

ABSTRACT
Bacterial translocation and lipopolysaccharide (LPS) leakage occur at a very early stage of liver fibrosis in animal models. We studied the role of LPS in hepatic stellate cell (HSC) activation and the underlying mechanisms in vitro and in vivo. Herein, we demonstrated that LPS treatment led to a dramatic increase in autophagosome formation and autophagic flux in LX-2 cells and HSCs, which was mediated through the AKT-MTOR and AMPK-ULK1 pathway. LPS significantly decreased the lipid content, including the lipid droplet (LD) number and lipid staining area in HSCs; pretreatment with macroautophagy/autophagy inhibitors or silencing ATG5 attenuated this decrease. Furthermore, lipophagy was induced by LPS through the autophagy-lysosomal pathway in LX-2 cells and HSCs. Additionally, LPS-induced autophagy further reduced retinoic acid (RA) signaling, as demonstrated by a decrease in the intracellular RA level and Rar target genes, resulting in the downregulation of Bambi and promoting the sensitization of the HSC's fibrosis response to TGFB. Compared with CCl4 injection alone, CCl4 plus LPS injection exaggerated liver fibrosis in mice, as demonstrated by increased Col1a1 (collagen, type I, α 1), Acta2, Tgfb and Timp1 mRNA expression, ACTA2/α-SMA and COL1A1 protein expression, and Sirius Red staining area, which could be attenuated by injection of an autophagy inhibitor. LPS also reduced lipid content in HSCs in vivo, with this change being attenuated by chloroquine (CQ) administration. In conclusion, LPS-induced autophagy resulted in LD loss, RA signaling dysfunction, and downregulation of the TGFB pseudoreceptor Bambi, thus sensitizing HSCs to TGFB signaling.
KEYWORDS: autophagy, BAMBI, hepatic stellate cell, liver fibrosis, LPS, retinoic acid signaling, TGFB
Introduction
Liver fibrosis represents an adaptive response to repeated chronic liver injuries, which are primarily caused by chronic viral hepatitis and steatohepatitis associated with either alcohol consumption or obesity.1, 2 Studies using animal models of liver fibrosis have demonstrated that myofibroblasts, which are not present in normal liver tissue, play a crucial role in hepatic fibrogenesis. Myofibroblasts are derived from 3 cellular sources, liver resident hepatic stellate cells (HSCs), portal fibroblasts and bone marrow-derived collagen-producing cells, and the composition of myofibroblasts varies depending on the etiology of liver fibrosis.3 However, activated HSCs are the predominant type of myofibroblasts observed during the progression of liver fibrosis.4 Quiescent HSCs, which are located in the space of Disse and store retinoids in lipid droplets (LDs), transform into an activated phenotype characterized by the induction of fibrotic markers and depletion of LDs. However, the mechanisms underlying HSC activation remain unclear. Therefore, elucidating these HSC activation events could promote new strategies for preventing and treating liver fibrosis.
Autophagy is an evolutionarily conserved process in which cytoplasmic constituents, including damaged and dysfunctional proteins and organelles, are delivered to lysosomes for degradation and recycling of the breakdown products.5 Macroautophagy (hereafter referred to as autophagy) involves the formation of double-membraned vesicles called autophagosomes and the subsequent fusion of autophagosomes with lysosomes for cargo recycling to maintain cellular homeostasis.6 Defects in autophagy have been closely associated with many human diseases, including cancer, infection and neurodegeneration.7-9 Recent studies have demonstrated that functional autophagy is involved in lipid clearance in hepatocytes, a process termed lipophagy.10-12 Previous studies have shown that autophagy plays a critical role in lipid clearance and fibrotic activity,13-15 and this effect is associated with the liberation of free fatty acids from LDs and subsequent mitochondrial β-oxidation, which provides energy to support HSC activation.16
Bacterial lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria that is among the strongest known inducers of inflammation, has been found to be associated with hepatic fibrogenesis through direct interactions with HSCs.17 During chronic liver injury caused by CCl4 injection or bile duct ligation (BDL), the plasma concentration of LPS is significantly elevated even at a very early stage due to changes in the intestinal mucosal permeability and increased bacterial translocation.17, 18 Consistent with these reports, one recent study observed that the transplantation of Gram-negative bacteria affects hepatic fibrogenesis after BDL.19 However, the molecular mechanism underlying the effects of LPS on HSC activation is poorly understood. Recent studies have shown that LPS induces autophagy in macrophages, linking 2 ancient processes, autophagy and innate immunity.20-22 Here, we hypothesized that the LPS regulates HSC activation through autophagy and retinoic acid signaling.
Results
LPS treatment induced autophagic markers in LX-2 cells
LX-2 cells were exposed to LPS at a concentration of 0, 0.1, 1, 10 or 100 ng/ml for 24 h, and the expression of the autophagy marker microtubule-associated protein 1 light chain 3B/LC3B, a mammalian homolog of yeast Atg8, was assessed. As shown in Fig. 1A, LPS treatment significantly increased the steady-state level of LC3B-II:LC3B-I in a concentration-dependent manner. Disruption of the biological effects of LPS by polymyxin B reversed the LPS-induced downregulation of SQSTM1/p62 (Fig. S1A). We also demonstrated that LPS dramatically increased LC3B-II expression and markedly reduced the expression of SQSTM1 in LX-2 cells pretreated with a combination of retinol and palmitic acid to stimulate lipid accumulation (Fig. S1B and S1C). Furthermore, LPS treatment failed to affect the cell viability, LX-2 cell death and cellular damage (Fig. S1D).
Figure 1.
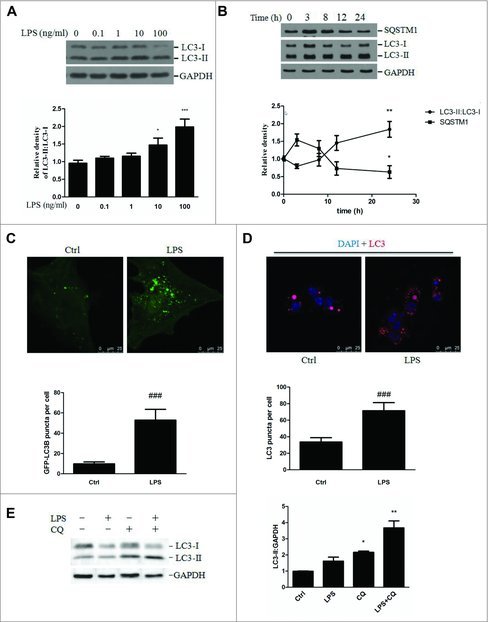
LPS-induced autophagosome formation in LX-2 cells. (A) Immunoblot analysis of the LC3 protein expression in LX-2 cells exposed to the indicated concentrations of LPS. The results from quantitative analyses of LC3 expression are shown in the lower panel. *p < 0.05, ***p < 0.001 compared with vehicle. (B) Immunoblot analysis of the LC3 and SQSTM1 proteins in LX-2 cells treated with LPS at various time points. The results from quantitative analyses are shown in the lower panel. *p < 0.05, **p < 0.01 compared with vehicle. (C) LX-2 cells were transfected with the plasmid GFP-LC3B for 24 h and then stimulated with LPS or vehicle for an additional 24 h. Representative images of GFP-LC3B puncta are shown. ###p < 0.001 versus vehicle. Scale bar: 25 μm. (D) LC3 staining was performed in LPS-treated HSCs and visualized by confocal microscopy. Representative images and the results of quantitative analyses of LC3-II puncta are shown in the upper and lower panels, respectively. ###p < 0.001 versus vehicle. Scale bar: 25 μm. (E) Freshly isolated MmHSCs were pretreated with CQ for 3 h and then stimulated with LPS for another 24 h. The upper panel depicts the LC3-II levels. The lower panel shows the results of quantitative analyses of LC3-II expression. *p < 0.05, **p < 0.01 compared with vehicle.
Next, we evaluated the time course of autophagy in response to LPS in LX-2 cells. LX-2 cells were treated with 100 ng/ml of LPS for 0, 3, 8, 12 and 24 h. The LC3B-II:LC3B-I expression ratio decreased at 3 h and then dramatically increased at 24 h, whereas SQSTM1 expression increased at 3 h and then significantly decreased at 24 h after LPS treatment (Fig. 1B). Taken together, these data demonstrated that LPS exposure at the concentration of 100 ng/ml led to a dramatic upregulation of autophagy after 24 h; therefore, this time point and dose were used in the subsequent experiments.
To further confirm that LPS induced autophagy in LX-2 cells, the endogenous LC3B level was determined using confocal microscopy. Incubation of LX-2 cells with LPS increased the formation of LC3B-II puncta compared with that in the control (Fig. S1E). Moreover, LX-2 cells were transfected with an exogenous GFP-LC3B plasmid and then treated with LPS or vehicle. A significant increase in GFP-LC3B puncta was observed in the LPS-treated LX-2 cells compared with the vehicle-treated cells, indicating the formation of autophagic vesicles (Fig. 1C). Similarly, a marked increase in endogenous LC3-II puncta was demonstrated in primary HSCs exposed to LPS (Fig. 1D).
LPS exposure promoted autophagic flux in LX-2 cells and HSCs
Increased autophagosome formation can be attributed to either induction of autophagy or, alternatively, the inhibition of late lysosomal fusion or degradation. To investigate whether LPS induces autophagic flux in stellate cells, chloroquine (CQ), which attenuates lysosomal acidification and thus blocks the turnover and degradation of autophagic cargo, was used. Incubation of cells with CQ resulted in significantly increased LC3B-II expression, and treatment with LPS+CQ revealed a further increase in LC3B-II expression, as shown in Fig. S3A. We also confirmed that LPS increased autophagic flux by transfecting LX-2 cells with the GFP-LC3 construct. As GFP-LC3 is delivered to lysosomes, the LC3 portion is sensitive to hydrolysis, whereas the GFP protein is relatively resistant. Therefore, the cleaved GFP level can be used to monitor autophagic flux. Incubation of LX-2 cells with LPS resulted in a marked increase in free GFP, whereas pretreatment with CQ attenuated the LPS-mediated upregulation of cleaved GFP, as shown in Fig. S3B. Furthermore, LX-2 cells were transfected with the dual fluorescent reporter mRFP-EGFP-LC3 and assessed for the number of red versus yellow puncta. As shown in Fig. S3C and S3D, LPS exposure increased the numbers of both yellow (autophagic vesicles) and red (autolysosomes) puncta. The yellow signal was generated by overlaying GFP fluorescence and RFP fluorescence, whereas the “red-only” puncta were generated from the RFP fluorescence. Similar results were also confirmed in primary HSCs pretreated with CQ followed by LPS (Fig. 1E). Taken together, our findings demonstrate that LPS exposure increased autophagic flux by increasing the initiation of autophagy.
The AKT-MTOR signaling pathway mediated LPS-induced autophagy
The AKT-MTOR cascade is considered a major negative regulator of autophagy in multiple types of cell.10, 23,24 Therefore, we examined whether this signaling pathway was also involved in LPS-induced autophagy. As shown in Fig. 2, LPS treatment led to significantly decreased phosphorylation of AKT at Thr308 and MTOR at Ser2448, and increased phosphorylation of AMPK at Thr172and ATG5 expression. It has been reported that cellular ceramide antagonizes LPS-mediated biological functions.25 Furthermore, phosphorylation of ULK1 at different sites by MTOR and AMPK is essential for autophagy induction. We next explored the effect of LPS on ULK1 phosphorylation and the possible role of ceramide in LPS-mediated autophagy induction. We observed a marked increase in ULK1 phosphorylation at Ser317 and a significant decrease in ULK1 phosphorylation at Ser757 in LX-2 cells treated with LPS, whereas pretreatment with C8 ceramide dramatically alleviated these effects (Fig. S4). Next, to investigate whether TLR4 was required for LPS-induced autophagy, LX-2 cells were transfected with a TLR4 siRNA and then treated with LPS. As shown in Figs. S5A and S5B, TLR4 knockdown had no impact on LPS-mediated MTOR, AKT and ULK1 phosphorylation. Consistent with these results, we showed that TLR4 knockdown also had no impact on LPS-induced LC3-II expression (Fig. S5C and S5D).
Figure 2.
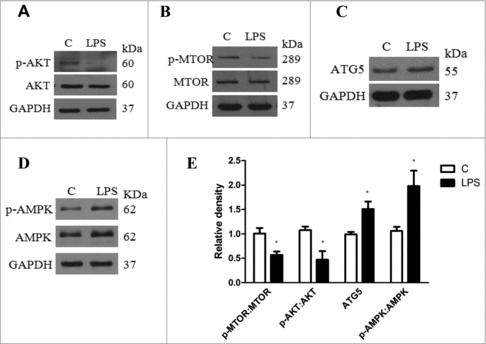
Expression of AKT-MTOR signaling proteins in LX-2 cells treated with LPS or vehicle for 24 h. (A-D) Representative western blots depicting total and phosphorylated (p- Thr308) AKT, total and phosphorylated (Ser2448) MTOR, total and phosphorylated (Thr172) AMPK and ATG5. (E) Quantitative analyses of the ATG5, MTOR and ATG5 expression ratios are shown. Data represent mean ± SEM. Open bar: control; closed bar: LPS group. *p < 0.05 versus control.
LPS stimulated HSC lipophagy
Because LD depletion is a salient feature of HSC activation and because lipophagy has been observed in hepatocytes both in vitro and in vivo,12 we explored whether the LPS-mediated induction of autophagy contributed to a reduction in lipid content in LX-2 cells. We observed a significant reduction in the lipid content of LX-2 cells pretreated with retinol and palmitic acid, followed by LPS treatment (Fig. S6A). LPS exposure also increased the amount of acridine orange staining, which is indicative of the formation of acidic organelles that characterizes autolysosomal activity (Fig. 3A). Furthermore, increased colocalization of LC3 and Bodipy following LPS treatment revealed a direct involvement of autophagy in LD metabolism (Fig. 3B). Additionally, we also observed an increased overlap between Bodipy 493 and LysoTracker Red staining in LX-2 cells treated with LPS, indicating the induction of lipophagy (Fig. S6B).
Figure 3.
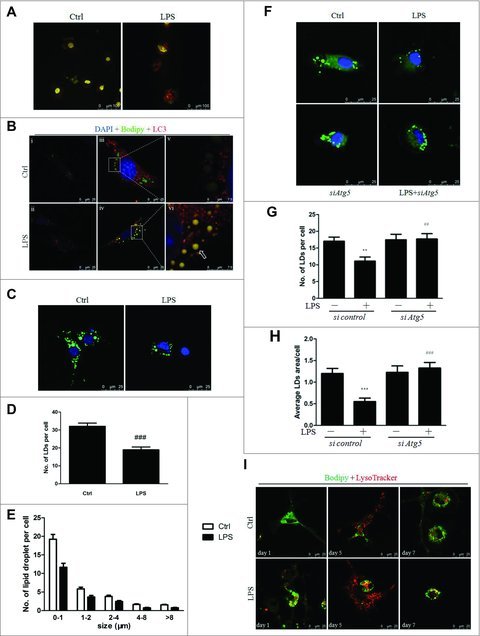
LPS modulated lipophagy via the autophagy-lysosomal pathway in LX-2 cells and HSCs. (A) AO staining showed that LPS increased lysosomal acidification in LX-2 cells. (B) LX-2 cells were pretreated with retinol + palmitic acid (iii-vi) or 0.2% ethanol (i, ii) overnight before LPS or vehicle stimulation for an additional 24 h. Representative images are shown, with (v) and (vi) showing the enlarged area of the white boxes enclosed in (iii) and (iv), respectively. Arrow indicates the colocalization of LDs and LC3. Scale bar: 25 μm for (i-iv) and 7.5 μm for (v-vi). (C-E) LDs staining (green) in freshly isolated HSCs treated with LPS. Nuclei were counterstained with DAPI (blue). Scale bar: 25 μm. Representative images (C) and results from the quantitative analysis of the number (D) and diameter distribution (E) of LDs are shown. ###p < 0.001 compared with control. Scale bar: 25 μm. (F-H) Freshly isolated HSCs were transfected with siAtg5 or scrambled siRNA for 24 h and then stimulated with LPS for another 24 h. LDs were stained with Bodipy. Representative images and quantitative results for the total number and area of LDs are shown in (F-H). Scale bar: 25 μm. (I) Representative images of LD staining (green) and lysosome (red) in isolated HSCs plated on dishes for 1, 5, and 7 d, followed by LPS or vehicle treatment of 24 h. Scale bar: 25 μm.
Next, to investigate whether LPS also induced lipophagy in primary Mus musculus (mouse) HSCs (MmHSCs), we treated MmHSCs with LPS for 24 h either immediately or on day 2 after isolation. LPS treatment resulted in significantly decreased lipid content (Fig. 3C-E), whereas pretreatment with the autophagy inhibitors CQ or LY294002 completely blocked the reduction of LDs, indicating the involvement of autophagy in LPS-induced lipid clearance (Fig. S7A-C and S7D-F). Additionally, transfecting HSCs with siAtg5 almost totally reversed the LPS-induced reduction of LDs (Fig. 3F-H). In contrast to the results for LX-2 cells, we did not observe any difference in the overlap between autophagosomes and LDs after LPS treatment in MmHSCs (Fig. S7G). However, the colocalization of lysosomes and LDs was significantly augmented in MmHSCs exposed to LPS (Fig. 3I).
LPS mediated the downregulation of Bambi through autophagy and the subsequent promotion of the HSC fibrotic response to TGFB signaling
Previous studies have shown that LPS downregulates the TGFB pseudoreceptor Bambi, thus sensitizing HSCs to TGFB activation.18,26,27 Here, we revealed that autophagy activation was involved in the LPS-mediated reduction of Bambi expression. First, the autophagy inhibitors CQ and bafilomycin A1 (BAF) significantly reversed the LPS-induced reduction of Bambi mRNA expression in primary HSCs (Fig. 4A). Second, treatment with the autophagy inducers rapamycin and Earle's balanced salt solution (EBSS) strongly decreased the BAMBI mRNA expression in LX-2 cells and HSCs (Fig. S8A). Furthermore, the marked decrease in BAMBI mRNA expression observed in LX-2 cells and HSCs in response to LPS was significantly reversed by ATG5 depletion. (Fig. 4B-D and Fig. S8B).
Figure 4.
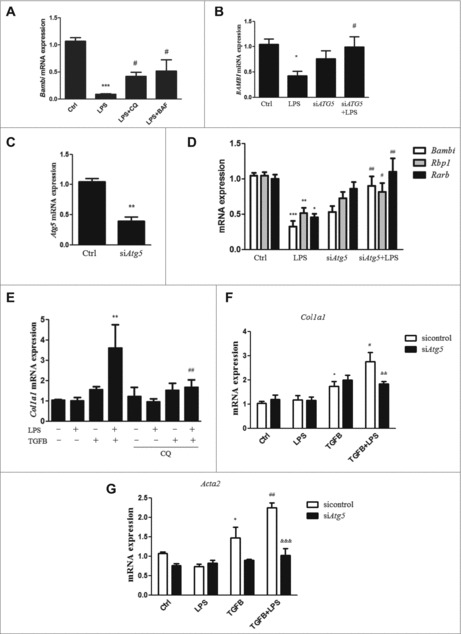
Role of autophagy in LPS-induced Bambi downregulation and HSC activation. (A) HSCs were pretreated with CQ and BAF for 3 h and subsequently treated with LPS for 24 h. Bambi mRNA expression is shown. ***p < 0.001 compared with control,#p < 0.05 compared with the LPS group. (B) BAMBI mRNA expression in LX-2 cells transfected with ATG5 siRNA and scrambled siRNA before LPS treatment. (C) The effect of siAtg5 knockdown was confirmed in HSCs. (D) Bambi, Rbp1 and Rarb mRNA expression in freshly isolated HSCs transfected with Atg5 siRNA and scrambled siRNA followed by LPS stimulation. (E) HSCs were pretreated with CQ before LPS treatment and then stimulated with TGFB. The mRNA expression of Col1a1 is shown. **p < 0.01 versus TGFB, ##p < 0.01 versus TGFB + LPS. (F-G) Col1a1 and Acta2 mRNA expression in HSCs transfected with siAtg5 or control siRNA and then stimulated with LPS followed by TGFB treatment. *p < 0.05 versus control, #p < 0.05, ##p < 0.01 versus TGFB, &&p < 0.01, &&&p < 0.001 versus TGFB+LPS.
To explore whether LPS-induced autophagy altered the sensitization of human LX-2 cells to TGFB, a genetic approach was used. ATG5 siRNA knockdown prevented the LPS-mediated promotion of fibrosis in the presence of TGFB (Fig. S8C-E), indicating that LPS-induced autophagy was involved in augmenting the fibrotic response to TGFB. Additionally, HSCs were pretreated with the autophagy inhibitor CQ and subsequently treated with LPS before TGFB stimulation. LPS treatment alone did not affect the mRNA expression of Col1a1 but did sensitize HSCs to TGFB stimulation; this effect was attenuated by CQ pretreatment (Fig. 4E). These results were further confirmed in HSCs transfected with siAtg5, in which LPS-mediated enhancement of Col1a1 and Acta2 expression in the presence of TGFB was significantly attenuated (Fig. 4F-G).
LPS-induced RA signaling dysfunction inhibited Bambi expression and promoted HSC activation
Previous studies have shown that there are similar amounts of retinyl esters and triglycerides contained in the LDs of HSCs.28,29 We investigated whether LPS-induced autophagy also contributed to retinoid depletion during HSC activation. Staining experiments showed a marked increase in retinyl ester and lysosome colocalization in LPS-treated MmHSCs compared with vehicle-treated cells, suggesting a possible role of autophagy in retinoid hydrolysis during HSC activation (Fig. S9). Additionally, LPS markedly reduced the mRNA expression of Rarb and Rarg in HSCs, and pretreatment with autophagy inhibitors restored the Rarb and Rarg expression without altering the Rara expression (Fig. 5A). By contrast, pretreatment with autophagy inhibitors dramatically suppressed the LPS-induced downregulation of RARA and RARG expression in LX-2 cells (Fig. S10A). Furthermore, ATG5 silencing abolished the LPS-mediated decrease in RARA expression in LX-2 cells (Fig. S10B). Next, treatment of HSCs with the autophagy inducer rapamycin and EBSS led to dysfunctional RA signaling, as indicated by a marked decrease in the expression of Rara, Rarb and Rarg mRNA, indicating a direct effect of autophagy on RA signaling (Fig. 5B). We also demonstrated that Atg5 depletion reversed LPS-mediated downregulation of the expression of the RAR target genes, Rbp1 and Rarb in HSCs (Fig. 4E). Direct quantitative measurement of RA showed a significantly reduced intracellular RA level in HSCs treated with LPS, while pretreatment with autophagy inhibitors almost completely prevented this effect (Fig. 5C). Similar colocalization of RA receptors (RARs) and lysosomes was observed in both LPS- and vehicle-treated HSCs (Fig. S11), thus excluding the possibility that RARs degradation contributed to the decreased induction of RAR target genes and confirming that the decreased RA concentration caused by LPS-mediated autophagy induction was responsible for the downregulation of RAR signaling.
Figure 5.
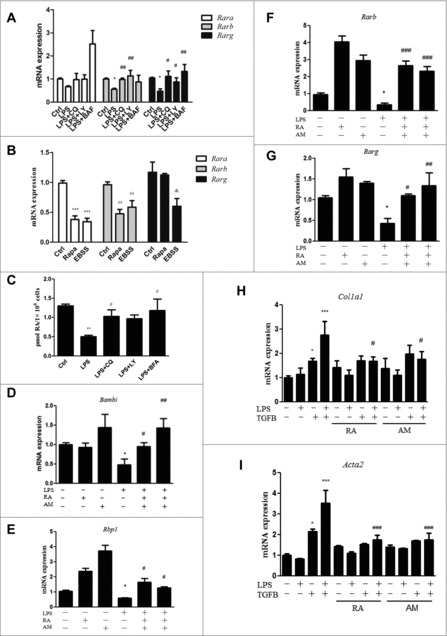
A decreased RA signal in HSCs was required for the LPS-induced downregulation of Bambi and enhancement of the HSC response to TGFB. (A) Rara, Rarb and Rarg mRNA expression in freshly isolated HSCs pretreated with CQ, LY and BAF followed by LPS stimulation. *p < 0.05 relative to control, #p < 0.05 and ##p < 0.01 relative to LPS. (B) Rara, Rarb and Rarg mRNA expression in freshly isolated HSCs treated with the autophagy inducers rapamycin and EBSS. ***p < 0.001, ##p < 0.01, andp < 0.05 versus the corresponding control. (C) Quantitative measurement of the intracellular RA concentration in HSCs pretreated with the autophagy inhibitors CQ, LY and BAF followed by LPS. (D-G) Bambi, Rbp1, Rarb and Rarg mRNA expression in HSCs pretreated with the RAR activators RA or Am580 before LPS stimulation. *p < 0.05 versus control, #p < 0.05, ##p < 0.01 and ###p < 0.001versus LPS. (H-I) Col1a1 and Acta2 mRNA expression in HSCs pretreated with RA and Am580, followed by LPS and TGFB treatments. *p < 0.05, ***p < 0.001 versus control, #p < 0.05 versus LPS+TGFB.
Next, to investigate the role of RA signaling in Bambi expression, HSCs and LX-2 cells were pretreated with the RAR activators RA or Am580, followed by LPS. A significant decrease in the expression of Bambi mRNA and protein was observed in both HSCs and LX-2 cells treated with LPS, and this effect could be totally abolished by RA or Am580 pretreatment (Fig. 5D and Fig. S10C). RA or Am580 pretreatment also markedly inhibited the LPS-induced decease in the expression of Rbp1, Rarb and Rarg mRNA (Fig. 5E-G). There were no significant changes in the expression of Rara mRNA (Fig. S10D). These results imply that RA signaling plays an upstream role in Bambi regulation in LPS-treated HSCs. To clarify the mechanisms by which RA signaling increased BAMBI expression, HSCs were treated with RA or Am580 in the presence or absence of cycloheximide (CHX), a protein synthesis inhibitor. Similar levels of Bambi mRNA induction were observed in HSCs treated with CHX, RA plus CHX or Am580 plus CHX (Fig. S12), indicating that RA signaling regulated Bambi expression at least in part through transcriptional effects. Decreased Bambi mRNA expression may be due to a decrease in either the intracellular RA concentration or Rars mRNA expression. To this end, we transfected HSCs with siRarb or scrambled siRNA. Rarb knockdown ameliorated the induction of Bambi mRNA expression by the RARs activator Am580 (Fig. S13), demonstrating that RA signaling regulated Bambi mRNA expression through RARs.
To determine whether restoring RA signaling through RA and Am580 treatment could inhibit the LPS-induced sensitization of HSCs to TGFB, freshly isolated HSCs were pretreated with RA or Am580, treated with LPS and then stimulated with TGFB. LPS plus TGFB markedly enhanced the mRNA expression of Col1a1 and Acta2 compared with TGFB treatment alone, and pretreatment with both RA and Am580 suppressed the enhancement (Fig. 5H-I). Overall, these data suggested that the decreased retinoid storage caused by LPS-induced autophagy in HSCs led to dysfunctional RA signaling, further reducing the expression of the TGFB pseudoreceptor BAMBI and increasing the sensitization of HSCs to TGFB.
Autophagy is involved in the LPS-mediated augmentation of the fibrotic response in CCl4-treated mice
To elucidate whether autophagy also plays a role in the LPS-mediated exaggeration of liver fibrosis in an animal model, mice were randomly assigned to 8 groups: control, LPS alone, CQ alone, LPS+CQ, CCl4 alone, CCl4+LPS, CCl4+CQ, and CCl4+LPS+CQ. Mice treated with CCl4 developed liver fibrosis as expected. Compared with CCl4, LPS plus CCl4 significantly increased the Sirius Red staining area (an indication of fibrosis) in liver sections, whereas co-treatment with CQ markedly inhibited this effect (Fig. 6A). Consistent with this finding, the hepatic mRNA expression levels of Col1a1, Acta2, Tgfb and Timp1 were significantly higher in LPS-treated mice than in controls in the CCl4-induced liver fibrosis model (Fig. 6B-C and Figs. S14A, S14B). This effect was dramatically ameliorated by CQ injection. Furthermore, the hepatic ACTA2 and COL1A1 protein expression was markedly increased in CCl4+LPS-treated mice compared with that in mice treated with CCl4 alone, whereas CQ co-treatment significantly suppressed the LPS-induced effect (Fig. 6D-E). Collectively, these data indicate an important role of autophagy in the LPS-aggravated fibrotic response of mice treated with CCl4.
Figure 6.
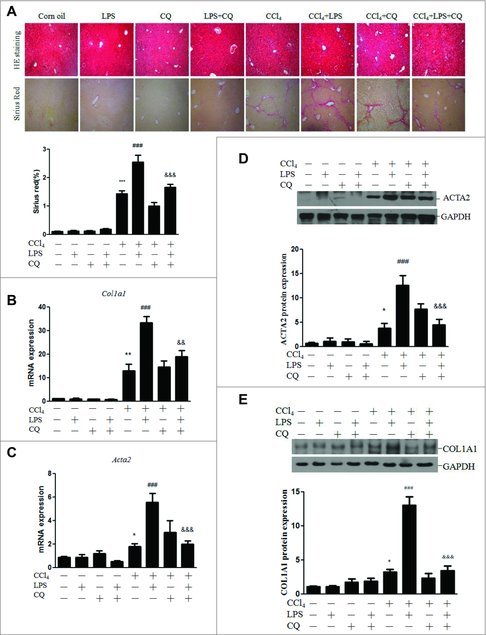
Autophagy contributed to the LPS-mediated enhancement of the fibrotic response in CCl4-treated mice. Mice were injected with CCl4 or maize oil for 4 wk. For the last 2 of these weeks, a subset of mice was treated with CQ as well. Thereafter, the mice were injected with LPS (n = 6–8/group). (A) Representative images of H&E (HE) and Sirius Red staining in liver sections and quantification of Sirius Red staining (lower panel). (B-C) Hepatic mRNA expression of Col1a1 and Acta2. (D) Representative blots of the hepatic ACTA2 protein expression, with quantification shown in the lower panel. (E) Representative blots of the hepatic COL1A1 protein expression, with quantification shown in the lower panel. *p < 0.05, **p < 0.01, ***p < 0.001 versus control. ##p < 0.01, ###p < 0.001 versus CCl4. &&p < 0.01, &&&p < 0.001 versus CCl4 + LPS.
LPS induced autophagy and decreased RA signaling in HSCs in vivo
To explore the effect of LPS on lipid storage and RA signaling in HSCs in vivo, mice were subjected to 3 intraperitoneal injections of CQ, followed by a single tail vein injection of LPS. After 4 h, HSCs were isolated and assessed to determine the lipid content and RAR target gene expression. As shown in Fig. 7A, the HSCs from saline-treated mice contained several large LDs, a characteristic feature of quiescent HSCs. LPS treatment of these mice resulted in the disappearance of large LDs and an increase in small LDs in the HSCs, whereas pretreatment with CQ restored the lipid content to a level similar to that of control. Moreover, the Bambi, Cyp26a1, Rbp1 and Rarg expression was dramatically decreased in the HSCs from LPS-treated mice relative to that of the control, whereas CQ pretreatment reversed this change (Fig. 7B-E). However, CQ treatment failed to reverse the LPS-induced downregulation of Rarb mRNA expression, and no significant difference in the expression of Rara mRNA was observed (Fig. S15). We also performed experiments determining whether RA depletion by LPS-induced autophagy occurred in HSCs isolated from mice injected with CQ followed by LPS. The results showed a remarkable reduction of the RA level in HSCs in response to LPS, whereas pretreatment with CQ partly restored the RA concentration (Fig. 7F).
Figure 7.
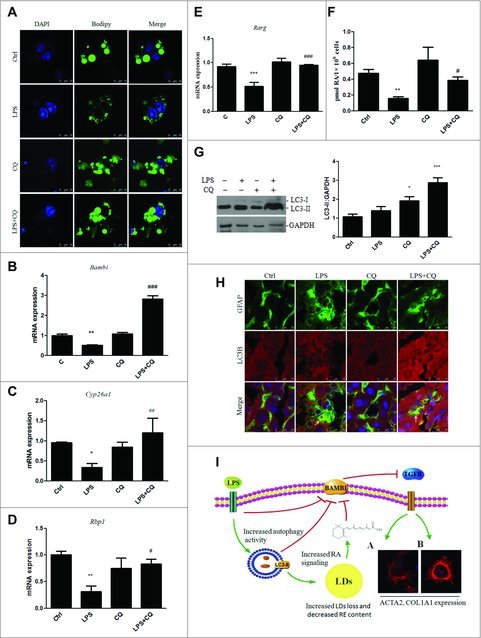
LPS induced autophagy, LD loss and decreased RA signaling in HSCs in vivo. Mice were injected with CQ before LPS administration. (A) LDs in HSCs were stained with Bodipy within 12 h after isolation. Scale bar: 10 μm. (B-E) mRNA expression of Bambi, Cyp26a1, Rbp1, and Rarg in isolated HSCs. *p < 0.05, **p < 0.01,***p < 0.001 versus control, #p < 0.05, ##p < 0.01, ###p < 0.001 versus the LPS group. (F) RA concentration was measured in HSCs in vivo. **p < 0.01 versus control, #p < 0.05 versus LPS. (G) Representative blots of LC3 expression are shown in HSCs, with quantification shown in the right panel. *p < 0.05, ***p < 0.001 compared with vehicle. (H) Double immunofluorescence staining of LC3 (red) and GFAP (green) in the liver. Arrows indicate the colocalization of GFAP and LC3. Scale bar: 25 μm. (I) A signal transduction diagram showing that LPS-induced autophagy mediated the LDs loss, decreased RA signaling and downregulated Bambi in HSCs, subsequently sensitizing them to TGFB-induced activation. TGs, triglycerides. RE, retinyl ester.
Next, to demonstrate that LPS induced autophagic flux in HSCs in vivo, mice were injected with CQ and then administered LPS, and HSCs were isolated. As shown in Fig. 7G, CQ alone blocked LC3-II turnover, with a further increase in the LC3-II level in the LPS+CQ group, suggesting that LPS stimulated autophagic flux in HSCs in vivo. We confirmed these findings in liver sections of mice injected with CQ followed by LPS. The liver sections were double immunostained for the autophagy marker LC3 and the HSC marker GFAP. The colocalization of GFAP with LC3-II was higher in the liver sections of LPS-treated mice, than in those of the control mice and was further increased by CQ pretreatment (Fig. 7H). Taken together, these results demonstrate that LPS can also induce autophagy in HSCs in vivo.
Discussion
In this study, we revealed a novel mechanism underlying the HSC activation mediated by LPS-induced autophagy. Treatment of HSCs with LPS induced autophagy that stimulated triglyceride and retinyl ester hydrolysis, which contributed to a reduction in the retinoid content and RA signaling. Furthermore, LPS downregulated the TGFB pseudoreceptor gene Bambi and sensitized HSCs to TGFB signaling, thus promoting HSC fibrosis in vitro and in vivo. The schematic of the signaling pathway in LPS-treated HSCs is shown in Fig. 7I.
MTOR (mechanistic target of rapamycin [serine/threonine kinase]), as a critical negative regulator of autophagy, phosphorylates ULK1 at Ser757, thus inhibiting ULK1, while AMPK promotes autophagy by directly phosphorylating and activating ULK1 at Ser317 and Ser777.30 The phosphoinositide 3-kinase-activated kinase AKT phosphorylates the MTOR repressor TSC2 (tuberous sclerosis 2), leading to MTOR activation and subsequent inhibition of autophagy activity.31 In keeping with these studies, our results demonstrated that LPS induced autophagy at least in part through suppressing AKT-induced ULK1 Ser757 phosphorylation by MTOR and stimulating ULK1 Ser317 phosphorylation by AMPK.
LPS upregulates autophagy in multiple animal tissues and cell lines. In human primary macrophages and the murine macrophage RAW264.7 cell line, LPS triggers autophagy via the TLR4 signaling pathway,21,22 and activation of autophagy by LPS in macrophages limits the inflammatory response in the livers of mice.32,33 Similarly, LPS stimulates autophagy in HL-1 cardiomyocytes, neonatal rat cardiomyocytes and mouse hearts.34 In the present study, we used multiple approaches to demonstrate that LPS stimulation promoted autophagy activity during HSC activation as assessed by increasing levels of LC3B-II and cleaved GFP-LC3B, thus enhancing the autophagic flux in vitro and in vivo.
Degradation of LDs via lysosomal lipases that have access to intracellular stores is termed lipophagy, which was first identified in mouse hepatocytes subjected to starvation.12,35 Thereafter, other studies also implicated autophagy in the LD degradation in hepatocytes treated with caffeine10 or thyroid hormone.11 As LD loss is an important hallmark of HSC activation, it is plausible to propose that LPS-induced autophagy might contribute to LD depletion in HSCs. As expected, we found that LPS caused a dramatic reduction in the LD content in LX-2 cells and primary HSCs; inhibiting autophagy using either pharmacological inhibitors or genetic ablation prevented this reduction. We further demonstrated that the autophagy-lysosomal pathway was involved in the LD depletion in LPS-mediated HSCs. We also found that LPS increased the colocalization between retinyl esters and lysosomes in HSCs, indicating that increased autophagy activity might be involved in retinyl ester hydrolysis in lysosomes. This might result in a reduction of basal retinyl esters and RA level in LX-2 cells and primary HSCs. We also examined the RA target genes, including Rbp1, Rara, Rarb and Rarg, as they are usually indicative of the RA level in cells.36,37 RA target genes were significantly decreased by LPS in HSCs, and this effect was reversed by autophagy inhibition in vitro and in vivo. Direct measurement of RA concentration in vitro and in vivo revealed that autophagy was involved in LPS-mediated reduction of RA levels. Pretreatment with RA and the RAR agonist Am580 reversed the LPS-induced reduction of Bambi and Rar expression, further demonstrating that LPS-induced autophagy contributed to the decreased RA signaling in HSCs. In agreement with our results, a previous study showed that autophagy was responsible for the maintenance of retinoid levels in the retinal pigment epithelium to support vision.38
One of the most important findings in our study is that LPS linked 2 salient features of HSC activation, LD depletion and enhanced Col1a1 and Acta2 expression, as presented in a diagrammatic sketch in Fig. 7G. Typically, LD loss occurs simultaneously with obvious upregulation of Col1a1 and Acta2 expression. Our results demonstrated that LPS treatment caused lipid loss in vitro and in vivo but did not affect the Col1a1 or Acta2 expression, suggesting that LPS-induced lipid loss may not be a prerequisite for Col1a1 and Acta2 expression. However, our findings did not indicate complete and total independence of the pathophysiological processes. A significant reduction in retinyl ester content during lipid loss leads to lower all trans-retinoic acid levels and downstream RAR signaling. BAMBI functions as a decoy type I receptor that negatively modulates TGFB-BMP- activin signaling by stably associating with type II receptors, which prevents the formation of active receptor complexes.39 Because RA signaling positively regulated BAMBI expression, the decrease in RA signaling resulted in reduced BAMBI expression, thereby exacerbating fibrosis in response to TGFB signaling in vitro and CCl4 administration in vivo. This study is the first to demonstrate that RA, or more accurately the RA signaling, decreases HSC activation only in the presence of TGFB through Bambi upregulation but does not induce or inhibit liver fibrosis. By contrast, a previous study reported that RA markedly reduced liver fibrosis in rats subjected to BDL,40 whereas other studies indicated that RA promoted HSC activation and liver fibrosis.37,41 These discrepancies may be due to the different experimental designs and HSC activation conditions.42,43
Importantly, we demonstrated that LPS decreased Bambi expression and sensitized HSCs to TGFB treatment by regulating autophagy. In other words, autophagy mediated the LPS-induced downregulation of BAMBI, thus making HSCs more susceptible to TGFB activation. Liver fibrosis often occurs with a concomitant increase in intestinal permeability and bacterial translocation. In particular, the Gram-negative bacterial product LPS leaks into the bloodstream and is delivered to the liver, where it activates HSCs. This process obviously exacerbates existing liver damage. Our study showed that autophagy inhibition likely attenuated LPS-induced HSC activation. We also hypothesized that any factors that promote intestinal permeability and bacterial translocation have the potential to increase the risk of enhanced liver fibrosis if another type of liver damage is present, such as chronic viral hepatitis infection or steatohepatitis associated with either alcohol consumption or obesity. Under physiologic conditions, HSC autophagy results in partial self-digestion to maintain energy homeostasis and recycle dysfunctional proteins and organelles. However, in pathophysiological situations, LPS, as a form of stress, causes increased autophagy in HSCs. In the hyperfunctional state, LPS-induced autophagy may impair BAMBI signaling, which promotes HSC fibrosis.
In summary, the present study demonstrates that LPS exposure promotes HSC fibrosis through increased autophagy activity and dysfunctional RA signaling. This novel mechanism underlying the LPS-induced fibrotic response of HSCs is associated with LDs loss and downregulation of the TGFB pseudoreceptor BAMBI.
Materials and methods
Reagents
Reagents were obtained as follows: LPS (L4524), chloroquine (C6628), LY294002 (L9908), rapamycin (V900930), acridine orange (AO; 318337), polymyxin B (P1004), Am580 (A8843) and all-trans retinoic acid (A2625) were from Sigma. Bafilomycin A1 (11038) was from Cayman. Antibodies to ATG5 (12994), LC3 (2775; recognizes LC3B and possibly other isoforms), p-MTOR (5536), MTOR (2972), p-AKT (13038), AKT (4685), p-ULK1 (S757; 14202), p-ULK1 (S317; 12753), ULK1 (8054), p-AMPK (2535) and AMPK (2532) were from Cell Signaling Technology. RARs (sc-773), GAPDH (sc-25778) and secondary anti-rabbit and anti-mouse antibodies (sc-2004; sc-2005) were from Santa Cruz Biotechnology. LC3 (ab48394), GFAP (3670), SQSTM1/p62 (ab109012), COL1A1/COLLAGEN I (ab34710) and GFP (ab290) antibodies were purchased from Abcam. ACTA2/α-SMA (A2547) was from Sigma. TGFB/TGF-β (240-B) was obtained from R&D System. Bodipy 493/503 (D3922), LysoTracker Red (L7528), Alexa Flour 546 anti-rabbit secondary antibody (A10040), Alexa Flour 633 anti-mouse secondary antibody (A21050), Prolong Gold Antifade Reagent (P36931), Lipofectamine 2000 (1668019), Lipofectamine RNAiMAX (13778150), CellLight lysosome-GFP (C10507), collagenase IV (17104019) and Earle's balanced salt solution (EBSS; 24010043) were from Life Technologies. ATG5 siRNA (sc41445), Atg5 siRNA (sc-41446), TLR4 siRNA (sc-40260) and scramble siRNA (sc-37007) were from Santa Cruz Biotechnology. Rarb siRNA (siQ0001) and non-target control siRNA was purchased from Ribobio. DNase I (10104159001) was from Roche. Pronase E (7433) was obtained from Milliopre. Mobile phase solvents acetonitrile, acetic acid, methanol, hexane and 2-propanol were from Fisher. BAMBI antibody (bs-12418) was obtained from Bioss. Other chemicals were purchased from Sigma unless otherwise specified.
Animals
All experimental protocols were approved by the animal care and use committee of Sun Yat-Sen University. Eight-wk-old Balb/c mice were purchased from the animal center of Sun Yat-Sen University and housed under controlled temperature and light conditions (20°C∼22°C and 12 h:12 h light:dark cycle), and were also allowed ad libitum access to water and diet. For induction of liver fibrosis, mice were randomly divided and administrated CCl4 (20% in maize oil) or maize oil at a dose of 0.5 μl/g body weight 2 times a wk for 4 wk, during which mice underwent CQ peritoneal injection or saline at a dose of 60 mg/kg 2 times a week in the last 2 wk. Finally, mice were subjected to intravenous LPS (10 mg/kg) and killed 4 h later.44 In other experiment, mice were intraperitoneally injected with CQ or saline daily for 3 d following LPS administration. Four h later, HSCs were isolated and assessed for lipid content, retinoic acid concentration, protein and mRNA expression.
HSC isolation and cell culture
HSCs were isolated from male Balb/c mice as described previously.45 Briefly, after in situ perfusion of the liver with 2-step pronase-collagenase digestion, HSCs were separated from other nonparenchymal cells by density-gradient centrifugation using OptiPrep (Axis-Shield, 1114542). HSCs were maintained in Dulbecco's modified Eagle's medium (Gibco, C11995500BT) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a humidified atmosphere with 5% CO2. Cell purity was identified by retinoid autofluorescence when excited by ultraviolet light and was found to be above 95% (Fig. S2). All treatments were initiated 12 h after isolation unless otherwise indicated. LX-2 cells were kindly provided by Dr. Scott Friedman and cultured in Dulbecco's modified Eagle's medium supplemented with 2% fetal bovine serum and 1% penicillin/streptomycin. All experiments were repeated at least 3 times.
Liver histology
Liver tissues were fixed with 10% buffered formalin for 24 h and embedded in paraffin. Five-µm-thick sections were cut and stained with hematoxylin and eosin, and Sirius Red (Sigma, 365548). Quantification of Sirius Red-positive area:total area was performed using ImageJ. At least 10 fields of each slide were measured.
Plasmid preparation and transfection
Plasmids GFP-LC3B (Addgene, 11546; deposited by Karla Kirkegaard) and mRFP-GFP-LC3B (Addgene, 21074; deposited by Tamotsu Yoshimori) were extracted by using PureLink HiPure plasmid midiprep kit (Invitrogen, K210004), dissolved into TE buffer (10 mM Tris-HCl, pH 8.0, 0.1 mM EDTA) and quantified by Biophotometer (Eppendorf, Biophotometer 6131). For transfection experiments, LX-2 cells were cultured on glass coverslips and transfected with 2 μg of plasmids. After being treated with LPS, cells were observed by confocal microscopy (Leica SP5) for exogenous GFP-LC3B and tandem fluorescent LC3B. Quantification of autophagosomes and autolysosomes dots were performed using ImageJ software.
Real-time quantitative polymerase chain reaction analysis
Total RNA was extracted from HSCs or LX-2 using Trizol reagent (Invitrogen, 15596026), according to the manufacturer's instructions. After reverse transcription, real-time PCR was performed to quantify mRNA expression using an ABI ViiA 7 real time PCR system (Applied Biosystems), and Powerup SYBR Green Master Mix (Applied Biosystems, A25778). Gene expression values were normalized against the housekeeping genes RNA18S. Fold changes were calculated using the delta delta Ct method. Gene-specific primers are described in Table S1.
Western blot
Total protein concentrations were determined using the BCA protein assay kit (Pierce, 23225). Equal amounts of total proteins were separated using SDS-PAGE and then transferred to PVDF membrane (Millipore, IPVH00010). After blocking with 5% nonfat milk in TBST (TBS + 0.1% Tween-20) for 1 h, the blots were incubated with primary antibodies to LC3B, SQSTM1, GAPDH, p-AKT, AKT, p-MTOR, MTOR, ATG5, p-AMPK, AMPK, p-ULK1, ULK1, BAMBI, COL1A1 and ACTA2 overnight followed by 3 washes in TBST for 5 min each. HRP-conjugated secondary anti-rabbit, and anti-mouse antibodies were applied for 1 h, followed by 3 washes in TBST for 5 min each. Blots were then incubated with ECL (Pierce, 32109). The intensity of bands was quantified using software ImageJ.
Immunocytochemistry and immunohistochemistry
After treatment, LX-2 or HSCs were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS; Boster, AR0030) for 10 min at room temperature followed by overnight incubation with endogenous anti-LC3 (1:200) at 4°C or Bodipy 493/503 (1:200) to stain lipid droplets at room temperature for 30 min. For LC3 staining, an Alexa Fluor 546 donkey anti-rabbit secondary antibody (1:250) was added to detect LC3 followed by 3 washes in PBS. Cells were mounted with ProLong Gold antifade reagent with DAPI (Life Technologies, P36931) before being observed with confocal microscopy. Quantification of LC3 puncta and number of lipid droplets were performed using software imageJ.
AO and LysoTracker Red DND 99 staining was performed as described previously. Briefly, cells were incubated with either 1 µg/mL of AO or 100 nM of LysoTracker Red for 15–30 min at 37°C followed by 3 PBS washes, and then immediately observed under confocal microscopy. HSCs were transduced with a LAMP1-eGFP construct according to the manufacturer's instruction (Life Technologies, C10507), then transfected with siAtg5 siRNA for 24 h, followed by LPS treatment. Cells were fixed, and immunostained with antibodies to RARs (1:100).
For immunohistochemistry, liver sections were fixed with 10% formaldehyde for 10 min followed by 3 washes with PBS. Then, sections were incubated with anti-GFAP (1:200) and anti-LC3B (1:300) at 4°C overnight. The next day, sections were washed with PBS and then incubated with Alexa Fluor 546 donkey anti-rabbit secondary antibody (1:250) or Alexa Fluor 633 goat anti-mouse secondary antibody (1:250) for 1 h. Cells were mounted with ProLong Gold antifade reagent with DAPI before being observed with confocal microscopy.
siRNA transfection
LX-2 cells were plated into 6-well plates to grow to approximately 80% confluency. Then cells were transfected with ATG5 siRNA, TLR4 siRNA or scramble siRNA (100 nM) using Lipofectamine 2000 (Life Technologies, 1668019) for 5 h before addition of normal medium. Thereafter, medium was removed and cells were incubated with LPS for 24 h followed by protein and mRNA detection. HSCs were transfected with siAtg5 siRNA (50 nM), Rarb siRNA (50 nM) or scramble siRNA using Lipofectamine RNAiMAX (Life Technologies, 13778150) for 24 h before other treatment.
HPLC
HSCs were quantified and retinoic acid was extracted as described previously.46 Reverse-phase chromatography was performed on an Agilent 1200 HPLC system equipped with a UV detector and analytical column (Symmetry C18, 5-μm particle size, Waters). The chromatographic conditions described previously were as follows:47 mobile phase, 57.5% acetonitrile-25% acetic acid (diluted to 2% with water)-17.5% methanol; flow rate, 0.8 ml/min, UV detection wavelength, 354 nm. Acitretin (Sigma, 44707) was used as an internal standard. All manipulations with light-sensitive retinoids were performed under red light.
Statistical analysis
All data are presented as the means ± standard error of the mean. Statistical analyses were performed using an unpaired Student t test for comparisons between 2 groups or one-way analysis of variance for more than 3 groups, which was followed by a comparison of 2 groups. The results were considered significantly different at p < 0.05.
Supplementary Material
Funding Statement
This study was supported by the National Basic Research Program of China (973 Program) under grant number: 2012CB517506 and Science and Technology Program of Guangzhou under grant number: 201604020002.
Abbreviations
- BAF
bafilomycin A1
- BAMBI
BMP and activin membrane-bound inhibitor
- BDL
bile duct ligation
- CHX
cycloheximide
- GFAP
glial fibrillary acidic protein
- HSCs
hepatic stellate cells
- LDs
lipid droplets
- LPS
lipopolysaccharide
- MAP1LC3B
microtubule-associated protein 1 light chain 3 β
- MmHSC
Mus musculus (mouse) HSC
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- RARA
retinoic acid receptor, α
- RARB
retinoic acid receptor, β
- RARG
retinoic acid receptor, gamma
- RBP1
retinol binding protein 1, cellular
- tfLC3
tandem fluorescent mRFP-GFP-LC3
- TGFB
transforming growth factor, β
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209-18. https://doi.org/10.1172/JCI24282. PMID:15690074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38-53. https://doi.org/10.1016/S0168-8278(02)00429-4. PMID:12591185 [DOI] [PubMed] [Google Scholar]
- [3].Xu J, Liu X, Koyama Y, Wang P, Lan T, Kim IG, Kim IH, Ma HY, Kisseleva T. The types of hepatic myofibroblasts contributing to liver fibrosis of different etiologies. Front Pharmacol. 2014;5:167. https://doi.org/10.3389/fphar.2014.00167. PMID:25100997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, Liu X, Xu J, Wang P, Paik YH, et al.. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U S A. 2014;111:E3297-305. https://doi.org/10.1073/pnas.1400062111. PMID:25074909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461-72. https://doi.org/10.1038/nrm4024. PMID:26177004 [DOI] [PubMed] [Google Scholar]
- [6].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313-26. https://doi.org/10.1016/j.cell.2010.01.028. PMID:20144757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Guo ML, Liao K, Periyasamy P, Yang L, Cai Y, Callen SE, Buch S. Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy. 2015;11:995-1009. https://doi.org/10.1080/15548627.2015.1052205. PMID:26043790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42. https://doi.org/10.1016/j.cell.2007.12.018. PMID:18191218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Arroyo DS, Gaviglio EA, Peralta RJ, Bussi C, Rodriguez-Galan MC, Iribarren P. Autophagy in inflammation, infection, neurodegeneration and cancer. Int Immunopharmacol. 2014;18:55-65. https://doi.org/10.1016/j.intimp.2013.11.001. PMID:24262302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sinha RA, Farah BL, Singh BK, Siddique MM, Li Y, Wu Y, Ilkayeva OR, Gooding J, Ching J, Zhou J, et al.. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology. 2014;59:1366-80. https://doi.org/10.1002/hep.26667. PMID:23929677 [DOI] [PubMed] [Google Scholar]
- [11].Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X, Privalsky ML, Cheng SY, Stevens RD, Summers SA, et al.. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest. 2012;122:2428-38. https://doi.org/10.1172/JCI60580. PMID:22684107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131-5. https://doi.org/10.1038/nature07976. PMID:19339967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhao J LFR, Guimar Es ELM, Dollé L, Mannaerts I, Najimi M, Sokal E, van Grunsven LA.. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353-60. https://doi.org/10.1016/j.jhep.2011.07.010. PMID:21803012 [DOI] [PubMed] [Google Scholar]
- [14].Deng J, Huang Q, Wang Y, Shen P, Guan F, Li J, Huang H, Shi C. Hypoxia-inducible factor-1alpha regulates autophagy to activate hepatic stellate cells. Biochem Biophys Res Commun. 2014;454:328-34. https://doi.org/10.1016/j.bbrc.2014.10.076. PMID:25450397 [DOI] [PubMed] [Google Scholar]
- [15].He W, Wang B, Yang J, Zhuang Y, Wang L, Huang X, Chen J. Chloroquine improved carbon tetrachloride-induced liver fibrosis through its inhibition of the activation of hepatic stellate cells: Role of autophagy. Biol Pharm Bull. 2014;37:1505-9. https://doi.org/10.1248/bpb.b14-00297. PMID:25177034 [DOI] [PubMed] [Google Scholar]
- [16].Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938-46. https://doi.org/10.1053/j.gastro.2011.12.044. PMID:22240484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fouts DE, Torralba M, Nelson KE, Brenner DA, Schnabl B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J Hepatol. 2012;56:1283-92. https://doi.org/10.1016/j.jhep.2012.01.019. PMID:22326468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324-32. https://doi.org/10.1038/nm1663. PMID:17952090 [DOI] [PubMed] [Google Scholar]
- [19].De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L, Mingarelli E, Facinelli B, Magi G, Palmieri C, et al.. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology. 2014;59:1738-49. https://doi.org/10.1002/hep.26695. PMID:23959503 [DOI] [PubMed] [Google Scholar]
- [20].Xu Y, Liu XD, Gong X, Eissa NT. Signaling pathway of autophagy associated with innate immunity. Autophagy. 2008;4:110-2. https://doi.org/10.4161/auto.5225. PMID:18059159 [DOI] [PubMed] [Google Scholar]
- [21].Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135-44. https://doi.org/10.1016/j.immuni.2007.05.022. PMID:17658277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Waltz P, Carchman EH, Young AC, Rao J, Rosengart MR, Kaczorowski D, Zuckerbraun BS. Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy. 2011;7:315-20. https://doi.org/10.4161/auto.7.3.14044. PMID:21307647 [DOI] [PubMed] [Google Scholar]
- [23].Guo R, Xu X, Babcock SA, Zhang Y, Ren J. Aldehyde dedydrogenase-2 plays a beneficial role in ameliorating chronic alcohol-induced hepatic steatosis and inflammation through regulation of autophagy. J Hepatol. 2015;62:647-56. https://doi.org/10.1016/j.jhep.2014.10.009. PMID:25457208 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [24].Zhou Y, Liang X, Chang H, Shu F, Wu Y, Zhang T, Fu Y, Zhang Q, Zhu JD, Mi M. Ampelopsin-induced autophagy protects breast cancer cells from apoptosis through Akt-mTOR pathway via endoplasmic reticulum stress. Cancer Sci. 2014;105:1279-87. https://doi.org/10.1111/cas.12494. PMID:25088800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jung JS, Shin KO, Lee YM, Shin JA, Park EM, Jeong J, Kim DH, Choi JW, Kim HS. Anti-inflammatory mechanism of exogenous C2 ceramide in lipopolysaccharide-stimulated microglia. Biochim Biophys Acta. 2013;1831:1016-26. https://doi.org/10.1016/j.bbalip.2013.01.020. PMID:23384839 [DOI] [PubMed] [Google Scholar]
- [26].Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Okada Y, Kurihara C, Irie R, Yokoyama H, et al.. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology. 2014;59:154-69. https://doi.org/10.1002/hep.26604. PMID:23832448 [DOI] [PubMed] [Google Scholar]
- [27].Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Usui S, Furuhashi H, Kimura A, Nishiyama K, et al.. Acyl-CoA:cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J Hepatol. 2014;61:98-106. https://doi.org/10.1016/j.jhep.2014.03.018. PMID:24657401 [DOI] [PubMed] [Google Scholar]
- [28].Shirakami Y, Lee SA, Clugston RD, Blaner WS. Hepatic metabolism of retinoids and disease associations. Biochim Biophys Acta. 2012;1821:124-36. https://doi.org/10.1016/j.bbalip.2011.06.023. PMID:21763780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Blaner WS, O'Byrne SM, Wongsiriroj N, Kluwe J, D'Ambrosio DM, Jiang H, Schwabe RF, Hillman EM, Piantedosi R, Libien J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim Biophys Acta. 2009;1791:467-73. https://doi.org/10.1016/j.bbalip.2008.11.001. PMID:19071229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132-41. https://doi.org/10.1038/ncb2152. PMID:21258367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vucicevic L, Misirkic M, Janjetovic K, Vilimanovich U, Sudar E, Isenovic E, Prica M, Harhaji-Trajkovic L, Kravic-Stevovic T, Bumbasirevic V, et al.. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011;7:40-50. https://doi.org/10.4161/auto.7.1.13883. PMID:20980833 [DOI] [PubMed] [Google Scholar]
- [32].Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, Tanaka KE, Czaja MJ. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271-84. https://doi.org/10.1080/15548627.2015.1009787. PMID:25650776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wan J, Ma J, Anand V, Ramakrishnan S, Roy S. Morphine potentiates LPS-induced autophagy initiation but inhibits autophagosomal maturation through distinct TLR4-dependent and independent pathways. Acta Physiol (Oxf). 2015;214:189-99. https://doi.org/10.1111/apha.12506. PMID:25850855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC, Gottlieb RA. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 2009;296:H470-9. https://doi.org/10.1152/ajpheart.01051.2008. PMID:19098111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shatz O, Holland P, Elazar Z, Simonsen A. Complex relations between phospholipids, autophagy, and neutral lipids. Trends Biochem Sci. 2016;41:907-23. https://doi.org/10.1016/j.tibs.2016.08.001. PMID:27595473 [DOI] [PubMed] [Google Scholar]
- [36].Kluwe J, Wongsiriroj N, Troeger JS, Gwak GY, Dapito DH, Pradere JP, Jiang H, Siddiqi M, Piantedosi R, O'Byrne SM, et al.. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut. 2011;60:1260-8. https://doi.org/10.1136/gut.2010.209551. PMID:21278145 [DOI] [PubMed] [Google Scholar]
- [37].O'Mahony F, Wroblewski K, O'Byrne SM, Jiang H, Clerkin K, Benhammou J, Blaner WS, Beaven SW. Liver X receptors balance lipid stores in hepatic stellate cells through Rab18, a retinoid responsive lipid droplet protein. Hepatology. 2015;62:615-26. https://doi.org/10.1002/hep.27645. PMID:25482505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, et al.. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154:365-76. https://doi.org/10.1016/j.cell.2013.06.012. PMID:23870125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lantero A, Tramullas M, Díaz A, Hurlé MA. Transforming growth factor-β in normal nociceptive processing and pathological pain models. Mol Neurobiol. 2012;45:76-86. https://doi.org/10.1007/s12035-011-8221-1. PMID:22125199 [DOI] [PubMed] [Google Scholar]
- [40].He H, Mennone A, Boyer JL, Cai SY. Combination of retinoic acid and ursodeoxycholic acid attenuates liver injury in bile duct-ligated rats and human hepatic cells. Hepatology. 2011;53:548-57. https://doi.org/10.1002/hep.24047. PMID:21274875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Okuno M, Moriwaki H, Imai S, Muto Y, Kawada N, Suzuki Y, Kojima S. Retinoids exacerbate rat liver fibrosis by inducing the activation of latent TGF-beta in liver stellate cells. Hepatology. 1997;26:913-21. https://doi.org/10.1053/jhep.1997.v26.pm0009328313. PMID:9328313 [DOI] [PubMed] [Google Scholar]
- [42].Lee Y, Jeong W. Retinoic acids and hepatic stellate cells in liver disease. J Gastroen Hepatol. 2012;27:75-9. https://doi.org/10.1111/j.1440-1746.2011.07007.x. PMID:22320921 [DOI] [PubMed] [Google Scholar]
- [43].Milliano MT, Luxon BA. Rat hepatic stellate cells become retinoid unresponsive during activation. Hepatol Res. 2005;33:225-33. https://doi.org/10.1016/j.hepres.2005.08.007. PMID:16253547 [DOI] [PubMed] [Google Scholar]
- [44].Affo S, Morales-Ibanez O, Rodrigo-Torres D, Altamirano J, Blaya D, Dapito DH, Millán C, Coll M, Caviglia JM, Arroyo V, et al.. CCL20 mediates lipopolysaccharide induced liver injury and is a potential driver of inflammation and fibrosis in alcoholic hepatitis. Gut. 2014;63:1782-92. https://doi.org/10.1136/gutjnl-2013-306098. PMID:24415562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc. 2015;10:305-15. https://doi.org/10.1038/nprot.2015.017. PMID:25612230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Van Merris V, Meyer E, De Wasch K, Burvenich C. Simple quantification of endogenous retinoids in bovine serum by high-performance liquid chromatography – diode-array detection. Anal Chim Acta. 2002;468:237-44. https://doi.org/10.1016/S0003-2670(02)00601-3 [Google Scholar]
- [47].Radaeva S, Wang L, Radaev S, Jeong WI, Park O, Gao B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol Gastrointest Liver Physiol. 2007;293:G809-16. https://doi.org/10.1152/ajpgi.00212.2007. PMID:17673545 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.