

The MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) conjugation system is a key functional module in macroautophagy (henceforth autophagy) for efficient formation of autophagosomes and selective degradation of autophagosomal cargoes through the lysosome. Recently, it has been appreciated that LC3 and homologs (LC3s) have roles outside of canonical autophagy. One such example is the presence of LC3s on the membrane of phagosomes containing certain pathogen-associated molecular patterns. In this process, termed LC3-associated phagocytosis (LAP), the LC3s modulate fusion between the phagosome and the lysosome. Adding to this theme, we have uncovered another crucial function of LC3s on the membranes of pathogen-containing vacuoles, where they recruit antimicrobial proteins to the LC3-marked membrane. We dubbed this process targeting by autophagy proteins (TAG). In our new study, TAG was identified to mediate a novel antiviral mechanism against the membranous shelters of viruses, known as viral replication complexes (RCs).
All known viruses with positive-sense RNA (+RNA) genomes reorganize cellular membranes to form their RCs. These structures are critical in the viral life cycle, shielding viruses from immune detection during genome replication. Although the formation of these RCs has been intensely studied, there have been few studies on how these structures are counteracted by the host immune system. While studying a potential role of autophagy in +RNA virus replication, we found that IFNG/IFN-gamma inhibits the RC of murine norovirus (MNV) and consequently its replication. Intriguingly, this IFNG-mediated inhibition of the MNV RC requires the ATG12–ATG5-ATG16L1 complex but not lysosomal degradation through canonical autophagy. The E3-like ligase complex, ATG12–ATG5-ATG16L1, localizes to the RC of MNV, suggesting that the LC3 homologs might be conjugated to the membrane of the MNV RC. However, it was unknown how the ATG12–ATG5-ATG16L1 complex functions in this lysosome-independent, IFNG-mediated inhibition of the MNV RC.
A clue came from a eukaryotic parasite, Toxoplasma gondii, which forms a parasitophorus vacuole (PV) from the host plasma membrane during its invasion and survives within this cytoplasmic vacuole. Upon activation of cells with IFNs, IFN-inducible GTPases are expressed and targeted to the PV membrane (PVM) of T. gondii. These dynamin-like GTPases oligomerize on the targeted membranes and disrupt the structure through vesiculation. Surprisingly, we and others found that the proper targeting of these GTPases to the PVM is dependent on the ATG12–ATG5-ATG16L1 complex but not canonical autophagy. Furthermore, we demonstrated that the ATG12–ATG5-ATG16L1 complex marks the PVM of T. gondii with LC3s and that the LC3s are necessary and sufficient to recruit the IFN-inducible GTPases to the LC3-marked membranes.
Based on this novel function of the LC3 conjugation system in cell-autonomous immune defense, we speculated that TAG may be responsible for the IFNG-mediated inhibition of the MNV RC. Indeed, in our new study, we found that the antiviral activity of IFNG is analogous to the role of IFNG in controlling T. gondii. The ATG12–ATG5-ATG16L1 complex is required to mark the MNV RC with LC3s, allowing proper targeting of IFN-inducible GTPases to the RC membrane. The targeting of RCs by the membranolytic GTPases leads to the subsequent disruption of these membrane structures.
The role of LC3s in TAG is fundamentally different from those in canonical autophagy and LAP, of which the endgame is fusion with the lysosome and degradation of vacuolar contents (Fig. 1). In fact, the end result of TAG in the IFN-activated cells is to release the vacuolar contents to the cytoplasm, contrary to canonical autophagy. Such a fundamental difference raises 2, if not more, important questions. First, what brings the ATG12–ATG5-ATG16L1 complex to the membranous shelters of pathogens? In canonical autophagy, WIPI2B (WD repeat domain, phosphoinositide interacting 2B) binds to phosphatidylinositol 3-phosphate (PtdIns3P) at the site of autophagosome formation and recruits the ATG12–ATG5-ATG16L1 complex to this membrane via its interaction with ATG16L1. In contrast, PtdIns 3-kinase activity is not required to bring the ATG12–ATG5-ATG16L1 complex to the membrane of pathogen shelters, suggesting a novel mechanism of recruitment independent of PtdIns3P. Because the ATG12–ATG5-ATG16L1 complex can be recruited onto the PVM of T. gondii within minutes post-infection, a rapid protein-protein and/or protein-lipid interaction-mediated recruitment is likely. In this regard, it is noteworthy that ATG5 can directly bind membranes with currently unknown binding specificity. Further, considering the relatively slow generation of viral RC at hours post-infection and the subsequent recruitment of the ATG12–ATG5-ATG16L1 complex to the RC, multiple different mechanisms may function in a pathogen-specific manner for the recruitment of the ATG12–ATG5-ATG16L1 complex, while converging at the same steps of LC3 conjugation and downstream effector recruitment.
Figure 1.
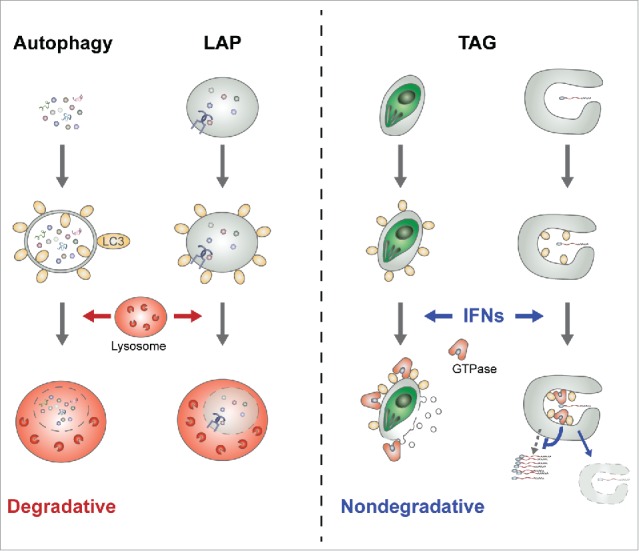
Degradative and nondegradative functions of the LC3 conjugation system. In canonical autophagy and LAP, LC3-marked autophagosomes and phagosomes, respectively, are fused to the lysosome and degraded (left). In contrast, during the TAG process, the membranous shelters of pathogens (such as protists or +RNA viruses) are marked with LC3 homologs and membranolytic IFN-inducible GTPases are targeted to the LC3-marked membranes upon their induction by IFNs (right). Consequently, the GTPase-targeted membrane structures are disrupted and the survival/replication of pathogens without functional shelters is inhibited.
Second, what brings the IFN-inducible GTPases to the LC3-marked membranes of pathogen shelters, instead of the LC3-marked autophagosomal membrane? Again, there is currently no clear answer to this question. It is noteworthy that LC3 marks the pathogen-associated membrane even in the absence of an IFN signal. Despite the presence of LC3 at these membranes, overexpression of the IFN-inducible GTPases without IFN is not sufficient for their proper targeting. These data suggest that the IFN signal may modify LC3, thereby affecting the interaction between the LC3 homologs and the GTPases on the target membranes. Thus, one potential mechanism is an IFN-mediated post-translational modification of LC3s on the membranous shelters of pathogens, and consequently specific recruitment of the GTPases to the modified LC3s. Alternatively, infection-dependent modification of the membrane signature (protein, lipid, or both) may cooperate with LC3s for specific recruitment of downstream effectors.
Our finding paves the way for future investigation on the antiviral mechanism against the RCs of +RNA viruses, which encompass one-third of all known viruses and include many medically important viruses such as norovirus and Zika virus. Our data also consolidate the immune-defense functions of the IFN-inducible GTPases against vacuolar pathogens, which now includes +RNA viruses as well as bacteria, protists, and fungi. Moreover, TAG in its essence reminds us of the fact that the LC3 homologs are ubiquitin-like proteins. As ubiquitin is not just a degradation signal for the conjugated proteins through the proteasome, LC3s are not just degradation signals for the conjugated membranes through the lysosome.
Declaration of potential conflicts of interest
No potential conflicts of interest were declared.
Funding
This work was supported by startup fund to S.H., and in part by the Brinson Foundation Junior Investigator Grant, Cancer Research Foundation, an Institutional Research Grant (IRG-58-004-53-IRG) from the American Cancer Society, the University of Chicago Digestive Diseases Research Core Center (NIDDK P30DK42086), the University of Chicago Comprehensive Cancer Center Support Grant (P30 CA14599), the Cancer Center Core facilities (DNA sequencing and genotyping, flow cytometry, integrated microscopy, and monoclonal antibody), and the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1 TR000430). S.B.B. was supported (in part) by NIH T32 GM007183 and H.M.B. was supported (in part) by NIH T32 AI007090.