

ABSTRACT
The imprinting of the mouse Peg3 domain is controlled through the Peg3-DMR, which obtains its maternal-specific DNA methylation during oogenesis. In the current study, we deleted an oocyte-specific alternative promoter, termed U1, which is localized 20 kb upstream of the Peg3-DMR. Deletion of this alternative promoter resulted in complete removal of the maternal-specific DNA methylation on the Peg3-DMR. Consequently, the imprinted genes in the Peg3 domain become biallelic in the mutants with maternal transmission of the deletion. Expression levels of the imprinted genes were also affected in the mutants: 2-fold upregulation of Peg3 and Usp29 and downregulation of Zim1 to basal levels. Breeding experiments further indicated under-representation of females among the surviving mutants, a potential sex-biased outcome from the biallelic expression of the Peg3 domain. Overall, the results suggest that U1-driven transcription may be required for establishing oocyte-specific DNA methylation on the Peg3 domain.
KEYWORDS: Peg3, DNA methylation, genomic imprinting, imprinting control region
Abbreviations
- APeg3
Antisense Peg3 gene
- CRISPR
Clustered regularly interspaced short palindromic repeats
- DMR
Differentially methylated region
- DNMT3A
DNA (cytosine-5)-methyltransferase 3A
- Gnas
Guanine nucleotide-binding protein G(s) subunit alpha
- ICR
Imprinting control region
- Peg3
Paternally expressed gene 3
- Sry
Sex-determining region Y protein
- Snrpn
Small nuclear ribonucleoprotein-associated protein N
- Usp29
Ubiquitin-specific protease 29
- YY1
Ying Yang 1
- Zim1
Zinc finger gene 1 imprinted
- Zim2
Zinc finger gene 2 imprinted
- Zim3
Zinc finger gene 3 imprinted
- Zfp264
Zinc finger protein 264
- Zac1
PLAG1 like Zinc finger 1
Introduction
Paternally expressed gene 3 (Peg3) is the founding member of the mammalian Peg3 imprinted domain, which covers an evolutionarily well-conserved 500-kb genomic region in human chromosome 19q13.4/proximal mouse chromosome 7.1 This domain contains paternally expressed Peg3, Usp29, APeg3, Zfp264, and maternally expressed Zim1, Zim2, Zim3.2 The Peg3 domain is controlled through an imprinting control region (ICR) termed the Peg3-differentially methylated region (DMR).3 The Peg3-DMR covers a 4-kb genomic region that harbors a bidirectional promoter for Peg3 and Usp29 and also the 1st intron region of Peg3 with a tandem array of 7 YY1 binding sites.4 According to recent results, deletion of this ICR resulted in global changes in the transcriptional levels and monoallelic expression patterns of the entire domain, confirming that this region is indeed a major controlling factor for the Peg3 domain.5 The Peg3-DMR obtains DNA methylation as a gametic signal during oogenesis, and maintains its maternal-specific methylation pattern throughout the lifetime of mammals. It is, however, currently unknown how this ICR obtains DNA methylation during oogenesis.
Results from the study of Gnas, Zac1, Snrpn, and Kcnq1 imprinted domains suggest that alternative promoters may be involved in the targeting process of DNA methylation to ICRs.6-10 In these imprinted domains, alternative promoters are functional during oogenesis, subsequently triggering transcription. Also, these alternative promoters are all located upstream of their ICRs, thus the transcription usually traverses through the ICRs during oogenesis. The passing of RNA polymerase II has been shown to be important for setting up oocyte-specific DNA methylation on the ICRs. Deletion of the alternative promoters or truncation of the alternative transcripts usually causes defects in the setting up of oocyte-specific DNA methylation on the ICRs.6,9 Recent studies further suggest that the recruitment of the de novo DNA methyltransferase DNMT3A to ICRs may require two particular histone modifications, H3K4me0 and H3K36me3, which are generated during the transcription elongation process by RNA polymerase II.11
Transcription of Peg3 also involves three alternative promoters, termed U1, U2, and U3, which are localized 20, 26, and 160 kb upstream of the Peg3-DMR, respectively.12 Among these, the alternative promoter U1 is functional in oocytes.9,12 This suggests that this alternative promoter may be involved in the DNA methylation of the downstream main promoter, Peg3-DMR. In the current study, we tested this possibility by generating and analyzing a mutant strain lacking the alternative U1 promoter. Our results confirm that this alternative promoter is indeed required for the establishment of oocyte-specific DNA methylation on the Peg3-DMR.
Results
Generation of a mutant allele by deletion of the alternative promoter U1
To test the predicted function of U1, we deleted the genomic region containing the U1 promoter using a CRISPR/Cas9-based strategy (Figure 1A). We designed two single-stranded guide RNA flanking the 1-kb genomic region of the U1 promoter, and subsequently tested the feasibility of this deletion strategy in vitro. After the initial confirmation, these two guide RNA were injected into 200 fertilized eggs of the C57BL/6J background along with Cas9 mRNA. We obtained 45 live mice from this initial set of injected eggs. Among these, 12 mice were shown to be properly targeted by the two guide RNA based on the results of PCR-based genotyping (Figure 1B). Ten of these 12 mice showed an almost identical and predicted size of PCR products, indicating proper deletion of the 1-kb region as designed; this set of deleted alleles was named U1 deletion regular (U1ΔR). In contrast, the other two mice showed a 20 bp larger and an 80 bp smaller deletion, named U1ΔL and U1ΔS, respectively. The exact deleted regions within these founder lines were further confirmed via individual Sanger sequencing (Sup_Fig 1). The exact boundaries of the deleted regions are slightly different between the individual founders since this CRISPR/Cas9-driven deletion scheme was based on non-homologous end joining repair mechanism. Among these founders, we selected five founders from the U1ΔR group for further breeding experiments. All five founders successfully transmitted their deleted alleles to the following generations without major effects; therefore, these five lines have been collectively regarded as a single line, the U1ΔR line, hereafter. Through this initial set of breeding experiments, we successfully established a deletion mutant line and also obtained several litters of F1 and F2 with both paternal and maternal transmission of the deleted allele. Using these mice, we first performed a series of RT-PCR detecting the transcript driven by U1 (Figure 1C). Besides oocytes, the U1 promoter is known to trigger transcription in the hypothalamus of adult mice.11 Also, the U1 promoter is allele-specific, only functional from the maternal allele.11 Thus, we prepared a set of total RNA from the hypothalamus of adult mice that had inherited the deleted allele maternally and paternally. As expected, the U1 transcript was detected in the hypothalamus of both wild type (WT) and mutants with paternal transmission (KO+/−-P), but not in mutants with maternal transmission (KO−/+-M) (Figure 1C). This observation confirmed that the deletion indeed abolished the transcriptional activity of the U1 promoter. In summary, we have successfully established a deletion mutant line targeting the U1 promoter using a CRISPR/Cas9-based strategy.
Figure 1.
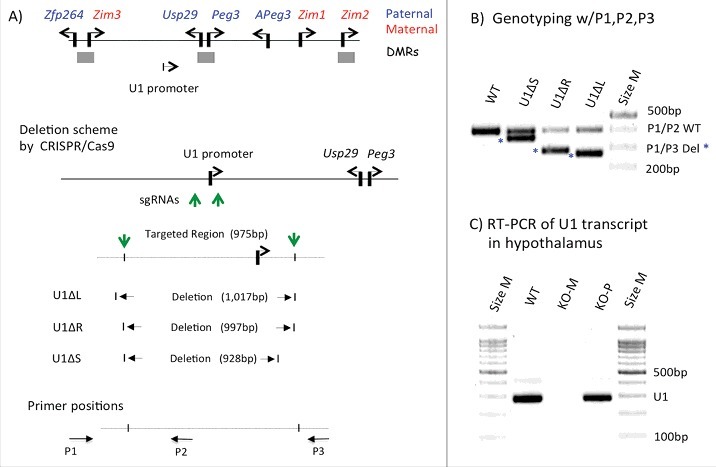
CRISPR/Cas9-based deletion of the U1 promoter. (A) Schematic representation of the Peg3 domain. Each imprinted gene is indicated with an arrow. Paternally and maternally expressed genes are indicated in blue and red, respectively. The three DMRs are indicated with gray boxes. The relative position of the U1 promoter is indicated with an arrow. The detailed genomic structure of U1 is also shown with the two single-stranded guide RNA for CRISPR/Cas9-based deletion, the positions of which are indicated with green vertical arrows. The CRISPR/Cas9-based deletion derived the three representative mutant alleles, and their deleted regions are shown. The arrows on the bottom indicate the three primers (P1, P2, P3) that were used for genotyping. (B) PCR-based genotyping of the deleted alleles. The three primers were used together for genotyping: the combination of P1 and P2 primers targets the wild type allele (403 bp in length), whereas the combination of P1 and P3 primers targets the deleted alleles (about 270 bp). (C) RT-PCR detecting the transcript driven by the U1 promoter. This series of analyses used a set of total RNA isolated from the hypothalamus of adult mice of three genotypes: WT, KO(−/+)-M, and KO(+/−)-P, the last two inheriting the deletion maternally and paternally, respectively.
Effects of U1 deletion on Peg3-DMR methylation
We next tested the potential effects of U1 deletion on the DNA methylation status of the Peg3-DMR (Figure 2). We prepared two sets of genomic DNA: DNA from mature eggs and DNA from somatic tissues. First, we isolated genomic DNA from two sets of 3-month-old females: heterozygotes (KO) and WT littermates. Isolated DNA was bisulfite converted,13 and then used for PCR amplification followed by cloning and sequencing. These series of DNA methylation analyses targeted two genomic regions: one within the Peg3-DMR and the other within the Peg10-DMR (as a control). As expected, the DNA methylation levels of the Peg10-DMR were close to 100% in both WT and KO samples (Figure 2A), confirming the purity of the isolated eggs as well as the feasibility of the approach. In contrast, DNA methylation levels of the Peg3-DMR were quite different between the two samples: 65% in WT vs. 7% in KO. The high levels of DNA methylation in WT were expected since the Peg3-DMR is methylated during oogenesis. In contrast, the significantly lower levels in KO indicated a target-specific outcome by the deletion of U1, thus confirming the predicted role of U1 during oogenesis. We repeated this series of experiments three times, including eggs set from the female homozygotes (Sup_Fig 3), and the results were overall similar to those shown in Figure 2A.
Figure 2.
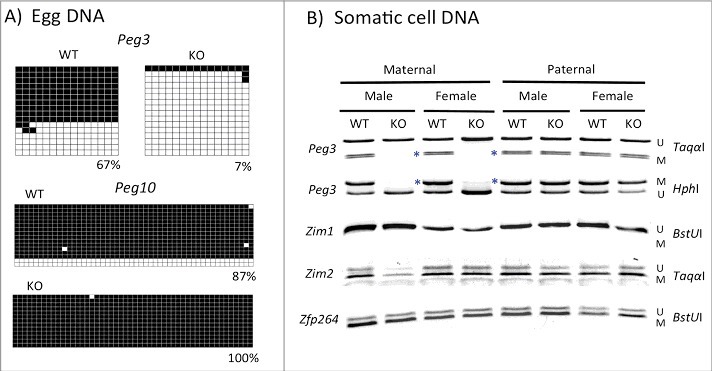
Effects of the U1 deletion on the DNA methylation levels of the Peg3-DMR. (A) A series of DNA methylation analyses were performed with DNA from mature eggs isolated from two sets of 3-month-old females: WT and KO. Bisulfite-converted DNA was amplified with two sets of primers targeting Peg3-DMR and Peg10-DMR. Amplified PCR products were subsequently analyzed by individual sequencing. Each square represents a CpG site with close and open squares indicating methylated and unmethylated CpG, respectively. (B) A similar series of DNA methylation analyses were performed on a set of somatic DNA isolated from tails of WT and KO of both sexes with maternal and paternal transmission of the U1 deletion. Amplified PCR products were analyzed by COBRA. Each target region was digested with enzymes that can differentiate the methylation status of the original DNA. The letter U or M indicates the unmethylation or methylation status, respectively, based on the digestion pattern by a given restriction enzyme. The blue * indicates the mutational effects of the U1 deletion on the Peg3-DMR: removal of maternal-specific DNA methylation.
We also performed DNA methylation analyses using DNA isolated from the tails of sets of WT and KO neonates of both sexes with maternal and paternal transmission of the deletion (Figure 2B). DNA was analyzed similarly as described above except that DNA methylation levels were measured using combined bisulfite restriction analysis (COBRA).14 KO mice with maternal transmission of the U1 deletion showed complete loss of DNA methylation on the Peg3-DMR (see lanes 2 and 4 in Figure 2B). We also tested several regions within the Peg3-DMR, which, again, showed the same outcome: no DNA methylation within the Peg3-DMR. This agrees with the results from mature eggs, showing very low levels of DNA methylation in KO (Figure 2A). We repeated this series of DNA methylation analyses using DNA isolated from F1 and F2 pups, totaling 48 carriers with maternal transmission: the majority of these pups (35/48 = 72.3%) showed no methylation on the Peg3-DMR (Sup_Fig 2), suggesting that the mutation is approximately 70% penetrant. In contrast, results from mice with paternal transmission of the U1 deletion showed no major changes in DNA methylation levels of the Peg3-DMR (see lanes 6 and 8 in Figure 2B). This is consistent with the fact that transcription by the U1 promoter is undetectable and non-functional during spermatogenesis; therefore, the paternal transmission of the deletion should not cause any effect. We also surveyed DNA methylation levels of the three other regions within the Peg3 domain, including the promoter of Zim1 and the DMRs of Zim2 and Zfp264/Zim3 (Figure 1A). However, we did not observe any changes in these regions in either set of samples, indicating no major effect on the DNA methylation levels of the other DMRs within the Peg3 domain. Overall, this series of DNA methylation analyses showed that deletion of U1 resulted in significant reduction in DNA methylation levels on the Peg3-DMR during oogenesis and, in addition, that the majority of the mutant mice inheriting the deletion maternally showed no methylation on the Peg3-DMR.
Effects of U1 deletion on the imprinting status of the Peg3 domain
The results described above were followed up with imprinting tests. For this series of analyses, we prepared hybrid pups derived from the reciprocal crossing between the KO of the C57BL/6J(B6) background and WT of the PWD/PhJ(PWD) background (Figure 3). Total RNA isolated from the neonatal heads of the hybrids were used for generating cDNA, which was subsequently used as template for amplifying several imprinted genes within the Peg3 domain. The results are summarized as follows. The majority of the genes within the Peg3 domain became biallelic in mutants with maternal transmission of the U1 deletion, including Peg3, Usp29, Zim1, and Zim2 (see lane 4 in Figure 3). Transcription of these genes was also derived equally from both alleles. Biallelic expression of Peg3 and Usp29 was expected, given no allele-specific DNA methylation was observed on their bidirectional promoter Peg3-DMR. Biallelic expression of the other genes, Zim1 and Zim2, is also consistent with the fact that the imprinting of these adjacent genes is also co-regulated through the Peg3-DMR as an ICR along with that of Peg3/Usp29. In contrast, paternal transmission of the U1 deletion did not cause any changes in the allele-specific expression patterns of the same set of genes, the exact same patterns of allele-specific expression were observed in WT and KO (compare lanes 5 and 6 in Figure 3). This agrees with the observation that paternal transmission of the U1 deletion has no effects on the DNA methylation of the Peg3-DMR (Figure 2B). Overall, this series of analyses demonstrated that the removal of allele-specific DNA methylation on the Peg3-DMR resulted in biallelic expression of the Peg3 domain.
Figure 3.
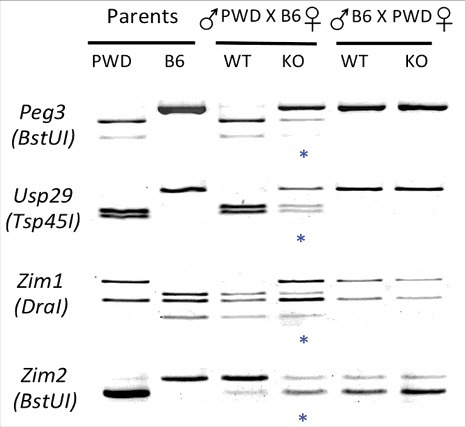
Effects of the U1 deletion on the imprinting status of the Peg3 domain. A series of imprinting tests were performed with total RNA isolated from neonatal heads of the F1 hybrid that had been prepared through the reciprocal crossing of heterozygotes with the B6 background and the breeding partners with the PWD/PhJ background. The products from RT-PCR were digested with a given restriction enzyme to differentiate parental alleles, which are shown as different-sized DNA fragments on gel images. The two columns on the left represent the digestion patterns for two parental strains for each gene; the two middle columns represent results from the F1 hybrid set with maternal transmission of the U1 deletion (female heterozygote with male PWD/PhJ); the two columns on the right represent the results from the F1 hybrid set with paternal transmission of the U1 deletion (female PWD/PhJ with male heterozygote). The blue * indicates the mutational effects of the U1 deletion on the imprinting status of genes: biallelic expression.
Effects of U1 deletion on the expression levels of the Peg3 domain
We also tested potential effects of the U1 deletion on the expression levels of the imprinted genes within the Peg3 domain (Figure 4). This series of expression analyses used a set of total RNA isolated from the neonatal heads of WT and KO of both sexes with maternal transmission of the U1 deletion. Isolated RNA was converted into cDNA, which was then used for qRT-PCR. The expression levels of Peg3 and Usp29 were significantly upregulated in both sexes: 2.5- and 2.8-fold in females and 1.9- and 2.5-fold in males, respectively (P value <0.001). In contrast, expression levels of Zim1 were significantly downregulated in both male and female KO, 3–5% of WT levels (P value <0.001) (Figure 4A). It is relevant to note that Zim1 is also normally expressed from the paternal allele, but at very low levels relative to the levels of the maternal allele.15 Thus, the biallelic expression of Zim1 observed in the hybrid KO might be an outcome of a scenario in which the significantly reduced expression levels of Zim1 from the maternal allele became similar to the basal levels of the paternal allele, thus showing the biallelic expression pattern (Figure 3). On the other hand, the expression levels of Zfp264 were not affected in either sex. We repeated a similar series of analyses using a set of total RNA isolated from the hypothalamus of adult mice (Figure 4B). The results were similar to those observed from neonatal heads, except that the levels of upregulation observed for both Peg3 and Usp29 were lower (1.5-fold) than those from neonatal heads. Overall, these series of analyses concluded that Peg3 and Usp29 were upregulated in the mutants with maternal transmission of the U1 deletion. This is consistent with the results showing loss of maternal-specific methylation of the Peg3-DMR and subsequent biallelic expression of Peg3 and Usp29.
Figure 4.
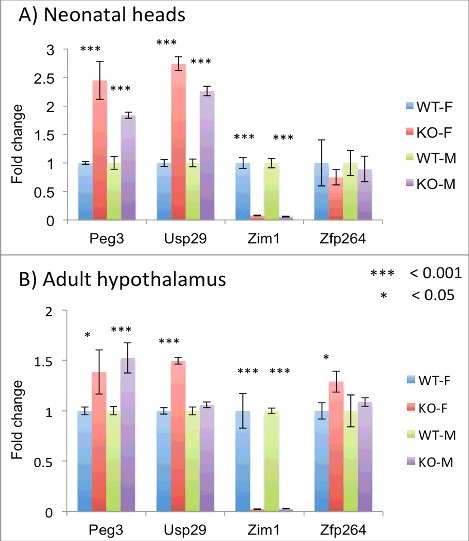
Effects of the U1 deletion on the expression levels of the Peg3 domain. Potential effects of the U1 deletion on the expression levels of the individual genes were analyzed using a set of total RNA isolated from neonatal heads (A) and adult hypothalamus (B) of WT and KO(−/+) mice with maternal transmission of the deletion. This series of qRT-PCR analyses tested four imprinted genes, Peg3, Usp29, Zim1, and Zfp264. The expression levels of each gene were first normalized with an internal control (β-actin), and the subsequent value was compared between KO and WT. The relative levels are presented with their standard deviation. This set of analyses was repeated using two biological replicates.
Effects of U1 deletion on survival and growth rates of the animals
Potential effects of the U1 deletion on the survival and growth rates of the animals were tested with four individual breeding experiments (Figure 5). Pups derived from these crosses were analyzed in terms of their genotype, sex, and weight at birth: First, the deletion of U1 did not cause any major effects on the growth rates of the surviving animals, showing similar average weights at neonatal and weaning ages among four individual genotypes: WT(+/+), Heterozygote(Het)(−/+), Het(+/−), and Homozygote (Hom)(−/−) (data not shown). This suggests that the U1 deletion and subsequent biallelic expression of Peg3/Usp29 may have minimal effects on the growth rates of the surviving animals. Second, the U1 deletion did not cause a significant impact on the litter size of the animals. The four breeding schemes showed similar litter sizes, ranging from 6.7 to 8.1. This indicates no obvious embryonic lethality associated with these breeding schemes, especially during the post-implantation stage. Third, the number of homozygotes carrying the U1 deletion were considerably smaller than the expected frequencies from the two breeding schemes: for the Het × Het crossing, 3 were observed while 6.75 were expected; for the Het × Hom crossing, 4 were observed while 23.5 were expected (P value <0.001) (Figure 5C and D). Fourth, the number of female heterozygotes with maternal transmission of the U1 deletion was lower than that of male heterozygotes (18 vs. 30, respectively) (Figure 5A). This became even more obvious when we compared only the numbers of pups showing no methylation on the Peg3-DMR (11 vs. 24, respectively; P value <0.05), which presumably have a double (2X) gene dosage of Peg3/Up29, as well as other imprinting defects. This implies that females might be more susceptible to the biallelic expression and subsequent imprinting defects of the Peg3 domain. A similar trend was also observed from the pups of the other breeding scheme, for example, the four homozygotes from the Het × Hom crossing (Figure 5D). The DNA methylation status on the Peg3-DMR in these homozygotes was variable between the two sexes. The two male homozygotes showed no DNA methylation, and were thus thought to have imprinting defects of the Peg3 domain. In contrast, the two remaining female homozygotes were still methylated with the U1 deletion, and were thus thought to have normal imprinting of the Peg3 domain. This suggests that females may be more susceptible to the imprinting defects of the Peg3 domain, in particular the 2X gene dosage of Peg3. In summary, these series of breeding experiments provided two new insights: (i) embryonic lethality is associated with the homozygosity of the U1 deletion; (ii) there is female-biased susceptibility to the imprinting defects of the Peg3 domain.
Figure 5.
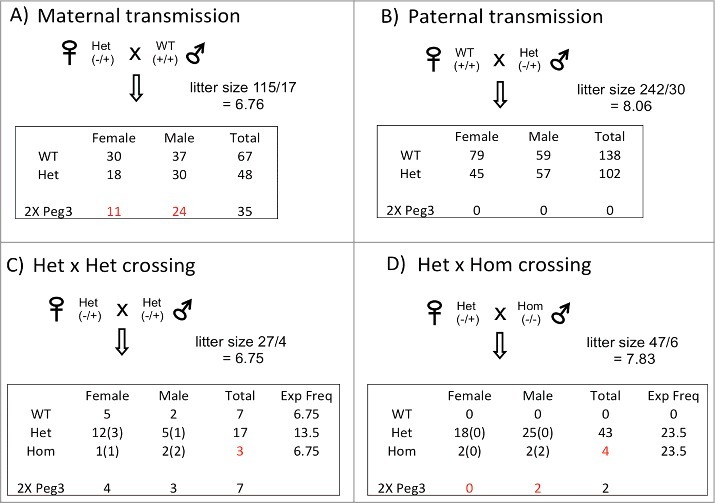
Effects of the U1 deletion on survival. Four individual sets of breeding experiments were performed to test potential effects of the U1 deletion on survival (A-D). The results from each breeding experiment were summarized with its average litter size, transmission ratios between individual genotypes, and sex ratios. The last row (2X Peg3) summarizes the numbers of heterozygote and/or homozygote mice presumably having a double dosage of Peg3 as well as the imprinting defects, which were measured by the DNA methylation status of the Peg3-DMR. Numbers in parentheses indicate the numbers of mice with 2X Peg3 and the imprinting defects in each genotype. Numbers in red indicate the unusual outcomes that deviate significantly from the expected frequencies.
Discussion
In the current study, we deleted the alternative promoter U1, which is highly active during oogenesis, to test its potential role in the establishment of DNA methylation on the Peg3-DMR. As predicted, deletion of this alternative promoter indeed caused complete removal of maternal-specific DNA methylation on the Peg3-DMR. As an outcome, the imprinted genes in the Peg3 domain became biallelically expressed in mutants with maternal transmission of the U1 deletion. The expression levels of imprinted genes were also affected in the mutants; we observed 2-fold upregulation of Peg3 and Usp29 and downregulation of Zim1 to basal levels (Figure 6). Breeding experiments further revealed that females were under-represented among the surviving mutants, a potential sex-biased outcome caused by the biallelic expression of the Peg3 domain. Overall, the current study suggests that U1-driven transcription may be required for establishing oocyte-specific DNA methylation on the Peg3 domain.
Figure 6.
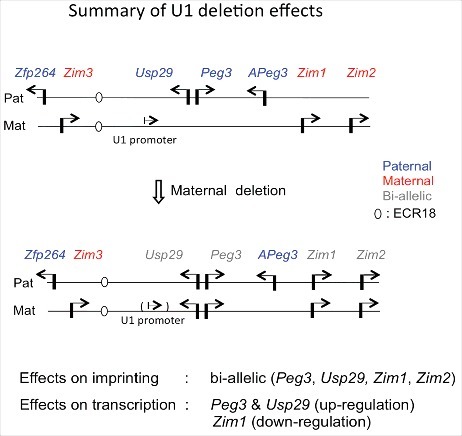
Summary of U1 deletion effects on imprinting and expression of the Peg3 domain. A schematic representation of the Peg3 domain is shown on the upper panel. Each imprinted gene is indicated with an arrow. Paternally and maternally expressed genes are indicated in blue and red, respectively. Biallelically expressed genes are indicated in gray. The position of ECR18, a potential enhancer, is indicated with an oval. Mutational effects by maternal transmission of the U1 deletion are summarized from results derived from total RNA isolated from neonatal heads of both sexes. Paternal transmission of the U1 deletion did not cause any major effects, thus omitted in this summary.
Deletion of the U1 alternative promoter resulted in complete removal of maternal-specific DNA methylation on the Peg3-DMR (Figure 2). This is in agreement with results from other imprinted domains, including the Gnas, Zac1, Snrpn, and Kcnq1 domains.6-10 Thus, oocyte-specific DNA methylation on the Peg3-DMR is likely established through transcription-driven recruitment of the de novo methyltransferase DNMT3A during oogenesis.11 The results from the current study, however, provide another insight that this transcription-mediated targeting process may not be the only mechanism necessary for de novo DNA methylation at the Peg3-DMR, since deletion of the U1 promoter was about 70% penetrant in abolishing DNA methylation imprint among mutant mice (Figure 5 and Sup_Fig 2). This transcription-mediated mechanism might be one of several mechanisms involved in the targeting process of DNA methylation to ICRs, and other unknown mechanisms might compensate the defects in the remaining 30% cases of de novo DNA methylation. This is further supported by the fact that the U1 transcript has not been detected in oocytes from other mammals, such as humans. In humans, the U1 promoter is, in fact, active in testis, so that several transcripts driven by the U1 promoter are detected in the Expressed Sequence Tags database. Consistent with this, the DNA sequence of the U1 promoter itself seems to have evolved very rapidly (data now shown), suggesting that U1 might have adapted to slightly different roles in individual species. Another possibility is that U1 might not be the only promoter triggering transcription in the mice oocyte. Two additional alternative promoters, U2 and U3, are localized in the 200-kb upstream region of the Peg3-DMR; however, these promoters have not been shown to be functional in oocytes so far.12 Overall, the current study, along with other studies, strongly suggests that a transcription-driven mechanism may be the main mechanism recruiting de novo DNA methylation to the ICRs.
Removal of the maternal-specific methylation on the Peg3-DMR resulted in biallelic expression of the majority of imprinted genes in the Peg3 domain (Figure 3 and 6). This observation is noteworthy. First, in previous genetic studies on genomic imprinting, genomic regions of the ICRs have been deleted to test their predicted functions.16,17 In this situation, it was difficult to determine whether genomic sequences or allele-specific DNA methylation were more critical for the functions of ICRs. According to the results from the U1 deletion, allele-specific DNA methylation on ICRs is more critical for the imprinting of the Peg3 domain. Second, the de-repression of Peg3 and Usp29 from the maternal allele appeared to coincide with the significantly downregulated maternal expression of Zim1 (Figure 3, 4, and 6). This coincidence might be reflecting the fact that two genes, Peg3 and Zim1, cannot be co-expressed in cis due to unknown reasons, which could also be the basis for the opposite imprinting.2 In the normal maternal allele, Peg3 is repressed through DNA methylation, thus Zim1 can be expressed. In contrast, the paternal allele of Zim1 cannot be expressed from the paternal allele due to the expression of Peg3. In the U1 mutants, Peg3 is now expressed from the de-repressed maternal allele, which in return causes the downregulation of Zim1 (Figure 6). The exact molecular mechanisms for this opposite imprinting are currently unknown, but it is most likely that these two genes may be competing for shared enhancers. One likely candidate for a shared enhancer is ECR18. This potential enhancer is localized 200 kb upstream of Peg3 (Figure 6), and has been shown to interact with several promoters within the Peg3 domain.18 It should be interesting to further characterize the detailed mechanisms of this opposite imprinting of Peg3 and Zim1 in the near future.
Mutants with maternal deletion of U1 provide several insights regarding the in vivo functions of the mammalian Peg3 domain. We have seen several cases for the complete loss of Peg3 gene dosage through mouse KO experiments or complete methylation on the PEG3-DMR in human cancers.2 However, we have never observed biallelic expression and subsequent double gene dosage of Peg3. Until now, mammals with double gene dosage of the Peg3 domain were assumed to be not viable. The results from the breeding experiments rather support this prediction in that females are extremely susceptible to the increased gene dosage of the Peg3 domain (Figure 5). Although animals with biallelic expression of Peg3 can be generated in the lab, these mutant animals are extremely unlikely to survive in the wild setting, since double dosage of the Peg3 domain compromises the survival of females. It is interesting to note that complete loss or zero dosage of Peg3, on the other hand, has been shown to cause similar effects on male, the opposite sex.5 Thus, it is reasonable to hypothesize that the optimum or tolerable range of gene dosage of Peg3 may be different in the two sexes. This is reminiscent of the hypothesis known as ‘intralocus sexual conflict,’ in which one sex favors higher gene dosage whereas the opposite sex favors lower dosage for a given locus.19 This evolutionary tug-of-war usually results in different expression levels for a given locus in each of the two sexes.19 Expression levels of Peg3 are indeed different between the two sexes, being higher in males than females,20,21 thus supporting the possibility that the mammalian Peg3 locus may have been subjected to intralocus sexual conflict.5 Taken together, the available data strongly suggest the presence of potential sexual conflicts over the Peg3 domain. It is thus likely that the Peg3 locus might have evolved not only to resolve parental conflicts but also to resolve sexual conflicts.
Materials and methods
Ethics statement
All the experiments related to mice were performed in accordance with National Institutes of Health guidelines for care and use of animals, and also approved by the Louisiana State University (LSU) Institutional Animal Care and Use Committee (IACUC), protocol #16-060.
Generation of the mutant allele deleting the U1 alternative promoter
The 1-kb genomic interval (mm10, chr7:6,750,918-6,751,939) surrounding the U1 promoter was deleted using a CRISPR/Cas9-based scheme described below. Two single-stranded guide RNA (5′-ATAACAAACGTGTTCAATCCAGGG-3′ and 5′-CCTTGGGTATAATACATAGAGGC-3′) along with Cas9 mRNA were injected into the 200 fertilized eggs that had been prepared from timed mating of C57BL/6J mice. These injected eggs were placed back to the uterus of the 6 pseudo-pregnant females for the full-term development, deriving 45 live pups. The subsequent mice were genotyped to confirm the proper deletion using the following three primers: P1, 5′-TAGCAAGGGAGAGGGCCTAG-3′; P2, 5′-GGAAGCCTCCATCCGTTTGT-3′; P3, 5′-AGCACAGCTAGAAATACACAGA-3′. Some of these founders have been used for establishing the main deletion line, U1ΔR, for the current study.
Mouse breeding
The current study used the following breeding schemes: the maternal and paternal transmission of the U1 deletion through mating the female and male heterozygotes individually with male and female wild-type littermates, the inter-crossing between two heterozygotes, and finally the crossing between the female heterozygote and the male homozygotes (Figure 5). One-day-old pups derived from these breeding experiments were analyzed in terms of sex, genotype, and body weight. Statistical significance of potential difference of litter size and average weight between the breeding experiments was tested using the Χ2 test. All the mice were housed at the Division of Lab Animal Medicine (DLAM) of LSU on a regular 12-12 dark-light cycle under a constant temperature of 70°F and 50% humidity. All animals were given ad libitum access to water and Rodent Diet 5001. Nursing females were with Mouse Diet 5015. Mice were euthanized by CO2 asphyxiation in accordance with the rules and regulations set forth by the IACUC. For genotyping, genomic DNA was isolated from either clipped ears or tail snips by incubating the tissues overnight at 55°C in the lysis buffer (0.1 M Tris-Cl, pH 8.8, 5 mM EDTA, pH 8.0, 0.2% SDS, 0.2 M NaCl, 20 μg/ml Proteinase K). Isolated DNA was subsequently genotyped using three primers, P1-P3, which were described above. Sex of the pups was determined by PCR using the following primer set: mSry-F (5′-GTCCCGTGGTGAGAGGCACAAG-3′) and mSry-R (5′-GCAGCTCTACTCCAGTCTTGCC-3′).
Expression analyses and imprinting tests
Total RNA was isolated from tissues of one-day-old heads or hypothalamus of adult mice using a commercial kit (Trizol, Invitrogen). Total RNA was then reverse-transcribed using the M-MuLV Kit (Invitrogen), and the subsequent cDNA was used as template for qRT-PCR. This analysis was performed with the iQ SYBR green supermix (Bio-Rad) using the ViiA™ 7 Real-Time PCR System (Life Technologies). All qRT-PCR reactions were carried out for 40 cycles under standard PCR conditions. The analyses of the results derived from qRT-PCR were described previously.22 Statistical significance of potential difference of expression levels of a given gene between two samples was tested with Student's t-test. The information regarding individual primer sequences and PCR conditions is available in the previous study.3,5 For the imprinting test, heterozygotes of the C57BL/6J background were reciprocally crossed with wild type mice of the PWD/PhJ background (Jackson Lab, Stock No. 004660). The F1 hybrid of these crossing was used for isolating total RNA. The polymorphisms and restriction enzymes used for each gene imprinting test are also available in the previous study.3,5
DNA methylation analysis
The current study used two types of genomic DNA for DNA methylation analyses. Mature eggs were isolated using the following method. In brief, three-month-old female mice were super-ovulated by a series of intraperitoneal hormonal injections. First, 0.1 ml pregnant mare serum gonadotropin (EMD Millipore™, Cat. No. 36-722-21000I) was injected. Second, 0.1 ml human chorionic gonadotropin (Sigma, Cat. No. C1063) was injected 48 hours later. Ampullae were removed 12 hours later by gross dissection and placed directly in PBS. Fat was trimmed away from ampullae under a dissection scope. The ampullae were then washed in PBS. Under the dissection scope, ampullae were torn open using a 27-gauge needle and egg sacks were transferred by mouth pipetting to HCZB solution with hyaluronidase (Sigma, Cat. No. H3506). After incubating for 30 minutes, oocytes were transferred by mouth pipetting into clean HCZB/hyaluronidase solution to separate them from somatic cells. This washing step was repeated 5 times to ensure the isolation of mature oocytes from somatic cells. Finally, the recovered oocytes were mouth-pipetted into the lysis buffer (Tris-HCl pH 8.1 100 mM, EDTA 5 mM, NaCl 200 mM, SDS 0.2%) supplemented with Proteinase K (NEB, Cat. No. P8107S). For DNA methylation analyses, genomic DNA from mature eggs or tails was treated with the bisulfite conversion protocol.13 Converted DNA was used as a template for PCR using the specific primers that were designed for amplifying each target region. The amplified products from mature eggs were individually cloned into a commercial vector and, on average, 16 clones for each PCR product were sequenced to assess final DNA methylation level. The sequence reads were trimmed and aligned using Clustal Omega. PCR clones were removed based on analysis of the phylogenetic tree of aligned sequences. Amplified products from tail DNA were analyzed by COBRA.14 Information regarding the sequences of oligonucleotides and PCR conditions for each target region is also available in the previous study.3
Supplementary Material
Funding Statement
This research was supported by the National Institute of Health (R01-GM066225 and R01-GM097074 to J.K.).
Acknowledgments
We would like to thank Drs. Jason Heaney and Denise Lanza at Baylor College of Medicine helping us for CRISPR/Cas9-based deletion of the U1 promoter. We also like to thank the members of JooKim Lab for their thoughtful feedback and discussion on the manuscript. This research was supported by the National Institute of Health (R01-GM066225 and R01-GM097074 to J.K.).
References
- 1.Kim J, Ashworth L, Branscomb E, Stubbs L. The human homolog of a mouse-imprinted gene, Peg3, maps to a zinc finger gene-rich region of human chromosome 19q13.4. Genome Res. 1997;7:532-40. doi: 10.1101/gr.7.5.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He H, Kim J. Regulation and function of the Peg3 imprinted domain. Genomic Inform. 2014;12:105-13. doi: 10.5808/GI.2014.12.3.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim J, Ekram MB, Kim H, Faisal M, Frey WD, Huang JM, Tran K, Kim MM, Yu S. Imprinting Control Region (ICR) of the Peg3 domain. Hum Mol Genet. 2012;21:2177-687. doi: 10.1093/hmg/dds092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim J, Kollhoff A, Bergmann A, Stubbs L. Methylation-sensitive binding of transcription factor YY1 to an insulator sequence within the paternally expressed imprinted gene, Peg3. Hum Mol Genet. 2003;12:233-45. doi: 10.1093/hmg/ddg028. [DOI] [PubMed] [Google Scholar]
- 5.He H, Perera BP, Ye A, Kim J. Parental and sexual conflicts over the Peg3 imprinted domain. Sci Rep. 2016;6:38136. doi: 10.1038/srep38136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chotalia M, Smallwood SA, Ruf N, Dawson C, Lucifero D, Frontera M, James K, Dean W, Kelsey G. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 2009;23:105-17. doi: 10.1101/gad.495809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith EY, Futtner CR, Chamberlain SJ, Johnstone KA, Resnick JL. Transcription is required to establish maternal imprinting at the Prader-Willi syndrome and Angelman syndrome locus. PLoS Genet. 2011;7:e1002422. doi: 10.1371/journal.pgen.1002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis MW, Brant JO, Kramer JM, Moss JI, Yang TP, Hansen PJ, Williams RS, Resnick JL. Angelman syndrome imprinting center encodes a transcriptional promoter. Proc Natl Acad Sci USA. 2015;112:6871-75. doi: 10.1073/pnas.1411261111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veselovska L, Smallwood SA, Saadeh H, Stewart KR, Krueger F, Maupetit-Méhouas S, Arnaud P, Tomizawa S, Andrews S, Kelsey G. Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol. 2015;16:209. doi: 10.1186/s13059-015-0769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh VB, Sribenja S, Wilson KE, Attwood KM, Hillman JC, Pathak A, Higgins MJ. Blocked transcription through KvDMR1 results in absence of methylation and gene silencing resembling Beckwith-Wiedemann syndrome. Development. 2017;144(10):1820-30. doi: 10.1242/dev.145136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stewart KR, Veselovska L, Kim J, Huang J, Saadeh H, Tomizawa S, Smallwood SA, Chen T, Kelsey G. Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev. 2015;29:2449-62. doi: 10.1101/gad.271353.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perera BP, Kim J. Alternative promoters of Peg3 with maternal specificity. Sci Rep. 2016;6:24438. doi: 10.1038/srep24438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22(15):2990-97. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong Z, Laird PW. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25(12):2532-34. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Lu X, Stubbs L. Zim1, a maternally expressed mouse Kruppel-type zinc-finger gene located in proximal chromosome 7. Hum Mol Genet. 1999;8:847-54. doi: 10.1093/hmg/8.5.847. [DOI] [PubMed] [Google Scholar]
- 16.Barlow DP, Bartolomei MS. Genomic imprinting in mammals. Cold Spring Harb Perspect Biol. 2014;6. doi: 10.1101/cshperspect.a018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011;3. doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiaville MM, Kim H, Frey WD, Kim J. Identification of an evolutionarily conserved cis-regulatory element controlling the Peg3 imprinted domain. PloS ONE. 2013;8:e75417. doi: 10.1371/journal.pone.0075417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonduriansky R, Chenoweth SF. Intralocus sexual conflict. Trends Ecol Evol. 2009;24:280-88. doi: 10.1016/j.tree.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Faisal M, Kim H, Kim J. Sexual differences of imprinted genes' expression levels. Gene. 2013;533:434-38. doi: 10.1016/j.gene.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perera BP, Kim J. Sex and tissue specificity of Peg3 promoters. PLoS ONE. 2016;11(10):e0164158. doi: 10.1371/journal.pone.0164158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase–polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270:41-49. doi: 10.1006/abio.1999.4085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.