

ABSTRACT
The blood brain barrier is a necessity for cerebral homeostasis and response to environmental insult, thus loss in functionality with age creates opportunities for disease to arise in the aged brain. Understanding how the barrier is developed and maintained throughout the earlier years of adult life can identify key processes that may have beneficial applications in the restoration of the aged brain. With an unprecedented increasing global aged population, the prevention and treatment of age-associated disorders has become a rising healthcare priority demanding novel approaches for the development of therapeutic strategies. The aging cardiovascular system has long been recognised to be a major factor in age-associated diseases such as stroke, atherosclerosis and cardiac arrest. Changes in the highly specialised cerebral vasculature may similarly drive neurodegenerative and neuropsychiatric disease.
KEYWORDS: ageing, biology of endothelial barriers, blood brain barrier, dementia, psychiatric illness, tight junctions
Introduction
The brain is intrinsically susceptible to long-term damage due to its reliance on an intact network of neurons coupled to a low rate of adult neurogenesis. Indeed, neuron proliferation is mainly limited to defined regions of neurogenesis in the adult brain, the dentate gyrus (DG) of the hippocampus1 and the subventricular zone (SVZ) of the lateral ventricles.2 Both of these regions have specialised vasculatures which support neural stem cell maintenance while providing cues for differentiation and migration during injury.3 It is well defined that the interplay between the cerebral vasculature and neurogenesis aids recovery from neural insult as well as physiological neurogenesis associated with learning4 and cognitive function. The cerebral vasculature also protects the brain from insult through the endothelial blood brain barrier (BBB). This vasculature plays a central role in the maintenance and regulation of the adult neural stem cell population and gives it a key role in processes involving neurodegeneration and cognitive impairment.5,6
Initial dye-injection experiments by Paul Ehrlich and Edwin Goldmann revealed the capacity of the CNS vasculature to retain and exclude tracer molecules, leading to the discovery of the BBB. The BBB is a dynamic tissue barrier that acts to protect and regulate the neural environment through restricted permeability and selective molecule transport from blood to brain and vice versa. This is achieved through the co-operative actions of cells within the neurovascular unit (NVU) including pericytes, astrocytes, neurons and endothelial cells (ECs).7 While gases and very small lipophilic compounds can freely cross the BBB, a highly specialised CNS endothelium establishes a size selective barrier limiting paracellular traffic and preventing the passive diffusion of molecules as small as 400 Dalton.8,9 Indeed, creating an increased dependence on active transport allows the BBB to physically regulate molecular traffic via the remodelling of intercellular EC tight junctions (TJs) or through luminal or abluminal transport protein localisation.10,11 This regulation of transport and junctional proteins permits the high nutrient influx required for cognitive function, such as increased luminal expression of the glucose transporter GLUT112. Simultaneously, endogenous and exogenous toxins are excluded from the brain parenchyma via abluminally enriched efflux proteins.13
Dysregulation or deterioration of BBB integrity with age is believed to be a compounding factor in neurodegenerative and age-associated diseases such as multiple sclerosis,14 Alzheimer's,15 schizophrenia16 and type 2 diabetes mellitus.17 Changes in TJ protein expression and normal NVU functionality have been identified at preclinical stages in animal models of neurodegenerative diseases18 pointing towards the BBB as an early target in the disease pathogenesis. The changes observed in early stages of neurodegenerative diseases may indeed be part of the pathology, however they equally may be physiologic age-related changes which instead render the brain susceptible towards subsequent disease-inducing insult. Autoimmunity and neuroinflammation are often central to the pathologies observed in age associated disorders, and while the concept of a healthy brain being “immune privileged” – resistant to all leukocyte invasion – has been shown to be incorrect,19 leukocyte transmigration across the BBB is tightly regulated and often aberrant in the aged brain. This review aims to summarize our current understanding of BBB development, maintenance and functionality as well as how these NVU regulated processes – neurogenesis, NVU homeostasis and leukocyte infiltration – are changed within the aging brain.
The neurovascular unit
Shown by classical chick-quail studies of CNS and gastrointestinal interchange,20 the development of the BBB is a process not intrinsic to cerebral ECs. Instead, the neural environment supplies the extracellular signalling required to induce CNS endothelial cell characteristics such as the high trans-endothelial electric resistance (TEER) produced by continuous intercellular TJs. The NVU then maintains these characteristics in the developed brain. However, during the development of the BBB a stepwise induction occurs to create a functional embryonic barrier which does not appear to require the fully assembled NVU.21 Understanding these critical pathways involved in establishing and maturing the early BBB, we can also find potential targets to monitor during the detrimental process of aging.
Endothelial cells
BBB development in the embryo begins at approximately embryonic day 12 (E12) with invading angioblasts forming a vascular plexus of immature blood vessels which progressively branches to produce a vascularised brain.22 Following the recruitment of pericytes to the immature BBB endothelium, functional TJ strands form at EC paracellular clefts polarising the monolayer and generating a tracer excluding barrier by E1821. The endothelial TJ is comprised of three principal families of proteins; the claudins,23 MARVEL/D324 and Ig-like junctional adhesion molecules25 (JAMs). The TJ complex is anchored to the cell cytoskeleton through the binding of protein C-terminal PDZ domains to zonula occludens (ZO) proteins, tethering the TJ complex to F-actin.26 ZO-1 and ZO-2 also regulate the TJ complex itself, binding modifiers of Rho GTPases such as shroom2 and cortactin to regulate cytoskeletal anchoring.27,28
Claudin-5, which when lost results in postnatal lethality,8 is the most abundant claudin family member found in the CNS vasculature, enriched at least 500-fold respective to other family members.29,30 Claudins contain four-transmembrane domains, generating two extracellular loops which dimerise with other TJ proteins. These two loops are believed to have discrete functions, ECL1 acts to regulate paracellular transport via pore formation while ECL2 drives the physical closure of the paracellular cleft.31 Occludin, a member of the MARVEL/D3 family, was the first TJ protein identified and is enriched at CNS TJs, but unlike Cldn5−/− mice occludin knockouts are viable.32 The MARVEL/D3 and claudin family members have been shown to regulate each other within the TJ, indicating a primarily organisational and regulatory role for the MARVEL/D3 proteins in the BBB.33
While the former TJ proteins are central to TJ modulation and formation, the JAM protein family has a central role in regulating immune cell traffic through the BBB. JAM proteins have been implicated in establishing the apical-basal EC axis through recruiting cell polarity complexes, and in facilitating leukocyte infiltration.34,35 The actions of JAM proteins are in concert with basolaterally located adherens junctions (AJs) comprised of cadherins such as vascular endothelial-cadherin, and nectins from the cell adhesion molecules (CAMs) family which further regulate leukocyte passage across the BBB.36 Endothelial cells of the BBB also selectively express leukocyte adhesion proteins to facilitate immune cell infiltration during disease and injury, these will be further discussed in the context of aging (see Fig. 1 for normal BBB overview).
Figure 1.
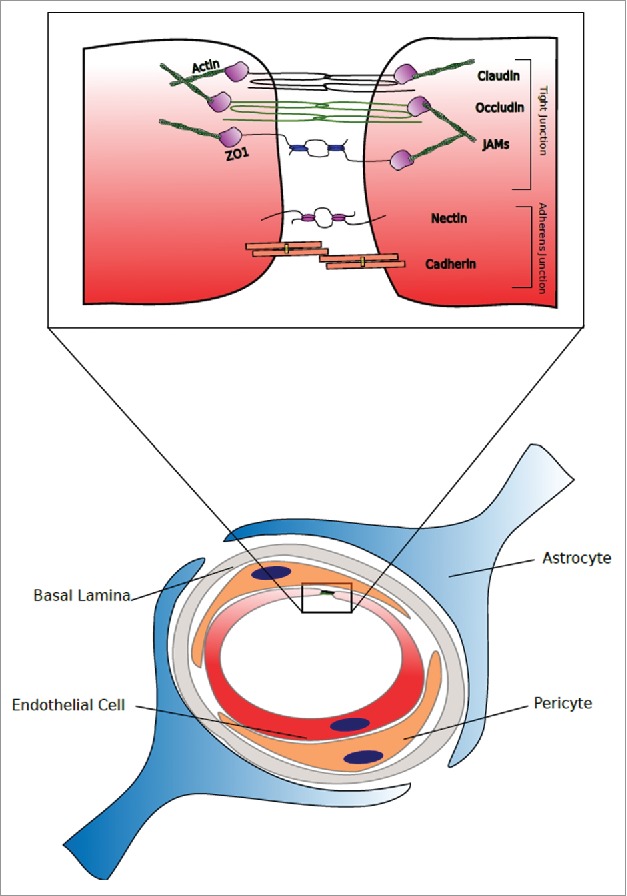
The Healthy Neurovascular Unit and Endothelial Tight junction. The normal/healthy tight junction is located at the apical periphery of two contacting endothelial cells or indeed at the point of contact of one endothelial encompassing the entire lumen of a vessel. The most enriched tight junction components in a normal/healthy brain endothelial cell tight junction are claudins-3, −5 and −12. Additionally, occludin and junctional adhesion molecule (JAM) are highly enriched at the blood brain barrier (BBB) compared to peripheral endothelial cells. Located below the tight junction is the adherens junction, comprising cadherins, catenins and nectin amongst others.
Pericytes
The arrival of pericytes at the developing endothelium correlates with TJ induction and reduced extravasation of tracer molecules,21 implicating this NVU component as one of the primary CNS inducers of the barrier. Blood vessels within the CNS have the highest pericyte to EC ratio in the body37 and this ratio has been shown to correlate with cerebral blood flow and barrier integrity.38 Pericytes preferentially make contact with cerebral capillaries at EC junctions while also positioning themselves at luminal faces of astrocytic end feet39; this advantageous localisation allows for the regulation of multiple aspects of the NVU such as capillary diameter, blood flow and end-feet guidance during barrier development.40
Endothelial platelet derived growth factor β (PDGFβ) has been shown to be induced by canonical Wnt/β-catenin signalling to drive pericyte recruitment through its receptor (PDGFRβ).41 Similarly, pericyte-derived glial-derived neurotrophic factor (GDNF) was found to be a potent stimulator of claudin-5 expression in both peripheral and cerebral ECs.42 With astrocytes arriving post-barriergenesis, the pericyte plays a critical role in establishing the BBB. Mice lacking either PDGFRβor its ligand die shortly after birth43 and through the use of hypomorphic alleles, research into the EC-pericyte dynamic has produced new insights into the role of pericytes within the NVU.42
Armulik et al. utilised the viable Pdgfβret/ret mouse expressing a variant of PDGFβ lacking its heparin sulphate proteoglycan binding ability and compared neurovascular changes against wild-type controls or Pdgfβ−/− mice with an EC-specific Pdgfβ knock-in construct (R26P).38 Initial findings showed pericyte coverage was reduced to 26% in the Pdgfβret/ret model, while the R26P+/− andR26P+/+ mice retained 40%and 72% respectively. Although TJ protein levels were unchanged, junction strand morphology in the Pdgfβret/ret and R26P+/− mice deviated from wild-type controls. The astrocyte-enriched water channel protein aquaporin 4 (AQP4) was also redistributed, potentially due to a loss in astrocyte end-feet guidance and polarisation. Immunohistochemical staining also revealed transcellular movement of albumin, Immunoglobulin-G (IgG) and injected 70 kDa dextran indicating an increased permeability in the absence of TJ expression differences. These injected tracers were observed to accumulated within the endothelium of Pdgfβret/ret and R26P+/− mice only, indicating transcellular transport across the BBB is affected under conditions of pericyte deficiency.
As will be discussed later in this review, pericyte dysfunction during ageing is as detrimental as in development, adding to the pro-inflammatory CNS environment and failing to maintain Aβ clearance as the brain ages. Understanding how these cells influence the development of the BBB may provide targets for restorative therapies to combat the effects of ageing.
Astrocytes – latecomers to the NVU and maintainers of the BBB
Astrocytes are glial cells within the CNS and project polarized end-feet to ensheath more than 90% of cerebral capillary surface area; these end-feet act to recycle neurotransmitters, provide nutrients and regulate local immune responses.44 Astrocytes are absent for nearly the duration of embryonic BBB development, appearing at E19 in the mouse and postnatally in the rat,21 however glial-like precursors may drive barriergenesis through Wnt and Sonic hedgehog (SHh) ligand secretion. The CNS specific mosaic of Wnt ligand expression may be a factor also in the unique endothelial and pericyte features seen in the NVU, driving elevated PDGFβ expression relative to peripheral endothelia and creating a GDNF-enriched environment for BBB induction and maintenance.
It is widely believed that the role of astrocytes within the NVU is the maintenance rather than establishment of the barrier; in adulthood, astrocytes secrete many of the ligands found within the developing CNS to preserve BBB characteristics. The Hedgehog (Hh) pathway involves ligand SHh binding to cell surface receptors such as Patched, de-repressing the G-coupled protein receptor smoothened (Smo) and activating downstream transcriptional changes through the Gli family of transcription factors. Within the CNS, Smo activity in ECs represses pro-inflammatory interleukin-8 (IL-8) and monocyte chemoattractant protein-1 (MCP1) production and upregulates TJ and AJ components. Treatment of EC monolayers with recombinant SHh or astrocyte-conditioned media induced downregulation of IL-8 and MCP-1; congruently an acute increase in BBB permeability and leukocyte transmigration is observed upon injection of mice with the Smo inhibitor cyclopamine.45 Developing mice with a conditional EC-restricted Smo deletion have reduced TJ expression, increased plasma protein leakage into the brain parenchyma and reduced astrocyte recruitment at later stages. SHh is exclusively sourced from astrocytes in the adult NVU, indicating their role in maintaining the signalling pathways established in the developing CNS. As well as their actions through the SHh pathway, astrocyte-secreted factors such as GDNF, apolipoprotein E and J (ApoE, ApoJ) and end-feet enriched transport proteins further regulate CNS ECs. These include AQP4, downregulated in paediatric CNS neoplasms with dysfunctional BBB46 and implicated in the clearance of soluble amyloid-β (Aβ)47; and excitatory amino acid transporters EAAT1 and EAAT2 which take up and recycle glutamate to prevent glutamate induced excitotoxic neuron death during stroke.48
The basal lamina & parenchymal basement membrane
The CNS vasculature has a dual set of extracellular basement membranes which are produced by the NVU and provide a cell scaffold, regulatory interface, and barrier against infiltrating cells.49,50
The basal lamina consists of endothelial and pericyte produced collagen IV, fibronectin, perlecan and lamins 4 and 5 which interact with integrins of infiltrating immune cells in the initial steps of leukocyte transmigration.50-52 The parenchymal basement membrane, which fuses with the basal lamina in cerebral microvessels,53 has a differing lamin profile and is enzymatically degraded via matrix-metalloproteases (MMPs) produced during leukocyte entry.54 The elasticity and thickness of these membranes changes over time, as does their susceptibility to degradation, which has detrimental effects in aging and in the response to CNS injury.55 The two-step process of crossing the BBB has been demonstrated in the experimental autoimmune encephalomyelitis (EAE) mouse model. Following clodronate treatment to deplete MMP-expressing macrophages, a transient accumulation of T-lymphocytes in the perivascular space could be induced where passage through the endothelium and EC basal lamina succeeds but fails upon reaching the parenchymal basement membrane.54 The central role played by MMPs in this process was further emphasised by obtaining the same results in untreated MMP2/MMP9 double knockout mice. This also has implications in age-associated neurodegenerative disease as the balance between MMPs and their inhibitors becomes upset in the aged CNS vascualture.55
The aging blood brain barrier
Aging is an intrinsic physiological process involving the decline of cell function leading ultimately to organ failure and organism death. Aberrant maintenance of cellular identity and replacement due to errors in DNA replication and repair,56 coupled with the reduced clearance of metabolism end-products, produces a stressed and vulnerable cellular environment.57,58 The mitochondrial volume in CNS ECs is twice that of non-BBB ECs, supplying the energy required for the complex BBB transport system.59 This has implications in the endothelium's susceptibility to aging-induced oxidative stress as increased reactive oxygen species (ROS) produced by the aging mitochondrial population affect the already oxygen-sensitive tissue. As well as the direct effects of aging on the NVU, BBB function in regulating the surrounding neural tissue also decreased with age. Stem cells are dependent on the surrounding microenvironment, or niche, for robust self-renewal and in the case of neural stem cells (NSCs) this involves a tight association with the NVU.5,60
The stem cell theory of aging proposes that a reduction in adult stem cell self-renewal and differentiation underlies the process of aging. As with all cells, adult stem cells fall victim to DNA damage and metabolic stress, reducing their ability to produce new differentiated cell populations. Hematopoietic stem cells isolated from aged or young individuals for example have reduced engraftment efficacy and skewed progenitor differentiation.61,62 The reduced rate of adult neurogenesis within the aging brain suggests a similar loss in stem cell function, activity and response to injury which may contribute to the onset of neurodegeneration.63,64 What is known so far about how the BBB changes with age comes from research into the age-associated pathologies seen in the brain. Although critical for understanding how neurodegenerative disease progresses, these data often are unable to distinguish between changes driving the disease and secondary changes brought about by the disease itself.
An environment primed for inflammation
In a healthy individual, deterioration of the BBB to a mild degree during aging, particularly in the hippocampus, is a physiological process that does not in itself induce a pathology.65 This mild dysfunction may predispose the aged brain to chronic inflammation or long-term damage in response to stress or injury, giving rise to age-associated neuropsychiatric and degenerative disease. Traumatic brain injury (TBI), for example, has a higher mortality rate in the elderly than in adult patients, indicating a robustness in youth which is lost with age.66 Berman et al. recently demonstrated the differential response of the BBB to controlled cortical impacts with age,55 finding that compared to adult controls, aged mice had a drastic increase in IgG extravasation post-impact. An imbalance between MMP9, MMP2 and their respective inhibitors TIMP1 and TIMP2 was identified as being causative in the aged mouse. Interestingly, the two MMPs underwent differing mechanisms of dysregulation; MMP9 expression was 13.4-fold higher in the injured aged brain independent of TIMP1, while MMP2 activity was elevated due to a 50% decrease in baseline TIMP2 expression exacerbating the injury response. Compounding the increase in TJ and basal lamina degrading enzyme activity, the injury-induced “recovery” increase in claudin-5 expression seen in the adult mouse was absent in the aged brain. Taken together this indicates a deficit in the aged BBB's response to injury, and although this was in the context of a cortical impact it hints towards an underlying loss of function that may lead to long-term barrier dysfunction.
In fact, the healthy aged BBB displays these degenerative pro-inflammatory characteristics in the absence of injury. Albumin at approximately 66.5 kDa is excluded by the intact BBB, however its leakage into the cerebrospinal fluid (CSF) increases during BBB dysfunction, allowing the CSF to serum albumin ratio to be utilised as a measure of barrier integrity. In the healthy aged brain, this ratio has been shown to robustly correlate with increasing age.67 This aged phenotype of the BBB was further expanded to include decreased endothelial occludin, vascular cell adhesion molecule-1 (VCAM1) and ZO-1 expression. Accompanying these changes were increases in the expression of pro-inflammatory glial fibrillary acidic protein (GFAP) in astrocytes, and upregulation of stress-sensitive heat shock chaperone protein GPR78 and cyclooxygenase-2 (COX2) in neurons.68 Elevated endothelial tumour necrosis factor alpha (TNFα) and peripheral IL-6 in the aged mice indicated that circulating signalling activators of the largely pro-inflammatory Nf-κB pathway may be the key underlying this age-related inflammation. Although accompanied by increased hippocampal and cortical IgG extravasation, there was a reported absence in the increased leukocyte infiltration characteristic of the majority of age-associated disorders.
An investigation into the cytokine response to TBI by Sandhir et al. revealed a similar neural environment primed for chronic inflammation in the aged mouse thalamus.69 Increased expression of interleukins IL-6, IL-1β, CCL5, TNFα, MCP1 and inducible nitric oxide synthase (iNOS) was recorded in the uninjured and injured aged brain relative to adult controls. Coupled with a reduction in interferon-γ, these data indicate an amplification of the “pro-inflammatory” Th1 response and corresponding reduction in “anti-inflammatory” Th2 response occurred post-TBI. A prolonged, 3.6-fold higher, TNFα upregulation following TBI was observed in the aged brain and the duration of cytokine increases were prolonged relative to the adult mouse.
Astrocyte regulation of the barrier has been shown to be disrupted by such cytokines, with IL-1β downregulating SHh expression and emphasising an already reduced responsivity to SHh stimulation due to aging.70 This increases astrocyte-derived cytokine and chemokine secretion, producing a leaky barrier pathology which facilitates immune cell infiltration in response to inflammation.71 These data suggest that in the aged CNS, responses to insult are more severe due to elevated baseline cytokine levels and sensitivity, with a corresponding increase in severity and long-term damage following injury (see Fig. 2 for aging BBB overview).
Figure 2.
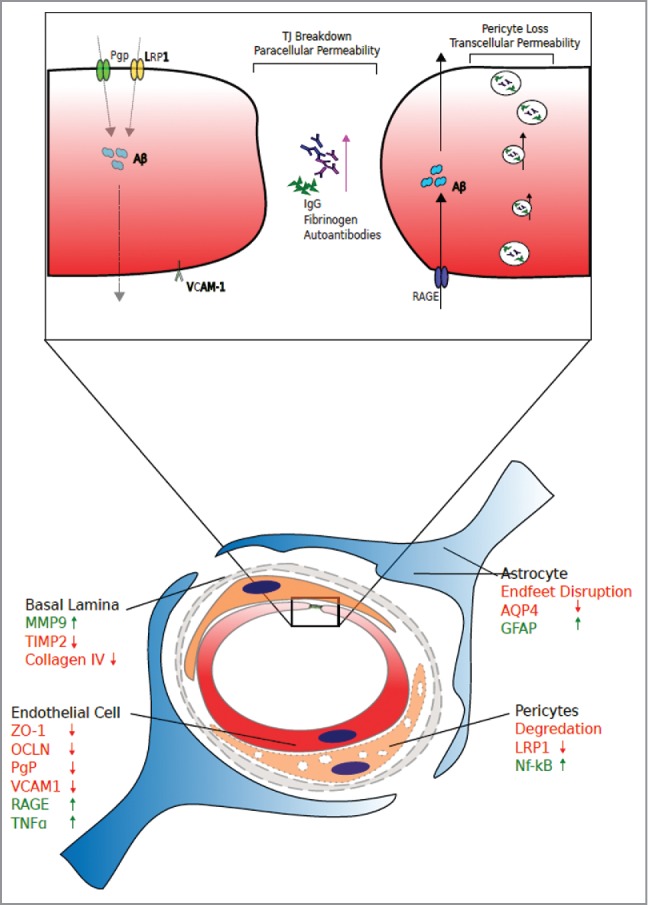
The Aged Neurovascular Unit and Aberrant Trans-Endothelial Transport. Tight junction breakdown is evident across a plethora of age associated neuropathies. Breakdown of the paracellular pathway can lead to extravasation of blood-borne material such as IgG, fibrinogen and autoantibodies into the delicate neural tissues. Additionally, pericyte loss and transcellular permeability can increase, with differential expression of basal lamina components matrix metalloproteinase-9 (MMP9), Tissue inhibitor of matrix metalloproteinase-2 (TIMP2) and collagen. Astrocyte components aquaporin-4 (AQP4) and glial fibrillary acidic protein (GFAP), pericyte components low density lipoprotein receptor-related protein-1 (LRP1) and pro-inflammatory nuclear factor kappa-B (Nf-kB). Endothelial molecules zonula-occludens-1 (ZO-1), occludin, P-glycoprotein (PgP), Vascular cell adhesion protein-1 (VCAM), receptor for advanced glycation end products (RAGE) and tumour necrosis factor-alpha (TNF-alpha).
An increasing reliance on ApoE and S100β signalling in the Aged CNS
An aspect of the CNS that makes it vulnerable to aging is its extensive cellular crosstalk. Failure of individual cell-cell interaction mechanisms is unlikely to induce the pathological effects of aging, however when the small effects of these changes are compounded upon by the effects of aging it may result in disease. This is seen in the case of both S100β and ApoE knockout mice, where age-related BBB deterioration is accelerated in the knockout compared to aged controls.
S100β is a calcium binding protein involved in cell differentiation72 and migration73 as well as the processes of learning and memory.74 A role for S100β has also been suggested in the pathologies of Azheimer's disease,75 schizophrenia76 as well as glutamate uptake and excitotoxicity.77 Using the S100β−/− mouse, Wu et al. identified an age-induced BBB phenotype which was accompanied by an increase in the passage of auto-antibodies into the brain.78 In the brains of 6, 9 but not 3-month old S100β−/− mice, extensive IgG leakage and IgG-neuron binding was observed, indicating that the compromised aged BBB facilitated the generation and entry of autoantibodies. Interestingly, the autoantibody binding profile differed between the 6 and 9 month old mice, suggesting that the antigen load escaping the CNS and driving antibody peripheral antibody production also changes with age. Accompanying the increase in barrier permeability were morphological changes in TJ folds, with knockout mice having discontinuous and flattened TJs. Maintained microglial activation was observed to precede TJ changes and the loss of barrier integrity, peaking as early as 3 months and potentially driving BBB breakdown via cytokine release. S100β binds the receptor for glycation end-products (RAGE) receptor on ECs, activating pro-inflammatory and migratory pathways through Nf-κB and Erk/Akt respectively that in the adult mouse may not be essential for CNS homeostasis.79,80 As the CNS becomes pro-inflammatory with age however, the deficit in NVU pro-inflammatory signalling may drive microglial activation instead leading to further barrier disruption.
As with S100β, ApoE also has an age-dependent impact on the BBB and TJ integrity, displaying isoform-specific effects correlating with Alzheimer's disease susceptibility alleles. In wild-type mice, there is a progressive increase in BBB permeability as the animal ages, however this is exacerbated by the loss of ApoE. In young (<40d) ApoE−/− mice, barrier integrity is identical to that of wild-type controls, yet when aged groups are compared there is a significant increase (3.7x) in the rate of barrier degradation in ApoE−/− animals.81 Bone-marrow chimeras revealed that both peripheral and CNS ApoE contribute to barrier maintenance over time, with ApoE−/− donor marrow inducing a phenotype intermediary of wild-type and knockout, and wild-type marrow only partially rescuing barrier preservation in knockouts. ApoE confers an allele-dependent susceptibility to Alzheimer's disease82 which is reflected in mouse models carrying knock-ins of the human alleles variants ApoE2, ApoE3 and ApoE4.83 Using primary ECs and pericytes co-cultured in astrocyte conditioned media, Nishitsuji et al. demonstrated that media produced from ApoE4, but not ApoE2 or ApoE3, astrocytes was unable to maintain TEERs above that of cells cultured in ApoE−/− media. At the molecular level, expression of TJ proteins was unchanged when ECs were grown in ApoE4 or ApoE−/− media. In the absence of TJ expression change there were reduced levels of phosphorylated protein kinase C-η (PKCη), a kinase which regulates TJs, and phosphorylated occludin. Replacing mouse ApoE with ApoE4 phenocopies the ApoE−/− mouse, lending support to the theory that the increased susceptibility for aged individuals carrying the ApoE4 allele is a product of aberrant BBB maintenance.
Pericytic low density lipoprotein receptor-related protein-1 (LRP1) binds ApoE and inhibits downstream cyclophillin A, preventing Nf-kB and subsequent MMP9 upregulation.84 On comparison between ApoE isoforms, ApoE4 was the sole isoform unable to induce this Nf-kB repression resulting in an increase in MMP9 and degradation of claudin-5, ZO-1, occludin, collagen IV. This targeting of ApoE to the pericyte further supports a vascular origin for age associated neurodegenerative disease and highlights the increasingly complex role of pericytes in BBB preservation. Similar to S100β, an unknown change in CNS homeostasis occurs in later stages of adulthood that triggers a dependency on functional ApoE signalling. Interestingly, LRP1 expression changes with age, discussed later in this section, which may result in a scarcity of functional ApoE-LRP1 signalling in the aged brain and further Nf-kB pathway activity.
Pericyte deterioration in the Aged CNS
As previously mentioned, pericytes are critical to the formation of the BBB in the developing brain and while astrocytes contribute greatly to the maintenance of the BBB, pericyte dysfunction has also been shown to reduce BBB integrity in an age dependent manner.85 As Pdgfrβ−/− mice are embryonic lethal, the role of pericytes in maintaining the BBB over age was investigated using heterozygous knockouts and the F7 line which express hypomorphic alleles in place of the wild-type gene. At 6 months both Pdgfrβ+/− and F7 mice display pericyte degeneration that co-localises with an apoptosing (TUNEL+) endothelium and stunted microvessel length. PDGFRβ staining negatively correlated with IgG extravasation and this correlation increased with age, indicating an increasing BBB dependency on pericyte function. Compared to wild-type controls, 8 month old F7 and Pdgfrβ+/− mice had a 17 and 10-fold increase in IgG extravasation respectively. At the same timepoint, vascular dysfunction possibly caused by the decreased TJ (claudin-5, ZO-1, occludin) and basement membrane (laminin, collagen IV) proteins resulted in an increase in thrombin, fibrinogen and plasmin passage into the brain. These invading components of the blood are neurotoxic at high levels and indeed neurodegeneration and cognitive deficits are seen in these pericyte-deficient mice. Neuronal death was observed in the hippocampal CA1 region of both pericyte-deficient mice was increasingly prevalent in the F7 mice, which have the lowest CNS vessel pericyte coverage. A vascular origin for this neurodegeneration was supported by the co-localisation of thrombin and TUNEL+ neurons, indicating that the BBB degeneration seen months prior to neurodegeneration allowed neurotoxic levels of peripheral macromolecules passage into the CNS. Interestingly, at 1 month both pericyte-deficient strains had novel object location and recognition scores comparatively to wild-type mice, indicating hippocampal function is preserved in early stages of BBB dysfunction. At 8 months however heterozygous and F7 mice scored much lower, with learning and memory function declining as neurodegeneration progresses.
These changes were followed by microglial activation at 16 months in PDGFRB+/− mice, with pro-inflammatory cytokine levels rising in the brain relative to aged controls. The stepwise degeneration of BBB maintenance and CNS damage may recapitulate the pathogenesis of age-associated neurodegenerative disease, with early stage dysfunction at the BBB leading to increased macromolecule entry and eventually neuron toxicity, cognitive decline and microglia driven neuroinflammation.
This deterioration shares many aspects with the developmental deficiencies observed in mouse models with low pericyte coverage and viability. As will be discussed later, pericyte deterioration also has a role in neural stem cell niche degeneration. These results highlight the importance of this cell type at the BB throughout life, it's primary induction role during development is preserved over time and such a critical role makes pericyte damage or death a strong factor in ageing and age-associated disease. Future work targeting the source of pericyte-deficiency induced phenotypes – developmental transcellular leakage, age-associated deterioration of the BBB and cerebral inflammation – may reveal central signalling pathways, conserved at all stages of life, for future therapeutic targeting.
A loss in transcellular clearance efficacy
Permeability increases at the BBB without paracellular TJ degeneration, and how it changes with age is currently underrepresented in the literature with a scarcity of publications reporting changing activity profiles with age. Transcellular transport across the BBB endothelium can occur via active, passive and receptor-mediated transport; the BBB endothelium expresses differing luminal and abluminal protein profiles to regulate the directionality of these transport proteins and their cargo. The ATP-binding cassette transporter Pgp, and Aβ-binding proteins RAGE and LRP1 have been documented to have changing expression and functionality with age. Pgp functionality can be measured using a 11C-labelled isotope of the Pgp substrate verapamil ([11C]verapamil) combined with positron emission tomography (PET) imaging.86 Earlier studies revealed increased retention of [11C]verapamil within elderly human subjects when compared to adult controls, and a follow-on study by van Assema et al. revealed that there were significant and sex-dependent changes in Pgp function during aging.87 Pgp functionality was significantly different in the youngest (20–30 yr) and eldest (55–70 yr) patient groups, but not between those and the middle group (40–50 yr). Interestingly, when the data was analysed by sex there were significant differences between all three of the male cohorts not observed in the female cohort.
With age being the greatest risk factor for Alzehimer's disease,88 an age-dependent alteration in the activities of Aβ transporting proteins may lead to the hallmark pathology of sterile Aβ plaque accumulation. Two studies utilising the Fischer 344/Brown Norway (F344/BN) rat hybrid model of aging have revealed age-dependent changes in Aβ transport proteins at the BBB as well as a potential interaction between these changes, Pgp and Aβ load. Silverberg et al. documented the changes in the soluble and insoluble Aβ isoforms, Aβ-40 and Aβ-42, in F344/BN rats from 3 to 30 months.89 Levels of Aβ-40 detected by ELISA rose gradually from 3 months, followed by a plateau from 12–20 month and a gradual decline; Aβ-42 rose at a slower pace, surpassing Aβ-40 at 12 months, however unlike Aβ-40 levels of the insoluble isoform kept rising until the 30-month cut off. In parallel to the increase in Aβ with age, expression of the Aβ influx transporter RAGE decreases from months 3–12, then undergoes a continuous increase until end of life. The timing of this increase corresponds with peak of Aβ-40 accumulation and the most rapid phase of Aβ-42 accumulation, with RAGE and Aβ-42 increasing in tandem until end of life.
A second study, using the same model, revealed decreased mRNA levels of Pgp and LRP-1, a pericytic Aβ uptake protein, past the 30-month mark.90 Interestingly, LRP1 transcript levels began to decrease by 12 months, corresponding with the previous changes seen in RAGE and Aβ accumulation.
These data create a picture of a dysfunctional Aβ clearance mechanism in the aged CNS. The dynamics of Pgp, RAGE and LRP1 expression fluctuations hint towards feedback loops as regulators of the transporters, providing a means to respond to the increased load of Aβ peptides with age. As seen in the above rat studies however, the balance of these loops may deteriorate with age, perhaps due to the cells within the NVU becoming overburdened with degradation-resistant Aβ1–42. As previously discussed, pericyte deficiency also increases the transcellular passage of macromolecules into the brain. It is possible that as the pericyte becomes overburdened with Aβ through LRP1 uptake, aberrant regulation of endothelial transcellular transport proteins results in a shift from Aβ efflux to influx.
The BBB in declining adult neurogenesis
Although limited, neurogenesis continues throughout adulthood to provide neurons in response to injury as well as to facilitate the neural growth involved in exercise, learning and memory.91 Neural stem cells (NSCs) undergo stepwise differentiation into migratory intermediates that travel from the two primary regions of the adult neurogenesis, the SVZ of the lateral ventricles and the DG of the hippocampus,1 to the olfactory bulb and hippocampus respectively where they further differentiate into functional neural subtypes. As with developmental neurogenesis, there is an intrinsic interplay between the CNS vasculature and adult NSC maintenance, development and migration. First observed in the higher vocal centre of adult songbirds, the adult NSC population resides within highly specialised vascular niches in the SVZ and SGZ where the components of the NVU regulate the activities of NSCs while the vasculature itself provides a migratory scaffold.92,93 This intimate relationship between the vasculature and the brain's ability to initiate neuron replacement provides an interface for peripheral factors to communicate with the NSC niche during disease and injury. The diverse NVU provides the complex mosaic of growth factors and signalling components to maintain the resident quiescent stem cell population, and inhibit over-proliferation and stem cell pool depletion. Pigment epithelium-derived factor (PEDF) secreted by both ECs and the ependymal cells of the ventricles acts to preserve NSC self-renewal through upregulating Notch-driven transcription,94 while EC-derived neurotrophin-3 represses NSC proliferation to preserve the multipotent reservoir through upregulation of the pro-quiescence factor endothelial nitric oxide synthase (eNOS) in the NSC niche.95 ECs also regulate NSCs through interactions between cell surface receptors EphrinB2, Jagged1 and their respective ligands Eph and Notch. Interactions between EphrinB2-Eph and Jagged1-Notch receptors requires cell-cell contact, inhibiting MAPK-driven cell cycle entry and cell differentiation respectively to regulate the adult NSC pool.5 In in vitro co-culture, EC-specific deletion of either ligand resulted in an acute increase in cell differentiation and division, followed by early depletion of NSC replicative potential. The requirement for this direct and intimate regulation demands an accommodating specialised vasculature within regions of adult neurogenesis while creating a sensitive system whereby BBB alterations can lead to niche disruption as in the case of the age-associated increase in EC-derived TGFβ1 driving NSC apoptosis.96 As the CNS ages and barrier function within the NVU homeostasis decreases, so does the rate of adult neurogenesis potentially contributing to the cognitive decline and neurodegenerative diseases associated with aging.63,64 Surprisingly, NSC maintenance appears to utilise TJ proteins expressed by the stem cells themselves.97 Both within the brain and in ex vivo culture NSCs grow in clusters, termed neurospheres in cultures, which express a range of TJ proteins until the onset of differentiation. Knock down of ZO-1 alone was sufficient to initiate NSC differentiation within these neurospheres, revealing a novel function of TJ components in regulating stem cell identity, likely though preserving cell-cell contacts within the 3D structures.
The SVZ lies beneath the ependyma of the lateral ventricles and above a specialised dense vascular network running parallel to the ventricle wall. This co-localisation allows for NSC sampling of both the CSF and the blood via protruding cell processes through the ependymal and endothelial cell monolayers.98 The SVZ niche itself deteriorates with age as blood vessel density and flow reduces, however the replicative potential of aged NSCs equals that of controls when isolated in vitro, pointing towards niche maintenance rather than stem cell dysfunction as the cause of reduced neurogenesis.99 The broad range of factors required for niche function creates a large target for age-associated deterioration to affect. Interestingly, the BBB of SVZ ECs have localised foci of increased permeability and reduced astrocyte and pericyte coverage,93 this may provide NSCs with increased peripheral cues via paracellular diffusion or create gaps for NSC extensions to sample the contents of the vasculature. It's possible that this steady state dysfunction of the BBB in the SVZ could lead to localised neurodegeneration following the increased susceptibility of the brain towards peripheral pro-inflammatory factors. In fact, although the SGZ lacks the leaky and exposed BBB observed in the SVZ niche it has been reported to degenerate with age due to BBB degeneration.65 Looking at cohorts of aged and adult individuals with mild cognitive impairment (MCI), BBB breakdown in two regions of the hippocampus (the dentate gyrus and region CA1) within the MCI cohort surpassed aged controls with normal cognition, while the adult cohort had the most intact hippocampal BBB. These results correlated with increased CSF levels of albumin and soluble PDGFRβ (sPDGFRβ) but not a rise in MMPs, cytokines or markers of endothelial cell death. In mouse models of pericyte cell death (Pdgfrβ+/−) as well as Alzheimer's (Tg2576), sPDGFRβ is increased compared to controls in the range of 200-fold and 500-fold respectively, giving rise to the hypothesis that pericyte cell death had occurred in the MCI cohort and may have thus affected hippocampal function and neurogenesis.
As is becoming increasingly evident, pericytes are susceptible to age-dependent deterioration at the BBB despite the initiating factor of pericyte cell death being unknown. Due to their location, these cells are likely to come into contact with and phagocytose infiltrating cytotoxic factors from the lumen, as these increase with age it may stress the lysosomal machinery of the cell and lead to cell death.
Conclusions
The complex and infinitesimal combinations of injuries, diseases and cerebral changes which occur throughout life results in a heterogeneous aged population presenting common disorders such as Alzheimer's, schizophrenia and dementia. Although the underlying causes for each of these disorders are likely to similarly drive pathogenesis, how the CNS becomes primed for disease onset is likely to be individual in nature with neurodegeneration being the product of many compounding effects of aging and underlying genetic predisposition. Different strategies to combat the effects of aging on the BBB and CNS include broad approaches such as the application of “young” rejuvenating circulatory factors, for example BMP11,100 as well as targeting specific key molecules involved in the processes discussed in this review. The BBB and NVU's involvement in age-associated disease as well as physiological CNS deterioration make the CNS vasculature ideal targets for drug action to preserve barrier functionality, robust injury response, stem cell maintenance and to combat pathologic cellular changes such as pericyte degeneration.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The Campbell laboratory in the Smurfit Institute of Genetics is funded by grants from Science Foundation Ireland (SFI) (grant no. 200667) and the Bright Focus Foundation (grant no. 204164).
References
- [1].Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319-35. doi: 10.1002/cne.901240303. PMID:5861717 [DOI] [PubMed] [Google Scholar]
- [2].Allen E. The cessation of mitosis in the central nervous system of the albino rat. J Comp Neurol. 1912;22:547-68. [Google Scholar]
- [3].Lin R, Cai J, Nathan C, Wei X, Schleidt S, Rosenwasser R, Iacovitti L. Neurogenesis is enhanced by stroke in multiple new stem cell niches along the ventricular system at sites of high BBB permeability. Neurobiol Dis. 2015;74:229-39. doi: 10.1016/j.nbd.2014.11.016. PMID:25484283 [DOI] [PubMed] [Google Scholar]
- [4].Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci. 1999;2:260-5. doi: 10.1038/6365. PMID:10195219 [DOI] [PubMed] [Google Scholar]
- [5].Ottone C, Parrinello S. Multifaceted control of adult SVZ neurogenesis by the vascular niche. Cell Cycle. 2015;14:2222-5. doi: 10.1080/15384101.2015.1049785. PMID:26115376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gómez-Gaviro MV, Lovell-Badge R, Fernández-Avilés F, Lara-Pezzi E. The vascular stem cell niche. J Cardiovasc Transl Res. 2012;5:618-30. doi: 10.1007/s12265-012-9371-x. PMID:22644724 [DOI] [PubMed] [Google Scholar]
- [7].Kim KJ, Filosa JA. Advanced in vitro approach to study neurovascular coupling mechanisms in the brain microcirculation. J Physiol. 2012;590:1757-70. doi: 10.1113/jphysiol.2011.222778. PMID:22310311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5–deficient mice. J Cell Biol. 2003;161:653-60. doi: 10.1083/jcb.200302070. PMID:12743111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207-17. doi: 10.1083/jcb.34.1.207. PMID:6033532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hoshi Y, Uchida Y, Tachikawa M, Inoue T, Ohtsuki S, Terasaki T. Quantitative atlas of blood-brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J Pharm Sci. 2013;102:3343-55. doi: 10.1002/jps.23575. PMID:23650139 [DOI] [PubMed] [Google Scholar]
- [11].Liu H, Li Y, Lu S, Wu Y, Sahi J. Temporal expression of transporters and receptors in a rat primary co-culture blood-brain barrier model. Xenobiotica. 2014;44:941-51. doi: 10.3109/00498254.2014.919430. PMID:24827375 [DOI] [PubMed] [Google Scholar]
- [12].Maher F, Vannucci SJ, Simpson IA. Glucose transporter proteins in brain. Faseb J. 1994;8:1003-11. PMID:7926364 [DOI] [PubMed] [Google Scholar]
- [13].Terasaki T, Hosoya KI. The blood-brain barrier efflux transporters as a detoxifying system for the brain. Adv. Drug Deliv. Rev. 1999;36:195-209. doi: 10.1016/S0169-409X(98)00088-X. PMID:10837716 [DOI] [PubMed] [Google Scholar]
- [14].Kwon EE, Prineas JW. Blood-brain barrier abnormalities in longstanding multiple sclerosis lesions. An immunohistochemical study. J Neuropathol Exp Neurol. 1994;53:625-36. doi: 10.1097/00005072-199411000-00010 [DOI] [PubMed] [Google Scholar]
- [15].Zenaro E, Piacentino G, Constantin G. The blood-brain barrier in Alzheimer's disease. Neurobiol Dis. 2017;107:41-56. doi: 10.1016/j.nbd.2016.07.007. PMID:27425887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun Z-Y, Wei J, Xie L, Shen Y, Liu S-Z, Ju G-Z, Shi J-P, Yu Y-Q, Zhang X, Xu Q, et al.. The CLDN5 locus may be involved in the vulnerability to schizophrenia. Eur Psychiatry. 2004;19:354-7. doi: 10.1016/j.eurpsy.2004.06.007. PMID:15363474 [DOI] [PubMed] [Google Scholar]
- [17].Starr JM, Wardlaw J, Ferguson K, MacLullich A, Deary IJ, Marshall I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2003;74:70-6. doi: 10.1136/jnnp.74.1.70. PMID:12486269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Morgan L, Shah B, Rivers LE, Barden L, Groom AJ, Chung R, Higazi D, Desmond H, Smith T, Staddon JM. Inflammation and dephosphorylation of the tight junction protein occludin in an experimental model of multiple sclerosis. Neuroscience. 2007;147:664-73. doi: 10.1016/j.neuroscience.2007.04.051. PMID:17560040 [DOI] [PubMed] [Google Scholar]
- [19].Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48-65. doi: 10.1111/j.1600-065X.2006.00441.x. PMID:16972896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: A study using quail-chick transplantation chimeras. Dev Biol. 1981;84:183-92. doi: 10.1016/0012-1606(81)90382-1. PMID:7250491 [DOI] [PubMed] [Google Scholar]
- [21].Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562-6. doi: 10.1038/nature09513. PMID:20944625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Risau W, Wolburg H. Development of the blood-brain barrier. Trends Neurosci. 1990;13:174-8. doi: 10.1016/0166-2236(90)90043-A. PMID:1693235 [DOI] [PubMed] [Google Scholar]
- [23].Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539-50. doi: 10.1083/jcb.141.7.1539. PMID:9647647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Occludin: A novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777-88. doi: 10.1083/jcb.123.6.1777. PMID:8276896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, et al.. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117-27. doi: 10.1083/jcb.142.1.117. PMID:9660867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M, Tsukita S. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006;126:741-54. doi: 10.1016/j.cell.2006.06.043. PMID:16923393 [DOI] [PubMed] [Google Scholar]
- [27].Etournay R, Zwaenepoel I, Perfettini I, Legrain P, Petit C, El-Amraoui A. Shroom2, a myosin-VIIa- and actin-binding protein, directly interacts with ZO-1 at tight junctions. J Cell Sci. 2007;120:2838-50. doi: 10.1242/jcs.002568. PMID:17666436 [DOI] [PubMed] [Google Scholar]
- [28].Katsube T, Takahisa M, Ueda R, Hashimoto N, Kobayashi M, Togashi S. Cortactin associates with the cell-cell junction protein ZO-1 in both Drosophila and mouse. J Biol Chem. 1998;273:29672-7. doi: 10.1074/jbc.273.45.29672. PMID:9792678 [DOI] [PubMed] [Google Scholar]
- [29].Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood-brain barrier transcriptome: A new resource for understanding the development and function of brain endothelial cells. PLoS One. 2010;5:1-16. doi: 10.1371/journal.pone.0013741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ohtsuki S, Yamaguchi H, Katsukura Y, Asashima T, Terasaki T. mRNA expression levels of tight junction protein genes in mouse brain capillary endothelial cells highly purified by magnetic cell sorting. J Neurochem. 2008;104:147-54. PMID:17971126 [DOI] [PubMed] [Google Scholar]
- [31].Krause G, Winkler L, Piehl C, Blasig I, Piontek J, Müller SL. Structure and function of extracellular claudin domains. Ann N Y Acad Sci. 2009;1165:34-43. doi: 10.1111/j.1749-6632.2009.04057.x. PMID:19538285 [DOI] [PubMed] [Google Scholar]
- [32].Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11:4131-42. doi: 10.1091/mbc.11.12.4131. PMID:11102513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cording J, Berg J, Kading N, Bellmann C, Tscheik C, Westphal JK, Milatz S, Gunzel D, Wolburg H, Piontek J, et al.. In tight junctions, claudins regulate the interactions between occludin, tricellulin and marvelD3, which, inversely, modulate claudin oligomerization. J Cell Sci. 2013;126:554-64. doi: 10.1242/jcs.114306. PMID:23203797 [DOI] [PubMed] [Google Scholar]
- [34].Ostermann G, Weber KSC, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. 2002;3:151-8. doi: 10.1038/ni755. PMID:11812992 [DOI] [PubMed] [Google Scholar]
- [35].Ebnet K, Aurrand-Lions M, Kuhn A, Kiefer F, Butz S, Zander K, Meyer zu Brickwedde M-K, Suzuki A, Imhof BA, Vestweber D. The junctional adhesion molecule (JAM) family members JAM-2 and JAM-3 associate with the cell polarity protein PAR-3: a possible role for JAMs in endothelial cell polarity. J Cell Sci. 2003;116:3879-91. doi: 10.1242/jcs.00704. PMID:12953056 [DOI] [PubMed] [Google Scholar]
- [36].Vorbrodt AW, Dobrogowska DH. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist's view. Brain Res Brain Res Rev. 2003;42:221-42. doi: 10.1016/S0165-0173(03)00177-2. PMID:12791441 [DOI] [PubMed] [Google Scholar]
- [37].Shepro D, Morel NM. Pericyte physiology. FASEB J. 1993;7:1031-8. PMID:8370472 [DOI] [PubMed] [Google Scholar]
- [38].Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al.. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557-61. doi: 10.1038/nature09522. PMID:20944627 [DOI] [PubMed] [Google Scholar]
- [39].Cuevas P, Gutierrez-Diaz JA, Reimers D, Dujovny M, Diaz FG, Ausman JI. Pericyte endothelial gap junctions in human cerebral capillaries. Anat Embryol (Berl). 1984;170:155-9. doi: 10.1007/BF00319000. PMID:6517350 [DOI] [PubMed] [Google Scholar]
- [40].Diaz-Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E, Martín-Vasallo P, Díaz-Flores J Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol. Histopathol. 2009;24:909-69. [DOI] [PubMed] [Google Scholar]
- [41].Reis M, Czupalla CJ, Ziegler N, Devraj K, Zinke J, Seidel S, Heck R, Thom S, Macas J, Bockamp E, et al.. Endothelial Wnt/β-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J Exp Med. 2012;209:1611-27. doi: 10.1084/jem.20111580. PMID:22908324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shimizu F, Sano Y, Saito K, Abe MA, Maeda T, Haruki H, Kanda T. Pericyte-derived glial cell line-derived neurotrophic factor increase the expression of claudin-5 in the blood-brain barrier and the blood-nerve barrier. Neurochem Res. 2012;37:401-9. doi: 10.1007/s11064-011-0626-8. PMID:22002662 [DOI] [PubMed] [Google Scholar]
- [43].Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242-5. doi: 10.1126/science.277.5323.242. PMID:9211853 [DOI] [PubMed] [Google Scholar]
- [44].Neuhaus J. Orthogonal arrays of particles in astroglial cells: quantitative analysis of their density, size, and correlation with intramembranous particles. Glia. 1990;3:241-51. doi: 10.1002/glia.440030403. PMID:2144504 [DOI] [PubMed] [Google Scholar]
- [45].Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonniere L, Bernard M, et al.. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727-31. doi: 10.1126/science.1206936. PMID:22144466 [DOI] [PubMed] [Google Scholar]
- [46].Hong CS, Ho W, Piazza MG, Ray-Chaudhury A, Zhuang Z, Heiss JD. Characterization of the blood brain barrier in pediatric central nervous system neoplasms. J Interdiscip Histopathol. 2016;21:4062-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, et al.. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. PMID:22896675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yi JH, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006;48:394-403. doi: 10.1016/j.neuint.2005.12.001. PMID:16473439 [DOI] [PubMed] [Google Scholar]
- [49].Tilling T, Korte D, Hoheisel D, Galla H-J. Basement Membrane Proteins Influence Brain Capillary Endothelial Barrier Function In Vitro. J Neurochem. 1998;71:1151-7. [DOI] [PubMed] [Google Scholar]
- [50].Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol. 2001;153:933-45. doi: 10.1083/jcb.153.5.933. PMID:11381080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, Robenek H, Tryggvason K, Song J, Korpos E, et al.. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. 2009;15:519-27. doi: 10.1038/nm.1957. PMID:19396173 [DOI] [PubMed] [Google Scholar]
- [52].Jucker M, Tian M, Norton DD, Sherman C, Kusiak JW. Laminin alpha 2 is a component of brain capillary basement membrane: reduced expression in dystrophic dy mice. Neuroscience. 1996;71:1153-61. doi: 10.1016/0306-4522(95)00496-3. PMID:8684619 [DOI] [PubMed] [Google Scholar]
- [53].Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol. 2007;28:5-11. doi: 10.1016/j.it.2006.11.007. PMID:17140851 [DOI] [PubMed] [Google Scholar]
- [54].Agrawal S, Anderson P, Durbeej M, Van Rooijen N, Ivars F, Opdenakker G, Sorokin LM. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. 2006;203:1007-19. doi: 10.1084/jem.20051342. PMID:16585265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Berman NEJ, Lee P, Kim J, Williams R, Sandhir R, Gregory E, Brooks WM. Effects of aging on blood brain barrier and matrix metalloproteases following controlled cortical impact in mice. Exp Neurol. 2012;234:50-61. doi: 10.1016/j.expneurol.2011.12.016. PMID:22201549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Vijg J. Somatic mutations and aging: a re-evaluation. Mutat Res. 2000;447:117-35. doi: 10.1016/S0027-5107(99)00202-X. PMID:10686308 [DOI] [PubMed] [Google Scholar]
- [57].Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci. 1994;91:10771-8. doi: 10.1073/pnas.91.23.10771. PMID:7971961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Semba RD, Nicklett EJ, Ferrucci L. Does Accumulation of Advanced Glycation End Products Contribute to the Aging Phenotype? J Gerontol Ser A. 2010;65A:963-75. doi: 10.1093/gerona/glq074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1:409-17. doi: 10.1002/ana.410010502. PMID:617259 [DOI] [PubMed] [Google Scholar]
- [60].Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramva N, Vincent P, Pumiglia K, Temple S. Endothelial Cells Stimulate Self-Renewal and Expand Neurogenesis of Neural Stem Cells. Science (80-). 2004;304:1338-40. doi: 10.1126/science.1095505 [DOI] [PubMed] [Google Scholar]
- [61].Pang WW, Price E A, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, Weissman IL. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci. 2011;108:20012-7. doi: 10.1073/pnas.1116110108. PMID:22123971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996;2:1011-6. doi: 10.1038/nm0996-1011. PMID:8782459 [DOI] [PubMed] [Google Scholar]
- [63].Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027-33. PMID:8604047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Daynac M, Morizur L, Chicheportiche A, Mouthon M-A, Boussin FD. Age-related neurogenesis decline in the subventricular zone is associated with specific cell cycle regulation changes in activated neural stem cells. Sci Rep. 2016;6:21505. doi: 10.1038/srep21505. PMID:26893147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, et al.. Blood-Brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296-302. doi: 10.1016/j.neuron.2014.12.032. PMID:25611508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Susman M, DiRusso SM, Sullivan T, Risucci D, Nealon P, Cuff S, Haider A, Benzil D. Traumatic brain injury in the elderly: increased mortality and worse functional outcome at discharge despite lower injury severity. J Trauma. 2002;53:219-23-4. doi: 10.1097/00005373-200208000-00004 [DOI] [PubMed] [Google Scholar]
- [67].Chen CPC, Chen RL, Preston JE. The influence of ageing in the cerebrospinal fluid concentrations of proteins that are derived from the choroid plexus, brain, and plasma. Exp Gerontol. 2012;47:323-8. doi: 10.1016/j.exger.2012.01.008. PMID:22532968 [DOI] [PubMed] [Google Scholar]
- [68].Elahy M, Jackaman C, Mamo JC, Lam V, Dhaliwal SS, Giles C, Nelson D, Takechi R. Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun Ageing. 2015;12:2. doi: 10.1186/s12979-015-0029-9. PMID:25784952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sandhir R, Puri V, Klein RM, Berman NEJ. Differential expression of cytokines and chemokines during secondary neuron death following brain injury in old and young mice. Neurosci Lett. 2004;369:28-32. doi: 10.1016/j.neulet.2004.07.032. PMID:15380302 [DOI] [PubMed] [Google Scholar]
- [70].Heimann G, Canhos LL, Frik J, Jäger G, Lepko T, Ninkovic J, Götz M, Sirko S. Changes in the Proliferative Program Limit Astrocyte Homeostasis in the Aged Post-Traumatic Murine Cerebral Cortex. Cereb Cortex. 2017;27:4213-28. doi: 10.1093/cercor/bhx112. PMID:28472290 [DOI] [PubMed] [Google Scholar]
- [71].Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B, Kawanokuchi J, Mizuno T, Takeuchi H, Suzumura A. Interleukin-1B Induces induces blood-brain barrier disruption by downregulating sonic hedgehog in astrocytes. PLoS One. 2014;9:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Raponi E, Agenes F, Delphin C, Assard N, Baudier J, Legraverend C, Deloulme J-C. S100B expression defines a state in which GFAP-expressing cells lose their neural stem cell potential and acquire a more mature developmental stage. Glia. 2007;55:165-77. doi: 10.1002/glia.20445. PMID:17078026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Brozzi F, Arcuri C, Giambanco I, Donato R. S100B Protein Regulates Astrocyte Shape and Migration via Interaction with Src Kinase: Implications for astrocyte development, activation, and tumor growth. J Biol Chem. 2009;284:8797-811. doi: 10.1074/jbc.M805897200. PMID:19147496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gerlai R, Wojtowicz JM, Marks a, Roder J. Overexpression of a calcium-binding protein, S100 beta, in astrocytes alters synaptic plasticity and impairs spatial learning in transgenic mice. Learn Mem. 1995;2:26-39. doi: 10.1101/lm.2.1.26. PMID:10467564 [DOI] [PubMed] [Google Scholar]
- [75].Mrak RE, Griffinb WST. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer's disease. Neurobiol Aging. 2001;22:915-22. doi: 10.1016/S0197-4580(01)00293-7. PMID:11754999 [DOI] [PubMed] [Google Scholar]
- [76].Pedersen A, Diedrich M, Kaestner F, Koelkebeck K, Ohrmann P, Ponath G, Kipp F, Abel S, Siegmund A, Suslow T. Memory impairment correlates with increased S100B serum concentrations in patients with chronic schizophrenia. Prog Neuro-Psychopharmacology Biol Psychiatry. 2008;32:1789-92. doi: 10.1016/j.pnpbp.2008.07.017 [DOI] [PubMed] [Google Scholar]
- [77].Tramontina F, Tramontina AC, Souza DF, Leite MC, Gottfried C, Souza DO, Wofchuk ST, Gonçalves CA. Glutamate uptake is stimulated by extracellular S100B in hippocampal astrocytes. Cell Mol Neurobiol. 2006;26:81-6. PMID:16633903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wu H, Brown EV, Acharya NK, Appelt DM, Marks A, Nagele RG, Venkataraman V. Age-dependent increase of blood-brain barrier permeability and neuron-binding autoantibodies in S100B knockout mice. Brain Res. 2016;1637:154-67. [DOI] [PubMed] [Google Scholar]
- [79].Valencia J V, Mone M, Zhang J, Weetall M, Buxton FP, Hughes TE. Divergent Pathways of Gene Expression Are Activated by the RAGE Ligands S100b and AGE-BSA. Diabetes. 2004;53:743 LP-751. doi: 10.2337/diabetes.53.3.743 [DOI] [PubMed] [Google Scholar]
- [80].Villarreal A, Seoane R, Torres AG, Rosciszewski G, Angelo MF, Rossi A, Barkert PA, Ramos AJ. S100B protein activates a RAGE-dependent autocrine loop in astrocytes: Implications for its role in the propagation of reactive gliosis. J Neurochem. 2014;131(2):190-205. doi: 10.1111/jnc.12790. PMID:24923428 [DOI] [PubMed] [Google Scholar]
- [81].Hafezi-Moghadam A, Thomas KL, Wagner DD. ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am J Physiol Cell Physiol. 2007;292:C1256-62. doi: 10.1152/ajpcell.00563.2005. PMID:16870825 [DOI] [PubMed] [Google Scholar]
- [82].Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small Gw al, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science (80-). 1993;261:921-3. doi: 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- [83].Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem. 2011;286:17536-42. doi: 10.1074/jbc.M111.225532. PMID:21471207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, et al.. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512-6. PMID:22622580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic B V. Pericytes Control Key Neurovascular Functions and Neuronal Phenotype in the Adult Brain and during Brain Aging. Neuron. 2010;68:409-27. doi: 10.1016/j.neuron.2010.09.043. PMID:21040844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Bauer M, Karch R, Neumann F, Abrahim A, Wagner CC, Kletter K, Müller M, Zeitlinger M, Langer O. Age dependency of cerebral P-gp function measured with (R)-[11C]verapamil and PET. Eur J Clin Pharmacol. 2009;65:941-6. doi: 10.1007/s00228-009-0709-5. PMID:19655132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Van Assema DME Lubberink M, Boellaard R, Schuit RC, Windhorst AD, Scheltens P, Lammertsma AA, Van Berckel BNM. P-glycoprotein function at the blood-brain barrier: Effects of age and gender. Mol Imaging Biol. 2012;14:771-6. doi: 10.1007/s11307-012-0556-0. PMID:22476967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013;80:1778-83. doi: 10.1212/WNL.0b013e31828726f5. PMID:23390181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Silverberg GD, Miller MC, Messier AA, Majmudar S, Machan JT, Donahue JE, Stopa EG, Johanson CE. Amyloid deposition and influx transporter expression at the blood-brain barrier increase in normal aging. J Neuropathol Exp Neurol. 2010;69:98-108. doi: 10.1097/NEN.0b013e3181c8ad2f. PMID:20010299 [DOI] [PubMed] [Google Scholar]
- [90].Osgood D, Miller MC, Messier AA, Gonzalez L, Silverberg GD. Aging alters mRNA expression of amyloid transporter genes at the blood-brain barrier. Neurobiol Aging. 2017;57:178-85. doi: 10.1016/j.neurobiolaging.2017.05.011. PMID:28654861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lugert S, Basak O, Knuckles P, Haussler U, Fabel K, Götz M, Haas CA, Kempermann G, Taylor V, Giachino C. Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell. 2010;6:445-56. doi: 10.1016/j.stem.2010.03.017. PMID:20452319 [DOI] [PubMed] [Google Scholar]
- [92].Louissaint A, Rao S, Leventhal C, Goldman SA. Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron. 2002;34:945-60. doi: 10.1016/S0896-6273(02)00722-5. PMID:12086642 [DOI] [PubMed] [Google Scholar]
- [93].Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, Garcia-Verdugo JM, Doetsch F. A Specialized Vascular Niche for Adult Neural Stem Cells. Cell Stem Cell. 2008;3:279-88. doi: 10.1016/j.stem.2008.07.025. PMID:18786415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Andreu-Agullo C, Morante-Redolat JM, Delgado AC, Farinas I. Vascular niche factor PEDF modulates Notch-dependent stemness in the adult subependymal zone. Nat Neurosci. 2009;12:1514-23. doi: 10.1038/nn.2437. PMID:19898467 [DOI] [PubMed] [Google Scholar]
- [95].Delgado AC, Ferron SR, Vicente D, Porlan E, Perez-Villalba A, Trujillo CM, D'Ocon P, Farinas I. Endothelial NT-3 delivered by vasculature and CSF promotes quiescence of subependymal neural stem cells through nitric oxide induction. Neuron. 2014;83:572-85. doi: 10.1016/j.neuron.2014.06.015. PMID:25043422 [DOI] [PubMed] [Google Scholar]
- [96].Pineda JR, Daynac M, Chicheportiche A, Cebrian-Silla A, Sii Felice K, Garcia-Verdugo JM, Boussin FD, Mouthon M-A. Vascular-derived TGF-β increases in the stem cell niche and perturbs neurogenesis during aging and following irradiation in the adult mouse brain. EMBO Mol Med. 2013;5:548-62. doi: 10.1002/emmm.201202197. PMID:23526803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Watters AK, Rom S, Hill JD, Dematatis MK, Zhou Y, Merkel SF, Andrews AM, Cena J, Potula R, Skuba A, et al.. Identification and Dynamic Regulation of Tight Junction Protein Expression in Human Neural Stem Cells. Stem Cells Dev. 2015;24:1377-89. doi: 10.1089/scd.2014.0497. PMID:25892136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, Goderie SK, Roysam B, Temple S. Adult SVZ Stem Cells Lie in a Vascular Niche: A Quantitative Analysis of Niche Cell-Cell Interactions. Cell Stem Cell. 2008;3:289-300.doi: 10.1016/j.stem.2008.07.026. PMID:18786416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Ahlenius H, Visan V, Kokaia M, Lindvall O, Kokaia Z. Neural Stem and Progenitor Cells Retain Their Potential for Proliferation and Differentiation into Functional Neurons Despite Lower Number in Aged Brain. J Neurosci. 2009;29:4408 LP-4419. doi: 10.1523/JNEUROSCI.6003-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, Chen JW, Lee RT, Wagers AJ, Rubin LL. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014;344:630-4. doi: 10.1126/science.1251141. PMID:24797482 [DOI] [PMC free article] [PubMed] [Google Scholar]