

ABSTRACT
Maintenance of stem cell plasticity is determined by the ability to balance opposing forces that control gene expression. Regulation of transcriptional networks, signaling cues and chromatin-modifying mechanisms constitute crucial determinants of tissue equilibrium. Histone modifications can affect chromatin compaction, therefore co-transcriptional events that influence their deposition determine the propensities toward quiescence, self-renewal, or cell specification. The Paf1 complex (Paf1C) is a critical regulator of RNA PolII elongation that controls gene expression and deposition of histone modifications, however few studies have focused on its role affecting stem cell fate decisions. Here we delineate the functions of Paf1C in pluripotency and characterize its impact in deposition of H2B ubiquitylation (H2BK120-ub) and H3K79 methylation (H3K79me), 2 fundamental histone marks that shape transcriptional regulation. We identify that H2BK120-ub is increased in the absence of Paf1C on its embryonic stem cell targets, in sharp contrast to H3K79me, suggesting opposite functions in the maintenance of self-renewal. Furthermore, we found that core pluripotency genes are characterized by a dual gain of H2BK120-ub and loss of H3K79me on their gene bodies. Our findings elucidate molecular mechanisms of cellular adaptation and reveal novel functions of Paf1C in the regulation of the self-renewal network.
KEYWORDS: Elongation, Embryonic stem cells, Pluripotency, Paf1 complex, Phf5a, Promoter-proximal pausing, Self-renewal network, RNA Polymerase II
Introduction
Cellular identity is characterized by distinct gene expression patterns and the mechanisms that regulate transcriptional activity are crucial to an organism's development.1 Alterations in cellular homeostasis often lead to disease due to aberrant regulation of chromatin.2 Therefore, elucidating the functions of cellular adaptation is paramount to address a range of issues including the role of transcription in normal development and its deregulation in disease.
The state of chromatin, which is defined as the wrapping of DNA with histones together with non-histone proteins, exerts pivotal effects on the establishment and maintenance of gene expression.3 Post-translational modifications of histones can directly affect chromatin assembly or compaction.4 Furthermore, they function as binding sites for effector proteins5,6 including additional chromatin-remodeling complexes,7 ultimately affecting transcriptional initiation and/or elongation.8,9 Several histone modifications may also act collaboratively in developmental transitions denoting their importance in chromatin regulation. Consistent with their fundamental role in cellular differentiation, many chromatin remodelers have been also implicated in human disease including cancer.10,11
Embryonic stem cells (ESCs) are derived from the inner cell mass of the pre-implantation blastocyst and are pluripotent defined by their ability to contribute to all tissues of the adult organism.12,13 ESCs can self-renew in vitro and are characterized by an open chromatin configuration.14,15 However, in differentiation conditions they commence cell specification programs and differentiated cells are marked by dynamic changes in chromatin state and global remodeling.16 These events ultimately result in a progressive transition to a more compact and repressive chromatin state. Pluripotent and differentiated cells are therefore an excellent tool to study cell plasticity changes and over the past years extensive work has resulted in comprehensive epigenome maps. However, a global understanding of the alterations in the state of chromatin and how these result in gene expression changes are far from complete.
The Paf1 transcriptional complex (Paf1C) is known to exert context-specific roles in modulation of gene expression.17,18 Paf1C is important for cell differentiation as it regulates RNA Polymerase II (RNA PolII) function and is therefore necessary in development.19–21 We have recently studied its roles in self-renewal, cellular reprogramming and differentiation and identified the PHD-finger protein 5a (Phf5a) as a potent modulator of Paf1C in pluripotency.22 Phf5a is highly expressed in pluripotent cells (ESCs and iPSCs), however, its expression levels are downregulated in differentiation. We demonstrated that Phf5a binds to the Paf1 complex (Paf1C) controlling its integrity and facilitates expression of pluripotency genes through Paf1C binding at their loci. The Paf1C therefore functions as a positive regulator of transcription in ESCs dictating RNA PolII elongation rates. Phf5a-depleted ESCs exhibit aberrant promoter-proximal pausing of self-renewal genes and decreased Ser2-RNA PolII phosphorylation, a hallmark of effective elongation.
Furthermore, Paf1C regulates deposition of histone modifications associated with transcription, as Ser2-phosphorylation and subunit integrity can influence their levels.17,18 At least 4 histone post-translational modifications are associated with Paf1C functions manifesting its importance for histone crosstalk. These include histone 3 lysine 4 methylation (H3K4me), histone 3 lysine 79 methylation (H3K79me), histone 3 lysine 36 methylation (H3K36me) and histone 2B lysine 120 ubiquitylation (H2BK120-ub). All of those modifications are primarily associated with coding regions of transcribed genes, underlying their involvement in RNA PolII elongation.23 Histone modifications are mainly considered a consequence of transcriptional activity, rather than a cause, suggesting a complex relationship with RNA PolII.24 In particular, the Paf1C-related histone modifications have been assigned co-transcriptional roles, regulating nucleosome dynamics during RNA PolII transcription and recruiting additional regulators of elongation and RNA processing.25–28 Paf1C has been recently implicated to function both as an activator22,29 and as a repressor30 of transcriptional activity, suggesting that its behavior may be altered by additional factors depending on the cellular context. However, how deposition of histone modifications occurs during elongation and how Paf1C regulates activation or repression of gene expression is a topic of intense investigation.
There are several differences regarding the role of Paf1C in these processes between lower and higher eukaryotes that can shed light on its diverse functions. In yeast, several Paf1C subunits are important for modification of histones and H2BK120 monoubiquitylation is important for histone crosstalk. Paf1C recruits the E2 ubiquitin-conjugating enzyme Rad6 and the E3 ligase Bre1 that monoubiquitylate H2B at lysine 123 (lysine 120 in mammals)31–33 and the Rtf1 subunit is required for H2BK120-ub.34 In addition, Paf1- and Rtf1-null yeast cells have lower levels of H3K4me3 and Set1 histone methyltransferase is absent from gene promoters.35,36 Furthermore, Ctr9, Paf1 and Cdc73 subunits are required for full levels of H3K36me3 by Set2 methyltransferase.37 Therefore in yeast, histone H2B monoubiquitination is considered a co-transcriptional event regulating H3K4 and H3K79 methylation by Set1 and Dot1 methyltransferases, respectively manifesting the importance of Paf1C in histone crosstalk.38–41
In mammals, however, the role of Paf1C in histone modification crosstalk is unclear, and although Paf1C positively affects deposition of elongation marks H3K36me and H3K79me in actively expressed genes, its function regarding H2BK120-ub might be different. Mammalian H2BK120-ub is deposited by RNF20 and RNF40, which are both homologs of yeast E3 ligase Bre1 and is preferentially enriched in the coding regions of genes.42–44 It marks active genes and was shown to play important roles in Hox gene activation.42 In stem cells, H2BK120-ub associates with highly transcribed genes, and its levels significantly increase upon differentiation.45,46 Inhibition of H2BK120-ub by RNF20/40 depletion or by mutation of lysine 120, impairs differentiation and inhibits lineage-specific gene upregulation.45 Strikingly, however, Paf1C is downregulated in ESC differentiation,22 whereas H2BK120-ub is increased, suggesting a possible negative regulation of this mark by Paf1C in pluripotency.
We have previously shown that loss of Phf5a can recapitulate Paf1C depletion resulting in the inability of Paf1C to bind its target genes.22 Phf5a depletion negatively affects elongation of pluripotency genes and results in efficient loss of Paf1C-associated H3K79 and H3K36 methylation on gene bodies of Paf1C targets. In contrast, the promoter-associated mark H3K4me3 was not affected upon Phf5a depletion, suggesting elongation-specific functions for Phf5a resulting in promoter-proximal RNA PolII pausing.47–49 Here we study the roles of Phf5a and Paf1C in H2BK120-ub and compare its deposition with other Paf1C-dependent histone modifications. We found that H2BK120 is increased in gene bodies of pluripotency-associated loci following shPhf5a depletion and Paf1C loss. Furthermore, direct comparison with H3K79 methylation reveals that Paf1C targets are characterized by a dual gain of H2BK120-ub and loss of H3K79me marks. Our results suggest an inhibitory role for Paf1C on H2BK120-ub deposition in pluripotency genes.
Results
To investigate the role of H2BK120 ubiquitylation in pluripotent cells and its association with Paf1C function we directly interrogated its occupancy using ChIP-sequencing in the presence or absence of Phf5a. We observed a global alteration in H2BK120-ub densities upon Phf5a silencing, supporting a critical role of Paf1C function for this histone mark (Fig. 1a). We identified approximately 4580 transcripts that gained H2BK120-ub and another 1350 that lost density of this mark after Phf5a depletion, consistent with its overall accumulation in stem cell differentiation.45,46 We next dissected peak localization and determined occupancy among transcription start sites (TSSs), coding regions (gene bodies), as well as upstream regulatory or intergenic regions. Consistent with regulation of active gene expression, we found that H2BK120-ub peaks fall almost exclusively (90%) within gene bodies (Fig. 1b).
Figure 1.
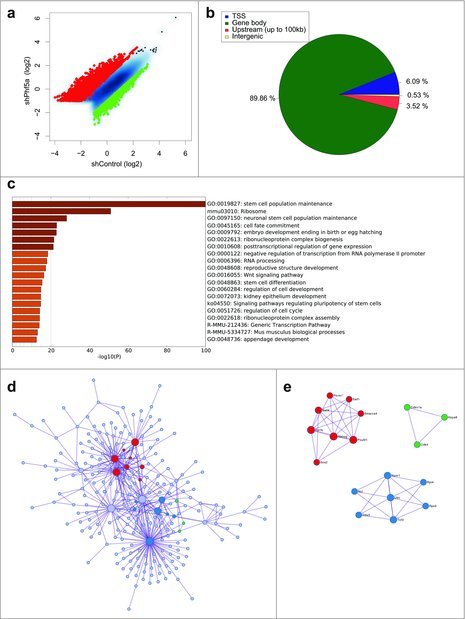
(a) Scatterplot of differentially regulated H2BK120-ub peaks in mouse ESCs following shControl or shPhf5a knockdown, respectively. Red: Transcripts that significantly gain H2BK120-ub densities. Green: Transcripts that significantly lose H2BK120-ub densities. (b) Binding profiles for genomic distribution of H2BK120-ub peaks (Transcription start site (TSS), Gene body, Upstream (up to 100kb) and intergenic) in ESCs, showing preferential (90%) binding within gene bodies. (c) Statistically enriched gene ontology terms and canonical pathways using Metascape (http://metascape.org) for all significantly altered H2BK120-ub transcripts after shPhf5a depletion. (d) Protein-protein interaction network of representative terms after hierarchical clustering analysis using MCODE.51 Each MCODE network node is assigned a unique color. Gene ontology enrichment analysis was applied to each MCODE network nodes to assign “meanings” to the network component. (e) Protein-protein interaction network of MCODE node components. Representative proteins include several core pluripotency factors that consist previously identified Paf1C targets.22
To assign functional characteristics on differentially H2BK120-ub bound transcripts, we performed gene ontology enrichment analysis and identified all statistically significant terms using Metascape (http://metascape.org).50 Interestingly, gene ontology (GO) analysis revealed signatures associated with stem cell identity and includes categories of genes that regulate stem cell maintenance, cell fate commitment, and embryo development, suggesting that H2BK120-ub plays pivotal roles in these processes (Fig. 1c). We further organized genes that gained and lost H2BK120-ub into interaction networks51 (Fig. 1d and e). Surprisingly, we observed that H2BK120-ub densities in the network of genes that regulate maintenance of self-renewal and embryonic morphogenesis increase after shPhf5a depletion. These include several core pluripotency genes such as Nanog, Pou5f1, Sox2, Esrrb, Sall4 and other previously identified Paf1C targets.22 This suggests that H2BK120-ub is paradoxically increased in the absence of Paf1C, in contrast to other histone modifications dependent on Paf1C function.
These results prompted us to further interrogate H2BK120-ub occupancy following shPhf5a depletion. Loss of Phf5a can recapitulate Paf1C depletion since it results in the inability of Paf1C to bind its target genes in chromatin (Fig. 2a). We directly compared deposition of H2BK120-ub among 3 gene categories that include actively expressed transcripts, housekeeping genes and Paf1C targets, respectively. We found that destabilization of Paf1C after loss of Phf5a results in a pronounced gain of H2BK120-ub on Paf1C targets compared with housekeeping or all other expressed genes (Fig. 2b). This suggests opposing functions of Paf1C in the deposition of H2BK120-ub compared with H3K79me or H3K36me, which also depend on Paf1C function in ESCs.22 For this reason, we reanalyzed profiles of additional histone marks associated with Paf1C function in the presence or absence of Phf5a. We performed the same analysis for H3K79 methylation and we found that Phf5a depletion results in a pronounced loss of H3K79me on Paf1 targets compared with housekeeping genes or all expressed transcripts (Fig. 2b). We further analyzed the occupancy profiles for these 2 histone marks on gene bodies, since both of those are associated with active elongation. We found that Phf5a depletion results in the differential regulation of H2BK120-ub and H3K79me on gene bodies of Paf1C targets and self-renewal genes (Fig. 2c). Since our findings directly implicate Paf1C to affect both H2BK120-ub gain and H3K79me loss on gene bodies, we directly investigated the overlap between the 2 histone marks. We found that most of Paf1C targets are characterized by the opposing regulation of these 2 histone marks (Fig. 2d).
Figure 2.
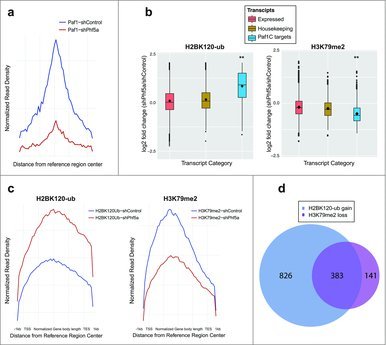
(a) Normalized read density profiles following Paf1 ChIP-sequencing centered on reference regions in the presence (blue) or absence (red) of Phf5a. (b) Box plots representing log2 fold change of normalized read density for H2BK120-ub, and H3K79me2 ChIP-sequencing in ESCs following shControl or shPhf5a silencing. Plots represent comparisons of all expressed transcripts in ESCs with housekeeping genes and also direct Paf1 targets around their gene bodies. **p<0.0001, non-parametric Wilcoxon signed rank test. (c) Normalized read density profiles around gene bodies, following ChIP-sequencing of H2BK120-ub and H3K79me2, on Paf1C targets and pluripotency genes in ESCs in the presence (blue) or absence (red) of Phf5a. (d) Venn diagram representing the numbers of genes in which H2BK120-ub densities are gained and H3K79me2 densities are lost, respectively, following shPhf5a depletion.
Last, we directly interrogated histone mark occupancy on several representative Paf1C targets and pluripotency genes before and after Phf5a loss. We found that many of those genes, including the ones directly associated with ESC self-renewal or with other gene ontology terms we found affected, exhibit both increased levels of H2BK120-ub and diminished levels of H3K79me in their gene bodies. Representative genes include key regulators of the self-renewal network, such as Nanog, Pou5f1, Sox2, Sall4, Myc, Esrrb, and others (Fig. 3). In contrast, we found that housekeeping genes, such as Cul1, Psmb2 and others, were not significantly affected (Fig. 3). These data are in agreement with our previous observations on elongation and strengthen our findings that Phf5a affects Paf1C functions.22 These include regulation of RNA PolII elongation and deposition of histone modifications on gene bodies. We conclude that Phf5a stabilizes Paf1C on its target genes in ESCs and that Paf1C directly represses H2BK120-ub deposition on Paf1C targets, in contrast to H3K79 methylation. Upon shPhf5a knockdown, deposition of H2BK120-ub is de-repressed and key genes that regulate self-renewal accumulate this mark.
Figure 3.
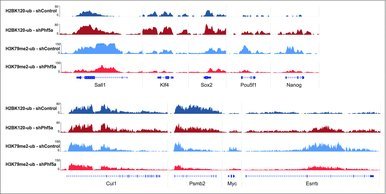
Genome browser snapshots of representative H2BK120-ub and H3K79me2 density ChIP-sequencing tracks on pluripotency genes or control loci. Dark blue: H2BK120-ub under conditions of shControl silencing. Dark red: H2BK120-ub under conditions of shPhf5a silencing. Light blue: H3K79me2 under conditions of shControl silencing. Light red: H3K79me2 under conditions of shPhf5a silencing.
Our findings support a new model of regulation of pluripotency-associated genes by Paf1C. Collectively our findings support that Paf1C targets are characterized by the following constellation of histone marks after shPhf5a loss: Gain of H2BK120-ub on gene bodies, coupled with loss of H3K79me and H3K36me, whereas the promoter-associated mark H3K4me remains stable.22 We conclude that the majority of Paf1C targets, which lose H3K79me, are also accompanied by a significant gain of H2BK120-ub.
Discussion
Transcription factor networks and chromatin conformation determine cell fate decisions in stem cells.52,53 Heritable patterns in development are set by different degrees of chromatin compaction ultimately establishing patterns of specialized gene expression. Post-translational modifications of histones impact gene expression by influencing chromatin structure, compaction and recruitment of additional modifiers or remodelers.54 In embryonic stem cells mechanisms that tip the balance between self-renewal and differentiation determine the degree of cell specification.55,56
In this study we demonstrate the association between H2B ubiquitylation and Paf1C in pluripotency and we contrast its functions with other histone modifications linked to transcriptional elongation. Although H2BK120-ub is a mark associated with actively expressing genes, we present evidence suggesting that the self-renewal network of embryonic stem cells is paradoxically characterized by absence of this mark. In contrast, aberrant loss of Paf1C from pluripotency genes, after perturbing Phf5a, leads to increased deposition of H2BK120-ub. We further find that additional Paf1C-associated histone marks, such as H3K79me, are decreased. Collectively we find that Paf1C targets are surprisingly characterized by a dual gain of H2BK120-ub and loss of H3K79me after Phf5a depletion.
Although H2B ubiquitylation has been shown to differentially regulate transcription of different sets of genes, our knowledge of their functions in pluripotent stem cells is limited. In contrast to lower eukaryotes, such as in yeast, where deposition of H2B ubiquitylation has been shown to directly require Paf1C,31,33,34 its regulation in mouse and human embryonic stem cells appears to be different. Several studies so far have provided evidence in support of a repressive role of Paf1C on H2BK120-ub in pluripotency: First, H2BK120Ub levels are low in self-renewal but they increase during initiation of differentiation.45,46 Second, Paf1C follows the opposite expression pattern, its levels are high in self-renewal but rapidly downregulated as cells differentiate.22 This suggests that Paf1C may hinder deposition of H2BK120Ub in self-renewal, however its functions might be de-repressed in differentiation in the absence of Paf1C. Functional variation between yeast and mammalian Paf1C might also be due to differences in subunit composition, since the subunit Wdr61 – a protein binding to Phf5a directly22 – is present in mammals but absent in yeast.42
Recent studies in human cells revealed how mammalian H2B ubiquitin ligases are involved in both transcriptional stimulation and repression of 2 distinct sets of genes.44,57 However, it remains obscure how they promote transcription in a set of genes, but not others. The enzymatic activity of RNF20/RNF40 has been suggested to limit the binding of Paf1C with TFIIS,44,57 shedding light into the underlying mechanisms, however the exact transcriptional stimulatory or repressive functions of H2BK120-ub and Paf1C remain to be investigated in additional cell types. Furthermore, opposing functions of RNF20 and USP44 in embryonic stem cells is suggested to fine-tune addition or removal of this mark.46
Our studies offer a direct overview of the control of core pluripotency factor circuitry by Paf1C and H2B-ub. We report genome-wide analysis for H2BK120-ub using ChIP-sequencing in pluripotent cells and we contrast its profiles after perturbing Paf1C binding on chromatin using Phf5a depletion. Besides providing further support that Phf5a knockdown can function as a surrogate for Paf1C depletion, we also show the feasibility of using pluripotent cells as a model system for studying Paf1C functions in chromatin modulation. Our results suggest that loss of Paf1C from its target genes triggers aberrant accumulation of H2BK120-ub, in contrast to H3K79me. In agreement with these findings core pluripotency genes are characterized by elevated RNA PolII pausing following shPhf5a knockdown, which leads to differentiation.22
Pluripotent stem cells are influenced by changes in gene expression, rendering them as an invaluable system to study transcriptional alterations during differentiation. The Paf1C acts as a positive modulator in transcriptional elongation and Phf5a can balance its functions in pluripotent cells. Distortion of the transcriptional equilibrium through shPhf5a knockdown triggers aberrant elongation stalling on Paf1C targets and changes in histone modifications. Our results provide further insight and suggest a repressive role for Paf1C on H2BK120-ub deposition in pluripotency genes. The importance for proper cell specification becomes apparent after perturbing gene expression circuitries that result in changes on chromatin, often leading to disease. The elucidation of mechanisms that offer novel insights into the origins of human development and disease can potentially lead to new therapeutic interventions.
Methods
Mouse ESC culture and inducible shRNA knockdown
Mouse ESCs were cultured under standard conditions as described previously.22,58 on gelatin-coated plates using recombinant LIF. For inducible knockdown, miR-30 hairpins against Control or Phf5a were cloned into a modified pColTGM vector59 containing a TRE-regulated RFP-miR-30, targeting the Col1a1 locus. Co-electroporation of modified pColTGM and pCAGS-FlpE recombinase in KH2 ESCs promotes integration and confers hygromycin resistance. ESCs were selected with hygromycin (140μg/mL daily for 10 days) and individual colonies were tested for efficient knockdown upon the addition of 2μg/mL doxycycline.
ChIP-sequencing library preparation
ChIP experiments were performed as described previously60,61 For H2BK120-ub, ESC nuclei were processed using micrococcal nuclease digestion and ChIP was performed with the anti-Ubiquityl-Histone H2B Antibody, clone 56 (Millipore, 05–1312). Libraries were prepared using Illumina standard protocols, including end repair, A-tailing, adaptor ligation (Illumina TruSeq system) and PCR amplification. AMPure XP beads (Beckman Coulter, A63880) were used for DNA cleaning in each step of the process.
Data sources and computational pipelines
Samples were run using Illumina HiSeq2000. Raw images were processed by CASAVA to remove the first and last bases and then they were used to generate sequence reads in fastq format. Raw reads were aligned against the mouse genome assembly mm10/GRCm38. Alignments were performed using Bowtie v.1.0.054 using the parameter –m 1 to report only unique alignments. MACS 2.0 was used for peak calling with the following parameter values: (a) –nomodel, (b) –broad, (c) –shiftsize = 200, (d) –q 0.05. The suite GenomicTools62 version 2.8.152 was used for genome binning, genomic annotations and the construction of occupancy profiles. For plotting, R version 3.2.0 was used (R Core Team (2016). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL: https://www.R-project.org), along with the VennDiagram package63 for the generation of Venn diagrams and ggplot264 for the generation of boxplots.
All ChIP-sequencing experiments were performed in triplicates and only the peaks present at least in 2 out of the 3 replicates were used for downstream analysis. The files with the aligned reads were converted to wig format using Genomic Tools 62 and then to bigwig format using the corresponding UCSC tool. The data are available on Gene Expression Omnibus (GEO): GSE92727 for H2BK120-ub ChIP-sequencing and GSE63974 for all other histone modification ChIP-sequencing experiments.
Peak characterization
Peaks were assigned to the following categories based on their genome-wide distribution: (a) Transcription start site (TSS): all peaks that fall within 1kb from TSS, (b) Gene body: it includes all peaks that fall within the 5’ UTR, the coding region of genes and the 3’ UTR, Upstream: all the peaks that fall up to 100kb from TSS, (d) All the peaks that fall within the remaining genomic loci. The peak characterization was performed using in-house scripts.
Gene ontology
Gene ontology, pathway and network analysis was performed based on ChIP-sequencing data by using the online tool Metascape (http://metascape.org).
Funding Statement
A.S. has been supported by the New York State Stem Cell Program (NYSTEM) of the New York State Health Department (Institutional NYU Stem Cell Training Grant C026880). I.A. is supported by the National Institutes of Health (RO1CA133379, RO1CA105129, RO1CA149655, 5RO1CA173636, 1RO1CA194923) and the NYSTEM program (NYSTEM-N11G-255). A. Heguy and the NYU Genome Technology Center is supported in part by the National Institutes of Health (NIH)/National Cancer Institute (NCI) grant P30CA016087–30. P.N. is supported by the National Cancer Institute (R00CA188293–02), the American Society of Hematology, the Zell Foundation, the St. Baldrick's Foundation, the Leukemia Research Foundation and the Chicago Region Physical Science-Oncology Center (CR-PSOC).
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank all members of the Aifantis laboratory for useful comments and discussions. We thank A. Heguy and the NYU Genome Technology Center for expertise with sequencing experiments. This work has used computing resources at the High Performance Computing Facility at the NYU Medical Center.
References
- [1].Perino M, Veenstra GJ. Chromatin control of developmental dynamics and plasticity. Dev Cell 2016; 38:610–20; PMID: 27676434; http://dx.doi.org/10.1016/j.devcel.2016.08.004 [DOI] [PubMed] [Google Scholar]
- [2].Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev 2005; 15:163–76; http://dx.doi.org/10.1016/j.gde.2005.01.005 [DOI] [PubMed] [Google Scholar]
- [3].Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 2014; 15:703–8; PMID: 25315270; http://dx.doi.org/10.1038/nrm3890 [DOI] [PubMed] [Google Scholar]
- [4].Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693–705; PMID: 17320507; http://dx.doi.org/10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- [5].Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol 2012; 19:1218–27; PMID: 23211769; http://dx.doi.org/10.1038/nsmb.2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol 2007; 14:1025–40; PMID: 17984965; http://dx.doi.org/10.1038/nsmb1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ho L, Crabtree GR. Chromatin remodelling during development. Nature 2010; 463:474–84; PMID: 20110991; http://dx.doi.org/10.1038/nature08911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Petesch SJ, Lis JT. Overcoming the nucleosome barrier during transcript elongation. Trends in genetics : TIG 2012; 28:285–94; PMID: 22465610; http://dx.doi.org/10.1016/j.tig.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell 2007; 128:669–81; PMID: 17320505; http://dx.doi.org/10.1016/j.cell.2007.01.033 [DOI] [PubMed] [Google Scholar]
- [10].Chakravarthi BV, Nepal S, Varambally S. Genomic and Epigenomic Alterations in Cancer. Am J Pathol 2016; 186:1724–35; PMID: 27338107; http://dx.doi.org/10.1016/j.ajpath.2016.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ntziachristos P, Abdel-Wahab O, Aifantis I. Emerging concepts of epigenetic dysregulation in hematological malignancies. Nat Immunol 2016; 17:1016–24; PMID: 27478938; http://dx.doi.org/10.1038/ni.3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981; 292:154–6; PMID: 7242681; http://dx.doi.org/10.1038/292154a0 [DOI] [PubMed] [Google Scholar]
- [13].Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A 1981; 78:7634–8; PMID: 6950406; http://dx.doi.org/10.1073/pnas.78.12.7634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell 2011; 145:835–50; PMID: 21663790; http://dx.doi.org/10.1016/j.cell.2011.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lessard JA, Crabtree GR. Chromatin regulatory mechanisms in pluripotency. Annu Rev Cell Dev Biol 2010; 26:503–32; PMID: 20624054; http://dx.doi.org/10.1146/annurev-cellbio-051809-102012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Young RA. Control of the embryonic stem cell state. Cell 2011; 144:940–54; PMID: 21414485; http://dx.doi.org/10.1016/j.cell.2011.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jaehning JA. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta 2010; 1799:379–88; PMID: 20060942; http://dx.doi.org/10.1016/j.bbagrm.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tomson BN, Arndt KM. The many roles of the conserved eukaryotic Paf1 complex in regulating transcription, histone modifications, and disease states. Biochim Biophys Acta 2013; 1829:116–26; PMID: 22982193; http://dx.doi.org/10.1016/j.bbagrm.2012.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Akanuma T, Koshida S, Kawamura A, Kishimoto Y, Takada S. Paf1 complex homologues are required for Notch-regulated transcription during somite segmentation. EMBO Rep 2007; 8:858–63; PMID: 17721442; http://dx.doi.org/10.1038/sj.embor.7401045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Langenbacher AD, Nguyen CT, Cavanaugh AM, Huang J, Lu F, Chen JN. The PAF1 complex differentially regulates cardiomyocyte specification. Dev Biol 2011; 353:19–28; PMID: 21338598; http://dx.doi.org/10.1016/j.ydbio.2011.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nguyen CT, Langenbacher A, Hsieh M, Chen JN. The PAF1 complex component Leo1 is essential for cardiac and neural crest development in zebrafish. Dev Biol 2010; 341:167–75; PMID: 20178782; http://dx.doi.org/10.1016/j.ydbio.2010.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Strikoudis A, Lazaris C, Trimarchi T, Galvao Neto AL, Yang Y, Ntziachristos P, Rothbart S, Buckley S, Dolgalev I, Stadtfeld M, et al.. Regulation of transcriptional elongation in pluripotency and cell differentiation by the PHD-finger protein Phf5a. Nat Cell Biol 2016; 18:1127–38; PMID: 27749823; http://dx.doi.org/10.1038/ncb3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Smolle M, Workman JL. Transcription-associated histone modifications and cryptic transcription. Biochim Biophys Acta 2013; 1829:84–97; PMID: 22982198; http://dx.doi.org/10.1016/j.bbagrm.2012.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tanny JC. Chromatin modification by the RNA Polymerase II elongation complex. Transcription 2014; 5:e988093; PMID: 25494544; http://dx.doi.org/10.4161/21541264.2014.988093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21:381–95; PMID: 21321607; http://dx.doi.org/10.1038/cr.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Campos EI. Reinberg Des: annotating chromatin. Ann Rev Genet 2009; 43:559–99; PMID: 19886812; http://dx.doi.org/10.1146/annurev.genet.032608.103928 [DOI] [PubMed] [Google Scholar]
- [27].Fuchs G, Oren M. Writing and reading H2B monoubiquitylation. Biochim Biophys Acta 2014; 1839:694–701; PMID: 24412854; http://dx.doi.org/10.1016/j.bbagrm.2014.01.002 [DOI] [PubMed] [Google Scholar]
- [28].Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 2012; 13:115–26; PMID: 22266761; http://dx.doi.org/10.1038/nrm3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yu M, Yang W, Ni T, Tang Z, Nakadai T, Zhu J, Roeder RG. RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science 2015; 350:1383–6; PMID: 26659056; http://dx.doi.org/10.1126/science.aad2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen FX, Woodfin AR, Gardini A, Rickels RA, Marshall SA, Smith ER, Shiekhattar R, Shilatifard A. PAF1, a Molecular Regulator of Promoter-Proximal Pausing by RNA Polymerase II. Cell 2015; 162:1003–15; PMID: 26279188; http://dx.doi.org/10.1016/j.cell.2015.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kim J, Roeder RG. Direct Bre1-Paf1 complex interactions and RING finger-independent Bre1-Rad6 interactions mediate histone H2B ubiquitylation in yeast. J Biol Chem 2009; 284:20582–92; PMID: 19531475; http://dx.doi.org/10.1074/jbc.M109.017442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wood A, Schneider J, Dover J, Johnston M, Shilatifard A. The Paf1 complex is essential for histone monoubiquitination by the Rad6-Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. J Biol Chem 2003; 278:34739–42; PMID: 12876294; http://dx.doi.org/10.1074/jbc.C300269200 [DOI] [PubMed] [Google Scholar]
- [33].Xiao T, Kao CF, Krogan NJ, Sun ZW, Greenblatt JF, Osley MA, Strahl BD. Histone H2B ubiquitylation is associated with elongating RNA polymerase II. Mol Cell Biol 2005; 25:637–51; PMID: 15632065; http://dx.doi.org/10.1128/MCB.25.2.637-651.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ng HH, Dole S, Struhl K. The Rtf1 component of the Paf1 transcriptional elongation complex is required for ubiquitination of histone H2B. J Biol Chem 2003; 278:33625–8; PMID: 12876293; http://dx.doi.org/10.1074/jbc.C300270200 [DOI] [PubMed] [Google Scholar]
- [35].Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, et al.. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell 2003; 11:721–9; PMID: 12667454; http://dx.doi.org/10.1016/S1097-2765(03)00091-1 [DOI] [PubMed] [Google Scholar]
- [36].Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 2003; 11:709–19; PMID: 12667453; http://dx.doi.org/10.1016/S1097-2765(03)00092-3 [DOI] [PubMed] [Google Scholar]
- [37].Chu Y, Simic R, Warner MH, Arndt KM, Prelich G. Regulation of histone modification and cryptic transcription by the Bur1 and Paf1 complexes. EMBO J 2007; 26:4646–56; PMID: 17948059; http://dx.doi.org/10.1038/sj.emboj.7601887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Briggs SD, Xiao T, Sun ZW, Caldwell JA, Shabanowitz J, Hunt DF, Allis CD, Strahl BD. Gene silencing: trans-histone regulatory pathway in chromatin. Nature 2002; 418:498; PMID: 12152067; http://dx.doi.org/10.1038/nature00970 [DOI] [PubMed] [Google Scholar]
- [39].Dover J, Schneider J, Tawiah-Boateng MA, Wood A, Dean K, Johnston M, Shilatifard A. Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J Biol Chem 2002; 277:28368–71; PMID: 12070136; http://dx.doi.org/10.1074/jbc.C200348200 [DOI] [PubMed] [Google Scholar]
- [40].Nakanishi S, Lee JS, Gardner KE, Gardner JM, Takahashi YH, Chandrasekharan MB, Sun ZW, Osley MA, Strahl BD, Jaspersen SL, et al.. Histone H2BK123 monoubiquitination is the critical determinant for H3K4 and H3K79 trimethylation by COMPASS and Dot1. J Cell Biol 2009; 186:371–7; PMID: 19667127; http://dx.doi.org/10.1083/jcb.200906005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 2002; 418:104–8; PMID: 12077605; http://dx.doi.org/10.1038/nature00883 [DOI] [PubMed] [Google Scholar]
- [42].Zhu B, Zheng Y, Pham AD, Mandal SS, Erdjument-Bromage H, Tempst P, Reinberg D. Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol Cell 2005; 20:601–11; PMID: 16307923; http://dx.doi.org/10.1016/j.molcel.2005.09.025 [DOI] [PubMed] [Google Scholar]
- [43].Kim J, Guermah M, McGinty RK, Lee JS, Tang Z, Milne TA, Shilatifard A, Muir TW, Roeder RG. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell 2009; 137:459–71; PMID: 19410543; http://dx.doi.org/10.1016/j.cell.2009.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shiloh Y, Shema E, Moyal L, Oren M. RNF20-RNF40: A ubiquitin-driven link between gene expression and the DNA damage response. FEBS Lett 2011; 585:2795–802; PMID: 21827756; http://dx.doi.org/10.1016/j.febslet.2011.07.034 [DOI] [PubMed] [Google Scholar]
- [45].Chen S, Li J, Wang DL, Sun FL. Histone H2B lysine 120 monoubiquitination is required for embryonic stem cell differentiation. Cell Res 2012; 22:1402–5; PMID: 22847742; http://dx.doi.org/10.1038/cr.2012.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fuchs G, Shema E, Vesterman R, Kotler E, Wolchinsky Z, Wilder S, Golomb L, Pribluda A, Zhang F, Haj-Yahya M, et al.. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol Cell 2012; 46:662–73; PMID: 22681888; http://dx.doi.org/10.1016/j.molcel.2012.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 2008; 322:1845–8; PMID: 19056941; http://dx.doi.org/10.1126/science.1162228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jonkers I, Kwak H, Lis JT. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife 2014; 3:e02407; PMID: 24843027; http://dx.doi.org/10.7554/eLife.02407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nechaev S, Adelman K. Promoter-proximal Pol II: when stalling speeds things up. Cell Cycle 2008; 7:1539–44; PMID: 18469524; http://dx.doi.org/10.4161/cc.7.11.6006 [DOI] [PubMed] [Google Scholar]
- [50].Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al.. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 2015; 18:723–35; http://dx.doi.org/10.1016/j.chom.2015.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform 2003; 4:2; PMID: 12525261; http://dx.doi.org/10.1186/1471-2105-4-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Loh KM, Lim B. A precarious balance: pluripotency factors as lineage specifiers. Cell Stem Cell 2011; 8:363–9; PMID: 21474100; http://dx.doi.org/10.1016/j.stem.2011.03.013 [DOI] [PubMed] [Google Scholar]
- [53].Thomson M, Liu SJ, Zou LN, Smith Z, Meissner A, Ramanathan S. Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell 2011; 145:875–89; PMID: 21663792; http://dx.doi.org/10.1016/j.cell.2011.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang Y, Wysocka J, Perlin JR, Leonelli L, Allis CD, Coonrod SA. Linking covalent histone modifications to epigenetics: the rigidity and plasticity of the marks. Cold Spring Harb Symp Quant Biol 2004; 69:161–9; PMID: 16117646; http://dx.doi.org/10.1101/sqb.2004.69.161 [DOI] [PubMed] [Google Scholar]
- [55].Strikoudis A, Guillamot M, Aifantis I. Regulation of stem cell function by protein ubiquitylation. EMBO Rep 2014; 15:365–82; PMID: 24652853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chen CY, Cheng YY, Yen CY, Hsieh PC. Mechanisms of pluripotency maintenance in mouse embryonic stem cells. Cell Mol Life Sci 2016; [Epub ahead of print] PMID:27999898; http://dx.doi.org/10.1007/s00018-016-2438-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Shema E, Kim J, Roeder RG, Oren M. RNF20 inhibits TFIIS-facilitated transcriptional elongation to suppress pro-oncogenic gene expression. Mol Cell 2011; 42:477–88; PMID: 21596312; http://dx.doi.org/10.1016/j.molcel.2011.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Buckley SM, Aranda-Orgilles B, Strikoudis A, Apostolou E, Loizou E, Moran-Crusio K, Farnsworth CL, Koller AA, Dasgupta R, Silva JC, et al.. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell 2012; 11:783–98; PMID: 23103054; http://dx.doi.org/10.1016/j.stem.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Premsrirut PK, Dow LE, Kim SY, Camiolo M, Malone CD, Miething C, Scuoppo C, Zuber J, Dickins RA, Kogan SC, et al.. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell 2011; 145:145–58; PMID: 21458673; http://dx.doi.org/10.1016/j.cell.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, et al.. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 2012; 18:298–301; PMID: 22237151; http://dx.doi.org/10.1038/nm.2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, Loizou E, Holmfeldt L, Strikoudis A, King B, et al.. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014; 514:513–7; PMID: 25132549; http://dx.doi.org/10.1038/nature13605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tsirigos A, Haiminen N, Bilal E, Utro F. GenomicTools: a computational platform for developing high-throughput analytics in genomics. Bioinformatics 2012; 28:282–3; PMID: 22113082; http://dx.doi.org/10.1093/bioinformatics/btr646 [DOI] [PubMed] [Google Scholar]
- [63].Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform 2011; 12:35; PMID: 21269502; http://dx.doi.org/10.1186/1471-2105-12-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wickham H. ggplot2: Elegant Graphics for Data Analysis. Use R 2009; 1–212; http://dx.doi.org/10.1007/978-0-387-98141-3 [Google Scholar]