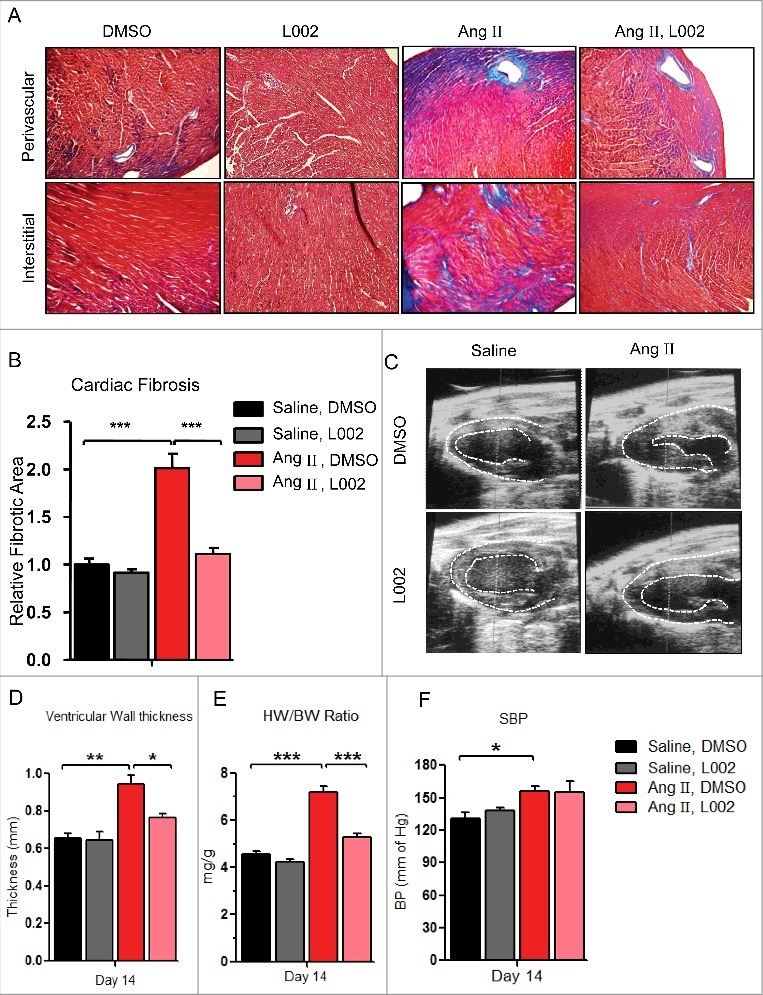
L002 reduces hypertension (HTN)-induced murine cardiac fibrosis and hypertrophy: (A) After two weeks of Ang II infusion and L002 treatment, cardiac sections from the four groups were stained with Masson's trichrome to identify collagen deposits. Upper panel shows perivascular deposition of collagen, while the lower panel identifies deposition of cardiac interstitial collagen. (B) Collagen deposition (perivascular + Interstitial) was quantified by ImagePro software program analysis, n = 7–12. (C) M-mode echocardiographic images showing thickness of left ventricular (LV) wall and LV diameters. (D) Quantification of the thickness of LV wall from echocardiography analysis, n = 4–12. (E) Post-mortem heart weight to body weight (HW/BW) were assessed in control and treatment groups. n = 7–12. (F) Shows systolic blood pressures (SBP) after two weeks of Ang II treatment as assessed by tail-cuff methods, n = 4–12. Data represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.005.
L002 reduces hypertension-associated renal fibrosis and pro-fibrogenic responses in renal cells
As hypertension also induces renal fibrosis and L002 effectively reduced hypertension-induced cardiac fibrosis and hypertrophy, we next investigated the effect of L002 on renal fibrogenesis. As elevated levels of FATp300 are documented in fibrotic kidneys,5 we hypothesized that L002 reduces Ang II-induced renal fibrosis. In agreement with prior reports,8,9 we observed that Ang II induces significant deposition of renal interstitial and perivascular collagen (Fig. 3A, B). Most importantly, co-treatment with L002 blocks Ang II-induced collagen deposition and renal fibrosis (Fig. 3A, B). Therefore, treatment of hypertensive mice with L002 targeting epigenetic regulator FATp300 suppresses both cardiac and renal fibrosis, which if left untreated leads to cardiac or renal failure, and early mortality.
Figure 3.
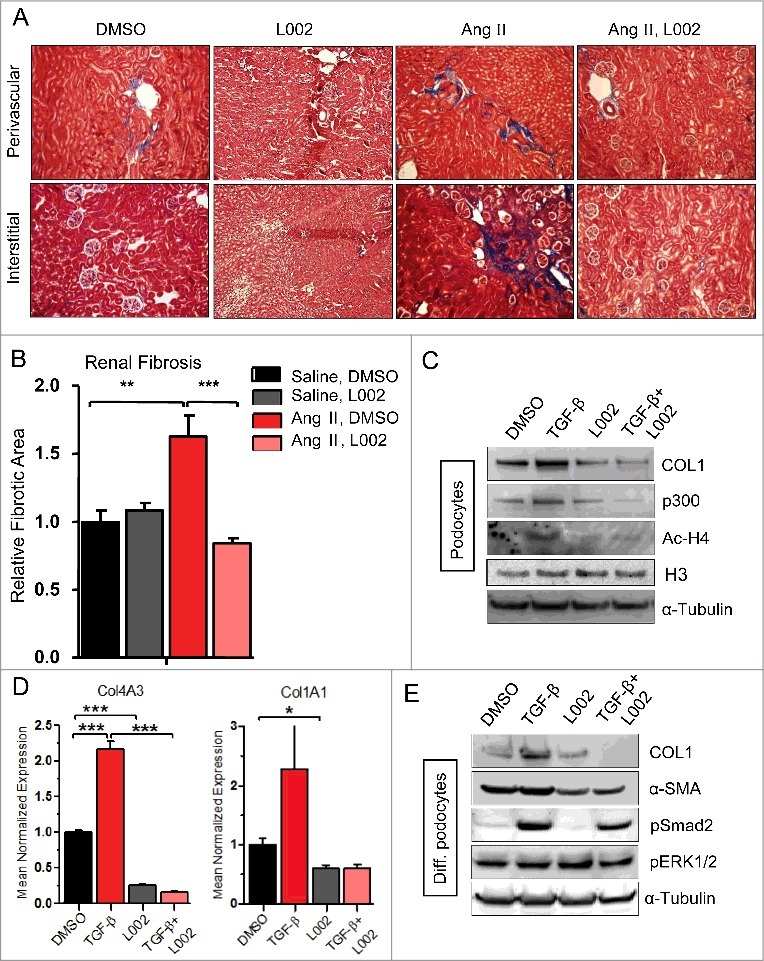
L002 reduces hypertension induced renal fibrosis and pro-fibrogenic responses in renal cells: (A) After two weeks of Ang II infusion and L002 treatment, renal sections were stained with Masson's trichrome to identify collagen deposits. Upper panel shows perivascular deposition of collagen, while the lower panel identifies interstitial collagen. (B) Collagen deposition (perivascular + Interstitial) was quantified by ImagePro software program analysis, n = 6–12. (C-E) Cultures of human (proliferating) podocytes and differentiated (diff.) podocytes were treated in triplicate with L002 or DMSO in the presence or absence of TGF-β. Total protein were isolated from three wells, pooled and processed for Western blot (C, E) using specific antibodies. Experiments were repeated twice. Total RNA were isolated from three wells and processed for qPCR in triplicate (D) using gene specific primers. Data presented as mean ± SEM *p < 0.05, **p< 0.01, ***p < 0.005.
To expand our in vivo observations, we next investigated the effects of L002 on renal cells in vitro. We observed that while TGF-β upregulates Type I collagen synthesis in the podocytes, L002 blunts its upregulation (Fig. 3C). Additionally, L002 also reduces TGF-β-enhanced transcription of COL4A3—major collagen isoform synthesized by podocytes— and basal and TGF-β-induced COL1A1 transcription (Fig. 3D). To further confirm our observations, we differentiated proliferating podocytes (culturing at 37°C for 2 weeks) to mimic adult podocytes and investigated the effects of L002. We observed that L002 blunts TGF-β-mediated upregulation of collagen and α-SMA expression in differentiated podocytes as well (Fig. 3E). As increased migration of podocytes accompany its effacement and many renal disorders,10 next we tested the effects of L002 on podocyte migration and observed that L002 blocks podocyte migration in the absence (Online Fig. 3) and presence of TGF-β (Fig. 4A). Further highlighting the critical role of FATp300 in renal fibrogenesis, inhibition of FATp300 by C646 also suppresses production of collagen and α-SMA by podocytes (Fig. 4B). The differential effect of C646 on TGF-β-induced α-SMA expression in human cardiac fibroblasts (Fig. 1D) and podocytes (Fig. 4B) appears to be cell type-specific.
Figure 4.
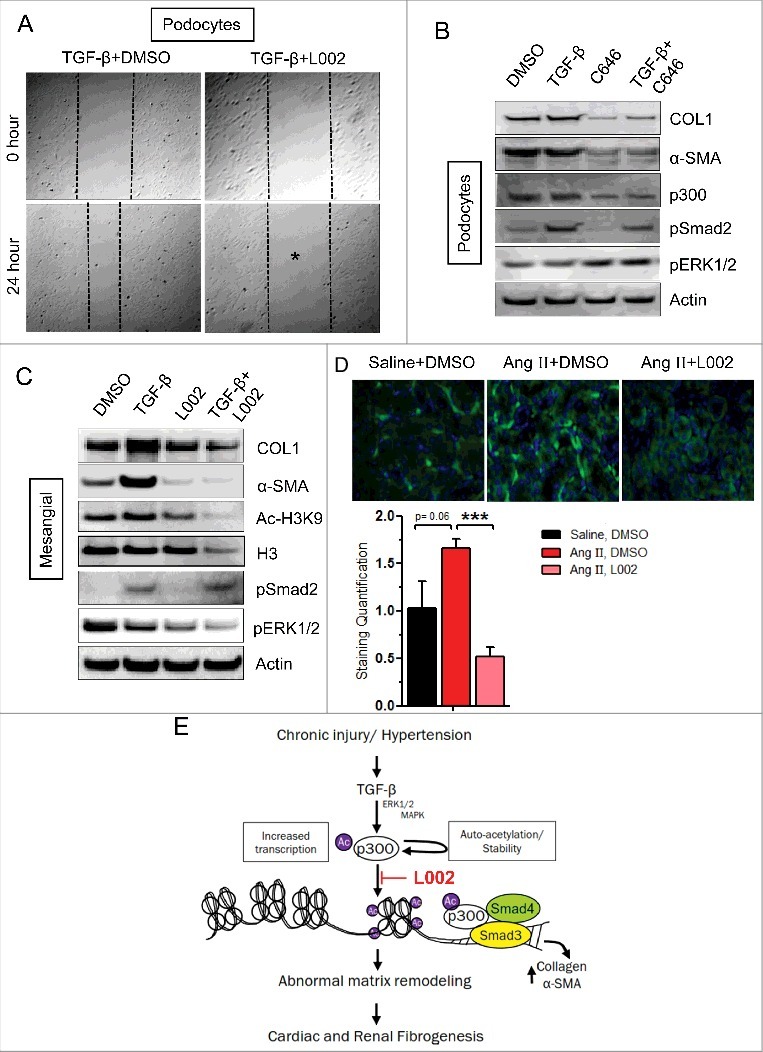
L002 reduces TGF-β-induced pro-fibrogenic responses in renal cells, and mechanistic insights. (A) For migration assay, podocytes were pretreated in triplicate with L002 or DMSO for 1 hour. Then scratch wounds were made in monolayer cultures, followed by TGF-β treatment for 24 h. Photographs were taken at 0 hour and 24 hours after treatment. (B-C) Cultures of human (proliferating) podocytes or mesangial cells were treated in triplicate with L002/ C646 or DMSO in the presence or absence of TGF-β. Total protein was isolated, pooled and processed for Western blotting. Experiments were repeated twice. (D) Effect of L002 on p300 levels. Renal sections from saline, Ang II and Ang II, L002 groups were immuno-stained with anti-p300 antibody and quantified. Saline (n = 3, 12–15 fields); Ang II (n = 5, 20–25 fields); Ang II, L002 (n = 5, 20–25 fields). Data represented as mean ± SEM. ***p < 0.001. (E) Model showing effect of L002 on fibrogenesis: Profibrogenic signals induce the levels of FATp300 due to increased transcription and auto-acetylation. FATp300 acetylates histones and transcription factors and augments matrix protein synthesis causing organ fibrogenesis. Small molecule L002 reduces FATp300 activity and blunts organ fibrogenesis.
To widen our understanding of L002 anti-fibrotic effects on the kidney, we next investigated if L002 affects TGF-β induced collagen production in the human mesangial cells. Mesangial cells play a critical supporting role in the renal parenchyma and when impaired, contribute to hypertensive renal fibrosis11. Similar to our results with the podocytes, we observed that L002 reduces TGF-β-mediated upregulation of collagen and α-SMA in mesangial cells (Fig. 4C). Concluding from our current observations and the specificity and the potency of L002, we predict that L002 or its analogs could form the structural framework for the development of novel anti-fibrotic therapies targeting druggable epigenetic regulator FATp300.
Mechanistic insights into the anti-fibrogenic effects of L002
Our data shows that in vivo administration of L002 reduces HTN-associated cardiac and renal fibrosis. In vitro, we demonstrate that the beneficial effects of L002 involve the reduction of profibrogenic processes viz. cellular differentiation, migration, proliferation and collagen production. Hence, we next aimed to identify the molecular mechanism(s) governing the anti-fibrotic effects of L002. Interestingly, we observed that L002—in addition to its FATp300 inhibitory activity—suppresses Ang II-mediated upregulation of p300 in renal interstitial fibroblasts (Fig. 4D). Similarly, L002 and C646 also suppress TGF-β-mediated upregulation of p300 in podocytes (Fig. 3C, 4B). Therefore, we propose that protective in vivo effects of L002 arise from both the inhibition of acetyltransferase activity and the downregulation of FATp300. It is well-documented that FATp300 acetylates histones, loosens the chromatin and promotes transcription of several pro-fibrogenic genes.3 Consequently, we observed that L002 mediated inhibition of FATp300 decreases acetylation of H4 in the podocytes (Fig. 3C) and H3K9 in the mesangial cells (Fig. 4C). Importantly, downregulation of FATp300 in kidney tissue by L002 was also associated with decreased number of Ac-H3K9 positive cells (Online Fig. 4). Similar to the “histone-inactivating” effect of L002, C646 also inhibited H3K9 acetylation in human cardiac fibroblasts (Fig. 1D and online Fig. 5A,B). Therefore, FATp300 inhibitors, L002 and C646 suppress specific histone acetylation and thereby repress transcription of major ECM proteins, including collagen. As Ang II transduces its profibrogenic signal through TGF-β-regulated angiotensin receptors (AT1/AT2), we next investigated the effect of FATp300 inhibitor L002 on expression of AT1 and AT2 receptors. Interestingly, we observed that while L002 inhibits TGF-β-induced expression of AT1 receptor (activator of profibrogenic signaling), L002 activates basal expression and partially rescues TGF-β-induced suppression of AT2 receptor (repressor of profibrogneic signaling) in human cardiac fibroblasts (Online Fig. 6). Lastly, pro-fibrogenic pathways are often interconnected and self-amplifying.2 As both canonical and non-canonical TGF-β signaling pathways contribute to fibrosis,12 we investigated whether incubation with L002 alters any component of the TGF-β signaling cascade. We noticed that L002 suppresses basal- and TGF-β-mediated phosphorylation of pSmad2 and pERK1/2 (canonical and non-canonical TGF-β signaling) in rat cardiac fibroblasts (Fig. 1C). Extending from in vitro observations, we propose that L002 ameliorates hypertensive cardiac fibrosis by suppressing FATp300-dependent TGF-β signaling and downstream collagen gene transcription. In contrast, the effect of L002 on renal TGF-β signaling appears to be cell-specific and limited, as L002 suppresses only the ERK1/2 MAPK pathway in human mesangial cells (Fig. 4C). However, both L002 and C646 failed to antagonize the levels of Smad and ERK phosphorylation in the podocytes (Fig. 3E, 4B). As FATp300 activity is also controlled by phosphorylation of its specific amino acids by different kinases,13 we asked whether L002 exerts any indirect influence on FATp300 phosphorylation. Equal amounts of proteins from DMSO and L002-treated HCFs, in the presence and absence of TGF-β, were immunoprecipitated with p300 antibody and immunoprecipitates were subjected to Western blot using phospho-serine/threonine and p300 antibodies. Results showed no detectable differences in p300 phosphorylation in control and L002-treated protein extracts relative to the levels of immunoprecipitated total p300 protein (data not shown). Thus these results collectively highlight the significance of L002-mediated suppression of factor acetyltransferase activity of p300 in amelioration of profibrogenic signaling and cardio-renal fibrogenesis.
Discussion
We previously demonstrated that FATp300 is an essential epigenetic regulator of profibrogenic signal-induced ECM protein synthesis. Further, FATp300 levels are elevated in myofibroblast-derived from resident fibroblasts and endothelial cells.5 Increased levels of FATp300 are also documented in several fibrotic tissues including dermal fibrosis, chronic glomerulonephritis, heart failure-associated myocardial fibrosis and transverse aortic constriction-induced murine cardiac fibrosis.5 These results predict a strong correlative and a causative link between the elevation of FATp300 and organ fibrosis. We propose that FATp300 is a biomarker of organ fibrosis and hypothesize that its inhibition in injured or stressed tissues is an ideal approach to ameliorate fibrosis-associated organ failure. Utilizing a high-throughput setup, Yang et al., recently identified L002 as a specific inhibitor (IC50 = 1.98 µmol/L) of FATp300 from a library of over 600,000 compounds.6 In this brief report, we investigate the effect of L002 in suppressing pro-fibrogenic signaling and organ fibrogenesis.
Our results demonstrate that L002 effectively suppresses cellular fibrogenic responses and organ fibrosis, both in vivo and in vitro. The beneficial effect of L002 likely involves three interconnected pathways (Fig. 4E). Firstly, as L002 is a potent, specific inhibitor of FATp300, it suppresses FATp300-mediated acetylation of histones and the subsequent transcription of AT1 receptor, collagen and α-SMA. In contrast, L002 stimulates basal expression of AT2 receptor and partially reverses TGF-β mediated suppression of AT2 receptor, a suppressor of profibrogenic signaling,14 indicating an additional mechanism operating in FATp300 inhibitor L002 mediated suppression of Ang II-induced fibrogensis. Secondly, L002 decreases Ang II/TGF-β-mediated upregulation of p300. This effect might stem from FATp300 activity inhibition as p300 acetylates itself,15 and the acetylated form may be more stable.16 The absence of acetylation renders the deacetylated p300 unstable and susceptible to ubiquitin-dependent proteasomal degradation.17 Additionally, suppression of FATp300 in renal tissue is associated with decreased number of Ac-H3K9 positive renal cells, consistent with our in vitro observations in podocytes and mesangial cells. Therefore, L002 suppresses both, the activity and elevation of FATp300 level. The obvious question is what could be the possible epigenetic mechanisms of antifibrogenic action of L002. Using chromatin immunoprecipitation (ChIP) assay, we previously demonstrated that TGF-β induces the levels of p300 and its recruitment to collagen gene promoter, associated with increased levels of acetylated H4 at the chromatin level and increased collagen transcription in fibroblasts.18,19 Therefore, it is reasonable to speculate that similar epigenetic mechanism operates during TGF-β-induced collagen synthesis in cardiac fibroblasts, podocytes and mesangial cells, and hence pharmacological inhibition of p300 acetyltransferase activity by L002 decreases collagen synthesis and organ fibrogenesis. However, a further detailed investigation is needed to understand the precise molecular mechanism(s) of L002 mediated suppression of FATp300 and subsequent epigenetic modulation of TGF-β or Ang II-induced profibrogenic signaling. Thirdly, we discovered that L002 surprisingly antagonizes canonical and non-canonical TGF-β signaling. However, this antagonism of TGF-β was cell-specific as L002 inhibited both Smad and ERK1/2 MAPK pathways in RCFs, while only inhibiting the ERK1/2 MAPK pathway in mesangial cells and no effect on both canonical and non-canonical pathway activation in podocytes. Therefore, present study clearly indicate that FATp300 is an ideal druggable target for fibrosis therapy and L002 exerts its major anti-hypertrophic and anti-fibrotic effects through suppression of essential epigenetic regulator FATp300 in both in vitro and in vivo models.
It is well established that cellular migration plays a vital role in fibrogenesis.2 Recent studies have highlighted that podocytes are mobile, as a cell-specific expression of GFP/YFP revealed simultaneous migrating podocytes in murine kidneys.20 Additionally, increased migration of podocytes has been documented during many renal pathologies.10,20 Our migration studies on cardiac fibroblasts and podocytes revealed that L002 decreases both basal and TGF-β-induced cellular migration. The migration-inhibitory role of L002 is however expected, as prior loss-of-function studies document an essential role of p300 in cellular migration.21 Our observation on suppression of cardiac fibroblast proliferation by pharmacological inhibition of FATp300 is consistent with the previous observation showing the decreased rate of proliferation of p300 null mouse embryonic fibroblasts compared to wildtype fibroblasts.22 To summarize, the inhibition of FATp300 by L002 reduces TGF-β-mediated cellular migration, proliferation, ECM protein synthesis (collagen, α-SMA) and p300 upregulation. Additionally, target specificity of L002 is evidenced by its ability to block overexpressed FATp300-induced collagen synthesis in cardiac fibroblasts. These in vitro observations predict a “therapeutic” role of L002 during the initiation and progression of murine, and possibly human, organ fibrosis.
Given our encouraging in vitro observations and lack of any apparent L002-induced cellular toxicity, we proposed that mice administered with L002 will be protected from hypertension-induced cardiac and renal fibrosis. Indeed, after two weeks of Ang II infusion, mice co-administered with L002 exhibited reduced cardio-renal fibrosis. Additionally, echocardiography study and post-mortem analysis suggested that L002 also reduces Ang II-induced cardiac hypertrophy. The present finding on amelioration of cardiac hypertrophy by pharmacological inhibition of p300 acetyltransferase activity corroborates with the previous studies which demonstrate that, while overexpressed FATp300 induces cardiomyocyte hypertrophy, FAT domain deleted mutant form of p300 inhibits hypertrophy.23,24 FATp300 exerts its pro-hypertrophic effect possibly through activation of transcription factors like GATA-4 that regulates expression of hypertrophic genes.23 Interestingly, despite its anti-hypertrophic and anti-fibrotic effects, L002, did not lower Ang II-mediated changes in blood pressure. While it is plausible that chronic (>4–6 weeks) treatment of L002 may reduce blood pressures by decreasing arterial remodeling, our results predict that a combination therapy of L002 and blood pressure lowering drugs is an attractive approach towards reducing hypertensive cardio-renal fibrosis.
Cardiovascular disease (CVD) and chronic kidney disease (CKD) are the leading causes of death in the United States.25 While aberrant and excessive organ fibrosis underlie the pathogenesis of both CVDs and CKDs, no effective anti-fibrotic therapy currently exists. Here we report a novel, fibrosis-impeding role of L002 which is a cell permeable, small molecule inhibitor of FATp300. Current results demonstrate—for the first time—that L002 suppresses pro-fibrogenic responses in cardiac fibroblasts, podocytes, and mesangial cells. Importantly, the fibrogenesis-suppressing effect of L002 also extends in vivo, as mice administered with L002 exhibit reduction in cardiac fibrosis, hypertrophy, and renal fibrosis. Further studies need to investigate the effect of L002 in other murine models of cardiac and renal fibrosis. Secondly, while we establish the therapeutic efficacy of L002 in cardiac and renal fibrosis, the role of L002 in pulmonary, dermal, hepatic fibrosis and hypertrophic scars should also be investigated. Potential animal toxicity is always a cause for concern with any new drug. While we did not observe any apparent toxic effects in vitro or in vivo, long-term studies with large animal cohorts are necessary to rule out adverse effects of L002, if any. Lastly, the utilization of L002 and C646 establishes the vital role of FATp300 in fibrogenesis and advocates the development and investigation of orally stable FATp300 inhibitors as anti-fibrotic agents.
Materials and methods
Experimental animals, implantation of minipump, induction of renal and cardiac fibrosis, FATp300 inhibitor treatment and organ collection: Wild-type mice on a C57BL/6 background were purchased from Jackson laboratory (Bar Harbor, ME) and were maintained. All mouse protocols were approved by the Animal Care and Use Committee of Northwestern University (Chicago, IL). Six weeks old wildtype C57BL/6 mice were used to generate Angiotensin II (Ang II)-induced cardiac fibrosis model. Mice were anesthetized using isoflurane (Baxter Healthcare, NJ, USA) and Ang II containing osmotic minipumps (Alzet) were implanted subcutaneously by mid-scapular skin incision. Then the incision was closed using wound clips.7 The control group animals received saline (n = 7) and the treatment group animals received Angiotensin II (Ang II, Bachem, Torrance, CA, USA; 1500 ng/kg/min for 2 weeks) filled into osmotic minipumps (model: Alzet-200; Durect Corp., CA) (n = 12) as described.7 Batches of mice received L002 (Sigma Chemicals, USA) 0.5 mg/mouse (∼20 μg/gm body weight) by intraperitoneal injection on every 3rd day in the absence and presence of Ang II (n = 6–9). Control group received equal volume of vehicle (DMSO). The mice were sacrificed and hearts and kidneys collected on day 15. Part of heart and kidney was fixed in formalin and stored for histochemical analysis. Other half of the organ was snap frozen and stored at −80°C and processed for biochemical analysis.
Effect of L002 on Ang II-induced pathological structures and functions of hearts. The blood pressure, heart rate and body weight in each group of mice were recorded in order to examine the effect of FATp300 inhibitor L002 on cardiac structure and function in response to Ang II. Trans-thoracic two-dimensional M-mode echocardiography was performed using Vevo 770 (VisualSonics, Toronto, Canada) equipped with a 30 MHz transducer. Echocardiographic studies were performed 2 weeks after pump implantation in mice. M-mode tracings were used to measure LV wall thickness, LV dimensions, LV volume and LV mass. The mean value of at least 3–5 cardiac cycles were used to determine the measurements for each animal treated with or without L002 according to Verma et al.26
Histology and Collagen Staining: Hearts were sectioned in the short axis along the mid ventricle. Kidneys were hemi-sectioned and fixed with 10% formalin. Paraffin-embedded heart and kidney tissues were subjected to microtome sectioning and processed for histological analysis.27 The levels of collagen deposition in hearts and kidneys were determined by Masson trichrome staining. Photographs were taken with an Olympus DP71 camera. To quantify the extent of fibrosis, heart and kidney stained sections from saline controls (n = 7), L002 (n = 6), Ang II (n = 12) and Ang II + L002 mice (n = 9) were analyzed with Image-Pro software (Media Cybernetics, Bethesda, MD).
Immunohistochemistry: Paraffin-embedded kidney tissue sections were deparaffinized, blocked with blocking buffer for 45 min and then incubated with primary antibody against p300 or Ac-H3K9 overnight at 4°C. The slides were washed and incubated with rabbit secondary antibody. Photographs were taken in either a fluorescence or a light microscope. Immunostained areas were determined with Image-Pro software (Media Cybernetics). The sum of 5–6 fields per mouse was used for statistical analysis.27
Cell Culture, Cell transfection, Cell treatment, Protein extraction and Western blot analysis: Conditionally immortalized human podocytes28 proliferate at a permissive temperature (33°C) mimicking an in vitro model of embryonic podocytes with proliferative capacity (mimicking embryonic podocytes). Podocytes were cultured in RPMI supplemented with Insulin-transferrin-sodium selenite (ITS) and 10% fetal bovine serum. Human podocytes were also differentiated by culturing at 37°C for 14 days before treatment. Rat and human cardiac fibroblasts were cultured in 10% FBS containing DMEM media. Human mesangial cells were cultured in MsGM (Mesangial Cell Growth Medium) supplemented with 10% FBS and 1% Penn/Strep. Cultures of podocytes, mesangial cells and cardiac fibroblasts in 12-well clusters were treated (in triplicate) with a small molecule inhibitor of FATp300, L002 (10 μM) (kind gift from Dr. Daiquing Liao, Univ. of Florida for an initial experiment) (Sigma Chemicals, MO) or vehicle in the presence or absence of TGF-β (10 ng/ml). To confirm the significance of acetyltransferase activity of p300 in profibrogenic responses, another inhibitor C646 (10 μM) (Sigma Chemicals, MO) was also used. Experiments were repeated two-three times. Following indicated period of incubation, cells from 3-wells were harvested and pooled. Whole cell lysates were prepared using RIPA lysis buffer with protease and phosphatase inhibitors (Sigma Chemicals, MO). For coupled immunoprecipitation-immunoblot analysis, equal amount of protein from control and L002 or L002, TGF-β treated human cardiac fibroblasts was used for immunoprecipitation with p300 antibody (Millipore, Billerica, MA) and immunoprecipitates were subjected to immunoblot analysis using p300 (Santa Cruz, CA) and phospho-serine/threonine antibody (Abcam, Cambridge, MA). To study the effect of L002 on overexpressed exogenous FATp300-induced collagen synthesis, human cardiac fibroblasts were transfected with empty vector pCI, wildtype FATp300 and FAT deleted p300 expression vector4 (1μg/well in 12-well cluster) in quadruplicate using Lipofectamine 3000 transfection kit (Invitrogen, Carlsbad, CA). After 24 h cells were treated either with DMSO or L002 for 48 h. Whole cell lysates were prepared. For Western blot analysis, equal amount of protein was loaded on 4–12% Tris-glycine gradient gel and run at 90 volts for 2 h at room-temperature. The separated proteins were transferred to PVDF membrane using iBlot transfer apparatus (Invitrogen, Grand Island, NY). The PVDF membranes were blocked with 10% fat free milk containing TBST for 2 h at room temperature. Then membranes were incubated with antibodies against Type I collagen (Southern Biotech, Alabama), α-SMA (Sigma, St Louis, MO), p300 (Santa Cruz, CA), pSmad2, pERK1/2, Ac-H4, Ac-H3K9 (Cell Signaling, MA), H3 (Novus Biologicals, CO), Actin (Abcam, Cambridge, MA), α-tubulin (GenScript, Piscataway, NJ) overnight at 4°C. The membranes were washed three times with TBST (0.05% Tween-20) for 60 minutes followed by incubation with HRP-tagged specific secondary antibodies. The membranes were washed with TBST and the signals were detected by ECL reagents and images were captured at BIORAD molecular imager ChemiDoc XRS system (BIORAD, Hercules, CA).
Quantitative qPCR analysis: Total RNA from cultured cells in the presence or absence of L002 or C646 and TGF-β2 were isolated and specific transcript level was determined by qPCR using gene specific primers. The messenger RNA (mRNA) levels of COL4 α3, α-SMA, COL1A1, AT1 receptor, AT2 receptor and GAPDH were quantified by complementary DNA synthesis using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) and quantitative polymerase chain reaction with SYBR Green SuperMix for IQ (Quanta Bioscience, Gaithersburg, MD). Q-gene software was used for quantification.
L002 on Human Cardiac Fibroblast (HCF) Proliferation: On day 0, 50,000 HCFs (Cell Applications Inc. (San Diego, CA)/well were seeded in 10% DMEM. After 6 hours, HCFs were pretreated with either L002 or DMSO for 1h followed by TGF-β for 48 h. After 24 h and 48 h of treatment, cells were typsinized and counted via Invitrogen countess automated cell counter.
Wound Healing Assay/Migration Assay: The effect of L002 on human cardiac fibroblast and podocyte migration was determined by scratch assay. Human cardiac fibroblasts and podocytes were grown to confluency. Scratch wounds were induced in monolayers using 200 μl pipette tips, followed by change of culture media to remove the detached cells from the scratches. The cells were immediately treated with L002 or vehicle DMSO for 1h followed by TGF-β treatment for 24 h. For each treatment conditions 3 wells were used. The wound areas were photographed at 0 and 24 h and the images were captured by phase contrast microscopy.
Statistical analysis: Data are presented as Mean ± SEM. The significance of differences between controls and experimental groups was estimated by t-test or 2-way ANOVA and a value of P < 0.05 by Student t test was considered statistically significant. Statistical analyses were performed with GraphPad Prism 3.0 (GraphPad Software Inc, San Diego, CA).
Funding Statement
This work was supported by grants from #1. American Heart Association (16GRNT31130010), #2. NIH-NHBLI (5R01HL051387-19 and 1P01HL108795) and #3. Gilead Research Scholars Program in Cardiovascular Disease (A123776).
Disclosure of Interest
No potential conflict of interest.
Acknowledgments
We thank Mesut Eren, PhD for his assistance and helpful discussion in this work. This work was supported by grants from #1. American Heart Association (16GRNT31130010), #2. NIH-NHBLI (5R01HL051387-19 and 1P01HL108795) and #3. Gilead Research Scholars Program in Cardiovascular Disease (A123776).
References
- 1.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524-529. doi: 10.1172/JCI31487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghosh AK, Quaggin SE, Vaughan DE. Molecular Basis of Organ Fibrosis: Potential Therapeutic Approaches. Exp Biol Med. 2013;238:461-481. doi: 10.1177/1535370213489441 [DOI] [PubMed] [Google Scholar]
- 3.Ghosh AK, Rai R, Flevaris P, Vaughan DE. Epigenetics in Reactive and Reparative Cardiac Fibrogenesis: The Promise of Epigenetic Therapy. J Cell Physiol. 2017;232:1941-1956. doi: 10.1002/jcp.25699 [DOI] [PubMed] [Google Scholar]
- 4.Ghosh AK, Yuan W, Mori Y, Varga J. Smad-dependent stimulation of Type I collagen gene expression in human skin fibroblasts by TGF-β involves functional cooperation with p300/CBP transcriptional coactivators. Oncogene. 2000;19:3546-3555. doi: 10.1038/sj.onc.1203693 [DOI] [PubMed] [Google Scholar]
- 5.Ghosh AK. FAT-Free p300 is Good for Scar-Free Tissue Repair. J Cell Biochem. 2014;115:1486-1489 doi: 10.1002/jcb.24820 [DOI] [PubMed] [Google Scholar]
- 6.Yang H, Pinello CE, Luo J, Li D, Wang Y, Zhao LY, Jahn SC, Saldanha SA, Chase P, Planck J, Geary KR, Ma H, Law BK, Roush WR, Hodder P, Liao D. Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Mol Cancer Ther. 2013;12:610-620. doi: 10.1158/1535-7163.MCT-12-0930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK, Vaughan DE. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation. 2016;133:291-301. doi: 10.1161/CIRCULATIONAHA.116.022627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitayama H, Maeshima Y, Takazawa Y, Yamamoto Y, Wu Y, Ichinose K, Hirokoshi K, Sugiyama H, Yamasaki Y, Makino H. Regulation of angiogenic factors in angiotensin II infusion model in association with tubulointerstitial injuries. Am J Hypertens. 2006;19:718-727. doi: 10.1016/j.amjhyper.2005.09.022. PMID:16814127 [DOI] [PubMed] [Google Scholar]
- 9.Lavoz C, Rodrigues-Diez R, Benito-Martin A, Rayego-Mateos S, Rodrigues-Diez RR, Alique M, Ortiz A, Mezzano S, Egido J, Ruiz-Ortega M. Angiotensin II contributes to renal fibrosis independently of Notch pathway activation. PLoS One. 2012;7:e40490. doi: 10.1371/journal.pone.0040490. PMID:22792351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arif E, Nihalani D. Podocytes as a therapeutic target. Ann Clin Exp Hypertension. 2013;1(1):1004. [Google Scholar]
- 11.Schnaper WH, Hayashida T, Hubchak SC, Poncelet AC. TGF-β signal transduction and mesangial cell fibrogenesis. AJP – Renal Physiology. 2003;284(2):F243-F252. doi: 10.1152/ajprenal.00300.2002 [DOI] [PubMed] [Google Scholar]
- 12.Ghosh AK. Factors involved in Type I collagen gene expression: implication in fibrosis. Exp Biol Med. 2002;227:301-314. doi: 10.1177/153537020222700502 [DOI] [PubMed] [Google Scholar]
- 13.Ghosh AK, Varga J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J Cell Physiol. 2007;213:663-671. doi: 10.1002/jcp.21162 [DOI] [PubMed] [Google Scholar]
- 14.Murphy AM, Wong AL, Bezuhly M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair. 2015;8:7. doi: 10.1186/s13069-015-0023-z. PMID:25949522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Black JC, Choi JE, Lombardo SR, Carey M. A mechanism for coordinating chromatin modification and preinitiation complex assembly. Mol Cell. 2006;23:809-818. doi: 10.1016/j.molcel.2006.07.018 [DOI] [PubMed] [Google Scholar]
- 16.Kim SH, Kang HJ, Na H, Lee MO. Trichostatin A enhances acetylation as well as protein stability of ERalpha through induction of p300 protein. Breast Cancer Res. 2010;12:R22. doi: 10.1186/bcr2562. PMID:20388208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuno A, Hori YS, Hosoda R, Tanno M, Miura T, Shimamoto K, Horio Y.. Resveratrol improves cardiomyopathy in dystrophin-deficient mice through SIRT1 protein-mediated modulation of p300 protein. J Biol Chem. 2013;288:5963-5972. doi: 10.1074/jbc.M112.392050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh AK, Bhattacharyya S, Wei J, Kim S, Barak Y, Mori Y, Varga J. Peroxisome proliferator- activated receptor-gamma abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J. 2009;23:2968-2977. doi: 10.1096/fj.08-128736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh AK, Bhattacharyya S, Lafyatis R, Farina G, Yu J, Thimmapaya B, Wei J, Varga J.. p300 is elevated in systemic sclerosis and its expression is positively regulated by TGF-β: epigenetic feed-forward amplification of fibrosis. J Invest Dermatol. 2013;133:1302-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hackl MJ, Burford JL, Villanueva K, Lam L, Suszták K, Schermer B, Benzing T, Peti-Peterdi J. Tracking the fate of glomerular epithelial cells in vivo using serial multiphoton imaging in new mouse models with fluorescent lineage tags. Nat Med. 2013;19:1661-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santer FR, Höschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT, Parson W, Meyers DJ, Cole PA, Culig Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther. 2011;10:1644-1655. [DOI] [PubMed] [Google Scholar]
- 22.Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361-372. [DOI] [PubMed] [Google Scholar]
- 23.Dai YS, Markham BE. p300 Functions as a coactivator of transcription factor GATA-4. J Biol Chem. 2001;276:37178-37185. [DOI] [PubMed] [Google Scholar]
- 24.Gusterson RJ, Jazrawi E, Adcock IM, Latchman DS. The transcriptional co-activators CREB-binding protein (CBP) and p300 play a critical role in cardiac hypertrophy that is dependent on their histone acetyltransferase activity. J Biol Chem. 2003;278:6838-6847. [DOI] [PubMed] [Google Scholar]
- 25.Bansal N. Evolution of Cardiovascular Disease During the Transition to End Stage Renal Disease. Semin Nephrol. 2017;37:120-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verma SK, Krishnamurthy P, Barefield D, Singh N, Gupta R, Lambers E, Thal M, Mackie A, Hoxha E, Ramirez V, Qin G, Sadayappan S, Ghosh AK, Kishore R. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-κB. Circulation. 2012;126:418-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh AK, Bradham WS, Gleaves LA, De Taeye B Murphy SB, Covington JW, Vaughan DE. Genetic deficiency of plasminogen activator inhibitor-1 promotes cardiac fibrosis in aged mice: involvement of constitutive transforming growth factor-beta signaling and endothelial-to- mesenchymal transition. Circulation. 2010;122:1200-1209. [DOI] [PubMed] [Google Scholar]
- 28.Saleem MA, O'Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630-638 [DOI] [PubMed] [Google Scholar]