

ABSTRACT
Accumulation of intracellular misfolded or damaged proteins is associated with both normal aging and late-onset degenerative diseases. Two cellular clearance mechanisms, the ubiquitin-proteasome system (UPS) and the macroautophagy/autophagy-lysosomal pathway, work in concert to degrade harmful protein aggregates and maintain protein homeostasis. Here we show that Mask, an Ankyrin-repeat and KH-domain containing protein, plays a key role in promoting autophagy flux and mitigating degeneration caused by protein aggregation or impaired UPS function. In Drosophila eye models of human tauopathy or amyotrophic lateral sclerosis diseases, loss of Mask function enhanced, while gain of Mask function mitigated, eye degenerations induced by eye-specific expression of human pathogenic MAPT/TAU or FUS proteins. The fly larval muscle, a more accessible tissue, was then used to study the underlying molecular mechanisms in vivo. We found that Mask modulates the global abundance of K48- and K63-ubiquitinated proteins by regulating autophagy-lysosome-mediated degradation, but not UPS function. Indeed, upregulation of Mask compensated the partial loss of UPS function. We further demonstrate that Mask promotes autophagic flux by enhancing lysosomal function, and that Mask is necessary and sufficient for promoting the expression levels of the proton-pumping vacuolar (V)-type ATPases in a TFEB-independent manner. Moreover, the beneficial effects conferred by Mask expression on the UPS dysfunction and neurodegenerative models depend on intact autophagy-lysosomal pathway. Our findings highlight the importance of lysosome acidification in cellular surveillance mechanisms and establish a model for exploring strategies to mitigate neurodegeneration by boosting lysosomal function.
KEYWORDS: ALS, autophagy, Drosophila model, FUS, lysosomal acidification, protein aggregation, tauopathy, ubiquitin-proteasome system, V-type ATPases
Introduction
Misfolded protein aggregates in and outside of cells in the central nervous system are pathological hallmarks of many neurodegenerative disorders including Alzheimer (AD), Parkinson (PD), Huntington (HD) diseases and amyotrophic lateral sclerosis (ALS). Interestingly, many of the aggregated proteins (such as MAPT (TAU) and APP for Alzheimer disease, SNCA/α-synuclein for Parkinson disease, HTT (Huntingtin) for Huntington disease, FUS, SOD1 and TARDBP/TDP-43 for ALS) can serve as seeds for “prion-like” spreading of the aggregation within and among cells.1 It is not entirely clear whether these aggregates are the causes or the results of progressive and cell-type-specific neurodegeneration. However, mounting evidence suggests that clearance and prevention of these toxic protein aggregates are beneficial for meliorating degeneration.2-4
Two major pathways collaborate in regulating intracellular protein degradation: the ubiquitin-proteasome system (UPS) and the autophagy-lysosomal system. Under the normal conditions, UPS serves as the primary route for rapid protein turnover while autophagy mainly degrades long-lived proteins and large cellular organelles under basal conditions and can be robustly induced in face of stresses such as starvation, organelle damage or accumulation of misfolded proteins. However when it comes to degradation of damaged proteins in diseased states, autophagy has been shown to play at least an equally important role as UPS.5 Many of the neurodegenerative disease-related proteins are delivered to autophagic vacuoles,6,7 and degraded by the autophagy pathway.8-10 Meanwhile, impairment of autophagy in the mouse brain causes neurodegeneration associated with ubiquitin-positive protein aggregation.11,12 These data suggest that UPS and autophagy are both indispensable in maintaining cellular protein homeostasis. Furthermore, recent studies indicate that UPS and autophagy pathways coordinate with each other to prevent accumulation of toxic protein aggregates, so that enhanced activity of one pathway can compensate if the other is compromised.5,13
Both UPS and autophagy degradation systems are complex processes consisting of chains of sequential events orchestrated by a large group of proteins. To understand their coordinated action, we need to identify novel players that are necessary and sufficient to mediate the compensatory function between the 2 systems. Here we show Mask, a conserved protein with Ankyrin repeats and a KH domain, as a novel and critical player in such a context. Initially identified as a modulator of receptor tyrosine signaling during Drosophila development,14 Mask has recently been shown to function as a cofactor of the Hippo pathway effector Yorkie and together they regulate target gene transcription with another transcription cofactor (Scalloped) during cell proliferation.15,16 The human ortholog of Mask, ANKHD1, is highly expressed in several cancer cell lines.17,18 We have recently reported that loss of mask function rescues the mitochondrial defects and muscle degeneration observed with pink1 and park mutants.19 In the present study, we show that in MAPT- and FUS-induced eye degeneration fly models, loss of Mask function enhances degeneration, while gain of Mask function suppresses degeneration. We demonstrate that by enhancing V-type ATPase expression, Mask promotes lysosome acidification and autophagic flux, and that Mask is necessary and sufficient to mediate a compensatory effect for partial loss of UPS function, to increase clearance of ubiquitinated proteins, and to protect against degeneration induced by aggregation-prone mutations.
Results
Mask activity suppresses eye degeneration induced by expressing MAPT and FUS in the photoreceptors
We recently reported that loss of mask function rescues pink1 and park/parkin mutant mitochondrial defects and muscle degeneration in flies.19 To test whether such a beneficial effect of mask knockdown is specific to the pink1 and park mutant background or a general phenomenon, we examined whether altering Mask levels may impact other forms of neurodegeneration. MAPT/TAU neurotoxicity plays a central role in the Alzheimer disease-frontotemporal dementia spectrum of disorders.20 Ectopic expression of human MAPT protein in fly neurons21 or fly eyes22 causes massive degeneration. When we coexpress the UAS-human MAPT transgene with UAS-control RNAi, UAS-mask RNAi or UAS-Mask transgenes, respectively, in fly eyes using the GMR-Gal4 driver, we found that coexpression of Mask significantly suppressed the degeneration while knocking down Mask enhanced the degeneration (Fig. 1B). Mutations of FUS (FUS RNA binding protein/Fused in sarcoma) are associated with familial forms of ALS.23,24 Ectopic expression of human ALS-causing mutant FUS proteins in flies causes neurodegeneration and early lethality.25 Similarly, we found that coexpression of Mask significantly suppressed the photoreceptor degeneration induced by human mutant FUS in the fly eyes, while coexpression of mask RNAi enhanced the degeneration phenotype (Fig. 1C).
Figure 1.
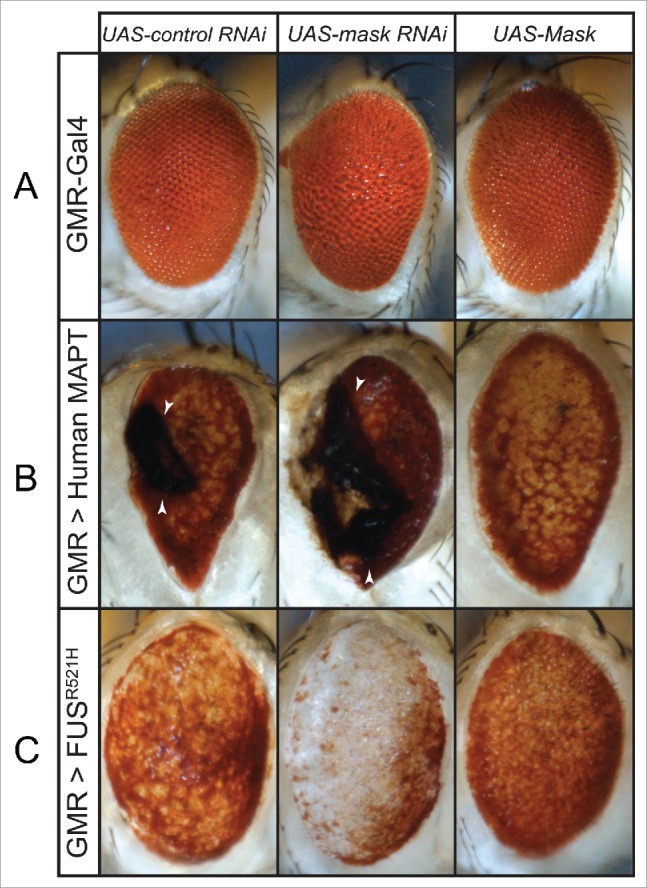
Mask suppresses eye degeneration induced by MAPT and FUS expression. (A) Light micrographs of d 3 adult eyes expressing UAS-control RNAi, UAS-mask RNAi or UAS-Mask under the control of the GMR-Gal4 driver. Notice that GMR-Gal4 caused a mild rough eye phenotype in control, Mask knockdown and overexpression. ((B)and C) Light micrographs of d 3 adult eyes expressing human MAPT (B) or human mutant FUSR521H (C) under the control of the GMR-Gal4 driver. For each of the 2 disease-linked transgenes, UAS-control RNAi, UAS-mask RNAi or UAS-Mask were coexpressed, respectively, in the fly eyes to evaluate how change in Mask activity modifies the degeneration defects. When coexpressed with control RNAi, MAPT induces necrotic scar (arrowheads) and FUSR521H induces patchy red pigmentation. Loss of mask function enhanced eye degeneration—indicated by increased necrotic scar in (B) and further loss of pigmentation in (C). Gain of mask function suppresses eye degeneration induced by both human-disease-linked transgenes.
Mask regulates abundance of ubiquitinated protein
In addition to modulating the FUS-induced eye phenotype, Mask also regulates FUS abundance in larval muscles. We found that coexpression of Mask with human FUSR521H mutant protein in larval muscles significantly reduced the protein abundance of FUSR521H in the nuclei (Fig. 2A and 2B). Although Mask knockdown did not alter nuclear abundance of FUSR521H, it clearly showed an increased presence of FUSR521H in the muscular cytoplasm (Fig. 2A and 2B). Abnormal cellular accumulation of MAPT and FUS have been the hallmarks of neurodegenerative diseases such as Alzheimer disease-frontotemporal dementia and ALS, and many of these cytoplasmic inclusions are also immunoreactive for ubiquitin.23,24 Given the ability of Mask to suppress the MAPT- and FUS-induced degeneration, and to regulate FUS abundance, we hypothesized that Mask activity may regulate the degradation of ubiquitinated proteins.
Figure 2.

Mask regulates the abundance of ubiquitinated proteins. (A) Projections of z-stack confocal images of HA-FUSR521H in larval muscle 12 coexpressing UAS-control RNAi, UAS-mask RNAi or UAS-Mask, driven by M12-Gal4. Note cytoplasmic localization of mutant FUS protein only in Mask-knockdown muscles. (B) Quantification of HA-FUSR521H intensity in muscle nuclei and cytoplasm. * P < 0.001, # P < 0.05, vs. control. (C) Representative confocal images of anti-FK-2 (mono- and poly-ubiquitin conjugation) staining in 3rd instar larval muscles of wild-type fly, and flies with muscle expression (24B-Gal4 driven) of UAS-mask RNAi, UAS-Mask, UAS-mCherry-Park, or UAS-mCherry-Park with UAS-mask RNAi, or UAS-Mask. (D) Quantification of the number and intensity of FK-2-positive puncta in larval muscles. ((E)to G) Western blot analysis of endogenous Mask protein, total ubiquitinated proteins (E), K48- (F) and K63-ubiquitin-conjugated proteins (G) in 3rd instar larval body walls with muscle expression of UAS-control RNAi or UAS-mask RNAi. (H) Quantification of the ubiquitin-conjugated proteins of the western blot analysis in E to G. Relative protein intensities were normalized to TUBB/β-tubulin. * P < 0.001.vs. Control RNAi. Scale bar: 10 µm.
To test this notion, we turned to fly larval muscles for their accessibility to study the underlying molecular and cellular mechanisms. We found that muscle-specific knockdown or overexpression of Mask altered the global level of ubiquitination, with Mask knockdown increasing the ubiquitination level while Mask overexpression reducing it, as measured by immunostaining of an antibody that recognizes both mono- and poly-ubiquitin chains (FK2) (Fig. 2C and 2D). Western blot analysis of total cell lysate of the larval body walls confirmed the enhanced total ubiquitination level by Mask knockdown (Fig. 2E). When the E3 ubiquitin ligase Park was overexpressed in the larval muscles, an elevated level of ubiquitination was observed (Fig. 2C and 2D), consistent with the ability of Park to change the landscape of the ubiquitinome and overall protein turnover.26,27 Muscle-specific coexpression of Park with mask RNAi further enhanced the ubiquitination level while coexpression with the UAS-Mask transgene significantly reduced the elevated ubiquitination level induced by Park (Fig. 2C and 2D). Thus, Mask protein negatively regulates both basal and Park-induced abundance of ubiquitin-conjugated proteins.
Mono- and poly-ubiquitin chains can be formed through many lysine residues within ubiquitin. K48-linked polyubiquitination acts as the canonical signal for targeting the substrate to the proteasome for degradation, while K63-linked ubiquitination plays a proteasome-independent role in the regulation of several cellular processes, including endocytosis, signal transduction, and DNA damage repair.28 We found that loss of mask function in muscle resulted in increase of both K48- and K63-linked ubiquitination (Fig. 2E-H), indicating that Mask is a key regulator controlling the overall abundance of ubiquitinated proteins in the cell.
Mask promotes autophagic flux but not UPS function
Mask may modulate the abundance of ubiquitinated proteins through promoting UPS function or autophagy activity, or both. To distinguish these possibilities, we first directly assayed autophagic flux in larval muscle by monitoring the degradation of a transgenic autophagosome marker protein mCherry-Atg8 a.29 When autophagosomes fuse with lysosomes, Atg8a is degraded, while the remaining free mCherry is more resistant to degradation. Therefore, the level of free mCherry can serve as an indicator of autophagic flux.29 In early third instar larvae, when the basal autophagy level is low, we detected no significant change of autophagic flux by manipulating Mask levels.19 However, at the wandering larval stage when autophagy level is high (see below), knocking down Mask in larval muscle significantly reduced autophagic flux while overexpression of Mask significantly increased autophagic flux, as shown by the relative amount of free mCherry proteins in the muscles (Fig. 3A and 3B), suggesting that Mask positively regulates autophagy. The nonprocessed form of Atg8a (often referred to as Atg8a-I) can be easily distinguished from the membrane-associated, faster migrating active form (Atg8a-II) in SDS-PAGE. We also quantified the amount of mCherry-Atg8a-II and found a significant increase in Mask-knockdown larval muscles and no significant change in Mask-overexpressing larval muscles (Fig. 3C). Such an increase in the amount of mCherry-Atg8a-II caused by mask loss of function indicates decreased autophagic degradation or increased autophagosome formation (see below), or both.30
Figure 3.

Mask promotes autophagy. (A) Western blot analysis of mCherry-Atg8a in larval muscles expressing 24B-Gal4-driven UAS-mCherry-Atg8a together with UAS-control RNAi, UAS-mask RNAi or UAS-Mask, at 114 h AEL. (B) Quantification of the levels of free mCherry as readout of autophagic flux. Intensities of free mCherry were normalized to those of TUBB/β-tubulin. (C) Quantification of relative mCherry-Atg8a-II intensity normalized to TUBB/β-tubulin. (D) Western blot analysis of endogenous Ref(2)P/p62 protein (indicated by arrowhead) levels in larval muscles expressing control RNAi or mask RNAi, with ACTA/α-actin serving as the loading control. (E) Quantification of relative Ref(2)P/p62 intensity normalized to ACTA. (F) Western blot analysis of MAPT(ΔC)-GFP in larval muscles expressing 24B-Gal4-driven UAS-MAPT(ΔC)-GFP together with UAS-Td-Tomato (as control), the UAS-control RNAi (as another control), the UAS-mask RNAi or UAS-Mask, at ∼114 h AEL. The bracket indicates MAPT(ΔC)-GFP. (G) Quantification of MAPT(ΔC)-GFP levels. Intensities of MAPT(ΔC)-GFP were normalized to TUBB/β-tubulin, and were shown as relative to control. (H) Pictures of pupae expressing no transgene, UAS-control RNAi, UAS-mask RNAi or UAS-Mask, with or without UAS-DTS5, under the control of muscle driver 24B-Gal4. Flies were cultured at 25oC. Arrows in the lower row indicate empty holes in the middle part of the pupae suggesting degeneration of muscle tissues. (I) Percentage of flies that successfully eclose from pupae were measured by dividing the number of empty pupae by the total number of pupae. Coexpression of only UAS-Mask drastically rescued the pupal lethality caused by UAS-DTS5 muscle expression. n = 8, * P < 0.001, compared with all the other DTS5 expression groups.
To further confirm that Mask is required for normal autophagy flux, we analyzed the endogenous Ref(2)P protein levels. Ref(2)P (SQSTM1/p62 in mammals) interacts with Atg8a and facilitates selective autophagic degradation of ubiquitinated proteins.31 Accumulation of Ref(2)P protein level is a reliable indicator of impaired autophagy degradation rate.29,30 Compared to control, larval muscles expressing mask RNAi showed a significant increase of endogenous Ref(2)P, suggesting that Mask is required for normal autophagy degradation (Fig. 3D and E). Overexpression of Mask in larval muscles did not change Ref(2)P protein level (Data not shown).
Previous studies show that a truncated MAPT protein (MAPT(ΔC)) is preferentially degraded through autophagy in mammalian cortical neurons and flies, as MAPT(ΔC) protein levels are significantly increased by loss of autophagy but not UPS.32 Therefore, MAPT(ΔC) can be used to study autophagy-dependent degradation. Targeted coexpression of MAPT(ΔC)-GFP with mask RNAi in larval muscles showed a significant increase of MAPT(ΔC)-GFP when compared with controls, while coexpression of MAPT(ΔC)-GFP with UAS-Mask significantly reduced the MAPT(ΔC)-GFP signal (Fig. 3F and G). These data suggested that Mask is capable of promoting clearance of ubiquitinated and misfolded proteins through enhancing autophagic degradation, which may contribute to its ability to suppress degeneration induced by toxic MAPT protein in the fly eyes.
We next used CL1-GFP,13 a fluorescent reporter of UPS function, to directly test whether Mask regulates UPS function. CL1-GFP is a fusion protein generated by introducing a degradation signal to GFP, which is rapidly degraded by the proteasome, therefore its levels reflect the functionality of the UPS.33 The fidelity of CL1-GFP in reporting UPS function was first validated using a temperature-sensitive proteasome mutant DTS7, as the CL1-GFP level significantly increased in the DTS7 mutant (Fig. S1A). In contrast, down- or upregulation of Mask showed no significant change in the strength of CL1-GFP fluorescent signal at the larva eye discs, compared with control, suggesting that Mask does not directly regulate UPS function (Fig. S1B).
We then assessed the impact of Mask-mediated autophagy on impaired UPS. DTS5 is another temperature-sensitive, dominant-negative mutation of the β6 subunit of the proteasome. At a nonpermissive temperature, this mutation significantly impairs the degradation functions of the proteasome.34 When we expressed DTS5 in Drosophila muscle, the fly developed normally from embryo to adult at 18oC (data not shown). However, at 25oC, substantial muscle degeneration occurred in these flies due to muscular proteasome impairment. We observed cessation of pupal development and lack of the middle thoracic portion of their bodies (Fig. 3H and I). Under this condition, coexpression of UAS-Mask almost completely suppressed the muscle degeneration and the associated lethality, as ∼80% of such flies showed normal pupal morphology and eclosed normally (Fig. 3H and I). Meanwhile, muscle-specific knockdown of Mask slightly enhanced the degenerative phenotype as the pupal size was further reduced when compared with control (Fig. 3H). Collectively, these data suggested that in the event of UPS impairment, Mask overexpression is sufficient to compensate the partial loss of UPS activity.
Mask promotes a late step of autophagy—autophagosomal degradation
Autophagy is a dynamic event involving sequential steps from induction of autophagy, autophagosome formation to its fusion with the lysosome and subsequent autophagosome degradation,35 and each step is highly regulated by multiple factors.36 Before we tested how Mask regulates autophagy in larval muscles, we first characterized the baseline autophagy landscape in developing wild-type larval muscles using a 24B-Gal4-driven GFP-mCherry dual-tagged Atg8a transgene as the marker.37,38 During autophagosome induction and formation, the dual-tagged Atg8a proteins are recruited to the phagophore membrane, and these structures, as well as completed autophagosomes, are positive for both green and red fluorescence. During autophagosomal degradation, autophagosomes fuse with lysosomes to form autolysosomes, GFP fluorescence is quenched due to its sensitivity to acidic environment in the lysosome, whereas the acid-resistant mCherry still emits red fluorescence. Therefore, autolysosomes are positive for only red fluorescence.39 Expression pattern and intensity of both GFP and mCherry autofluorescent signals in larval muscles were analyzed at 6 developmental stages from very early (∼84 h after egg laying [AEL]) to very late (120 h AEL) third instar. We identified 2 types of puncta: one group with relatively bigger size but irregular shapes is localized closely to the muscle nuclei, and emits only red fluorescence, which likely represents autolysosomes; the other group with a smaller size and spherical shape distributes sporadically in the muscle cytoplasm, and emits both red and green fluorescence, which likely represents autophagosomes. Very few autophagosomes and autolysosomes can be detected before 108 h AEL. Soon after, more dynamic change in autophagy occurs in the second half of the third instar larval stage. The number of autophagosomes peaks at about 114 h AEL, while the number and size of perinuclear autolysosomes reach their plateau at about 117 h AEL. The autolysosomes at 114 and 117 h AEL show very strong red fluorescence and faint green fluorescence, possibly representing rapid fusion of autophagosome with lysosome and transient state of degradation. At 120 h AEL (late wandering stage), only small, red-only, late-stage autolysosomes are observed in muscle cytoplasm. Meanwhile, all muscle nuclear GFP-mCherry-Atg8a proteins are released to the cytoplasm (Fig. S2). These data together suggested that there is an abrupt burst of autophagy when larvae enter wandering stage (from ∼114 h to 120 h AEL), consistent with the previous findings on a programmed burst of autophagy induced by ecdysone signaling in fat bodies of wandering-stage larvae.40 Such a burst was also accompanied by a sudden release of Atg8a proteins from nuclei to the cytosol, consistent with recent findings that deacetylation and nuclear release of Atg8a (whose mammalian orthologs include the MAP1LC3/LC3 family) play central roles in controlling the level of autophagy.41
We next tested whether change of Mask activity alters the landscape of autophagy during larval muscle development. At mid-third instar (stage 96 and 108 h AEL), when control larval muscles showed very few autophagosomes in the cytoplasm, substantially more autophagosomes are accumulated in Mask-knockdown muscles (Fig. 4A, B and 4F), these data are consistent with our previous finding that loss of mask function enhanced the colocalization of Atg8a-positive autophagosomal puncta and mitochondria at early third instar larval muscles.19 While Mask-knockdown muscles showed diffused localization of autolysosomes, Mask-expressing muscles showed significantly more and bigger autolysosomes around the nuclei (Fig. 4A, B and E). At stage 114 h AEL, when autophagosomes reached their highest presence at control muscles, Mask-knockdown muscles showed much stronger autophagosomal GFP signaling and weaker autolysosomal signal than control, while Mask-expressing muscles continued to show enhanced size and number of autolysosomes (Fig. 4C, E and F). At the peak of the autophagy burst in larval muscles (120 h AEL), the control muscles showed exclusively small and red-only late-stage autolysosomes in the cytoplasm and no nuclear accumulation of GFP-mCherry-Atg8a, while Mask-knockdown muscles still showed dual-tagged-Atg8a labeled puncta with both green and red fluorescence, suggesting defective degradation of autophagosomes after autolysosome formation (Fig. 4D and F). Meanwhile Mask-expressing muscles showed red-only autolysosomes with a greater level of intensity for mCherry fluorescence than that of the controls, suggesting enhanced lysosome-mediated degradation (Fig. 4D and E). While nuclear GFP-mCherry-Atg8a proteins were absent in control and Mask-expressing muscles, they were clearly detectable in the Mask-knockdown muscles (Fig. 4D), suggesting reduced usage of the Atg8a pool in the nuclei under mask loss of function.
Figure 4.

Mask promotes autophagy flux by enhancing degradation of autophagosomes. (A to D) Projections of z-stack confocal images of GFP-mCherry-Atg8a in larval muscle coexpressing UAS-control RNAi, UAS-mask RNAi or UAS-Mask in 4 indicated developmental stages (from 96 to 120 h AEL). Arrow heads indicate autophagosomes and arrows indicate autolysosomes. Dotted lines denote muscle nuclei. Note that Mask knockdown boosted formation of autophagosomes, and prevented perinuclear localization of autolysosomes at early stages, and blocked the degradation of autophagosomes and nuclear release of Atg8a at the peak of autophagy (120 h AEL). Also note that Mask overexpression in muscle enhanced the size and strength of autolysosomes across the developmental stages. (E and F) Quantification of relative autolysosome intensity (E) and number of autophagosomes (F) as described in (A)to D. * P < 0.001, ** P < 0.05, compared with control RNAi. # P < 0.005, ## P < 0.01, ### P < 0.05, comparing mask RNAi to control RNAi. n ≥ 4. (G) Representative autofluorescence confocal images of GFP-mCherry-Atg8 a expressed in the muscle of wild type, mask null-mutant mask10.22/Df or mask null-mutant plus muscle-driven UAS-Mask. Note that mask mutant larval muscle showed defects very similar to those of the muscle-specific knockdown of mask at the same developmental stage, and that muscle expression of UAS-Mask completely rescued the phenotype, and due to the high-level of Mask expression, showed much stronger presence of autolysosomes. Scale bar: 10 µm.
To further confirm that Mask regulates autophagosome degradation in larval muscles, we analyzed the pattern of dual-tagged Atg8a in mask null-mutant larval muscles, as well as muscle-specific rescue using the same UAS-Mask transgene. At 114 h AEL, mask10.22/Df mutant larval muscles showed almost identical pattern of dual-tagged Atg8a puncta (positive for both green and red) as Mask knockdown. At the same time, muscle expression of UAS-Mask in mask10.22/Df mutant larvae not only rescued the phenotype, but also showed an enhanced mCherry red fluorescence due to the high-level expression of the UAS-Mask transgene (Fig. 4G). Collectively, our time-course analysis suggested that loss of mask function does not prevent the formation of autophagosomes (induction), however it suppresses the lysosome-mediated degradation of autophagosomes (flux), and that overexpression of Mask is sufficient to enhance degradation of autophagosomes in larval muscles.
Mask promotes autophagosome degradation through enhancing lysosomal function
The impaired degradation of autophagosomes caused by loss of mask function can result from either failure in the fusion of autophagosome with lysosome, or impaired lysosome function. To distinguish these 2 possibilities, we first used a lysosomal marker, GFP-LAMP1, to assess lysosomal function in Mask-knockdown and Mask-overexpressing larval muscles. The GFP moiety of the GFP-LAMP1 fusion protein is located in the lysosomal lumen while the LAMP1 moiety is integrated in the lysosomal membrane. Therefore, less GFP signal will be detected in functioning lysosomes, whereas more GFP signal will be detected in defective lysosomes.42 We found that the intensity of GFP signal increased substantially in Mask-knockdown muscles, while the same signal was hardly detectable in Mask-overexpressing muscles when compared with control (Fig. 5A and 5B). The change of GFP-LAMP1 protein levels in larval muscles in response to Mask activity was confirmed by western blot analysis of GFP-LAMP1 (Fig. 5C). These results together indicated that lysosomal function is decreased in mask loss of function and increased in mask gain of function. Second, we found colocalization between mCherry-Atg8a (autophagosomes) and GFP-LAMP1 (lysosomes) in Mask-knockdown muscles (Fig. 5D), suggesting that autophagosome-lysosome fusion events occur in Mask-knockdown muscles. Third, we found substantial colocalization of autophagosomes and ubiquitin-conjugated proteins (by FK2 immunostaining) in Mask-knockdown larval muscles (Fig. 5E), suggesting that loss of mask function does not impair recognition and encircling of ubiquitinated protein aggregates by autophagosomes, but rather prevents the degradation of the protein contents inside autophagosomes. These findings together suggested Mask positively regulates lysosomal function. To directly test this notion, we stained dissected larval muscles with a fluorescent dye LysoTracker Red (DND-99) that preferentially accumulates in vesicles with acidic pH. Indeed, at both early and late third instar larvae, we observed an enhanced LysoTracker Red signal in Mask-overexpressing larval muscles and a much weakened LysoTracker Red signal in Mask-knockdown larval muscles (Fig. 5F). Together, these data strongly suggested that Mask is necessary and sufficient to promote lysosomal acidification, and by doing so it enhances lysosomal function.
Figure 5.

Mask positively regulates lysosomal function. (A) In vivo assessment of lysosomal function using the lysosomal activity sensor GFP-LAMP1 in Mask-knockdown and -overexpressing larval muscles. Representative confocal images of GFP immunostaining were captured. Compared with control, knocking down Mask dramatically increased GFP expression, indicating a failure of protein degradation. Mask overexpression substantially reduced GFP expression, indicating efficient GFP degradation. (B) Quantification of GFP-LAMP1 mean intensity. P < 0.001, compared with control RNAi. (C) Western blot analysis of GFP-LAMP1 in larval muscles expressing 24B-Gal4-driven UAS-GFP-LAMP1 together with UAS-control RNAi, UAS-mask RNAi or UAS-Mask. TUBB/β-tubulin and GFP were blotted in the same membrane. Note that GFP was fused with the transmembrane domain and cytoplasmic tail of LAMP1. Relative GFP-LAMP1 intensity was quantified by normalizing to TUBB/β-tubulin. (D) Mask knockdown does not alter autophagosome-lysosome colocalization. Representative single-layer confocal images of GFP and mCherry autofluorescence were captured in larval muscles expressing GFP-LAMP1 and mCherry-Atg8a with UAS-control RNAi or UAS-mask RNAi. Arrowheads indicate patchy colocalization of GFP-LAMP1 and mCherry-Atg8a in control. Arrows indicate many puncta that are positive for both GFP-LAMP1 and mCherry-Atg8a. (E) Mask knockdown showed increased colocalization between autophagosomes and ubiquitinated proteins. Representative single-layer confocal images of anti-GFP (green) and anti-FK2 (red) staining in larval muscles expressing mCherry-Atg8a with control or mask RNAi. Arrows indicate FK2-positive puncta in the control muscle that are not positive for mCherry-Atg8a. Arrowheads indicate many puncta that are positive for both FK2 and mCherry-Atg8 a. (F) LysoTracker Red staining of dissected larval muscles from early (108 h AEL) and late (120 h AEL) third instar larvae expressing UAS-control RNAi, UAS-mask RNAi or UAS-Mask. Mask knockdown decreased while Mask expression increased the strength of the pH-sensitive dye. Scale bar: 10 µm.
Mask promotes lysosomal acidification by enhancing the expression of V-type ATPases in a TFEB-independent manner
Lysosomes generate and maintain their acidic pH environment via a proton-pumping V-type ATPase (V-ATPase), a large enzymatic complex consisting an 8-subunit catalytic domain (V1), a 6-subunit membrane-spanning domain (V0) and 2 accessory subunits. To further explore the mechanism through which Mask regulates lysosomal acidification, we tested whether Mask regulates the expression of V-ATPase subunits. We identified 3 Drosophila lines, Vha55::YFP (V1 B subunit), Vha13::GFP (V1 G subunit) and VhaSFD::GFP (V1 H subunit), in protein trap databases that each expresses a YFP or GFP-fused V-ATPase subunit from its genomic locus.43,44 Therefore, the amount of these GFP (or YFP) tagged proteins will reflect their endogenous expression levels. We found that protein levels of all 3 V1 subunits show significant reduction in Mask-knockdown larval muscles, and significant increase in Mask-overexpressing larval muscles (Fig. 6A and B). To further support this notion, we performed western blots on the V1B subunit (Vha55::YFP) by dissecting out larval muscle cells from larval body wall preparation (to remove the high-level expression of Vha55::YFP in larval epidermis). Mask knockdown showed significantly reduced Vha55::YFP, while Mask overexpression showed significantly increased Vha55::YFP (Fig. 6C). To further confirm these results, we chose VhaSFD::GFP to analyze its expression in mask null-mutant larval muscles. mask10.22/Df mutant larval muscles show a very similar decrease of VhaSFD expression as Mask knockdown, and such a decrease is completely rescued by expression of the UAS-Mask transgene in muscles (Fig. 6D). These data suggested that Mask is necessary and sufficient to promote the expression of 3V-ATPase V1 subunits, which contributes to the enhanced lysosomal acidification by Mask.
Figure 6.

Mask functions as a positive and TFEB-independent regulator of V-ATPase V1 subunit VhaSFD, Vha13 and Vha55 expression. (A) Representative confocal images of anti-GFP staining in 3rd instar larval body wall of flies with genomic GFP/YFP trapped in the loci of vhaSFD, vha13 or vha55. For each protein trap line, we analyzed control (24B-Gal4 driver only), or muscle-driven UAS-mask RNAi, or UAS-Mask, respectively. (B) The mean GFP/YFP intensity of each genotype was quantified by selecting random body wall muscle 6 areas in of segment A2. Mask knockdown significantly reduced, while Mask overexpression significantly increased, GFP/YFP intensity in all 3 protein trap lines, comparing with control. * P < 0.001. (C) Western blot analysis of Vha55::YFP in Vha55::YFP protein trapped larval muscle expressing UAS-control RNAi, UAS-mask RNAi or UAS-Mask. Wild-type larval muscle sample was loaded to show nonspecific GFP bands. Note that larval muscles were dissected out from larval bodywall preparation. Relative Vha55::GFP intensity was normalized to ACTA and quantified. (D) Representative confocal images of anti-GFP staining in 3rd instar larval body wall of flies with genomic GFP trapped in the loci of VhaSFD in the background of wild type (WT), mask10.22/Df mutant, or mask10.22/Df plus muscle-specific expression of UAS-Mask, with quantification of VhaSFD::GFP mean intensity. Note that muscular expression of Mask rescued the phenotype and showed a level of VhaSFD expression even higher than wild type due to the strong expression level of UAS-Mask transgene. * P < 0.001, comparing with WT. Scale bar: 10 µm. (E) Representative confocal images and quantification of Vha16–1::GFP in Vha16–1 GFP-trapped larval muscles of control (24B-Gal4), mask knockdown (24B-Gal4-driven UAS-mask RNAi) or Mask overexpression (24B-Gal4-driven UAS-Mask). (F) Representative confocal images and quantification of GFP in 4MBox-GFP larval muscles of control (24B-Gal4), mask knockdown (24B-Gal4 driven UAS-mask RNAi) or Mask overexpression (24B-Gal4-driven UAS-Mask). * P < 0.001, vs. control. Note that trachea and NMJs in all representative images serve as internal control of the V-ATPase subunits expression.
The Drosophila genome contains 5 genes (vha16–1 to vha16–5) encoding V0 c subunits. We obtained a GFP-trapped line of vha16–1. Our data indicated that knockdown or upregulation of Mask in larval muscle does not affect the expression levels of Vha16–1 (Fig. 6E). It was reported that Mitf, the sole homolog of the Mit family of transcription factors (including TFEB) in flies, regulates the transcription of Vha16–1.42 Together these data suggest that Mask does not affect the transcriptional activity of Mitf. To directly test this notion, we examined whether Mask regulates the expression of a Mitf reporter gene that carries 4 tandem Mitf-binding motifs (4Mbox-GFP). Our data showed that knocking down Mask does not significantly alter the expression of the reporter gene (Fig. 6F). Overexpression of Mask reduced, rather than enhanced, the 4Mbox-GFP expression, possibly due to the increased level of autophagy in larval muscle (Fig. 6F). Therefore, Mask may regulate the expression of V-ATPase subunits through a TFEB-independent mechanism.
Mask-mediated rescue of the DTS5 phenotype and MAPT- and FUS-induced degeneration requires normal autophagy and lysosome function
To understand how Mask suppresses the phenotype associated with MAPT and FUS overexpression in fly eyes (Fig. 1), we tested whether alteration of Mask levels in photoreceptors changes autophagy and lysosomal function. When mCherry-Atg8a or GFP-LAMP1 reporters were expressed in the photoreceptor cells, mask-RNAi-mediated knockdown showed no effects on autophagic flux or lysosomal function in the eyes (Fig. 7A and B). However, Mask overexpression significantly enhanced autophagic flux as indicated by increased levels of free mCherry (Fig. 7A), and lysosomal function as indicated by reduced levels of GFP-LAMP1 (Fig. 7B). Similar effect was also observed in adult brains, where pan-neuronal expression of MASK led to a higher level of autophagic flux (Fig. S4). Next, we tested the genetic interaction between Mask and the autophagy pathway. Although autophagy is primarily cytoprotective,45 excessive upregulation of autophagy in fly eyes can lead to photoreceptor degeneration. Eye-specific overexpression of Atg1, a protein essential for the initiation of autophagosome, caused severe eye degeneration (Fig. 7C). We tested whether alteration of Mask levels can modify this phenotype. We found no significant rescue of the eye phenotype by eye-specific knockdown of mask, but a significant enhancement of the eye degeneration by eye-specific overexpression of the UAS-Mask transgene (Fig. 7C). These data are consistent with our finding that Mask expression in fly eyes enhances the autophagy-lysosomal pathway. Moreover, they also suggest that the Mask- mediated rescue of eye degeneration in the MAPT and FUS disease models is not due to some intrinsic beneficial effect of eye-specific Mask overexpression, but rather autophagy-lysosome-mediated clearance of toxic proteins.
Figure 7.
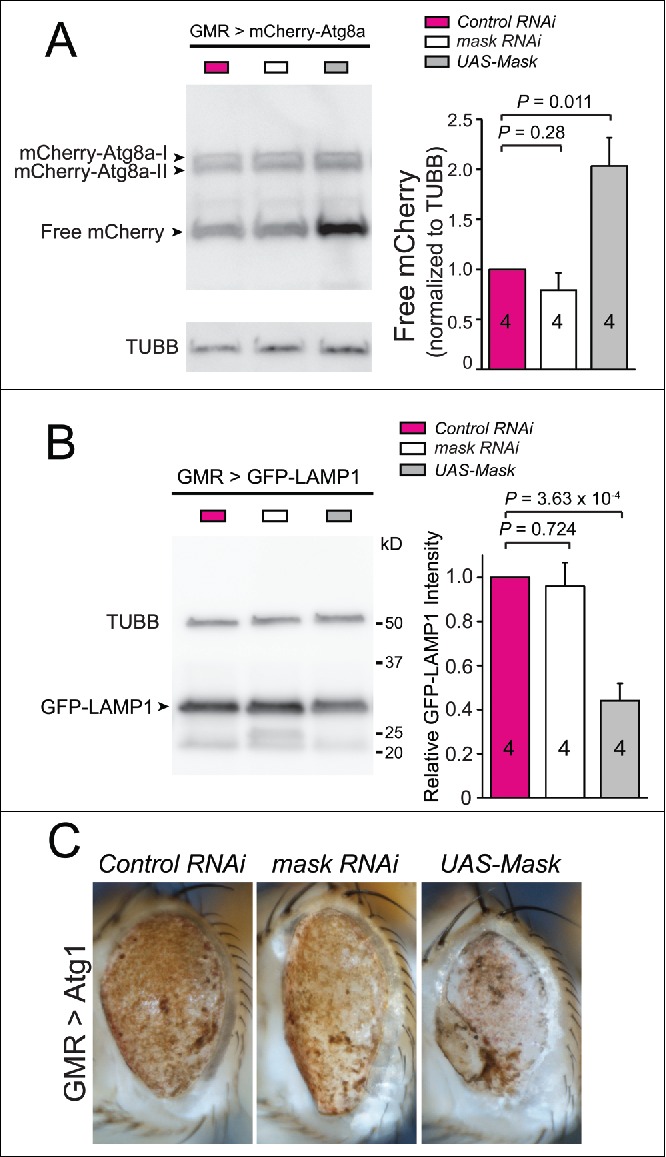
Overexpression of Mask in fly eyes enhances autophagy and lysosomal function. (A) Western blot analysis of mCherry-Atg8a in adult fly eyes expressing GMR-Gal4-driven UAS-mCherry-Atg8 a together with UAS-control RNAi, UAS-mask RNAi or UAS-Mask. Quantification of the levels of free mCherry, which were normalized to those of TUBB/β-tubulin. (B) Western blot analysis of GFP-LAMP1 in adult fly eyes expressing GMR-Gal4-driven UAS-GFP-LAMP1 together with UAS-control RNAi, UAS-mask RNAi or UAS-Mask. Quantification of the levels of GFP-LAMP1, which were normalized to those of TUBB/β-tubulin. (C) Mask overexpression enhances fly eye degeneration induced by Atg1 overexpression. Light micrograph of fly eyes expressing UAS-Atg1 with UAS-control RNAi, UAS-mask RNAi or UAS-Mask. Atg1 overexpression induced severe eye degeneration indicated by liquefied eye surface and patchy pigmentation. Mask coexpression enhanced the degeneration phenotype as most of fly eyes showed necrotic scars in addition to pigmentation loss.
To further test this notion in vivo, we examined genetically whether the intact function of autophagy and lysosome is required for the Mask-mediated rescue of the UPS mutant phenotype and MAPT and FUS-induced eye degeneration. The rescue of DTS5-induced lethality by the UAS-Mask transgene shown in Fig. 3H and I is a very quantitative assay. When heterozygous mutations of essential genes in autophagy (atg1−/+, atg2−/+ and atg18KG03090/+) or lysosome acidification (vha68–2EP/+) were introduced to the same rescuing paradigm, the ability of the UAS-Mask transgene to rescue the muscle degeneration and lethality is substantially reduced (Fig. 8A). Similarly, heterozygous mutations in the autophagy pathway (atg2 and atg18) as well as V-type ATPase (vha55) dominantly suppressed Mask-mediated rescue of the eye degeneration induced by human MAPT and FUS overexpression (Fig. 8B-D). Together, these data indicated that even partial loss of autophagy and lysosome function impairs Mask's ability to compensate the compromised UPS and to rescue the degeneration induced by different human disease proteins.
Figure 8.
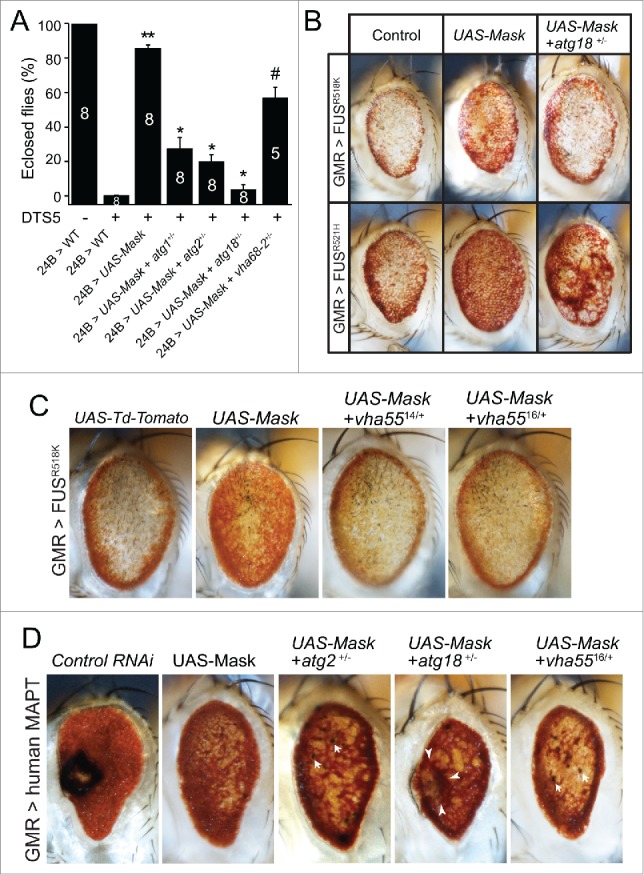
Normal autophagy-lysosome function is required for Mask-mediated rescue of the DTS5 phenotype and MAPT- and FUS-induced degeneration. (A) Quantification of Mask-muscle-expression-mediated rescue of DTS5-induced lethality. Heterozygous mutations in atg1(unc-5125), atg2EP3697, atg18KG03090, or vha68–2EP2346 dominantly suppressed the Mask-mediated rescue of lethality to various degrees. ** P < 0.001, comparing to DTS5 24B>WT; * P < 0.001, # P < 0.01, comparing to Mask-expression-mediated rescue. (B) Heterozygous mutation of atg18KG03090 dominantly suppressed Mask-mediated rescue of the eye degeneration induced by expression of human FUSR518K and FUSR521H mutant proteins. (C) Light micrograph of fly eyes expressing UAS-FUS R518 K with UAS-Td-Tomato (as control), UAS-Mask, or UAS-Mask with heterozygous vha5514 or vha5516 mutants. Note that the 2 vha55 mutants each dominantly suppressed Mask-mediated increase of eye pigment. (D) Light micrograph of fly eyes expressing UAS-human MAPT with UAS-control RNAi, UAS-Mask, or UAS-Mask with heterozygous atg2EP3697, atg18KG03090 or vha5516 mutations. Note that all 3 heterozygous mutations dominantly inhibited the Mask-mediated suppression of eye degeneration induced by human MAPT. Arrows indicate small necrotic spots, and arrow heads mark a collapsed eye region with dying tissue.
Discussion
Autophagy, an evolutionarily conserved cellular mechanism that preserves metabolic homeostasis during nutrient unavailability, is traditionally regarded as a selfeating degradative process with limited selectivity. However, mounting evidence suggests that both micro- and macro-autophagy can play cytoprotective roles to specifically target damaged and toxic organelles and proteins for clearance under pathological conditions.46-48 The mechanism of selective autophagy is unclear. There is some evidence that autophagy receptors can recognize ubiquitin-dependent and ubiquitin-independent signals for selective degradation.47-49 Autophagy is a multistep process including nucleation, autophagosome formation and fusion with lysosomes,36,50 and each step can be regulated to enhance degradation of damaged cellular components. Recently, research has emerged showing TFEB as a potent regulator of the autophagy-lysosomal pathway,51,52 whose activation can promote lysosomal function and mitigate disease in a range of neurodegenerative disorders.53 Here we show that Mask acts in a TFEB-independent manner to boost the expression of V-ATPase subunits. Our study provides novel evidence that lysosome function is not only required for the normal clearance of ubiquitinated and misfolded proteins, but its activity can also be boosted potential through enhanced lysosomal acidification, to mitigate cellular degeneration caused by toxic protein aggregation.
Mask is well positioned to regulate lysosome-mediated clearance of ubiquitinated and misfolded proteins. As a positive regulator of several V-type ATPase V1 subunits expression, Mask function is necessary and sufficient to promote lysosomal acidification and autophagosome degradation in a cell-autonomous manner. When the UPS function is impaired, increased Mask expression is sufficient to increase autophagic flux, which in turn compensates the partial loss of the proteasome-mediated degradation (Fig. 3H and I). Interestingly, even when UPS function is intact, levels of Mask activity impact the abundance of UPS-dependent (K48) and -independent (such as K63) ubiquitin-conjugated proteins, suggesting that autophagy and lysosome-mediated degradation plays an important role for basal protein homeostasis. Under pathological conditions such as UPS inactivation or excessive accumulation of disease proteins, upregulation of Mask activity substantially suppressed the cellular degeneration phenotypes in both muscles and photoreceptors, potentially through Mask-mediated increase of autophagy and lysosome activities and subsequent degradation of harmful protein aggregates, as suggested by our biochemical and genetic analyses. In support of this notion, upregulation of Mask promotes autophagic flux in larval muscles (Fig. 3A), adult eyes (Fig. 7A) and adult brains (Fig. S4).
Our work in the Drosophila model organism yielded new insight into Mask-mediated cellular protective mechanisms that regulate lysosomal function in normal and stressed conditions caused by misfolding-prone disease proteins or impaired UPS. Such mechanisms may provide a therapeutic approach for the treatment of a group of neurodegenerative disorders caused by intracellular inclusions.
Materials and methods
Fly stocks
Flies were maintained at 25 °C on standard food unless otherwise indicated. UAS-Mask and UAS-mCherry-Park were described previously.19 The following stocks were obtained from the Bloomington Drosophila Stock Center: UAS-control-RNAi [TRiP.JF01147], UAS-mask-RNAi [TRiP.HMS01045], UAS-DTS5, UAS-DTS7, UAS-GFP-mCherry-Atg8 a, UAS-GFP-LAMP1, UAS-Atg1, 24B-Gal4 GMR-Gal4, M12-Gal4, maskDf317, vha68–2EP2864, atg2EP3697, atg18KG03090, vha5514, vha5516, VhaSFD::GFP (vhaSFDG00259) and Vha13::GFP (vha13CA07644). Vha55::YFP (vha55CPTI002645) and Vha16–1::GFP (vha16–1G00007) is from Kyoto Stock Center. Other strains used in this study were UAS-Wild-type-FUS, UAS-FUSR521H, UAS-FUSR518K,25 atg1(unc-5125),54 UAS-human MAPT,22 UAS-CL1-GFP,13 UAS-mCherry-Atg8a (From Tom Neufeld), UAS-MAPT(ΔC)-GFP,32 4Mbox-GFP,42 UAS-Td-Tomato (a gift from Richard Daniels), and mask.10.22,14
Immunocytochemistry
Larval muscles were dissected in ice-cold phosphate-buffered saline (PBS; 137 mM NaCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, pH 7.4) and fixed in 4% paraformaldehyde for 30 min. For the detection of autofluorescence from proteins tagged with fluorescent molecules, the fixed tissues were washed with PBS, and mounted for imaging. For immunocytochemistry, the fixed tissues were stained following standard procedures. The primary antibodies used were: mouse anti-ubiquitin at 1:1000 (Millipore, Clone FK2, 04–263), rabbit anti-mCherry at 1:1000 (Clontech, 632496), Alexa Fluor 488-conjugated rabbit anti-GFP at 1:1000 (Life Technologies, A21311), rat anti-HA at 1:1000 (Roche, 11867431001). The following secondary antibodies were used: Cy3-conjugated goat anti-rabbit IgG at 1:1000 (Jackson ImmunoResearch, 111–165–144), Dylight 488 conjugated anti-mouse IgG at 1:1000 (Abcam, ab150113).
LysoTracker Red Staining
Larval muscles were dissected in PBS, incubated in 1 µM LysoTracker Red DND 99 (Molecular Probes, L7528) for 2 min, washed with PBS, fixed in 4% paraformaldehyde for 2 min, and mounted in PBS. Confocal imaging was performed right after the mounting to capture the red fluorescence signal.
Confocal and light microscopy
Single-layer or z-stack confocal images were captured on a Nikon C1 confocal microscope (Melville, NY). Unless otherwise noted, images shown in the same figure were acquired using the same gain from samples that had been simultaneously fixed and stained. All larval muscle images were acquired at muscle 6 in segments A3. The pupae and adult eye images were captured using a Nikon Digital Sight DS-Fi1 camera (Melville, NY) mounted on a Nikon AZ100 light microscope (Melville, NY). For each eye picture, 3 to 4 images with sequential focal points were taken to cover the entire eye, and these images were then stacked into a single image using the Zerene Stacker software. All quantifications of intensity were performed using NIS-Elements (version 3.0).
Anti-Ref(2)P antibody
To generate anti-Ref(2)p antibody, cDNA sequence encoding full-length Ref(2)p protein from EST clone GH06306 was fused in-frame into pET28a vector. His-tagged Ref(2)p protein was expressed from E. coli and the purified protein was used to immunize rabbits. The specificity of the antibodies was validated for its ability to specifically recognize the endogenous Ref(2)P protein that is present in wild-type adult flies but absent from homozygous ref(2)p mutants (c03993 line) (Fig. S3).
Western blots
Western blots were performed according to standard procedures. The following primary antibodies were used: rabbit anti-Mask (1:1,000),14 mouse anti-mCherry antibody (1:1000, Novus Biologicals, clone 1C51, NBP1–96752), mouse anti-ubiquitin (1:1000, Millipore, clone FK2, 04–263), rabbit anti-K48-Ub (1:1000, Cell Signaling Technology, clone D9D5, mAb #8081), rabbit anti-K63-Ub (1:500, Millipore, clone Apu3, 05–1308), rabbit anti-GFP (1:1000, Life Technologies, A11122), rabbit anti-Ref(2)P (1:10,000), mouse anti-ACTA/α-actin (1: 5000, JLA20) and mouse anti-TUBB/β-tubulin (1:1,000, E7) were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at the University of Iowa, Department of Biology, Iowa City, IA. HRP-conjugated anti-rabbit or HRP-conjugated anti-mouse secondary antibodies were used at 1:10000. Data were collected using Luminescent Image Analyzer LAS-3000 (FUJIFILM, Valhalla, NY) and quantified using Multi Gauge (FUJIFILM, Valhalla, NY).
Statistical analysis
Statistical analysis was performed and graphs were generated in Origin (Origin Lab, Northampton, MA). Each sample was compared with other samples in the group (more than 2) using a pertinent post-hoc test in ANOVA, or with the other sample in a group of 2 using a Student t test. All histograms are shown as mean ± SEM.
Supplementary Material
Abbreviations
- ACTA
actin α
- AEL
after egg laying
- ALS
amyotrophic lateral sclerosis
- DTS
dominant temperature-sensitive
- FUS
fused in sarcoma
- GFP
green fluorescent protein
- MAPT
microtubule associated protein tau
- PBS
phosphate-buffered saline
- TFEB
transcription factor EB
- TUBB
tubulin β
- UPS
ubiquitin-proteasome system
- V-ATPase
vacuolar-type ATPase
- YFP
yellow fluorescent protein
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Michael Simon, Udai Pandey, Yogesh Wairkar, George Jackson and Tom Neufeld for sharing reagents, Bloomington Stock center for other fly stocks. Mouse anti-ACTA and anti-TUBB antibodies were obtained from and Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. We also would like to thank Ryan Labadens for editorial assistance.
Funding
This work is supported by a Research Enhancement Fund from LSUHSC School of Medicine and an investigator initiated starter project grant from ALS (17-IIP-348) to C.W.
References
- [1].Polymenidou M, Cleveland DW. The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 2011;147:498-508. doi: 10.1016/j.cell.2011.10.011. PMID:22036560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162-7. doi: 10.1073/pnas.0230450100. PMID:12634421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087-93. doi: 10.1016/S0896-6273(03)00787-6. PMID:14687544 [DOI] [PubMed] [Google Scholar]
- [4].Kasturirangan S, Boddapati S, Sierks MR. Engineered proteolytic nanobodies reduce Abeta burden and ameliorate Abeta-induced cytotoxicity. Biochemistry (Mosc). 2010;49:4501-8. doi: 10.1021/bi902030m [DOI] [PubMed] [Google Scholar]
- [5].Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta. 2008;1782:691-9. doi: 10.1016/j.bbadis.2008.10.002. PMID:18930136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749-57. doi: 10.1093/hmg/ddg074. PMID:12651870 [DOI] [PubMed] [Google Scholar]
- [7].Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20:7268-78. PMID:11007884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107-17. doi: 10.1093/hmg/11.9.1107. PMID:11978769 [DOI] [PubMed] [Google Scholar]
- [9].Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009-13. doi: 10.1074/jbc.M300227200. PMID:12719433 [DOI] [PubMed] [Google Scholar]
- [10].Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292-5. doi: 10.1126/science.1101738. PMID:15333840 [DOI] [PubMed] [Google Scholar]
- [11].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885-9. doi: 10.1038/nature04724. PMID:16625204 [DOI] [PubMed] [Google Scholar]
- [12].Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al.. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880-4. doi: 10.1038/nature04723. PMID:16625205 [DOI] [PubMed] [Google Scholar]
- [13].Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al.. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859-63. doi: 10.1038/nature05853. PMID:17568747 [DOI] [PubMed] [Google Scholar]
- [14].Smith RK, Carroll PM, Allard JD, Simon MA. MASK, a large ankyrin repeat and KH domain-containing protein involved in Drosophila receptor tyrosine kinase signaling. Development. 2002;129:71-82. PMID:11782402 [DOI] [PubMed] [Google Scholar]
- [15].Sansores-Garcia L, Atkins M, Moya IM, Shahmoradgoli M, Tao C, Mills GB, Halder G. Mask is required for the activity of the Hippo pathway effector Yki/YAP. Curr Biol. 2013;23:229-35. doi: 10.1016/j.cub.2012.12.033. PMID:23333314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sidor CM, Brain R, Thompson BJ. Mask proteins are cofactors of Yorkie/YAP in the Hippo pathway. Curr Biol 2013;23:223-8. doi: 10.1016/j.cub.2012.11.061. PMID:23333315 [DOI] [PubMed] [Google Scholar]
- [17].Dhyani A, Machado-Neto JA, Favaro P, Saad ST. ANKHD1 represses p21 (WAF1/CIP1) promoter and promotes multiple myeloma cell growth. Eur. J. Cancer (Oxford, England: 1990). 2015; 51:252–9 [DOI] [PubMed] [Google Scholar]
- [18].Machado-Neto JA, Lazarini M, Favaro P, Franchi GC Jr, Nowill AE, Saad ST, Traina F. ANKHD1, a novel component of the Hippo signaling pathway, promotes YAP1 activation and cell cycle progression in prostate cancer cells. Exp Cell Res. 2014;324:137-45. doi: 10.1016/j.yexcr.2014.04.004. PMID:24726915 [DOI] [PubMed] [Google Scholar]
- [19].Zhu M, Li X, Tian X, Wu C. Mask loss-of-function rescues mitochondrial impairment and muscle degeneration of Drosophila pink1 and parkin mutants. Hum Mol Genet. 2015;24:3272-85. doi: 10.1093/hmg/ddv081. PMID:25743185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dermaut B, Kumar-Singh S, Rademakers R, Theuns J, Cruts M, Van Broeckhoven C. Tau is central in the genetic Alzheimer-frontotemporal dementia spectrum. Trends Genetics. 2005;21:664-72. doi: 10.1016/j.tig.2005.09.005. PMID:16221505 [DOI] [PubMed] [Google Scholar]
- [21].Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711-4. doi: 10.1126/science.1062382. PMID:11408621 [DOI] [PubMed] [Google Scholar]
- [22].Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509-19. doi: 10.1016/S0896-6273(02)00706-7. PMID:12062036 [DOI] [PubMed] [Google Scholar]
- [23].Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al.. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205-8. doi: 10.1126/science.1166066. PMID:19251627 [DOI] [PubMed] [Google Scholar]
- [24].Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al.. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208-11. doi: 10.1126/science.1165942. PMID:19251628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lanson NA Jr, Maltare A, King H, Smith R, Kim JH, Taylor JP, Lloyd TE, Pandey UB. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum Mol Genet. 2011;20:2510-23. doi: 10.1093/hmg/ddr150. PMID:21487023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372-6. doi: 10.1038/nature12043. PMID:23503661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A. 2013;110:6400-5. doi: 10.1073/pnas.1221132110. PMID:23509287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Adhikari A, Chen ZJ. Diversity of polyubiquitin chains. Dev Cell. 2009;16:485-6. doi: 10.1016/j.devcel.2009.04.001. PMID:19386255 [DOI] [PubMed] [Google Scholar]
- [29].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222. doi: 10.1080/15548627.2015.1100356. PMID:26799652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nagy P, Varga A, Kovacs AL, Takats S, Juhasz G. How and why to study autophagy in Drosophila: it's more than just a garbage chute. Methods. 2015;75:151-61. doi: 10.1016/j.ymeth.2014.11.016. PMID:25481477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131-45. doi: 10.1074/jbc.M702824200. PMID:17580304 [DOI] [PubMed] [Google Scholar]
- [32].Rui Y-N, Xu Z, Patel B, Chen Z, Chen D, Tito A, David G, Sun Y, Stimming EF, Bellen HJ, et al.. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17:262-75. doi: 10.1038/ncb3101. PMID:25686248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Neefjes J, Dantuma NP. Fluorescent probes for proteolysis: tools for drug discovery. Nat Rev Drug Discov. 2004;3:58-69. doi: 10.1038/nrd1282. PMID:14708021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schweisguth F. Dominant-negative mutation in the beta2 and beta6 proteasome subunit genes affect alternative cell fate decisions in the Drosophila sense organ lineage. Proc Natl Acad Sci U S A. 1999;96:11382-6. doi: 10.1073/pnas.96.20.11382. PMID:10500185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861-73. doi: 10.1101/gad.1599207. PMID:18006683 [DOI] [PubMed] [Google Scholar]
- [36].Ktistakis NT, Tooze SA. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016;26:624-35. doi: 10.1016/j.tcb.2016.03.006. PMID:27050762 [DOI] [PubMed] [Google Scholar]
- [37].DeVorkin L, Gorski SM. Monitoring autophagy in Drosophila using fluorescent reporters in the UAS-GAL4 system. Cold Spring Harb Protoc. 2014;2014:967-72. doi: 10.1101/pdb.prot080341. PMID:25183817 [DOI] [PubMed] [Google Scholar]
- [38].Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjorkoy G, Johansen T, Rusten TE, Brech A, Baehrecke EH, Stenmark H. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010;190:523-31. doi: 10.1083/jcb.201002035. PMID:20713604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452-60. doi: 10.4161/auto.4451. PMID:17534139 [DOI] [PubMed] [Google Scholar]
- [40].Rusten TE, Lindmo K, Juhasz G, Sass M, Seglen PO, Brech A, Stenmark H. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell. 2004;7:179-92. doi: 10.1016/j.devcel.2004.07.005. PMID:15296715 [DOI] [PubMed] [Google Scholar]
- [41].Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, Liu B, Chang C, Zhou T, Lippincott-Schwartz J, et al.. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57:456-66. doi: 10.1016/j.molcel.2014.12.013. PMID:25601754 [DOI] [PubMed] [Google Scholar]
- [42].Zhang T, Zhou Q, Ogmundsdottir MH, Moller K, Siddaway R, Larue L, Hsing M, Kong SW, Goding CR, Palsson A, et al.. Mitf is a master regulator of the v-ATPase, forming a control module for cellular homeostasis with v-ATPase and TORC1. J Cell Sci. 2015;128:2938-50. doi: 10.1242/jcs.173807. PMID:26092939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gleixner EM, Canaud G, Hermle T, Guida MC, Kretz O, Helmstadter M, Huber TB, Eimer S, Terzi F, Simons M. V-ATPase/mTOR signaling regulates megalin-mediated apical endocytosis. Cell Rep. 2014;8:10-9. doi: 10.1016/j.celrep.2014.05.035. PMID:24953654 [DOI] [PubMed] [Google Scholar]
- [44].Morin X, Daneman R, Zavortink M, Chia W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci U S A. 2001;98:15050-5. doi: 10.1073/pnas.261408198. PMID:11742088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682-95. doi: 10.1016/j.cell.2011.07.030. PMID:21884931 [DOI] [PubMed] [Google Scholar]
- [46].Zaffagnini G, Martens S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016; 428:1714-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Khaminets A, Behl C, Dikic I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 2016;26:6-16. doi: 10.1016/j.tcb.2015.08.010. PMID:26437584 [DOI] [PubMed] [Google Scholar]
- [48].Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53:167-78. doi: 10.1016/j.molcel.2013.12.014. PMID:24462201 [DOI] [PubMed] [Google Scholar]
- [49].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279-96. doi: 10.4161/auto.7.3.14487. PMID:21189453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395-8. doi: 10.1038/26506. PMID:9759731 [DOI] [PubMed] [Google Scholar]
- [51].Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al.. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473-7. PMID:19556463 [DOI] [PubMed] [Google Scholar]
- [52].Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al.. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429-33. doi: 10.1126/science.1204592. PMID:21617040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Martini-Stoica H, Xu Y, Ballabio A, Zheng H. The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 2016;39:221-34. doi: 10.1016/j.tins.2016.02.002. PMID:26968346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wairkar YP, Toda H, Mochizuki H, Furukubo-Tokunaga K, Tomoda T, Diantonio A. Unc-51 controls active zone density and protein composition by downregulating ERK signaling. J Neurosci. 2009;29:517-28. doi: 10.1523/JNEUROSCI.3848-08.2009. PMID:19144852 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.