

ABSTRACT
Cyclin D1 and cyclin E1, as vital regulatory factors of G1-S phase cell cycle progression, are frequently constitutive expressed and associated with pathogenesis and tumorigenesis in most human cancers and they have been regarded as promising targets for cancer therapy. In this study, we established NVP-BEZ235, a potent dual kinase inhibitor, could induce neuroblastoma cells proliferation inhibition without apoptosis activation. Moreover, we showed NVP-BEZ235 could induce neuroblastoma cells arrested at G0/G1 phase accompanied with significant reduction of the cyclin D1 and E1 proteins in a dose dependent manner at nanomole concentration. Additionally we found that GSK3β was dephosphorylated and activated by NVP-BEZ235 and then triggered cyclin D1 and cyclin E1 degradation through ubiquitination proteasome pathway, based on the evidences that NVP-BEZ235 induced downregulation of cyclin D1 and cyclin E1 were obviously recovered by proteasome inhibitor and the blockade of GSK3β contributed to remarkable rescue of cyclin D1 and cyclin E1. Analogous results about its anti-proliferation effects and molecular mechanism were observed on neuroblastoma xenograft mouse model in vivo. Therefore, these results indicate that NVP-BEZ235-induced cyclin D1 and cyclin E1 degradation, which happened through activating GSK3β, and GSK3β-dependent down-regulation of cyclin D1 and cyclin E1 should be available for anticancer therapeutics.
KEYWORDS: cyclin D1, cyclin E1, NVP-BEZ235, GSK3β, neuroblastoma
Introduction
Neuroblastoma is the most common extracranial solid tumor of childhood that derives from the peripheral sympathetic nervous system and accounts for 15% of childhood cancer deaths.1-3 It presents heterogeneous clinical features, such as at the early age of onset, spontaneous regression in infancy and the high risk of metastatic disease at diagnosis.4,5 The 10-year survival rate of high-risk neuroblastoma patients is as low as 20% despite decades of the considerable advances in diagnostic methods and the development in therapeutic treatment.6 Due to major obstacles like MYCN oncogene amplifications and germline or somatic activating ALK mutations, chemotherapy resistance and poor prognosis subsequently occur to patients.7 Therefore, the identification and validation of novel therapeutic agents remain an effective strategy to further improve survival and long-term quality of life of patients. Evidences have been shown that PI3K/Akt/mTOR pathway is often constitutively activated and correlate with excessive proliferation and drug resistance in neuroblastoma.8-10 NVP-BEZ235 is a synthetic small molecular compound that potently and reversibly inhibits the catalytic activities of PI3K and mTOR by competing at their ATP-binding sites.11 NVP-BEZ235 is the most effective small molecule inhibitor to date and has been a particularly attractive compound with the traits of low dose, well tolerance, notable effect and more selectivity, compared with other dual inhibitors. Multiple researches have demonstrated the prominent inhibitory efficacy of NVP-BEZ235 on the proliferation in a wide variety of human malignant tumors in vitro and preclinical animal models and are currently in phase I/II clinical trials for advanced solid tumors.12,13
It is generally believed that cell cycle arrest triggered by NVP-BEZ235 via antagonizing the PI3K/mTOR signaling pathway plays a pivotal role in its potent cytostatic effects.14 The activities of cyclins /cyclin-dependent kinases (CDKs) are involved in normal cell cycle progression. D- and E-type cyclins, which mediate G0-G1 phase cell cycle progression, form a complex with their specific catalytic CDKs kinases and then phosphorylate the retinoblastoma protein (pRb) that leads to release of E2F and promote G1/S transition by transcriptional activation of S-phase genes.15 In the whole of cyclin D and cyclin E isoforms, the overexpression of cyclin D1 and cyclin E1 is most frequently associated with pathogenesis of most human cancers, including neuroblastoma.16 Previous observations have revealed that elevated levels of cyclin D1 or cyclin E1 appear to be related to some diseases including those of the breast, esophagus, bladder and lung, and various therapeutic agents, such as retinoic acid, gambogic acid and resveratrol, have been identified to induce them down-regulation.15,17-19 Many researches have reported that PI3K/mTOR signal pathway plays an important role in the up-regulation of cyclin D1 and cyclin E1. Although several papers have reported that NVP-BEZ235 could down-regulate cyclin D1 or cyclin E1, the specific molecular mechanism are not yet clear.20,21 It is worthwhile to further investigate detailed molecular mechanism about the down-regulation of NVP-BEZ235-induced cyclin D1 and cyclin E1 in NB cells.
In this work, we investigated the antiproliferation effects of NVP-BEZ235 on neuroblastoma and the molecular mechanisms. We showed that GSK3β involved in NVP-BEZ235-induced cyclin D1 and cyclin E1 degradation in vitro and in vivo.
Results
NVP-BEZ235 inhibited the proliferation of neuroblastoma cells
To determine the effect of the dual PI3K/mTOR inhibitor NVP-BEZ235 on the proliferation of human NB cells, SH-SY5Y and SK-N-MC were treated with varying concentrations of NVP-BEZ235 (0, 10, 25, 50, 100, 200, 500 and 1000 nM) for 24 h and then cell viability was measured by CCK-8 assay. We showed that NVP-BEZ235 decreased the cell viability of both cell lines in a dose-dependent manner (Fig. 1A and C). We then detected the expression of PI3K/mTOR pathway related proteins in both cell lines treated with NVP-BEZ235. As shown in Fig. 1B and D, the phosphorylation level of Akt, p70S6K, mTOR and 4E-BP1 was significantly declined by NVP-BEZ235. Moreover, the number of both cells under the microscope and MCM2 protein levels related to cell growth were also persistently decreased in both cell lines treated with NVP-BEZ235 (Fig. 1E, F, G and H).
Figure 1.
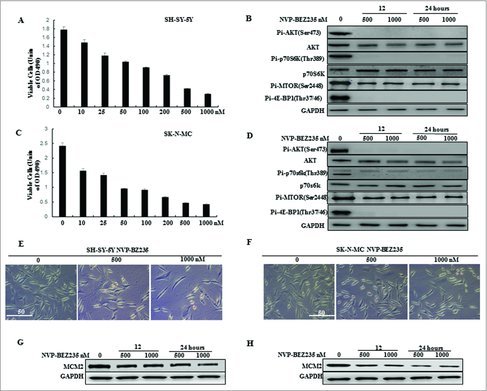
NVP-BEZ235 inhibited PI3K/AKT/mTOR pathway and proliferation of neuroblastoma cells. (A, C) SH-SY5Y and SK-N-MC cells were treated with 0, 10, 25, 50, 100, 200, 500 and 1000 nM NVP-BEZ235 for 24 h. Cell viability was determined by CCK-8 assay. (B, D) Western blot analysis for the expression of p-AKT, AKT, p-p70S6K, p70S6K, p-MTOR, p-4E-BP1 in SH-SY5Y and SK-N-MC cells treated with 500 and 1000 nM NVP-BEZ235 for 12 h and 24 h. (E, F) Morphological changes in SH-SY5Y and SK-N-MC cells treated with NVP-BEZ235 (500, 1000 nM) for 24 h. (G, H) Western blot analysis for the expression of M CM2 in SH-SY5Y and SK-N-MC cells treated with 500 and 1000 nM NVP-BEZ235 for 12 h and 24 h.
NVP-BEZ235 induced G0/G1 cell cycle arrest but not apoptosis in neuroblastoma cells
To further investigate whether NVP-BEZ235-induced proliferation inhibition was associated with an induction of apoptosis, acridine orange/ethidium bromide (AO/EB) fluorescence were performed and apoptotic related hallmark, PARP and caspase 3 cleavages were measured by immunoblotting on NB cells cultured in the presence of NVP-BEZ235. AO was able to infiltrate into the viable cells, and the nuclei were stained a bright green color. For the integrity of the cell plasma membrane, EB was unable to infiltrate into the cells which remained alive or were at the early stage of apoptosis, while the late apoptotic cells or dead cells had EB inside and the nuclei were stained a bright red color.22 Our results revealed that 500 nM and 1000 nM NVP-BEZ235 did not induce apoptosis in either cell line, as suggested by the absence of bright red color stained cells (Fig. 2A and B), PARP, caspase 3 cleavages (Fig. 2C and D) and sub-G1 fraction DNA histograms (Fig. 2E and F).
Figure 2.
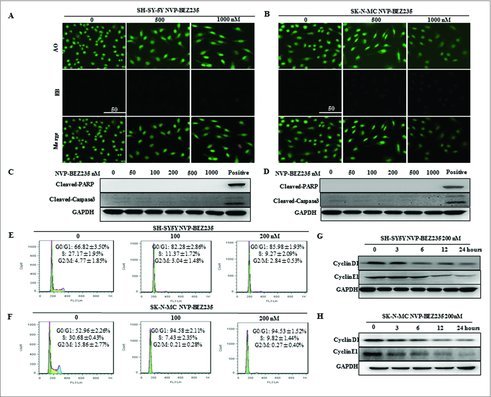
NVP-BEZ235 induced G0/G1 cell cycle arrest but not apoptosis in neuroblastoma cells. (A, B) Morphological changes in SH-SY5Y and SK-N-MC cells treated with NVP-BEZ235 (500, 1000 nM) for 24 h followed by AO/EB staining (magnification, 100 ×). Normal cells showed green fluorescence by acridine orange (AO) staining, apoptotic cells showed yellow-orange fluorescence by merge of ethidium bromide (EB) staining and AO staining. (C, D) Western blot analysis for cleaved caspase-3 and cleaved PARP in SH-SY5Y and SK-N-MC cells treated with serial NVP-BEZ235 for 24 h. (E, F) SH-SY5Y and SK-N-MC cells were treated with NVP-BEZ235 (100, 200 nM) for 24 h. Cell cycles were detected by flowcytometry analysis which determining cellular DNA content with hypotonic PI solution. (G, H) SH-SY5Y and SK-N-MC cells were treated with NVP-BEZ235 (200 nM) for the indicated times. Cyclin D1 and cyclin E1 proteins were performed by western blot analysis.
As apoptosis could not have accounted for the potent inhibitory effect of NVP-BEZ235 on NB cell growth, we next analyzed the effect of NVP-BEZ235 on cell cycle progression in NB cells by flow cytometry using PI staining of DNA content. Exposed to 100 nM and 200 nM NVP-BEZ235 for 24 h, there was a significant increase in the percentage of SH-SY5Y cells in the G0/G1 phase (82.28% ± 2.86% 100 nM, 85.98% ± 1.93% 200 nM) compared with the control group (66.82% ± 3.50%) (Fig. 2E). The G0/G1 cycle arrest was also detected in SK-N-MC cells (Fig. 2F). To further investigate correlation NVP-BEZ235-induced G1 cell cycle arrest with some important G1-phase regulatory proteins in NB cells, the expression of endogenous cyclin D1 and cyclin E1 were detected when SH-SY5Y and SK-N-MC cells are treated with 200 nM NVP-BEZ235 for various time. Cyclin D1 and cyclin E1 gover the restriction point of G1 to S phase progression and their inhibition or degradation lead to G1 phase arrest.23 As indicated by Fig. 2G and H, there was a remarkable reduction or gradual decrease in cyclin D1 and cyclin E1 protein levels in both cell lines. Based on the above results, we concluded that NVP-BEZ235 exerts notable G0/G1 cell cycle arrest in NB cell through the down-regulation of several key G1-phase regulatory proteins.
NVP-BEZ235 down-regulates cyclin D1 and cyclin E1 levels through affecting proteins stability
To elucidate if NVP-BEZ235 mediated down-regulation of cyclin D1 and cyclin E1 expression by transcriptional or post-transcriptional regulation, their mRNA levels were evaluated by Real time-PCR. As shown in Fig. 3A and B, the mRNA levels of cyclin D1 and cyclin E1 were not altered in the presence of 200 nM NVP-BEZ235. These indicate that NVP-BEZ235 may affect the proteins stability of cyclin D1 and cyclin E1 that were detected via CHX-chase analysis. As showed in Fig. 3C-J, NB cells were co-treated with 10 μg/ml CHX for different time followed by pre-treatment 200 nM NVP-BEZ235 for 1 h. NVP-BEZ235 obviously attenuated half-life of proteins in both cells. These data suggested that NVP-BEZ235 reduces cyclin D1 and cyclin E1 protein stability and down-regulates their proteins.
Figure 3.
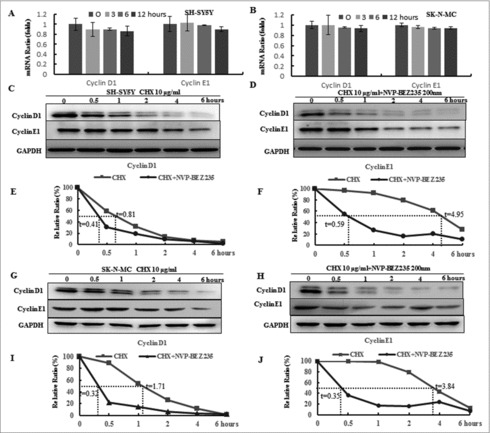
NVP-BEZ235 decreased cyclin D1 and cyclin E1 levels through enhancing their protein degradation. (A, B) SH-SY5Y and SK-N-MC cells were incubated with NVP-BEZ235 for the indicated time periods. Cyclin D1 and cyclin E1 mRNA levels were determined by Q-RT-PCR. (C-J) SH-SY5Y and SK-N-MC cells were pre-treated with vehicle or 200 nM NVP-BEZ235 for 1 h, then adding 10 μg/ml CHX for another various times and cyclin D1 and cyclin E1 protein expression levels were detected by immunoblotting and quantified by the densitometry analysis and normalized against β-Actin expression.
The ubiquitin proteasome pathway involved in NVP-BEZ235-induced the degradation of cyclin D1 and cyclin E1
To explore which pathway is necessary for the down-regulation of cyclin D1 and cyclin E1, protein degradation's effect is worthwhile to be considered. Firstly, lysosomal pathway inhibitors CQ was used to pre-treated for the SH-SY5Y and SK-N-MC cells for 1 h, followed by co-incubation 200 nM NVP-BEZ235 for another 12 h and we found that the levels of cyclin D1 and cyclin E1 proteins were not restored in both cells compared with NVP-BEZ235 single treatment (Fig. 4A-D). Then to investigate whether the 26S proteasome involved in NVP-BEZ235-induced down-regulation of cyclin D1 and cyclin E1, thus the SH-SY5Y and SK-N-MC cells were pre-incubated in the presence of the 26S proteasome inhibitor MG132 for 1 h, followed by co-incubation 200 nM NVP-BEZ235 for another 12 h. NVP-BEZ235-induced reduction of cyclin D1, E1 was abolished by adding MG132 into medium (Fig. 4E-H), compared to the treatment of NVP-BEZ235 alone.
Figure 4.
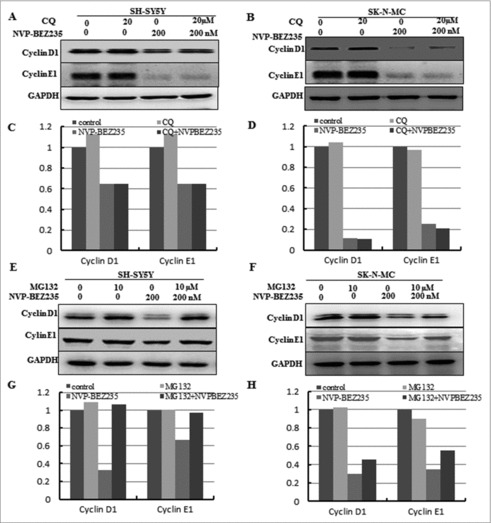
The ubiquitin proteasome pathway involved in NVP-BEZ235- induced the degradation of cyclin D1 and cyclin E1. (A-D) SH-SY5Y and SK-N-MC were pretreated for 1 h with autophagy inhibitor 20 μM CQ and subsequently treated for another 12 hours with 200 nM NVP-BEZ235. Western blot analyzed cyclin D1 and cyclin E1. (E-H) SH-SY5Y and SK-N-MC cells were pretreated for 1 h with 10 μM proteasome inhibitor (MG132) and subsequently treated for another 12 hours with 200 nM NVP-BEZ235 and lysates were probed for cyclin D1 and cyclin E1.
GSK3β is required for NVP-BEZ235- induced degradation of cyclin D1 and cyclin E1
Researchers have reported when PI3K/mTOR signal pathway was activated, the activity of GSK3β would be decreased 20,24. It is needed to elucidate whether NVP-BEZ235 involved in cyclin D1 and cyclin E1 down-regulation in a GSK3β-dependent way. Firstly, we investigate the GSK3β effect under NVP-BEZ235 treatment. NB cells were incubated with NVP-BEZ235 0, 3, 6 and 12 h, respectively. As shown in Fig. 5A and B, the expression of total GSK3β were not perturbed and the phosphorylated GSK3β (p-GSK3β) were down-regulated in a time-dependent manner in the SH-SY5Y and SK-N-MC cells treated by NVP-BEZ235. To assess whether GSK3β involved in NVP-BEZ235-mediated cyclin D1 and cyclin E1 degradation, we used GSK3β inhibitor LiCl and GSK3β specific siRNA. As shown in Fig. 5C and D, SH-SY5Y and SK-N-MC cells pretreated with LiCl for 1 h and then co-incubated with 200 nM NVP-BEZ235 for 12 h, we found that LiCl suppressed NVP-BEZ235-caused cyclin D1 and cyclin E1 degradation. And in Fig. 5E and F, GSK3β protein expressions were effectively knocked down by siRNA and contributed to restoration of cyclin D1 and cyclin E1 levels in NVP-BEZ235-treated SH-SY5Y and SK-N-MC cells. Moreover, depletion of GSK3β by siRNA significantly attenuated NVP-BEZ235-induced G0/G1 arrest in SH-SY5Y and SK-N-MC cells (Fig. 5G and H). These data suggest that NVP-BEZ235-induced cyclin D1 and cyclin E1 degradation and G0/G1 arrest are mediated by the activity of GSK3β.
Figure 5.
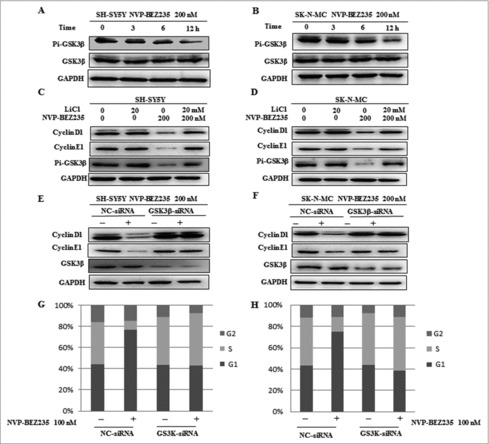
GSK3β is required for NVP-BEZ235- induced degradation of cyclin D1 and cyclin E1. (A and B) SH-SY5Y and SK-N-MC cells were incubated with NVP-BEZ235 for the indicated time periods. Western blot analyzed p-GSK3β and GSK3β. (C, D) SH-SY5Y and SK-N-MC were pretreated with GSK3β inhibitor LiCl for 1 h and then incubated with 200 nM NVP-BEZ235 for 12 h. Western blot analyzed cyclin D1 and cyclin E1. (E and F) SH-SY5Y and SK-N-MC were transfected with the siRNA targeting GSK3β for 48 h and then treated with 200 nM NVP-BEZ235 for another 12 h. The cells were lysed and analyzed by immunoblotting against cyclin D1 and cyclin E1. (G and H) SH-SY5Y and SK-N-MC cells were transfected with GSK3β siRNA, followed by NVP-BEZ235 treatment. The cell cycle distribution was measured by flow cytometric analysis.
NVP-BEZ235 down-regulates cyclin D1 and cyclin E1 levels via reducing p-GSK3β in NB xenograft tumor
NB xenograft mouse model were generated to assess the pathophysiological role of NVP-BEZ235 in vivo and further determine the regulation of cyclin D1, E1 and GSK3β. Compared with vehicle mice, tumor growth was impaired in NVP-BEZ235 -treated group as shown in Fig. 6A and B. In agreement with the blockage of cell cycle in vitro, NVP-BEZ235 also reduced the levels of cyclin D1, E1 in xenograft tumors, accompanied by decrease of p-GSK3β (Fig. 6C and D). We next verified NVP-BEZ235 could accumulate ubiquitinated cyclin D1, E1, which is consistent with the consecutive proteasomal degradation of cyclin D1, E1 in tumor tissues cell lysates (Fig. 6E and F) by immunoprecipitation assay.
Figure 6.
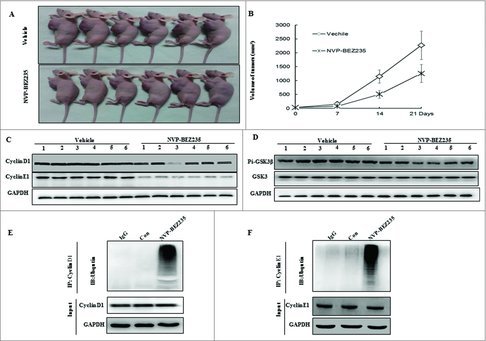
NVP-BEZ235 induced cyclin D1 and cyclin E1 ubiquitin-dependent proteasomal degradation through reducing p-GSK3β in NB xenograft tumor. (A) Treatment was initiated when the average tumor size reached 50 mm3. Nude Mice with SH-SY5Y xenografts were administered vehicle or NVP-BEZ235 (20 mg/kg/day). After 3-week consecutive treatment, the mice were sacrificed. (B) Tumor volumes were measured every week. (C and D) Tumor extracts were lysed and analyzed by immunoblotting against cyclin D1, cyclin E1, p-GSK3β and GSK3β. (E and F) Tumor extracts were immunoprecipitated (IP) by anti-cyclin D1 and cyclin E1 followed by immunoblotting with anti-Ub (top) and anti-cyclin D1 and cyclin E1 (bottom).
Discussion
PI3K/Akt/mTOR signaling pathways are critical for controlling normal cellular functions and the abnormal activation of this pathway play an important role in progression, metastasis, and chemoresistance in various types of cancer, including advanced neuroblastoma.10,25,26 NVP-BEZ235, as a potent dual PI3K and mTOR kinase inhibitor, presents potent antitumor effects via multiple mechanisms, including disturbing cell cycle and differentiation, promoting apoptosis, autophagy, angiogenesis and EMT in preclinical model of most human cancer, but the effect of NVP-BEZ235 has rarely been reported in neuroblastoma. In this report, we demonstrated NVP-BEZ235 failed to induce apoptosis. It induced cyclin D1 and cyclin E1 degradation and G0/G1 arrest in a GSK3β-dependent manner.
As is well-known that phosphorylation of 4E-BP and P70S6K facilitate assembly of the translational initiation complex with subsequent translation of proteins responsible for the progression of the cell from G0/G1 to S phase, such as cyclin D1 and cyclin E1.27-28 It is proved for the first time that NVP- BEZ235 inhibited neuroblastoma cells proliferation through blocking cell cycle progression at G0/G1 phase accompanied by the decrease of cyclin D1 and cyclin E1 protein expression in neuroblastoma cells. The up-regulation of cyclin D1 and cyclin E1 are hallmarks of poor prognosis in various human cancers including those of the breast, esophagus, bladder and lung.15,17,29 It was believed that aberrant degradation system appears to be responsible for the increased levels of cyclin D1 and cyclin E1.30,31 Consistent with this, there was a remarkable reduction in cyclin D1 and cyclin E1 proteins levels whereas their mRNA levels were not obviously changed in NVP-BEZ235-treated NB cells. And CHX-chase analysis further authenticated NVP-BEZ235 obviously attenuated half-life of these proteins and promoted their degradation. This manifested the significance of enhancing cyclin D1 and cyclin E1 degradation by NVP-BEZ235-induced in NB cells.
Prior reports have demonstrated that cyclin D1, E1 was modulated ubiquitin-dependent proteasomal degradation system which labeled target proteins for destruction by the 26S proteasome through three critical enzymes, including E1 ubiquitin-activating enzyme, E2 ubiquitin-activating enzyme and E3 ligase.32,33 We also affirm the notion that proteasomal degradation is sufficient to regulate cyclin D1 and cyclin E1 coupled with the fact that proteasome inhibitors can recover their reduction in NVP-BEZ235-treated neuroblastoma cells. Likewise, NVP-BEZ235 could accumulate ubiquitinated cyclin D1 and cyclin E1 in NB xenograft tumor cells lysates. Cyclin D1 is phosphorylated at Thr-286 and/or T288 by GSK-3β or Mirk/Dyrk1b, promoting cyclin D1 nuclear export and subsequent rapid ubiquitin-dependent degradation within the cytoplasm.16,34 The phosphorylation of cyclin E located at Thr380 which is thought to be the result of cyclin E/Cdk2 autocatalytic activity and GSK3β mediated is critical for ubiquitination.17 Above observations illustrate GSK-3β is indispensable for the turnover of cyclin D1 and cyclin E1. And GSK3β is negatively regulated by the PI3K/Akt/mTOR and can be inactivated by phosphorylation.35,36 In our study, we presented that NVP-BEZ235 dephosphorylates of GSK-3β, resulting in its activation. Likewise, similar results were also observed in vivo experiments. Our subsequent experiments elaborate inhibition of GSK-3β by its inhibitor LiCl or specific siRNA leads to abolishment of cyclin D1 and cyclin E1 degradation induced by NVP-BEZ235. In addition, blockade of GSK-3β strikingly rescued NVP-BEZ235-induced G0/G1 arrest which highlights the importance of the GSK3β-dependent catabolism of cyclin D1 and cyclin E1 in NB cells.
Taken together, these results clearly indicated that NVP-BEZ235 can enhance the degradation of cyclin D1, E1 via ubiquitin proteasome pathway in a GSK3β-dependent manner thus preserving the neuroblastoma cells in G0/G1 status and growth inhibition. Therefore, the therapeutic ablation of p-GSK3β, cyclin D1 and cyclin E1 may be critical targets for the prevention and treatment of neuroblastoma cells.
Materials and methods
Reagent, cell lines and cell culture
NVP-BEZ235 was purchased from Novartis (Selleck, China) and dissolved in DMSO to a stock concentration of 1 mM. CQ, MG132 were obtained from Sigma-Aldrich and dissolved in sterile H2O to a stock concentration of 20 mM and 10 mM respectively. CHX was purchased from Sigma (Sigma-Aldrich, USA) and dissolved in ethanol a stock concentration of 100 mg/ml. The stock solutions were wrapped in foil and maintained at −20°C.
SH-SY5Y, SK-N-MC were obtained from the Shanghai Institutes for Biological Sciences (Shanghai, China) and were cultured in RPMI 1640 medium, containing 10% fetal bovine serum (FBS) (Gemini, Australia) and 1% penicillin-streptomycin(Gibco, USA), and under standard culture conditions (37˚C and 5% CO2). Cells in the logarithmic phase of growth were used in all experiments.
Cell Counting Kit-8 assay
The cell proliferation was assessed by using Cell Counting Kit-8 reagent (Dojindo Molecular Technologies, Japan). SH-SY5Y and SK-N-MC cells were seeded in 96-well culture plates at 5 × 103 cells/well in triplicate and allowed to settle overnight, and treated with serial concentration of NVP-BEZ235 (0, 10, 25, 50, 100, 200, 500, 1000 nM) of in 200 μl medium for 24 h. The cells were then treated with CCK-8 solution and incubated for 2 h according to the CCK-8 kit instructions. The absorbance was measured at 450 nm using a microplate reader (Bio-Rad, Hercules, CA, USA). Each experiment was performed at least 3 times independently.
Acridine orange/ethidium bromide (AO/EB) fluorescence
NB cells were cultivated in a 24-well plate exposed to various concentrations (0, 500 and 1000 nM) of NVP-BEZ235 for 24 h in a humidified incubator (37˚C, 5% CO2). After removing the culture solution and washing three times with PBS, the cells were stained with 200 μl mixture of (100 μg/ml) acridine orange (AO) and (100 μg/ml) ethidium bromide (EB) (Solarbio, China) with the 1:1 AO to EB, and then incubated the plate for 3 min in the incubator. These cells were visualized by a fluorescence microscope (Nikon Eclipse Ti, Japan).
Cell cycle by flow cytometric analysis
NB cells in exponential growth were seeded in 6-well plate at a density of 5 × 105 per well for 24 h attachment, then they were treated with NVP-BEZ235 (0,100,200 nM) for 24 h. For cell cycle analysis, cells were digested with 0.25% trypsin and centrifuged at 2000 g for 5 minutes and washed three times with PBS and fixed by ice-cold 75% (v/v) ethanol at −20°C for 2 h, then after washing twice with PBS, the cells were resuspended with 50 μl PI solution (PI 50 μg/ml and RNase A 100 μg/ml). After incubating for 15 minutes at room temperature in the dark at then cell cycle distribution were analyzed by FACS can flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Western blot analysis
Cultured cells or mouse tumor tissues were homogenized on ice 1 × RIPA lysis buffer containing phosphatase and protease inhibitor cocktail(Roche, Swiss) and the protein concentration was measured by BCA Protein Assay Kit(Takara, Japan). Equal amounts (50 μg) of isolated proteins were separated by SDS polyacrylamide gel electrophoresis and transferred to the PVDF membrane. Membranes were blocked in 5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20 (TBST, pH7.4) for 1 h at room temperature, and incubated overnight at 4˚C a dilution of 1:1000 with primary antibodies for phospho-AKT(Ser473), AKT, phospho-p70s6k(Thr389), p70s6k, phospho-MTOR(Ser2448), phospho-4E-BP1(Thr37/46), MCM2, GAPDH, cleaved caspase-3, cleaved PARP, cyclin D1, cyclin E1, total and phosphorylated GSK3β (all from Cell Signaling Technology). Then the membranes were probed for 2 h at room temperature with species-specific HRP-conjugated rabbit anti-mouse IgG (Cell Signaling Technology, USA) at a dilution of 1:10000. Immunoreactive bands were identified by Immobilon Western chemiluminescent HRP substrate (Millipore Co., USA). The GAPDH is a loading control and the gray value of the protein brands were quantified by Image J software.
Immunoprecipitation Assays
After washing in cold PBS, Cells or tissues were lysed in 500 μl ice-cold RIPA lysis buffer(50 mM Tris-HCL pH 7.4, 1 mM EDTA, 0.5% Sodium Deoxycholate, 1%NP-40, 0.1%SDS, and 150 mM NaCl containing protease and phosphatase inhibitors(complete EDTA-free; Roche Applied Science, Mannheim Germany) on ice for 30 minutes. Total cell lysates were clarified by centrifugation at 12,000g for 15min at 4˚C, and were divided into two parts, one (400 μl, 500 μg) used for the immunoprecipitation assays and the other (50 μl) for total protein analysis. Lysates supernatants was incubated with 3 μg of anti-ubiquitin or rabbit IgG overnight at 4˚C with rotation, followed by preclearing with 50% protein A/G agarose slurry for 2 h and then the complexes were incubated with protein A/G agarose for 4 h at 4˚C. After washing 5 times with lysis buffer by centrifugation at 5,000 rpm for 5 min, samples were eluted in 2xSDS loading buffer by boiling for 8 min and subjected to immunoblot analysis with anti-cyclin D1 or anti-cyclin E1 or anti-ubiquitin antibody.
Quantitative real-time PCR
Total cellular RNA was extracted from NB cell lines using RNA Isolation Kit (Roche, Basel, Switzerland). The reaction was performed in Micro Amp Optical 96-well plates with SYBR Premix Ex Taq™ II (Takara, Japan) in Real-Time PCR Detection System according to manufacture instructions. Related gene expression was assessed after normalization by housekeeping gene human β-Actin. Primers were designed and synthesized by Sangon Biotech Co. Ltd (Shanghai, China). β-actin (Forward:5´-TCGTGCGTGACATTAAGGAG-3´ and Reverse: 5´-GTCAGGCAGCTCGTAGCTCT-3´), cyclin D1 (Forward: 5´-GCTGCGAAGTGGAAACCATC-3´ and Reverse: 5´-CCTCCT TCTGCACACATTTGAA-3´), cyclin E1 (Forward: 5´- GCCAGCCTTGGG ACAATAATG-3´ and Reverse: 5´-CTTGCACGTTGAGTTTGGGT-3´).
RNA interference
Small interfering RNA (siRNA) specific to human GSK3β and negative control siRNA were synthesized by GenePharma. Sequences are as follows: Negative control, 5′-UUCUCCGAACGUGUCACGUTT-3′.SiRNA1:5′-GGGCAGUGAUUCUGUUUAAUU-3′, SiRNA2:5′-GGGCCUUAUAUACUCUAAAUU-3′. SiRNA1 and SiRNA2 were equally mixed. Both cells were transfected with mixed siRNA using Lipofectamine-3000 (Life Technologies, USA) according to the manufacturer's protocol.
Nude mouse xenograft model of NB cancer
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Shanghai Jiao Tong University. Five- to six-week old female athymic (nu/nu) mice were housed under pathogen-free conditions in micro-isolator cages with laboratory chow and water ad libitum. SH-SY5Y cells at 5 × 106 suspended in 100 μl PBS were injected s.c. into the flank region of nude mice. When size of local tumors reached certain ranges (∼50 mm3), the mice were randomized into two groups according to tumor volumes and body weights for the following treatments: vehicle control (DMSO), NVP-BEZ235 (20 mg/kg/day, ip.). Tumor volumes were measured using caliper measurements once every 5 days and calculated with the formula: V = a2 × b × 0.4, where “a” is the smallest diameter and “b” is the diameter perpendicular to “a”. The body weight, feeding behavior, and motor activity of each animal were monitored as indicators of general health. Mice were euthanized after 3 weeks of interventions and tumor tissues were excised and fixed in 10% buffered formalin and embedded in paraffin.
Statistical analysis
Each sample was analyzed in triplicate, and experiments were repeated three times. In all figures, error bars are standard deviations. Statistical analyses were performed by Microsoft Office Excel 2007(Microsoft, Albuquerque, NM, USA) and Statistica version 10.0 (StatSoft, Tulsa, OK, USA). Differences between mean values were evaluated by the unpaired t-test. Differences were considered statistically significant at P < 0.05.
Funding Statement
This work was supported in part by the National Natural Science Foundation of China (81172322, 81302006), Shanghai Municipal Education Committee (13ZZ089), Science and Technology Committee of Shanghai (14401901500), Science and Technology Committee of Baoshan District (12-E-2), NO.3 People's Hospital affiliated to Shanghai Jiao-Tong University School of Medicine (sy2013-008).
Abbreviations
- 4E-BP1
eukaryotic initiation factor 4E-binding protein 1
- AKT
protein kinase B
- ALK
anaplastic lymphoma kinase
- CHX
Chlorhexidine
- CQ
chloroquine
- DMSO
dimethyl sulfoxide;GSK3β, glycogen synthase kinase-3β
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- MCM2
Minichromosome maintenance protein2
- mTOR
mammalian target of rapamycin
- NB cells
neuroblastoma cells
- p70s6k
ribosomal protein S6 kinase, 70kDa
- PARP
Poly (ADP-ribose) polymerase
- PI-3K
phosphatidylinositol 3 kinase
Declaration of interest statement
The authors enounce no conflict of interest in this work.
References
- [1].Duong C, Yoshida S, Chen C, Barisone G, Diaz E, Li Y, Beckett L, Chung J, Antony R, Nolta J, et al.. Novel targeted therapy for neuroblastoma: Silencing the MXD3 gene using siRNA. Pediatr Res. 2017;82:527–535. https://doi.org/ 10.1038/pr.2017.74. PMID:28419087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Olsen RR, Otero JH, Garcia-Lopez J, Wallace K, Finkelstein D, Rehg JE, Yin Z, Wang YD, Freeman KW. MYCN induces neuroblastoma in primary neural crest cells. Oncogene. 2017;36:5075–5082. https://doi.org/ 10.1038/onc.2017.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397-411. https://doi.org/ 10.1038/nrc3526. PMID:23702928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106-20. https://doi.org/ 10.1016/S0140-6736(07)60983-0. PMID:17586306 [DOI] [PubMed] [Google Scholar]
- [5].Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, Weiss WA. Neuroblastoma. Nat Rev Dis Primers 2016;2:16078. https://doi.org/ 10.1038/nrdp.2016.78. PMID:27830764 [DOI] [PubMed] [Google Scholar]
- [6].Thole TM, Lodrini M, Fabian J, Wuenschel J, Pfeil S, Hielscher T, Kopp-Schneider A, Heinicke U, Fulda S, Witt O, et al.. Neuroblastoma cells depend on HDAC11 for mitotic cell cycle progression and survival. Cell Death Dis. 2017;8:e2635. https://doi.org/ 10.1038/cddis.2017.49. PMID:28252645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang Y, Wang L, Guan S, Cao W, Wang H, Chen Z, Zhao Y, Yu Y, Zhang H, Pang JC, et al.. Novel ALK inhibitor AZD3463 inhibits neuroblastoma growth by overcoming crizotinib resistance and inducing apoptosis. Sci Rep. 2016;6:19423. https://doi.org/ 10.1038/srep19423. PMID:26786851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Westhoff MA, Faham N, Marx D, Nonnenmacher L, Jennewein C, Enzenmuller S, Gonzalez P, Fulda S, Debatin KM. Sequential dosing in chemosensitization: targeting the PI3K/Akt/mTOR pathway in neuroblastoma. PLoS One. 2013;8:e83128. https://doi.org/ 10.1371/journal.pone.0083128. PMID:24391739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xie G, Wang Z, Chen Y, Zhang S, Feng L, Meng F, Yu Z. Dual blocking of PI3K and mTOR signaling by NVP-BEZ235 inhibits proliferation in cervical carcinoma cells and enhances therapeutic response. Cancer Lett. 2017;388:12-20. https://doi.org/ 10.1016/j.canlet.2016.11.024. PMID:27894954 [DOI] [PubMed] [Google Scholar]
- [10].Seitz C, Hugle M, Cristofanon S, Tchoghandjian A, Fulda S. The dual PI3K/mTOR inhibitor NVP-BEZ235 and chloroquine synergize to trigger apoptosis via mitochondrial-lysosomal cross-talk. Int J Cancer. 2013;132:2682-93. https://doi.org/ 10.1002/ijc.27935. PMID:23151917 [DOI] [PubMed] [Google Scholar]
- [11].Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511-26. https://doi.org/ 10.1038/onc.2008.246. PMID:18794885 [DOI] [PubMed] [Google Scholar]
- [12].Bhende PM, Park SI, Lim MS, Dittmer DP, Damania B. The dual PI3K/mTOR inhibitor, NVP-BEZ235, is efficacious against follicular lymphoma. Leukemia. 2010;24:1781-4. https://doi.org/ 10.1038/leu.2010.154. PMID:20703254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cao P, Maira SM, Garcia-Echeverria C, Hedley DW. Activity of a novel, dual PI3-kinase/mTor inhibitor NVP-BEZ235 against primary human pancreatic cancers grown as orthotopic xenografts. Br J Cancer. 2009;100:1267-76. https://doi.org/ 10.1038/sj.bjc.6604995. PMID:19319133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gobin B, Battaglia S, Lanel R, Chesneau J, Amiaud J, Redini F, Ory B, Heymann D. NVP-BEZ235, a dual PI3K/mTOR inhibitor, inhibits osteosarcoma cell proliferation and tumor development in vivo with an improved survival rate. Cancer Lett. 2014;344:291-8. https://doi.org/ 10.1016/j.canlet.2013.11.017. PMID:24333720 [DOI] [PubMed] [Google Scholar]
- [15].Masamha CP, Benbrook DM. Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of cyclin E2 in ovarian cancer cells. Cancer Res. 2009;69:6565-72. https://doi.org/ 10.1158/0008-5472.CAN-09-0913. PMID:19638577 [DOI] [PubMed] [Google Scholar]
- [16].Alao JP. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. https://doi.org/ 10.1186/1476-4598-6-24. PMID:17407548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Moroy T, Geisen C. Cyclin E. Int J Biochem Cell Biol. 2004;36:1424-39. https://doi.org/ 10.1016/j.biocel.2003.12.005. PMID:15147722 [DOI] [PubMed] [Google Scholar]
- [18].Kuwajerwala N, Cifuentes E, Gautam S, Menon M, Barrack ER, Reddy GP. Resveratrol induces prostate cancer cell entry into s phase and inhibits DNA synthesis. Cancer Res. 2002;62:2488-92. PMID:11980638 [PubMed] [Google Scholar]
- [19].Liu HX, Ly I, Hu Y, Wan YJ. Retinoic acid regulates cell cycle genes and accelerates normal mouse liver regeneration. Biochem Pharmacol. 2014;91:256-65. https://doi.org/ 10.1016/j.bcp.2014.07.003. PMID:25087568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Daniel C C, Matthew B C, David J P, Michael Collins M, Ghebremichael M, Atkins MB, Signoretti S, Mier JW. The Efficacy of the Novel Dual PI3-Kinase/mTOR Inhibitor NVPBEZ235 Compared to Rapamycin in Renal Cell Carcinoma. Clin Cancer Res. 2010;16:3628-38. https://doi.org/ 10.1158/1078-0432.CCR-09-3022. PMID:20606035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Duan Y, Tian L, Gao Q, Liang L, Zhang W, Yang Y, Zheng Y, Pan E, Li S, Tang N. Chromatin remodeling gene ARID2 targets cyclin D1 and cyclin E1 to suppress hepatoma cell progression. Oncotarget. 2016;7:45863-75. https://doi.org/ 10.18632/oncotarget.10244. PMID:27351279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang LD, Liu Z, Liu H, Ran DM, Guo JH, Jiang B, Wu YL, Gao FH. Oridonin enhances the anticancer activity of NVP-BEZ235 against neuroblastoma cells in vitro and in vivo through autophagy. Int J Oncol. 2016;49:657-65. PMID:27278249 [DOI] [PubMed] [Google Scholar]
- [23].Gladden AB, Diehl JA. Cell cycle progression without cyclin E/CDK2: Breaking down the walls of dogma. Cancer Cell. 2003;4:160-2. https://doi.org/ 10.1016/S1535-6108(03)00217-4. PMID:14522248 [DOI] [PubMed] [Google Scholar]
- [24].Gwak H, Kim Y, An H, Dhanasekaran DN, Song YS. Metformin induces degradation of cyclin D1 via AMPK/GSK3beta axis in ovarian cancer. Mol Carcinog. 2017;56:349-58. https://doi.org/ 10.1002/mc.22498. PMID:27128966 [DOI] [PubMed] [Google Scholar]
- [25].Altomare DA, Wang HQ, Skele KL, De Rienzo A, Klein-Szanto AJ, Godwin AK, Testa JR. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene. 2004;23:5853-7. https://doi.org/ 10.1038/sj.onc.1207721. PMID:15208673 [DOI] [PubMed] [Google Scholar]
- [26].Moon du G, Lee SE, Oh MM, Lee SC, Jeong SJ, Hong SK, Yoon CY, Byun SS, Park HS, Cheon J. NVP-BEZ235, a dual PI3K/mTOR inhibitor synergistically potentiates the antitumor effects of cisplatin in bladder cancer cells. Int J Oncol. 2014;45:1027-35. https://doi.org/ 10.3892/ijo.2014.2505. PMID:24969552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ching CB, Hansel DE. Expanding therapeutic targets in bladder cancer: The PI3K/Akt/mTOR pathway. Lab Invest. 2010;90:1406-14. https://doi.org/ 10.1038/labinvest.2010.133. PMID:20661228 [DOI] [PubMed] [Google Scholar]
- [28].Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960-76. https://doi.org/ 10.1016/j.cell.2017.02.004. PMID:28283069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang H, Zhang X, Geng L, Teng L, Legerski RJ. Artemis regulates cell cycle recovery from the S phase checkpoint by promoting degradation of cyclin E. J Biol Chem. 2009;284:18236-43. https://doi.org/ 10.1074/jbc.M109.002584. PMID:19423708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mandal S, Freije WA, Guptan P, Banerjee U. Metabolic control of G1-S transition: cyclin E degradation by p53-induced activation of the ubiquitin-proteasome system. J Cell Biol. 2010;188:473-9. https://doi.org/ 10.1083/jcb.200912024. PMID:20176921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shao J, Sheng H, DuBois RN, Beauchamp RD. Oncogenic Ras-mediated cell growth arrest and apoptosis are associated with increased ubiquitin-dependent cyclin D1 degradation. J Biol Chem. 2000;275:22916-24. https://doi.org/ 10.1074/jbc.M002235200. PMID:10781597 [DOI] [PubMed] [Google Scholar]
- [32].Yang Y, Wu J, Cai J, He Z, Yuan J, Zhu X, Li Y, Li M, Guan H. PSAT1 regulates cyclin D1 degradation and sustains proliferation of non-small cell lung cancer cells. Int J Cancer. 2015;136:E39-50. https://doi.org/ 10.1002/ijc.29150. PMID:25142862 [DOI] [PubMed] [Google Scholar]
- [33].Sukamporn P, Rojanapanthu P, Silva G, Zhang X, Gritsanapan W, Baek SJ. Damnacanthal and its nanoformulation exhibit anti-cancer activity via cyclin D1 down-regulation. Life Sci. 2016;152:60-6. https://doi.org/ 10.1016/j.lfs.2016.03.038. PMID:27018445 [DOI] [PubMed] [Google Scholar]
- [34].Barbash O, Egan E, Pontano LL, Kosak J, Diehl JA. Lysine 269 is essential for cyclin D1 ubiquitylation by the SCF(Fbx4/alphaB-crystallin) ligase and subsequent proteasome-dependent degradation. Oncogene. 2009;28:4317-25. https://doi.org/ 10.1038/onc.2009.287. PMID:19767775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yu XJ, Han QB, Wen ZS, Ma L, Gao J, Zhou GB. Gambogenic acid induces G1 arrest via GSK3beta-dependent cyclin D1 degradation and triggers autophagy in lung cancer cells. Cancer Lett. 2012;322:185-94. https://doi.org/ 10.1016/j.canlet.2012.03.004. PMID:22410463 [DOI] [PubMed] [Google Scholar]
- [36].Mulholland DJ, Dedhar S, Wu H, Nelson CC. PTEN and GSK3beta: Key regulators of progression to androgen-independent prostate cancer. Oncogene. 2006;25:329-37. https://doi.org/ 10.1038/sj.onc.1209020. PMID:16421604 [DOI] [PubMed] [Google Scholar]