

ABSTRACT
Aminoglycosides are toxic to sensory hair cells (HCs). Macroautophagy/autophagy is an essential and highly conserved self-digestion pathway that plays important roles in the maintenance of cellular function and viability under stress. However, the role of autophagy in aminoglycoside-induced HC injury is unknown. Here, we first found that autophagy activity was significantly increased, including enhanced autophagosome-lysosome fusion, in both cochlear HCs and HEI-OC-1 cells after neomycin or gentamicin injury, suggesting that autophagy might be correlated with aminoglycoside-induced cell death. We then used rapamycin, an autophagy activator, to increase the autophagy activity and found that the ROS levels, apoptosis, and cell death were significantly decreased after neomycin or gentamicin injury. In contrast, treatment with the autophagy inhibitor 3-methyladenine (3-MA) or knockdown of autophagy-related (ATG) proteins resulted in reduced autophagy activity and significantly increased ROS levels, apoptosis, and cell death after neomycin or gentamicin injury. Finally, after neomycin injury, the antioxidant N-acetylcysteine could successfully prevent the increased apoptosis and HC loss induced by 3-MA treatment or ATG knockdown, suggesting that autophagy protects against neomycin-induced HC damage by inhibiting oxidative stress. We also found that the dysfunctional mitochondria were not eliminated by selective autophagy (mitophagy) in HEI-OC-1 cells after neomycin treatment, suggesting that autophagy might not directly target the damaged mitochondria for degradation. This study demonstrates that moderate ROS levels can promote autophagy to recycle damaged cellular constituents and maintain cellular homeostasis, while the induction of autophagy can inhibit apoptosis and protect the HCs by suppressing ROS accumulation after aminoglycoside injury.
KEYWORDS: aminoglycosides, apoptosis, autophagic flux, autophagosome, hair cell protection, lysosome, oxidative stress
Introduction
Hearing loss is one of the most common sensory disorders in humans and is a serious concern globally. Ototoxic drug-induced hair cell (HC) damage is one of the main causes of sensorineural hearing loss. Chemotherapeutics (e.g., cisplatin), aminoglycosides (e.g., neomycin), and loop diuretics (e.g., furosemide) are the major classes of ototoxic drugs. While the molecular events resulting in ototoxicity are complex, the accumulation of reactive oxygen species (ROS) plays a key role in the process of HC death by activating multiple apoptotic pathways.1-4 ROS are a byproduct of cellular metabolism or exposure to xenobiotic agents,5 and ROS are harmful when their concentration exceeds the capacity of the cell to repair itself. Thus, cell survival requires a balance between oxidative stress and antioxidant defense, and ROS accumulation leads to cell death.6,7
Previous studies have shown that ROS have the ability to induce cellular defense pathways such as autophagy.8,9 Autophagy is an orderly degradation and recycling mechanism that disassembles unnecessary or dysfunctional cellular components through a highly regulated process in all eukaryotic cells.10,11 This process initially involves the formation of a double membrane autophagosome that surrounds the organelle or other cytoplasmic components that are targeted for destruction.12,13 The autophagosomes travel to lysosomes and subsequently fuse with the lysosomes to form an autolysosome in which the cytoplasmic contents are degraded via acidic lysosomal hydrolases.14 MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) proteins (mammalian subfamily of orthologs of yeast Atg8) are ubiquitin-like proteins associated with the process of autophagy. An LC3-I protein can be conjugated with phosphatidylethanolamine to form LC3-II, which is located in the autophagosome membrane,15 and this makes LC3 a suitable marker for autophagy activity in cells.16
Autophagy is known to be a general cellular response to starvation or stress and to play an important role in cell survival in many diseases, but whether the activation of autophagy increases or decreases the rate of cell death in these diseases is still being debated.17-19 Autophagy plays a causative role in autophagic cell death, and excessive activation of autophagy can contribute to apoptotic cell death through degradative processes such as cardiac myocyte death during ischemia/reperfusion. In addition, excessive activation of autophagy can also promote pathological changes such as the development of hepatic fibrosis.20-23 The activation of autophagy has also been shown to be beneficial during various pathological and physiological states. Autophagy can protect the cells against nutrient deprivation, it can remove damaged organelles, it acts as a defense against microbial infection, and it plays a protective role in metabolic diseases, cardiac diseases, and neurodegenerative diseases (including Parkinson, Alzheimer, Huntington, and Creutzfeldt-Jakob diseases).21 In addition, there are some diseases, such as ischemic stroke and cancer, for which it is still not well established as to whether autophagy plays a protective or destructive role.24,25 Thus, fully understanding autophagy is necessary for the treatment of many human diseases.
Autophagy is an important cell survival process,26,27 but it is also implicated in cell death processes.28 Early studies suggested that autophagy serves as a cell survival mechanism in some pathological processes via its suppressive role in necroptosis and PARP-mediated cell death during unfavorable growth conditions or cellular stress.29,30 In retinal ganglion cells, autophagy plays an important role in suppressing apoptosis, and it has been observed that activation of autophagy can promote retinal ganglion cell survival and that deletion of autophagy can reduce cell survival during optic nerve degeneration.31 However, whether the activation of autophagy is a proapoptotic or antiapoptotic factor in response to aminoglycoside-induced HC injury in the cochlea remains uninvestigated. Therefore, understanding the relationship between autophagy and ROS, and understanding the role of autophagy in HC survival, might have therapeutic implications for the treatment of ototoxic drug-induced hearing loss.
In this study, we determined the autophagy activity by measuring the autophagic flux (including the expression of the autophagy marker LC3B-II), the colocalization of the autophagy markers LC3B and SQSTM1/p62, the degradation of the SQSTM1/p62 protein, and the expression of the mRFP-GFP-LC3B reporter; by performing the autophagy substrate experiment; and by transmission electron microscope (TEM) imaging. To explore the role of autophagy in HC survival, rapamycin was used to activate autophagy and 3-methyladenine (3-MA) or the knockdown of autophagy-related proteins (including ATG5, BECN1 and ATG7) were used to inhibit autophagy before neomycin or gentamicin treatment,32,33 and we assessed the influence of autophagy on the level of oxidative stress and on HC survival. Furthermore, the antioxidant N-acetylcysteine (NAC) was used to verify that autophagy mediates its protective effect by suppressing ROS accumulation. Together, our results suggest that autophagy might be a new therapeutic target for the prevention of aminoglycoside-induced HC death.
Results
Autophagy is increased in cochlear HCs after neomycin or gentamicin damage
We dissected the cochleae from postnatal d (P)3 mice and cultured them with different neomycin concentrations (0.2 mM, 0.5 mM, 1 mM, and 2 mM) for different exposure times (6 h and 24 h). Western blots showed that the 0.5 mM neomycin treatment of both 6 h and 24 h significantly increased the expression of LC3B (Fig. 1A and B, P < 0.05, n = 3). TEM images showed that there were significantly more autophagic vacuoles (double membrane-bound autophagosomes) and autolysosomes (containing lysosomal membrane proteins and enzymes) after 0.5 mM neomycin treatment of 6 h compared with the controls, which confirmed the occurrence of autophagy and autophagosomes in the explant cultured cochleae (Fig. 1C and D, P < 0.01, n = 3). GFP-LC3B mice were used to confirm the increase in autophagy after neomycin exposure. The cochleae were dissected from P3 GFP-LC3B mice and immunolabeled with the HC marker MYO7A (myosin VIIA) after culturing the cochleae with 0.5 mM neomycin for 6 h and 24 h. Quantification of the LC3B puncta in each HC showed that the numbers of LC3B puncta were significantly increased in HCs after both 6 h and 24 h neomycin treatments compared with the controls (Fig. 1E and F, P < 0.01, n = 6).
Figure 1.

Increased autophagy in cochlear HCs after neomycin treatment. (A) Western blotting using total cochlear homogenates showed changes of LC3B-II expression in the cochleae treated with different concentrations of neomycin (0.2 mM, 0.5 mM, 1 mM, and 2 mM) and different exposure times (6 h and 24 h). GAPDH served as the sample loading control, n = 3. (B) Quantification of the western blot in (A). (C) Transmission electron microscope (TEM) analysis to evaluate autophagy in cochlear HCs. The numbers of autophagic vacuoles and autolysosomes (arrows in pictures) were significantly increased after neomycin treatment compared with the control, n = 3. (D) Quantification of the results in C. (E) Immunofluorescence staining with MYO7A antibody in the cochleae from GFP-LC3B mice. The GFP-LC3B puncta were significantly increased with neomycin treatment, n = 6. (F) Quantification of the GFP-LC3B punctum number in E. For all experiments, *P < 0.05, **P < 0.01, ***P < 0.001.
To test whether these findings can be generalized to other aminoglycosides, we used gentamicin, which is also ototoxic, to treat the explant cultured cochleae. Western blots showed that the gentamicin treatment significantly increased the expression of LC3B-II (Fig. S1A and B, P < 0.05, n = 4). The cochlear immunolabeling results showed that when treated with 0.5 mM gentamicin for 6 h and 24 h, the number of LC3B puncta in each HC was significantly greater than in the undamaged controls (Fig. S1C and D, P < 0.001, n = 6). TEM images also showed that there were significantly more autophagic vacuoles and autolysosomes after 0.5 mM gentamicin treatment of 24 h compared with the controls (Fig. S1E and F, P < 0.001, n = 3). Together, these results demonstrated that autophagy is activated in cochlear HCs after neomycin or gentamicin damage.
Autophagy is increased in the HEI-OC-1 cells after neomycin or gentamicin damage
HEI-OC-1 cells express several molecular markers of cochlear HCs, including CALB1 (calbindin 1), CALM1 (calmodulin 1), ATOH1/MATH1 (atonal bHLH transcription factor 1), MYO7A, and SLC26A5/PRES (solute carrier family 26, member 5).34,35 First, we examined the expression of the autophagosome marker LC3 (LC3B) in HEI-OC-1 cells after treatment with different neomycin concentrations (0.5 mM, 1 mM, 2 mM, and 5 mM) for different exposure times (6 h and 24 h) and a 24 h recovery period. Western blots showed that the expression of LC3B-II increased as the neomycin concentration and the exposure time increased (Fig. 2A and B, P < 0.05, n = 3). We found that 2 mM neomycin treatment of 24 h led to the highest level of LC3B-II in HEI-OC-1 cells. Thus, we used this neomycin treatment condition for the following experiments in HEI-OC-1 cells.
Figure 2.

Increased autophagy in HEI-OC-1 cells after neomycin treatment. (A) Western blot showing the changes in LC3B-II expression in the HEI-OC-1 cells treated with different concentration of neomycin (0.5 mM, 1 mM, 2 mM, and 5 mM) and different exposure times (6 h and 24 h), n = 3. (B) Quantification of the western blot in (A). (C) TEM analysis to evaluate the presence of autophagy in HEI-OC-1 cells. The numbers of autophagic vacuoles and autolysosomes (arrows in pictures) were significantly increased after neomycin treatment compared with the control, n = 3. (D) Immunofluorescence staining with anti-LC3B antibody in HEI-OC-1 cells after neomycin injury, n = 4. (E) Quantification of the results in (C). (F) Quantification of the LC3B fluorescent puncta in (D). (G) Western blots with anti-LC3B antibody after neomycin and Baf treatment, n = 4. (H) Quantification of the western blot result in (G). (I) Immunofluorescence staining with anti-LC3B and anti-SQSTM1/p62 antibodies in HEI-OC-1 cells. The colocalization puncta (arrows in pictures) appeared after neomycin treatment. (J) Western blots with anti-SQSTM1/p62 antibody revealed a significant decrease in SQSTM1/p62 after neomycin treatment and significant increase after Baf treatment, n = 3. (K) Quantification of the western blot result in (J). (L) Transfection with mRFP-GFP-LC3B plasmids in HEI-OC-1 cells, n = 4. (M) Quantification of the results in (L). (N) HEI-OC-1 Cells were transfected with NHTT-150Q-EGFP and treated with neomycin, n = 4. For all experiments, *P < 0.05, **P < 0.01, ***P < 0.001.
We used TEM analysis to confirm the occurrence of autophagy and to explore the morphology of the autophagosome. In this experiment, the neomycin group was treated with 2 mM neomycin for 24 h and allowed to recover for 24 h. The neomycin-treated group had significantly more autophagic vacuoles (double membrane-bound autophagosomes) and autolysosomes (containing lysosomal membrane proteins and enzymes) compared with the control group (Fig. 2C and E, P < 0.001, n = 3).
We also used immunofluorescence staining with anti-LC3B antibodies in HEI-OC-1 cells to confirm the occurrence of autophagy after neomycin treatment. Quantification of the LC3B puncta in each HEI-OC-1 cell showed that the numbers of LC3B puncta were significantly increased in the cytoplasm after neomycin treatment compared with the control cells (Fig. 2D and F, P < 0.001, n = 4), suggesting that neomycin injury could enhance autophagosome synthesis in HEI-OC-1 cells. Bafilomycin A1 (Baf) is a V-ATPase inhibitor that inhibits autophagosome degradation by blocking the fusion of autophagosomes with lysosomes, and this allows us to monitor autophagosome synthesis.36,37 Immunohistochemistry and western blot experiments showed that the numbers of LC3B puncta and the LC3B-II protein expression level were both increased in HEI-OC-1 cells cotreated with neomycin and Baf compared with control cells treated with Baf alone (Fig. 2D, F to H, P < 0.05, n = 4), indicating that neomycin injury could still enhance autophagosome synthesis even when autophagosome-lysosome fusion was inhibited by Baf. We then examined the formation of autophagosome-lysosome complexes by analyzing the colocalization of the autophagy markers LC3B and SQSTM1/p62, which can interact with ubiquitin and trigger the degradation of proteins in the proteasome or lysosome.38 We found that the number of puncta that colocalized with these 2 markers in the cytoplasm was significantly increased after neomycin treatment (Fig. 2I). To confirm our finding that neomycin injury can enhance autophagosome–lysosome fusion, we performed a western blot assay to validate the degradation of the autophagic substrate SQSTM1/p62 protein. We found that the protein level of SQSTM1/p62 was significantly decreased in the HEI-OC-1 cells after neomycin treatment compared with control cells (Fig. 2J and K, P < 0.05, n = 3), again suggesting that neomycin injury could enhance autophagosome–lysosome fusion. We further examined the SQSTM1/p62 level when autophagosome–lysosome fusion was blocked by Baf. We found no change in the SQSTM1/p62 level in HEI-OC-1 cells cotreated with neomycin and Baf compared with control cells treated with Baf alone (Fig. 2J and K, P < 0.05, n = 3).
To better study the effect of neomycin injury on autophagic flux, HEI-OC-1 cells were transfected with the mRFP-GFP-LC3B plasmid as a reporter of autophagic flux.39 Because the GFP signal is pH sensitive, this method can be used to quantify the numbers of autophagosomes and autolysosomes. The yellow puncta represent the overlap of red and green puncta, indicating the autophagosomes that have not fused with the lysosomes, while the red puncta correspond to autolysosomes.39,40 We found a significant increase in the number of both autophagosomes and autolysosomes in neomycin-treated cells compared with control cells (Fig. 2L and M, P < 0.01, n = 4). We further examined the viability of cells that were transfected with EGFP-tagged NHTT-150Q (truncated N-terminal HTT [huntingtin] protein with 150 glutamine repeats), which is a known autophagy substrate.41,42 The GFP fluorescence intensity was significantly decreased after neomycin treatment compared with control cells (Fig. 2N, P < 0.001, n = 4), indicating that low concentrations of neomycin might trigger the cytoprotective effects of autophagy and prevent cell death. Taken together, all of these results demonstrated that neomycin injury could enhance both autophagosome synthesis and autophagosome–lysosome fusion in HEI-OC-1 cells.
To validate whether these results can be generalized to other aminoglycosides, we treated the HEI-OC-1 cells with different concentrations of gentamicin for 6 h or 24 h. Western blots showed that 0.5 mM gentamicin treatment of 24 h led to the greatest increase in LC3B-II expression (Fig. S2A to D, P < 0.001, n = 3). Thus, we used this gentamicin treatment condition for the following experiments in HEI-OC-1 cells. The immunolabeling results showed that the numbers of LC3B puncta in cells treated with 0.5 mM gentamicin for 24 h were significantly greater than the undamaged controls (Fig. S2E and G, P < 0.001, n = 4). When transfected with the mRFP-GFP-LC3B plasmid, we found a significant increase in the number of both autophagosomes and autolysosomes in gentamicin-treated cells compared with control cells (Fig. S2F and H, P < 0.001, n = 4). TEM images also showed that there were significantly more autophagic vacuoles and autolysosomes after 0.5 mM gentamicin treatment of 24 h compared with the controls (Fig. S2I and J, P < 0.001, n = 3).
The induction of autophagy affects cochlear HC survival after neomycin or gentamicin damage
Cochleae were dissected from P3 mice and pretreated with the autophagy activator rapamycin or the autophagy inhibitor 3-MA for 6 h before neomycin treatment. The cultures were then treated with 0.5 mM neomycin for 6 h or 24 h together with rapamycin or 3-MA. The tissues were collected for immunofluorescence staining. To confirm that the expression of LC3B-II was affected by the autophagy activator and inhibitor, we took advantage of the GFP-LC3B mice to show LC3B puncta and autophagosome formation. We found that the numbers of LC3B puncta were significantly increased in HCs after 6 h and 24 h neomycin exposure (Fig. 3A and B, P < 0.01, n = 6). The results also showed that treatment with rapamycin resulted in an increase in the number of LC3B puncta in HCs, while treatment with 3-MA markedly reduced the number of LC3B puncta in HCs compared with the neomycin-only group (Fig. 3A and B, P < 0.05, n = 6).
Figure 3.

Rapamycin and 3-MA affected both the induction of autophagy and HC survival in the cochlea after neomycin damage. (A) Immunofluorescence staining with the anti-MYO7A antibody in the cochleae from GFP-LC3B mice after different treatments, n = 6. (B) Quantification of the number of GFP-LC3B puncta in (A). The number of GFP-LC3B puncta was significantly increased in the rapamycin-pretreatment group and significantly decreased in the 3-MA-pretreatment group after neomycin treatment compared with neomycin treatment alone, n = 6. (C) Quantification of the MYO7A-positive HCs in Figure S4. Pretreatment with rapamycin promoted HC survival from the apical to the basal turn of the cochlea compared with neomycin exposure alone. In contrast, 3-MA accelerated HC apoptosis after neomycin injury, n = 6. Scale bars: 10 μm. For all experiments, *P < 0.05, **P < 0.01, ***P < 0.001.
We also tested the changes of LC3B puncta in HCs when treated with 0.5 mM gentamicin for 6 h or 24 h. We found that the numbers of LC3B puncta were significantly increased in gentamicin-treated HCs compared with the controls (Fig. S3A and B, P < 0.001, n = 6). Treatment with rapamycin also resulted in an increase in the number of LC3B puncta in HCs. In contrast, the number of LC3B puncta was significantly decreased in 3-MA–treated HCs compared with the gentamicin-only group (Fig. S3A and B, P < 0.001, n = 6).
We then assessed the effects of increased or decreased autophagy on HC survival by counting the MYO7A-positive HC number in 150 µm lengths in all 3 turns of the cochlea after neomycin damage. Immunofluorescence and cell counting indicated that HC loss was increased in all 3 turns of the cochlea after treatment with 0.5 mM neomycin for 24 h, and this loss was significantly increased after treatment with the autophagy inhibitor 3-MA (Fig. 3C and Fig. S4, P < 0.01, n = 6). In contrast, treatment with the autophagy inducer rapamycin significantly attenuated the neomycin-induced HC loss (Fig. 3C and Fig. S4, P < 0.05, n = 6). In addition, we found that autophagy also played an important role in HC survival after gentamicin damage. Activating autophagy significantly increased the number of surviving HCs, while inhibiting autophagy significantly decreased the number of surviving HCs after treatment with 0.5 mM gentamicin for 24 h (Fig. S5, P < 0.05, n = 6).
Inhibition and activation of autophagy affect autophagosome formation and cell survival in HEI-OC-1 cells following neomycin or gentamicin damage
We examined the effects of rapamycin (which is an autophagy activator), 3-MA (which is an autophagy inhibitor), and knockdown of the autophagy-related proteins ATG5, BECN1, and ATG7 (which results in the inhibition of LC3-II formation), on autophagosome formation in HEI-OC-1 cells after 2 mM neomycin treatment of 24 h. The western blot data showed that after neomycin treatment the expression of ATG5, BECN1, and ATG7 were all increased, and the expression levels of ATG5, BECN1, and ATG7 were all significantly reduced after siRNA transfection with or without neomycin treatment (Fig. S6, P < 0.05, n = 3). To confirm that the expression of LC3B-II was affected by the autophagy inhibitor and activator, we performed immunofluorescence staining with anti-LC3B antibodies. Quantification of the fluorophores showed that treatment with 3-MA or knockdown of ATGs resulted in decreased autophagosome formation after neomycin injury. In contrast, treatment with rapamycin markedly stimulated the formation of autophagosomes in the cytoplasm (Fig. 4A and B, P < 0.01, n = 4).
Figure 4.

Inhibition and activation of autophagy affect autophagosome formation and cell survival in HEI-OC-1 cells following neomycin damage. (A) Images of immunolabeled LC3B (green) in HEI-OC-1 cells, n = 4. (B) Quantification of the LC3B fluorescent puncta in (A). (C) Cells were transfected with mRFP-GFP-LC3B plasmids and treated with different drugs. Yellow dots indicate autophagosomes and red dots indicate autolysosomes, n = 4. (D) Quantification of the LC3B fluorescent puncta in (C). (E) After treatment with neomycin, the numbers of live cells in 7 groups (undamaged, neomycin alone, rapamycin-pretreatment, 3-MA-pretreatment, Atg5 siRNA, Becn1 siRNA, and Atg7 siRNA) were determined with the CCK-8 kit, n = 4. For CCK-8 experiments, the values for the normal controls were set to 1. Scale bars: 5 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
We then analyzed the autophagic flux with the mRFP-GFP reporter system after different treatments. We found that the numbers of both autophagosomes and autolysosomes were significantly increased after neomycin treatment compared with the controls (Fig. 4C and D, P < 0.01, n = 4). The addition of rapamycin resulted in a significantly greater increase in both autophagosomes and autolysosomes compared with the neomycin treatment alone group, and the formation of autophagosomes and autolysosomes was significantly inhibited when treated with 3-MA or after knockdown of ATGs (Fig. 4C and D, P < 0.05, n = 4). We used the CCK-8 kit to determine the HEI-OC-1 cell numbers when autophagy was activated or inhibited after neomycin injury. The cell number was significantly increased when cultured with rapamycin and was significantly decreased when cultured with 3-MA or after knockdown of ATGs (Fig. 4E, P < 0.05, n = 4).
To validate whether these results can be generalized to other aminoglycosides, we measured the LC3B puncta and the autophagic flux after 0.5 mM gentamicin treatment of 24 h in the presence of the autophagy activator or inhibitor. Knockdown of ATGs significantly decreased the formation of autophagosomes and autolysosomes, whereas rapamycin significantly enhanced the formation of autophagosomes and autolysosomes compared with the gentamicin treatment alone group (Fig. S7A to D, P < 0.05, n = 4). In addition, we found that activating autophagy significantly increased the HEI-OC-1 cell number, while inhibiting autophagy significantly decreased the cell number after gentamicin injury (Fig. S7E, P < 0.01, n = 3).
Autophagy modulates neomycin-induced apoptosis in cochlear HCs
In this study, we wanted to investigate the role of autophagy in neomycin-induced HC injury, and previous studies revealed that cleaved-CASP3 (caspase 3) and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) can be used as markers of cell apoptosis induced by aminoglycosides.43-45 Here, cleaved-CASP3 and TUNEL staining was performed to detect the apoptotic cochlear HCs after 3MA or rapamycin treatments. Immunofluorescence staining showed that the numbers and proportions of cleaved-CASP3-positive cells (6.4 ± 1.52) and TUNEL-positive cells (6.25 ± 0.96) per 150 µm cochlear length of the middle turn in the neomycin-treated groups were significantly greater than the undamaged controls (1.2 ± 0.45 cleaved-CASP3-positive cells and 3.75 ± 2.96 TUNEL-positive cells) (Fig. 5A to D, P < 0.05, n = 3). Moreover, the 3MA-pretreated cochleae had significantly more cleaved-CASP3-positive cells (13.33 ± 1.15) and TUNEL-positive cells (10.0 ± 1.63) compared with the neomycin-only group (Fig. 5A to D, P < 0.05, n = 3). In contrast, when autophagy was activated by rapamycin, the numbers and proportions of cleaved-CASP3-positive cells (3.8 ± 1.1) and TUNEL-positive cells (3.5 ± 0.8) were significantly reduced (Fig. 5A to D, P < 0.01, n = 3). The phenotypes of the apical and basal turns in the cochlea were similar to that of the middle turn (Fig. S8A to F, P < 0.05, n = 3), but the severity of the HC injury in the apical turn of the cochlea was less than in the middle and basal turns after the neomycin treatments.
Figure 5.

Autophagy modulates the injury severity in cochlear HCs after neomycin exposure. (A) Immunofluorescence staining with TUNEL and MYO7A in the middle turn of the cochlea after different treatments, n = 3. (B) Immunofluorescence staining for cleaved-CASP3 and MYO7A in the middle turn of the cochlea after different treatments. (C and D) Quantification of the numbers of TUNEL and MYO7A double-positive, as well as cleaved-CASP3 and MYO7A double-positive cells. The numbers and proportions of cleaved CASP3-positive cells and TUNEL-positive cells in the neomycin-treated groups were significantly greater than the undamaged controls. Moreover, the numbers of apoptotic cells were significantly increased by 3-MA and decreased by rapamycin, n = 3. (E) The mRNA levels of 8 apoptosis-related genes were analyzed by qRT-PCR after treatment with neomycin, n = 4. (F) qRT-PCR analysis of the apoptosis-related gene expression in the 3-MA-pretreatment group and rapamycin-pretreatment group after neomycin injury, n = 4. For qRT-PCR experiments, the values for the normal controls were set to 1. Scale bars: 20 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
We also performed a quantitative real-time PCR (qRT-PCR) experiment to explore the expression of apoptosis-related genes in the cochlea after 3-MA or rapamycin treatment. After neomycin treatment, the expression of the proapoptotic genes Casp3, Casp9, and Casp8 was significantly increased compared with the undamaged control, and the expression of the antiapoptotic gene Bcl2 (B cell leukemia/lymphoma 2) was significantly decreased compared with controls (Fig. 5E, P < 0.05, n = 4). In addition, the autophagy inhibitor 3-MA significantly upregulated the expression of the proapoptotic genes Casp3, Casp9, and Casp8 and reduced the expression of the antiapoptotic gene Bcl2 (Fig. 5F, P < 0.05, n = 4). The expression of the proapoptotic genes Bax (BCL2-associated X protein), Casp3, Casp9, and Casp8 was significantly decreased when the cochleae were treated with the autophagy activator rapamycin (Fig. 5F, P < 0.05, n = 4).
Autophagy affects apoptosis in HEI-OC-1 cells after neomycin damage
In this experiment, we investigated the role of autophagy in neomycin-induced cell death in HEI-OC-1 cells through upregulation or downregulation of autophagosome formation. HEI-OC-1 cells were pretreated with 3-MA or rapamycin before exposure to neomycin. The cells were then treated with 2 mM neomycin for 24 h and allowed to recover in culture medium for another 24 h together with 3-MA or rapamycin. In the ATG knockdown group, the cells were treated with 2 mM neomycin for 24 h and allowed to recover another 24 h after being transfected with siRNA directed against Atg5, Becn1, or Atg7. We used propidium iodide to label the dead cells and ANXA5/Annexin V to label the cells undergoing apoptosis. After neomycin treatment, the proportions of both dead and apoptotic cells were significantly increased (4.47 ± 0.44% and 13.2 ± 2.2% dead and apoptotic cells, respectively) compared with the undamaged control (0.3 ± 0.19% and 0.05 ± 0.04% dead and apoptotic cells, respectively) (Fig. 6A and B, P < 0.01, n = 4). We also found that the proportion of apoptotic cells after treatment with rapamycin (5.2 ± 0.35%) was significantly reduced compared with the neomycin-only group (Fig. 6A and B, P < 0.01, n = 4). The proportion of dead cells (3.25 ± 0.88%) was also reduced when treated with rapamycin, but the difference was not statistically significant. In contrast, the proportion of dead and apoptotic HEI-OC-1 cells (5.53 ± 0.18% and 48.4 ± 2.88%, respectively) treated with 3-MA and the apoptotic proportion in the ATG knockdown group (52.93 ± 1.54%, 46.19 ± 4.77%, and 50.88 ± 3.24% for the Atg5 siRNA, Becn1 siRNA, and Atg7 siRNA groups, respectively) were markedly increased compared with the neomycin-only group (Fig. 6A and B, Fig. S9A, D and E, P < 0.05, n = 4).
Figure 6.
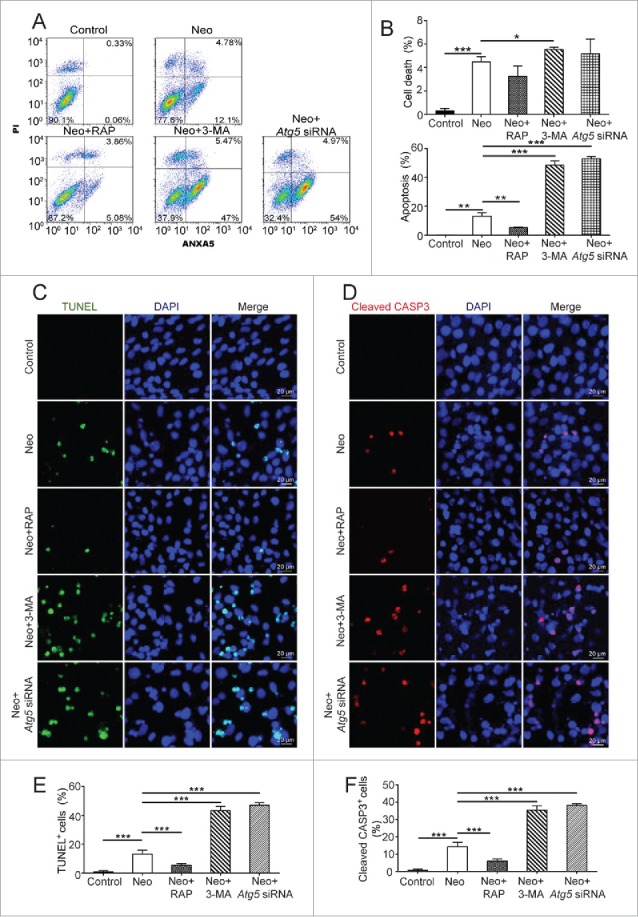
Autophagy affects cell death and apoptosis in HEI-OC-1 cells after neomycin exposure. (A) Apoptosis analysis by flow cytometry after different treatments. The lower left quadrants, lower right quadrants, and upper right quadrants of the images represent live cells, early apoptotic cells, and late apoptotic cells, respectively, n = 4. (B) Quantification of the flow cytometry data. The proportions of dead cells and early apoptotic cells after neomycin treatment were significantly increased compared with the undamaged groups. In addition, the dead and apoptotic proportions could be increased by 3-MA and ATG5 knockdown, and the apoptotic proportions could be reduced by rapamycin. (C) TUNEL and DAPI double staining showing the apoptotic HEI-OC-1 cells after different treatments, n = 4. (D) Cleaved-CASP3 and DAPI double staining confirmed the apoptotic cells after different treatments, n = 4. (E and F) Quantification of the numbers of TUNEL and DAPI double-positive, as well as cleaved-CASP3 and DAPI double-positive cells in (C and D), respectively.
Next, we performed cleaved-CASP3 and TUNEL staining to detect the apoptotic HEI-OC-1 cells after ATG knockdown or 3-MA or rapamycin treatments. Immunofluorescence staining showed that the proportions of cleaved-CASP3-positive (14.24 ± 2.57%) and TUNEL-positive cells (13.22 ± 2.7%) in the neomycin-treated groups were significantly greater than the undamaged controls (0.57 ± 0.31% and 0.83 ± 0.52%, respectively) (Fig. 6C to F, P < 0.001, n = 4). The ATG knockdown and 3-MA–treated cells had significantly more cleaved-CASP3-positive (38.11 ± 1.08%, 35.38 ± 6.58%, 38.95 ± 4.29%, and 35.4 ± 2.53% for the Atg5 siRNA, Becn1 siRNA, Atg7 siRNA, and 3-MA groups, respectively) and TUNEL-positive cells (46.02 ± 1.82%, 32.67 ± 4.72%, 39.28 ± 3.05%, and 43.3 ± 2.87% for the Atg5 siRNA, Becn1 siRNA, Atg7 siRNA, and 3-MA groups, respectively) compared with the neomycin-only group (Fig. 6C to F and Fig. S9B, C, F and G, P < 0.001, n = 4). In contrast, when autophagy was activated by rapamycin, the proportions of cleaved-CASP3-positive (5.95 ± 1.22%) and TUNEL-positive cells (5.48 ± 1.33%) were significantly reduced (Fig. 6C to F, P < 0.001, n = 4). Western blot showed that the protein expression of cleaved-CASP3 and cleaved-PARP1 (poly[ADP-ribose] polymerase 1) in HEI-OC-1 cells was significantly increased after neomycin treatment compared with the undamaged controls (Fig. 7A to D, P < 0.01, n = 4). The protein expression levels of both cleaved-CASP3 and cleaved-PARP1 were significantly reduced when treated with the autophagy activator rapamycin, and they were significantly increased when autophagy was inhibited by ATG knockdown (Fig. 7A to D, P < 0.05, n = 4). The qRT-PCR results showed that the expression of proapoptotic marker genes, including Casp8, Bax, and Casp3, was significantly higher in the neomycin-treated groups, and the expression of the antiapoptotic gene Bcl2 was significantly lower than controls (Fig. 7E, P < 0.05, n = 3). The autophagy inhibitor 3-MA and ATG5 knockdown significantly upregulated the expression of the proapoptotic marker genes and reduced the expression of the antiapoptotic genes (Fig. 7F, P < 0.05, n = 3). The expression of proapoptotic marker genes was significantly decreased and the expression of the antiapoptotic gene Bcl2 was significantly increased when the cells were treated with the autophagy activator rapamycin (Fig. 7F, P < 0.05, n = 3).
Figure 7.
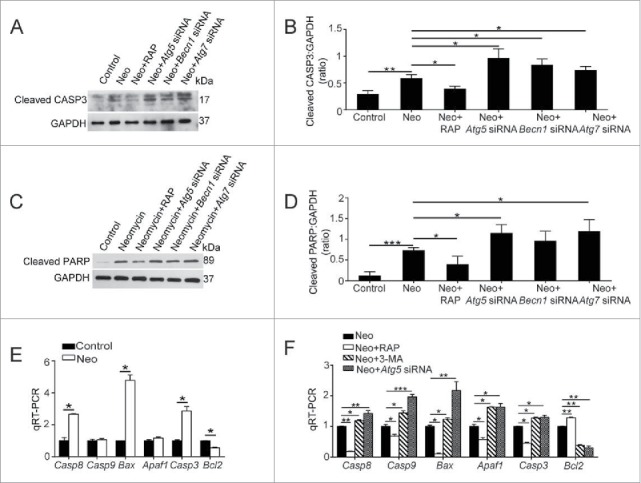
Autophagy affects the levels of apoptosis-related genes and proteins in HEI-OC-1 cells after neomycin exposure. (A) Western blots with anti-cleaved-CASP3 antibody revealed that the amount of cleaved-CASP3 is significantly increased after neomycin treatment. In addition, the amount could be increased by ATG knockdown and could be reduced by rapamycin, n = 4. (B) Quantification of the western blot in (A). (C) Western blots with anti-cleaved-PARP1 antibody, n = 4. (D) Quantification of the western blot in (C). (E) The mRNA levels of proapoptotic genes and antiapoptotic genes were analyzed by qRT-PCR after treatment with neomycin, n = 3. (F) qRT-PCR analysis of the apoptosis-related gene expression in the ATG5 knockdown group, 3-MA-pretreatment group, and rapamycin-pretreatment group after neomycin injury, n = 3. For qRT-PCR experiments, the values for the normal controls were set to 1. Scale bars: 20 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
After treatment with 0.5 mM gentamicin for 24 h, we also found that apoptotic cells were significantly increased compared with the undamaged controls (Fig. S10A to D, P < 0.05, n = 4). Moreover, the apoptotic cells were significantly decreased when autophagy was activated by rapamycin and were significantly increased when autophagy was reduced through knockdown of ATG expression (Fig. S10A to D, P < 0.05, n = 4).
Autophagy attenuates neomycin-induced oxidative stress in cochlear HCs
To determine the relationship between autophagy and oxidative stress in cochlear HCs, we dissected and cultured the cochleae from P3 mice and then treated them with neomycin together with 3-MA or rapamycin. We used Mito-SOX Red, which is a redox fluorophore that selectively detects mitochondrial superoxide, to evaluate mitochondrial ROS levels in neomycin-treated cochleae. Quantification of Mito-SOX Red colocalization with MYO7A showed that the ROS levels were increased in the middle turn of the cochlea after neomycin treatment compared with the undamaged controls (Fig. 8A and B, P < 0.05, n = 4). We then assessed the effects of increased autophagy on ROS levels using the autophagy stimulator rapamycin or the autophagy inhibitor 3-MA. After neomycin exposure, treatment with rapamycin markedly reduced the ROS levels in cochlear HCs, and treatment with 3-MA significantly increased ROS levels compared with the neomycin-only group (Fig. 8A and B, P < 0.05, n = 4). The phenotypes of the apical and basal turns in the cochlea were similar to that in the middle turn (Fig. S11A to C, P < 0.05, n = 4). Moreover, we found that the oxidative stress in the apical turn in the cochlea was lower than that in middle and basal turns after neomycin treatments. These findings are consistent with those shown in Fig. 5 and Fig. S8.
Figure 8.

Autophagy attenuates oxidative stress in cochlear HCs after neomycin injury. (A) Immunofluorescence staining with Mito-SOX and anti-MYO7A antibodies in the middle turn of the cochlea after different treatments, n = 4. (B) Quantification of the numbers and proportions of Mito-SOX and MYO7A double-positive cells in (A). The numbers and proportions of Mito-SOX-positive cells in the neomycin-treated groups were significantly greater than the undamaged controls. Moreover, the neomycin-induced oxidative stress was significantly increased by 3-MA and reduced by rapamycin, n = 4. (C) The mRNA levels of genes related to redox reactions were analyzed by qRT-PCR after neomycin treatment. The results showed that neomycin downregulated the expression of 5 genes (Sod1, Gsr, Glrx, Nqo1, and Tmx3), n = 3. (D) qRT-PCR analysis of the redox-related gene expression in the 3-MA-pretreatment group and rapamycin-pretreatment group after neomycin injury, n = 3. For qRT-PCR experiments, the values for the normal controls were set to 1. Scale bars: 20 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
Next, we performed a qRT PCR experiment to analyze the mRNA expression of redox-related genes in cochlear HCs after neomycin treatment. We found that the expression of 5 important antioxidant genes, including Sod1 (superoxide dismutase 1), Tmx3 (thioredoxin-related transmembrane protein 3), Nqo1 (NAD[P]H dehydrogenase, quinone 1), Gsr (glutathione reductase), and Glrx (glutaredoxin), was significantly decreased in cochlear HCs after neomycin treatment compared with undamaged cochleae (Fig. 8C, P < 0.05, n = 3). We then measured the expression of these genes after treatment with rapamycin and 3-MA. We found that the expression of 3 antioxidant genes (Sod1, Gsr, and Glrx) was significantly upregulated (Fig. 8D, P < 0.05, n = 3). The expression of Tmx3 and Cat in the rapamycin-treated groups was also increased, but the difference was not statistically significant compared with the neomycin-only group. In contrast, treatment with 3-MA significantly downregulated 2 antioxidant genes (Tmx3 and Nqo1) (Fig. 8D, P < 0.05, n = 3). The expression of Sod1 and Cat (catalase) was also decreased, but the difference was not statistically significant compared with the neomycin-only group.
Autophagy modulates oxidative stress in HEI-OC-1 cells after neomycin injury
The preceding experiments showed that autophagy can affect the apoptosis and cell survival of HEI-OC-1 cells after neomycin injury, and we hypothesized that the effect of autophagy on apoptosis in HEI-OC-1 cells might depend on oxidative stress. To test our hypothesis, we assessed the effects of autophagy-related protein (ATG5, BECN1, and ATG7) knockdown and 3-MA and rapamycin treatment on ROS levels in HEI-OC-1 cells. Mito-SOX Red was used to evaluate mitochondrial ROS levels in neomycin-treated HEI-OC-1 cells. Immunohistochemistry and flow cytometry showed that the ROS levels were increased after neomycin treatment compared with the undamaged controls (Fig. 9A, B, and C, P < 0.001, n = 4). After neomycin injury, the ROS levels were significantly increased with the 3-MA treatment and knockdown of ATG5, BECN1, and ATG7, while rapamycin significantly reduced the neomycin-induced ROS accumulation in HEI-OC-1 cells compared with the neomycin-only group (Fig. 9A to C and Fig. S12A to C, P < 0.05, n = 4).
Figure 9.

Autophagy modulates oxidative stress in HEI-OC-1 cells after neomycin injury. (A) Five different groups of HEI-OC-1 cells were labeled using the Mito-SOX staining kit, n = 4. (B) Flow cytometry data confirmed the results in (A), n = 4. (C) Quantification of the results of flow cytometry in (B). The ROS levels were significantly increased after neomycin treatment compared with the undamaged groups. In addition, the autophagy inducer rapamycin significantly reduced the ROS levels and both the autophagy inhibitor 3-MA and knockdown of ATG5 significantly increased the ROS levels. (D) The mRNA levels of 6 genes related to redox reactions were analyzed by qRT-PCR after neomycin treatment, n = 3. (E) qRT-PCR analysis of the redox-related gene expression in the ATG5 knockdown group, 3-MA-pretreatment group, and rapamycin-pretreatment group after neomycin injury, n = 3. For qRT-PCR experiments, the values for the normal controls were set to 1. Scale bars: 50 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
To further verify our findings, we performed a qRT PCR experiment to analyze the mRNA expression of 6 redox-related genes. We found that the expression of 5 important antioxidant genes, including Sod1, Nqo1, Tmx3, Gsr, and Glrx, was significantly decreased in HEI-OC-1 cells after neomycin treatment compared with undamaged cells (Fig. 9D, P < 0.05, n = 3). In addition, treatment with rapamycin significantly upregulated the expression of 4 antioxidant genes (Sod1, Cat, Gsr, and Tmx3) while treatment with 3-MA significantly reduced the expression of 5 antioxidant genes (Sod1, Cat, Nqo1, Tmx3, and Gsr) and ATG5 knockdown reduced the expression of 4 antioxidant genes (Sod1, Cat, Nqo1, and Gsr) compared with the neomycin-only group (Fig. 9E, P < 0.05, n = 3).
Furthermore, we investigated whether the damaged and ROS-producing mitochondria after neomycin treatment could be eliminated by autophagy. According to recent studies, effective mitophagy can be assessed by detecting the reduced levels of mitochondrial proteins, including the outer mitochondrial membrane protein TOMM20 (translocase of outer mitochondrial membrane 20 homolog [yeast]) and the inner mitochondrial membrane protein COX4I1/COX IV (cytochrome c oxidase subunit 4I1), during long-term culture.46,47 Therefore, we treated HEI-OC-1 cells with 2 mM neomycin for 24 h or 48 h and performed immunofluorescence and western blot experiments to detect the protein levels of TOMM20 and COX4I1. In contrast to the loss of TOMM20 and COX4I1 in classic mitophagy,46,47 we did not observe any statistically significant change in these mitochondrial protein levels in our experiments (Fig. S13A to E, n = 3). Taken together, our results show that the mitochondria were not eliminated by autophagy (mitophagy) in HEI-OC-1 cells during neomycin treatment.
Antioxidant treatment successfully rescues the increased HC loss and apoptosis induced by autophagy inhibitor after neomycin injury
To further determine whether autophagy mediates its protective effects by suppressing ROS levels, the cochleae were treated with the antioxidant NAC, which is a reduced glutathione provider and a direct scavenger of reactive oxygen intermediates, and then exposed the cochleae to neomycin. Immunohistochemistry showed that the ROS levels were significantly reduced in both the neomycin+NAC and neomycin+NAC+3-MA groups (Fig. 10A and B, P < 0.001, n = 4), suggesting that NAC treatment could efficiently reduce the ROS level.
Figure 10.

Antioxidant treatment promotes the survival of cochlear HCs after neomycin injury. (A) Immunofluorescence staining with Mito-SOX and MYO7A in the middle turn of the cochlea after different treatments, n = 4. (B) Quantification of the numbers of Mito-SOX and MYO7A double-positive cells in (A). (C) Quantification of the MYO7A-positive cells in (A). (D) Immunofluorescence staining for cleaved-CASP3 and MYO7A in the middle turn of the cochlea after different treatments, n = 4. (E) Immunofluorescence staining for TUNEL and MYO7A in the middle turn of the cochlea after different treatments, n = 4. (F and G) Quantification of the numbers of positive cells in (Dand E). The neomycin-induced oxidative stress and apoptosis in HCs were significantly reduced after pretreatment with NAC. Scale bars: 20 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
Next, we assessed HC survival after neomycin treatment by counting the number of MYO7A-positive HCs in 150-µm lengths of the cochlea. Compared to the neomycin-only group, the HC number was sharply decreased in the neomycin+3-MA group (Fig. 10A and C, P < 0.001, n = 4), and compared with the neomycin+NAC group the HC number in the neomycin+NAC+3-MA group was not significantly decreased (Fig. 10A and C, n = 4). Immunolabeling for the apoptotic markers TUNEL and cleaved-CASP3 also showed that compared with the neomycin-only group, the numbers of cleaved-CASP3 and MYO7A, as well as TUNEL and MYO7A double-positive cells were significantly increased in the neomycin+3-MA group (Fig. 10D to G, P < 0.001, n = 4). Compared to the neomycin+NAC group, the number of double-positive cells in the neomycin+NAC+3-MA group was not significantly different (Fig. 10D to G, n = 4). These results demonstrated that NAC successfully rescued the increased HC loss and apoptosis induced by the autophagy inhibitor 3-MA after neomycin injury, and this suggests that autophagy protects against neomycin-induced HC loss by suppressing ROS accumulation.
Antioxidant treatment successfully rescues the increased cell death and apoptosis induced by autophagy inhibition in HEI-OC-1 cells
To determine whether autophagy mediates its cytoprotective effects by suppressing ROS accumulation in HEI-OC-1 cells, we pretreated the HEI-OC-1 cells with the antioxidant NAC. Immunohistochemistry and flow cytometry showed that the ROS levels were significantly reduced in all NAC-pretreated groups compared with the neomycin-only group (Fig. 11A to C, P < 0.01, n = 4). Flow cytometry results showed that compared with the neomycin-only group, both dead and apoptotic cells in the neomycin+3-MA group and neomycin+Atg5 siRNA group were significantly increased (Fig. 11D and E, P < 0.05, n = 3). Compared to the neomycin+NAC group, the ratio of dead and apoptotic cells in the neomycin+NAC+3-MA group and neomycin+NAC+Atg5 siRNA group was not significantly changed (Fig. 11D and E, n = 3). Immunolabeling with the apoptotic markers TUNEL and cleaved-CASP3 also showed that compared with the neomycin-only group significantly more cleaved-CASP3 and DAPI, as well as TUNEL and DAPI double-positive cells were observed in the neomycin+3-MA group and neomycin+Atg5 siRNA groups (Fig. 12A to C, P < 0.001, n = 4). Compared to the neomycin+NAC group, the rate of cleaved-CASP3 and DAPI, as well as TUNEL and DAPI double-positive cells in the neomycin+NAC+3-MA and neomycin+NAC+Atg5 siRNA groups was not significantly changed (Fig. 12A to C, n = 4). These results demonstrated that NAC successfully rescued the increased cell death and apoptosis induced by 3-MA treatment or ATG5 knockdown after neomycin injury, thus suggesting that autophagy prevents neomycin-induced cell death and apoptosis in HEI-OC-1 cells by suppressing ROS accumulation.
Figure 11.

Antioxidant treatment rescues the cell death and apoptosis in HEI-OC-1 cells after neomycin injury. (A) Six different groups of HEI-OC-1 cells were labeled with the Mito-SOX staining kit, n = 4. (B) Flow cytometry data confirmed the results in A, n = 4. (C) Quantification of the results of flow cytometry in B. The ROS levels were significantly decreased after pretreatment with NAC. (D) Analysis of apoptotic HEI-OC-1 cells by flow cytometry after pretreatment with NAC, n = 3. (E) Quantification of the flow cytometry data. The proportions of dead and apoptotic cells in the ATG5 knockdown groups and 3-MA-pretreatment groups significantly decreased when the increased ROS level was blocked by NAC.
Figure 12.
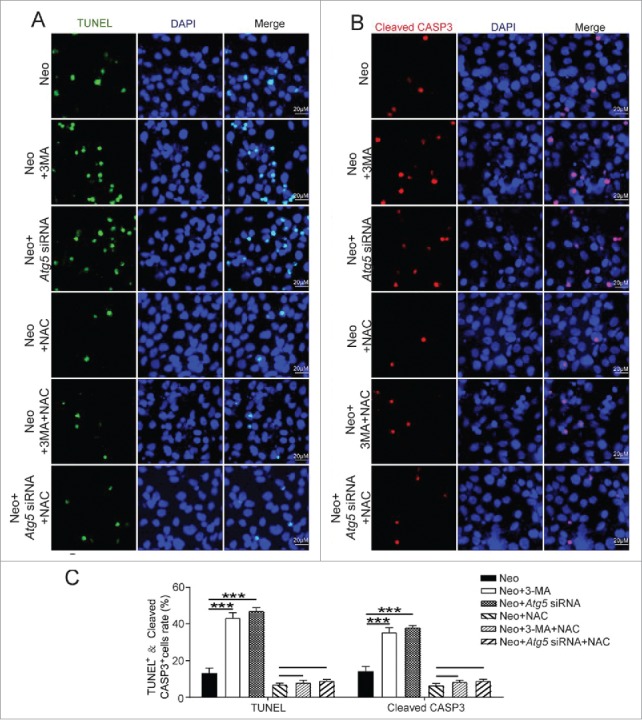
Antioxidant treatment promotes the survival of HEI-OC-1 cells after neomycin injury. (A) TUNEL and DAPI double staining and (B) cleaved-CASP3 and DAPI double staining showed the number of apoptotic HEI-OC-1 cells after the different treatments, n = 4. (C) Quantification of the numbers of TUNEL and DAPI double-positive, as well as cleaved-CASP3 and DAPI double-positive cells in (A and B), respectively. The apoptotic cells were significantly decreased after pretreatment with NAC. For flow cytometry quantification experiments, the values for the normal controls were set to 1. For all experiments, *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Aminoglycoside ototoxicity is typically associated with bilateral sensorineural hearing loss, because the sensory HCs are susceptible to aminoglycoside-induced cytotoxicity and are not able to regenerate once damaged.45,48 Although recent studies reported that mouse cochleae have very limited HC regeneration ability in neonatal ages, this limited spontaneous HC regeneration is not able to recover the hearing ability once HCs are damaged by aminoglycosides, and adult mice completely lose this HC regeneration ability.49-54 Ototoxic agents induce hearing loss by triggering apoptosis in auditory sensory HCs, and their primary target is the mitochondria.55-61 Abnormal mitochondrial respiration leads to the production of high levels of ROS, which subsequently activate multiple apoptotic pathways, such as the FAS-FASLG/FASL, NFKB/NF-κB, and TP53/TRP53/p53 pathways.62-64 The activation of autophagy has been reported to prevent cells from dying,66 but it also plays a more destructive role in many diseases.67 The relationship between autophagy and apoptosis has been extensively studied, but the interplay between them is still not well understood.68 Autophagy can be classified into nonselective bulk autophagy and selective autophagy, which degrades specific targets. Selective autophagy includes, but is not limited to, pexophagy,69 mitophagy,70 and reticulophagy.71 We know that cells have evolved protective mechanisms to scavenge ROS and maintain cellular redox homeostasis, and studies have shown that autophagy can protect cells by removing dysfunctional mitochondria and reducing the cellular ROS levels.73-75 This process is called mitophagy because it is mitochondria-selective autophagy that functions in the clearance of damaged mitochondria.72,76-78 In the current study we show that mitochondria are not eliminated by selective autophagy such as mitophagy in neomycin-treated HEI-OC-1 cells (Fig. S13), indicating that autophagy may protect the HCs by suppressing ROS accumulation and apoptosis in our model.
In this study, we fully explored the exact role of autophagy in neomycin or gentamicin-induced injury in cochlear HCs and in HC-like HEI-OC-1 cells. We found that the induction of autophagy is markedly increased after neomycin or gentamicin injury both in the HCs of explant culture and in HEI-OC-1 cells (Figs. 1 and 2). In addition, by measuring the autophagic flux (including the expression of the autophagy marker LC3B-II and the mRFP-GFP-LC3B reporter), the colocalization of the autophagy markers LC3B puncta and SQSTM1/p62, and the degradation of the SQSTM1/p62 protein, and by performing TEM imaging, and the autophagy substrate experiment, we found that neomycin injury could enhance both autophagosome synthesis and autophagosome–lysosome fusion in HEI-OC-1 cells. The increase in LC3B-II expression in the cochlea was not as great as in HEI-OC-1 cells, and we believe that this is due to the presence of many other cell types in the cochlea, such as supporting cells, that are not sensitive to neomycin. Also, with low concentrations of neomycin treatment, autophagy increased as the neomycin concentrations and exposure times increased; however, the high-concentration neomycin treatment decreased autophagy activity compared with the low-concentration neomycin treatment (Fig. 1). We hypothesized that autophagy might play a protective role against neomycin injury up to the point where the damage goes beyond the ability of the cell to rescue itself. The levels of ROS and the rates of apoptosis were significantly increased in the HCs after treatment with neomycin (Figs. 8 and 9), which indicated that ROS accumulation might contribute to the induction of autophagy.
Enhancing or blocking autophagy with autophagy activators or inhibitors can help us to understand autophagic mechanisms under oxidative stress. Rapamycin is a natural product with potent antifungal and immunosuppressive activities that has been widely reported to induce autophagy both in vivo and in vitro.79,80 The phosphatidylinositol-3-kinase inhibitor 3-MA was the first autophagy inhibitor to be identified and is the most widely used autophagy inhibitor.81 ATG5 is an important protein associated with phagophore formation, and deletion of ATG5 results in the complete absence of LC3-II.82,83 BECN1 (in yeast also known as Vps30/Atg6) is a core subunit of the phosphatidylinositol-3-kinase complex that mediates the formation of phosphatidylinositol 3-phosphate, which plays an important role in autophagosome initiation and maturation.84,85 ATG7 (ubiquitin E1-like activating enzyme) is a key protein involved in the ubiquitin-like conjugation systems of LC3 and ATG12 in the expansion of phagophore membranes.86-88 In our study, we found that the levels of LC3B-II were significantly increased and the levels of ROS were significantly decreased when HEI-OC-1 cells and cochleae were pretreated with rapamycin before neomycin exposure. In contrast, when the HEI-OC-1 cells and cochlear HCs were pretreated with the autophagy inhibitor 3-MA or knockdown of ATGs (ATG5, BECN1, and ATG7), the expression of LC3B-II was decreased and neomycin-induced ROS levels were increased (Fig. 3, Fig. 4, Fig. 8, and Fig. 9). Moreover, we found that treatment with rapamycin significantly increased the expression of 4 crucial antioxidant genes (Sod1, Cat, Gsr, and Tmx3), while autophagy inhibition markedly reduced the expression of 5 antioxidant genes (Sod1, Cat, Nqo1, Tmx3, and Gsr) in HEI-OC-1 cells after neomycin injury (Fig. 9). In cochlear explant cultures, treatment with rapamycin significantly enhanced the expression of 3 crucial antioxidant genes (Sod1, Gsr, and Glrx), while 3-MA pretreatment markedly reduced the expression of 2 antioxidant genes (Tmx3 and Nqo1) (Fig. 8). Together, these results indicated that there is a balance between oxidative stress and autophagy in sensory HCs and suggested that a suitable concentration of neomycin causes low levels of oxidative stress that activate autophagy. Autophagy attenuates the oxidative damage by regulating the activity of the oxidative stress-related signaling pathways. Downregulation of autophagy reduces the expression of antioxidant genes and leads to elevated ROS levels, which further contribute to mitochondrial dysfunction and apoptosis after neomycin injury.
Previous studies have shown that the accumulation of ROS triggers mitochondrial depolarization and initiates apoptosis.89 In this study, we showed that activation of autophagy contributes not only to reducing ROS levels, but also to promoting cell survival both in the HEI-OC-1 cell line and in cochlear HCs. We first showed that downregulation of autophagy dramatically increases cell death and apoptosis both in HEI-OC-1 cells and in cochlear HCs after neomycin injury, while rapamycin significantly reduces cell death and apoptosis after neomycin injury (Fig. 5, Figs. 6 and 7). We then performed a qRT-PCR experiment to analyze the expression of proapoptotic and antiapoptotic genes when autophagy was enhanced or reduced after neomycin injury. We found that expression of proapoptotic genes was significantly decreased and the expression of antiapoptotic genes was significantly increased in rapamycin-treated groups after neomycin injury, while the expression of proapoptotic genes was significantly increased and the expression of antiapoptotic genes was significantly decreased in the autophagy inhibition groups. These results suggested that autophagy plays an important role in regulating apoptotic pathways. Interestingly, the expression of Bcl2, which has long been known as an inhibitor of both apoptosis and autophagy,90,91 could also be regulated with an autophagy inducer or autophagy inhibitor. The result of the qRT-PCR experiment in HEI-OC-1 cells showed that Bcl2 expression could be significantly induced by the autophagy-inducer rapamycin. This phenomenon might be explained by the fact that Bcl2 plays an inhibitory role in preventing excess autophagy, which was found to be harmful to cell survival when autophagy was activated by rapamycin.
To validate whether the results after neomycin treatment can be generalized to other aminoglycosides, we treated the explant cultured cochleae and HEI-OC-1 cells with gentamicin, which is also ototoxic. We found that the expression of LC3B-II and the numbers of LC3B puncta were also increased in both cochlear HCs and HEI-OC-1 cells after gentamicin injury (Fig. S1 and S2). We also measured the effect of gentamicin on autophagic flux and found a significant increase in both autophagosomes and autolysosomes in gentamicin-treated HEI-OC-1 cells (Fig. S2). We found that the inhibition and activation of autophagy affects autophagosome formation and cell survival in both cochlear HCs and HEI-OC-1 cells following gentamicin damage (Fig. S3, S5 and S7). Our data also showed that inhibition and activation of autophagy affect apoptosis in HEI-OC-1 cells after gentamicin injury (Fig. S10). Together, these results demonstrated that our findings for neomycin can be generalized to other aminoglycosides.
As previously reported, autophagy can eliminate the increased mitochondria after damage in several tissues, including liver, muscle, and neuronal tissue.46,47,92-94 The next question is whether induction of autophagy can directly eliminate the damaged and ROS-producing mitochondria to protect HEI-OC-1 cells during neomycin treatment. In human primary cells (fibroblasts and IMR-90 cells) and immortalized cells (HeLa cells and KP-4 cells), activated mitophagy significantly enhances the clearance of damaged mitochondria.46,47 However, in this study we found that neither the outer mitochondrial membrane protein TOMM20 nor the inner mitochondrial membrane protein COX4I1 were decreased after 2 mM neomycin treatment of 24 h or 48 h compared with undamaged controls (Fig. S13), suggesting that autophagy might not directly target the damaged and ROS-producing mitochondria for degradation in HEI-OC-1 cells. Based on this, we speculated that autophagy might function downstream of mitochondrial dysfunction and protect cells against ROS accumulation and apoptosis signaling-induced cell toxicity and cell death during neomycin injury. This is likely because autophagy is induced by various stress conditions such as ROS and starvation, not just by mitochondrial dysfunction.95,96
Under normal physiological conditions, the ROS level is kept within a certain range due to the balance between ROS production and scavenging. To confirm our preceding findings showing the relation between ROS and autophagy, we used NAC, which is an ROS scavenger, to treat the cells before neomycin exposure.97 We found that NAC could successfully rescue the 3-MA or ATG5 knockdown-induced increase of apoptosis and cell death both in HEI-OC-1 cells and in cochlear HCs. These findings further demonstrated the relationship between oxidative stress and autophagy and suggested that autophagy mediates its protective effects by suppressing ROS accumulation.
In summary, this study shows that neomycin or gentamicin injury activates autophagy both in HEI-OC-1 cells and cochlear HCs. We report for the first time that there is a balance between oxidative stress and autophagy in sensory HCs after neomycin or gentamicin injury and that autophagy plays an important role in HC survival after neomycin or gentamicin injury. Our results suggest that moderate ROS levels can activate autophagy to recycle damaged cellular constituents and maintain cellular homeostasis, and the induction of autophagy after neomycin or gentamicin injury can inhibit apoptosis and protect the HCs by suppressing ROS accumulation. Our findings provide new insights into the interplay between autophagy and oxidative stress and suggest potential therapeutic strategies for the amelioration of drug-induced ototoxicity through autophagy activation.
Materials and methods
Mice and genotyping
GFP-LC3B mice (Jackson Laboratory, 027139) were obtained from the Jackson Laboratory. The genotyping for these transgenic mice with PCR was performed according to the Jackson Laboratory recommendations. All animal procedures were performed according to the protocols approved by the Animal Care and Use Committee of Southeast University and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used and to prevent their suffering.
Plasmid constructs
The NHTT-150Q-EGFP plasmids were kind gifts from Dr. Zheng Ying (Soochow University, China). NHTT-150Q-EGFP has been used to examine the cell viability of N-terminal-truncated huntingtin, which is a known autophagy substrate.41,42 The recombinant lentivirus vector mRFP-GFP-LC3B was produced by Hanbio Biotechnology (HB-AP2100001). A punctum with both GFP and mRFP fluorescence indicates an autophagosome that has not fused with a lysosome. Because the GFP signal is sensitive to the acidic environment when the autophagosome fuses with a lysosome, the mRFP punctum without GFP corresponds to an autolysosome.39,40
Cell cultures and tissue cultures
HEI-OC-1 cells were cultured at 33°C with 10% CO2 in DMEM containing 10% FBS and 100 IU/ml penicillin (Sigma-Aldrich, A0166).34 The cells were subcultured at 80% confluence using 0.25% trypsin/EDTA (Life Technologies, 25200056). When cells were cultured to a suitable density, we removed the serum and washed them with PBS 3 times. Neomycin (Sigma-Aldrich, N-18) was then added at a final concentration of 2 mM in serum-free medium to damage the HEI-OC-1 cells after preculturing in DMEM for 24 h, and 2 mM NAC (Sigma-Aldrich, A7250) was used to inhibit the ROS accumulation. Cochleae were dissected from P3 mice and cultured as previously reported,98 and neomycin (0.5 mM) was added for 24 h to damage the HCs. After neomycin was removed, the tissues were allowed to recover in serum-free medium for 24 h. Gentamicin (Sigma-Aldrich, E003632) was used at a final concentration of 0.5 mM for 24 h to damage both the HEI-OC-1 cells and the explant cultured cochleae.
Drug administration
3-MA (Sigma-Aldrich, M9281) and rapamycin (LC Laboratories, AY 22989) were used as the autophagy inhibitor and autophagy inducer, respectively. The cell line or explant cultured tissue was pretreated with 5 mM 3-MA or 0.1 µM rapamycin for 6 h, and neomycin was added for the following experiments. Baf (Sigma-Aldrich, B1793), which can block the fusion of autophagosomes with lysosomes,36,37 was used to monitor autophagosome synthesis. The cells were cotreated with Baf (100 nM) and neomycin for 24 h.
Real-time PCR
Total RNA was extracted from HEI-OC-1 cells or whole cochleae with ExTrizol Reagent (Protein Biotechnology, PR910) and reverse transcribed to cDNA by using cDNA Synthesis kits (Thermo Fisher Scientific, K1622) according to the manufacturer's protocol. Total DNA was extracted with the DNA and RNA Isolation Kit (Qiagen, Dusseldorf, 80204). The qRT-PCR was performed on an Applied Biosystems CFX96 real-time PCR system (Bio-Rad, Hercules, CA, USA) using the FastStart Universal SYBR Green (Rox) qRT-PCR Master Mix (Roche Life Science, 04913850001). Validated primers were designed for each targeted mRNA or DNA. qRT-PCR conditions were an initial denaturing step of 15 sec at 95°C followed by 40 cycles of 15 sec denaturation at 95°C, 60 sec annealing at 60°C, and 20 s extension at 72°C. The mRNA expression values were normalized to the mRNA expression of Actb and Gapdh. The results were calculated using the comparative cycle threshold (ΔΔCt) method.
Immunohistochemistry
The immunohistochemistry was performed using the Fast ImmunoCytoChemistry® Staining Kit (Protein Biotechnologies, BPICC30–1KT). Anti-cleaved-CASP3 antibody (Cell Signaling Technology, 9664S), anti-cleaved-PARP1 antibody (Cell Signaling Technology, 9544S), Mito-SOX Red (Life Technologies, M36008), anti-LC3B antibody (Santa Cruz Biotechnology, sc-16755), anti-SQSTM1/p62 antibody (Abcam, ab109012), anti-ATG5 antibody (Cell Signaling Technology, 12994), anti-BECN1 antibody (Cell Signaling Technology, 3495), anti-ATG7 antibody (Cell Signaling Technology, 8558S), anti-MYO7A antibody (Proteus Bioscience, 25–6790), anti-TOMM20 antibody (Proteintech, 11802–1-AP), and DAPI (Sigma-Aldrich, D9542) were used to analyze apoptotic cells, detect autophagy, measure ROS, stain HCs, measure mitochondrial number, and stain nuclei, respectively.
Mito-SOX was used for measuring ROS levels in live cells following the manufacturer's instructions. A TUNEL kit (Roche, 11684817910) was used to detect apoptotic cells according to the manufacturer's instructions. Briefly, the samples were incubated with 4% polyoxymethylene (Sigma-Aldrich, 158127) for 1 h and permeabilized with 0.5% Triton X-100 (Sigma-Aldrich, X100) for 1 h. They were then incubated with primary antibodies for 10 h (4°C) at a dilution of 1:400 to 1:1000. The cells were washed 3 times with PBST (1 × PBS [Wisent, 311–010-CL] with 0.1% Triton X-100) and incubated for 1 h (37°C) with secondary antibody (Abcam, ab150073, ab150075, ab150074, ab150105, ab150107) and DAPI. The cells were imaged with a confocal microscope (LSM700; Zeiss, Heidenheim, Germany).
Quantification of the immunofluorescence signals
The GFP-LC3B fluorescent puncta were quantified from the original confocal images. For cochlear tissue culture, we counted the GFP-LC3B fluorescent puncta in each HC, and we counted all of the HCs in 50 µm cochlear lengths to get an average number of GFP-LC3B fluorescent puncta per HC for each cochlea. At least 6 independent cochleae were counted. For HEI-OC-1 cell culture, we counted the GFP-LC3 fluorescent puncta in each HEI-OC-1 cell, and we counted 50 HEI-OC-1 cells to get an average number of GFP-LC3B fluorescent puncta per HEI-OC-1 cell for each culture experiment. At least 3 independent culture replicates were counted.
The immunolabeling intensity of antibodies was quantified from the original confocal images taken with the same lens under identical conditions and equal parameter settings for laser gains and photomultiplier tube gains. The samples from the different groups were processed in parallel and immunolabeled with identical solutions. The fluorescence was quantified with the ImageJ software, and the fluorescence intensity was then normalized to DAPI fluorescence intensity to further improve the accuracy of the measurement. The final relative fluorescence was quantified by normalizing the ratio of the average fluorescence of the drug-treated groups to the average fluorescence of the untreated groups.
Cell number analysis
After incubating HEI-OC-1 cells for 24 h in 96-well plates at 2,000 cells/well with 3 replicates, different drugs were added (controls received a similar volume of DMEM). Cell numbers were counted with the CCK-8 Cell Counting Kit (Protein Biotechnology, CC201) at different time points after incubation. After whole organ cultured tissues were treated with different drugs, the immunostained cells were quantified per 150 µm length of the cochlea in all 3 turns. The numbers of positive cells were counted in equal lengths from the apical to the basal turns of the cochlea.
Electron microscopy
HEI-OC-1 cells and cochleae were collected and immediately fixed in 2.5% glutaraldehyde (Sigma-Aldrich, G5882) for 24 h and then in 1% osmic acid (Sigma-Aldrich, O5500) for 1 to 2 h, dehydrated with acetone (Sinopharm Chemical Reagent, u1006801), and embedded in araldite CY 212 (TAAB, E009). The ultrathin sections were stained with alcoholic uranyl acetate (Polysciences, 6159–44–0) and alkaline lead citrate (Sigma-Aldrich, 15326), washed gently with distilled water, and observed with a JEM 1230 transmission electron microscope (JEOL Ltd, Tokyo, Japan).
Flow cytometry
Mito-SOX was used to analyze ROS production. The HEI-OC-1 cells were treated with different drugs then trypsinized and collected by centrifugation at 60 g for 5 min and resuspended in solution containing Mito-SOX for 10 min followed by washing with PBS and analysis by flow cytometry (FACSCanto, BD, San Jose, CA, USA). If required, the cells were pretreated with NAC to lower the ROS level.
An ANXA5 (Annexin V) kit (BD Biosciences, 556547) was used for apoptosis analysis. The HEI-OC-1 cells were treated with different drugs, trypsinized, collected by centrifugation at 60 g for 5 min, and then washed twice with PBS and resuspended in 1 × binding buffer at a concentration of 1 × 106 cells/ml. ANXA5-FITC and propidium iodide were added and gently mixed with the cells and incubated for 10 to 30 min at room temperature in the dark. The cells were analyzed by flow cytometry as soon as possible, and all experiments were repeated at least 3 times.
Western blot
The HEI-OC-1 cell line and tissues from the cochleae were lysed with ice-cold RIPA Lysis Buffer (Protein Biotechnology, PP109) plus Phosphatase Inhibitor Cocktails (Roche, 04693132001). Protein concentrations were measured using a BCA Protein Quantification Kit (Protein Biotechnology, PP202) according to the manufacturer's instructions using GAPDH as the reference protein. LC3B-II was monitored using anti-LC3B rabbit polyclonal antibody (Santa Cruz Biotechnology, sc-28266), and GAPDH was measured using a mouse monoclonal antibody (Abcam, ab8245). Peroxidase-conjugated goat anti-rabbit (or anti-mouse) immunoglobulin G (Abcam, ab6789, ab6721) was used as the secondary antibody. The proteins were bound to polyvinylidene fluoride membranes and detected using a SuperSignal West Dura chemiluminescent substrate kit (Thermo Scientific, 34075) according to the manufacturer's instructions. Semiquantification of the western blot result was performed using ImageJ software to measure the intensities of the bands. The band densities were normalized to background, and then the relative optical density ratio was calculated by comparison to the housekeeping gene GAPDH. Each experiment was repeated more than 3 times.
Statistical analysis
All of the data are shown as the mean ± S.D, and all experiments were repeated at least 3 times. Statistical analyses were conducted using Microsoft Excel and GraphPad Prism6 software. For the whole organ explant culture experiments, n represents the number of independent cochlea, and for all HEI-OC-1 cell culture experiments, n represents the number of independent culture replicates. Two-tailed, unpaired Student t tests were used to determine statistical significance when comparing 2 groups, and one-way ANOVA followed by a Dunnett multiple comparisons test was used when comparing more than 2 groups. A value of P < 0.05 was considered to be statistically significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding Statement
This work was supported by grants from the National Key Research Development Program of China (2017YFA0103900, 2015CB965000, 2017YFA0103903), the National Natural Science Foundation of China (Nos. 81622013, 81771019, 81771013, 81570913, 81470692, 81371094, 81500790, 81570921, 31500852, 31501194, 81670938), the Jiangsu Province Natural Science Foundation (BK20150022, BK20140620, BK20150598, BK20160125), the Science and Technology Commission of Shanghai Municipality (15pj1401000), the Yingdong Huo Education Foundation, the Boehringer Ingelheim Pharma GmbH, the Fundamental Research Funds for the Central Universities, and the Project of Invigorating Health Care through Science Technology and Education.
Abbreviations
- ATG
autophagy-related
- Baf
bafilomycin A1
- Bax
Bcl2-associated X protein
- Bcl2
B-cell leukemia/lymphoma 2
- BECN1
Beclin 1, autophagy related
- CASP
Casp caspase; Cat, catalase
- COX4I1
cytochrome c oxidase subunit 4I1
- Glrx
glutaredoxin
- Gsr
glutathione reductase
- HCs
hair cells
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MYO7A
myosin VIIA
- NAC
N-acetylcysteine
- NHTT
truncated N-terminal huntingtin protein
- Nqo1
NAD(P)H dehydrogenase, quinone 1
- PARP1
poly (ADP-ribose) polymerase 1
- qRT PCR
quantitative real-time PCR
- ROS
reactive oxygen species
- Sod1
superoxide dismutase 1
- TEM
transmission electron microscope
- Tmx3
thioredoxin-related transmembrane protein 3
- TOMM20
translocase of outer mitochondrial membrane 20 homolog (yeast)
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- 3-MA
3-methyladenine
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Sugahara K, Rubel EW, Cunningham LL. JNK signaling in neomycin-induced vestibular hair cell death. Hearing Res. 2006;221:128-35. doi: 10.1016/j.heares.2006.08.009. PMID:17005344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tabuchi K, Nishimura B, Nakamagoe M, Hayashi K, Nakayama M, Hara A. Ototoxicity: mechanisms of cochlear impairment and its prevention. Curr Medicinal Chem. 2011;18:4866-71. doi: 10.2174/092986711797535254. PMID:21919841 [DOI] [PubMed] [Google Scholar]
- [3].Niwa K, Matsunobu T, Kurioka T, Kamide D, Tamura A, Tadokoro S, Satoh Y, Shiotani A. The beneficial effect of Hangesha-shin-to (TJ-014) in gentamicin-induced hair cell loss in the rat cochlea. Auris, Nasus, Larynx. 2016;43(5):507-13. doi: 10.1016/j.anl.2015.12.012. PMID:26797463 [DOI] [PubMed] [Google Scholar]
- [4].Choung YH, Taura A, Pak K, Choi SJ, Masuda M, Ryan AF. Generation of highly-reactive oxygen species is closely related to hair cell damage in rat organ of Corti treated with gentamicin. Neuroscience. 2009;161:214-26. doi: 10.1016/j.neuroscience.2009.02.085. PMID:19318119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Navarro-Yepes J, Burns M, Anandhan A, Khalimonchuk O, del Razo LM, Quintanilla-Vega B, Pappa A, Panayiotidis MI, Franco R. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxidants Redox Signal. 2014;21:66-85. doi: 10.1089/ars.2014.5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Filomeni G, De ZD, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differentiation. 2014;22:377-88. doi: 10.1038/cdd.2014.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Franco R, Cidlowski JA. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differentiation. 2009;16:1303-14. doi: 10.1038/cdd.2009.107 [DOI] [PubMed] [Google Scholar]
- [8].Vernon PJ, Tang D. Eat-me: autophagy, phagocytosis, and reactive oxygen species signaling. Antioxidants Redox Signal 2013;18:677-91. doi: 10.1089/ars.2012.4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yuan H, Wang X, Hill K, Chen J, Lemasters J, Yang SM, Sha SH. Autophagy attenuates noise-induced hearing loss by reducing oxidative stress. Antioxidants Redox Signal. 2015;22:1308-24. doi: 10.1089/ars.2014.6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kobayashi S. Choose delicately and reuse Adequately: the newly revealed process of autophagy. Biol Pharmaceutical Bulletin. 2015;38:1098-103. doi: 10.1248/bpb.b15-00096 [DOI] [PubMed] [Google Scholar]
- [11].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Burman C, Ktistakis NT. Autophagosome formation in mammalian cells. Seminars Immunopathol. 2010;32:397-413. doi: 10.1007/s00281-010-0222-z. PMID:20740284 [DOI] [PubMed] [Google Scholar]
- [13].Cesen MH, Pegan K, Spes A, Turk B. Lysosomal pathways to cell death and their therapeutic applications. Exp Cell Res 2012;318:1245-51. doi: 10.1016/j.yexcr.2012.03.005. PMID:22465226 [DOI] [PubMed] [Google Scholar]
- [14].Lee HK, Marzella L. Regulation of intracellular protein degradation with special reference to lysosomes: role in cell physiology and pathology. Int Rev Exp Pathol. 1994;35:39-147. PMID:7860191 [PubMed] [Google Scholar]
- [15].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al.. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2003;22:4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542-5. doi: 10.4161/auto.4600. PMID:17611390 [DOI] [PubMed] [Google Scholar]
- [17].Meijer AJ, Codogno P. Autophagy: regulation and role in disease. Critical Rev Clin Lab Sci. 2009;46:210-40. doi: 10.1080/10408360903044068 [DOI] [PubMed] [Google Scholar]
- [18].Levine B, Kroemer G. Autophagy in the Pathogenesis of disease. Cell. 2008;132:27-42. doi: 10.1016/j.cell.2007.12.018. PMID:18191218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010;140:313-26. doi: 10.1016/j.cell.2010.01.028. PMID:20144757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Clarke PGH. Developmental cell death: morphological diversity and multiple mechanisms. Anatomy Embryol. 1990;181:195-213. doi: 10.1007/BF00174615 [DOI] [PubMed] [Google Scholar]
- [21].Yu L, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500-2. doi: 10.1126/science.1096645. PMID:15131264 [DOI] [PubMed] [Google Scholar]
- [22].Ryter SW, Mizumura K, Choi AM. The impact of autophagy on cell death modalities. Int J Cell Biol. 2014;2014:502676–. doi: 10.1155/2014/502676. PMID:24639873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Czaja MJ, Ding WX, Donohue TM, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, James Ou JH, et al.. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131-58. doi: 10.4161/auto.25063. PMID:23774882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen W, Sun Y, Liu K, Sun X. Autophagy:a double-edged sword for neuronal survival after cerebral ischemia. Neural Regeneration Res. 2014;9:1210-6. doi: 10.4103/1673-5374.135329. PMID:25206784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69-79. doi: 10.1038/cr.2013.161. PMID:24323045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell 2008;15:344-57. doi: 10.1016/j.devcel.2008.08.012. PMID:18804433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004-10. doi: 10.1038/nrm2529. PMID:18971948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy 2011;7:457-65. doi: 10.4161/auto.7.5.14226. PMID:21150268 [DOI] [PubMed] [Google Scholar]
- [29].Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al.. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025-40. doi: 10.1128/MCB.25.3.1025-1040.2005. PMID:15657430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shen HM, Codogno P. Autophagy is a survival force via suppression of necrotic cell death. Exp Cell Res. 2012;318:1304-8. doi: 10.1016/j.yexcr.2012.02.006. PMID:22366289 [DOI] [PubMed] [Google Scholar]
- [31].Rodríguezmuela N, Germain F, Mariño G, Fitze PS, Boya P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differentiation. 2012;19:162-9. doi: 10.1038/cdd.2011.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kalinec GM, Webster P, Lim DJ, Kalinec F. A cochlear cell line as an in vitro system for drug ototoxicity screening. Audio Neurotol. 2003;8:177-89. doi: 10.1159/000071059. PMID:12811000 [DOI] [PubMed] [Google Scholar]
- [33].Yang YP, Hu LF, Zheng HF, Mao CJ, Hu WD, Xiong KP, Wang F, Liu CF. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacologica Sinica. 2013;34:625-35. doi: 10.1038/aps.2013.5. PMID:23524572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kalinec GM, Webster P, Lim DJ, Kalinec F. A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol Neurotol. 2003;8:177-89. doi: 10.1159/000071059 [DOI] [PubMed] [Google Scholar]
- [35].Jeong HJ, Hong SH, Park RK, Shin T, An NH, Kim HM. Hypoxia-induced IL-6 production is associated with activation of MAP kinase, HIF-1, and NF-kappaB on HEI-OC1 cells. Hearing Res. 2005;207:59-67. doi: 10.1016/j.heares.2005.04.003. PMID:15913932 [DOI] [PubMed] [Google Scholar]
- [36].Shacka JJ, Klocke BJ, Shibata M, Uchiyama Y, Datta G, Schmidt RE, Roth KA. Bafilomycin A1 inhibits Chloroquine-induced death of Cerebellar Granule neurons. Mol Pharmacol. 2006;69:1125-36. doi: 10.1124/mol.105.018408. PMID:16391239 [DOI] [PubMed] [Google Scholar]
- [37].Schalkwyk DAV, Chan XWA, Misiano P, Gagliardi S, Farina C, Saliba KJ. Inhibition of Plasmodium falciparum pH regulation by small molecule indole derivatives results in rapid parasite death. Biochem Pharmacol. 2010;79:1291-9. doi: 10.1016/j.bcp.2009.12.025. PMID:20067768 [DOI] [PubMed] [Google Scholar]
- [38].Zhou L, Wang H, Chen D, Gao F, Ying Z, Wang G. p62/Sequestosome 1 Regulates Aggresome formation of Pathogenic Ataxin-3 with expanded Polyglutamine. Int J Mol Sci. 2014;15:14997-5010. doi: 10.3390/ijms150914997. PMID:25158237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452-60. doi: 10.4161/auto.4451. PMID:17534139 [DOI] [PubMed] [Google Scholar]
- [40].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445-544. doi: 10.4161/auto.19496. PMID:22966490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805-11. doi: 10.1038/nn.2575. PMID:20581817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Qin X, Wang H, Hao Z, Cheng F, Hu Q, Feng G, et al.. TDP-43 loss of function increases TFEB activity and blocks autophagosome–lysosome fusion. Embo J. 2016;35:121-142. doi: 10.15252/embj.201591998. PMID:26702100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Matsui JI, Ogilvie JM, Warchol ME. Inhibition of caspases prevents ototoxic and ongoing hair cell death. J Neurosci Official J SocietyNeurosci. 2002;22:1218-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].He Y, Yu H, Cai C, Shan S, Chai R, Li H. Inhibition of H3K4me2 Demethylation Protects Auditory Hair Cells from Neomycin-Induced Apoptosis. Mol Neurobiol. 2014;52:1-10. doi: 10.3389/fncel.2014.00248. PMID:25092125 [DOI] [PubMed] [Google Scholar]
- [45].Sun S, Sun M, Zhang Y, Cheng C, Waqas M, Yu H, He Y, Xu B, Wang L, Wang J, et al.. In vivo overexpression of X-linked inhibitor of apoptosis protein protects against neomycin-induced hair cell loss in the apical turn of the cochlea during the ototoxic-sensitive period. Frontiers Cell Neurosci. 2014;8:248. PMID:25278835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309-14. doi: 10.1038/nature14893. PMID:26266977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Correia-Melo C, Ichim G, Tait SWG, Passos JF. Depletion of mitochondria in mammalian cells through enforced mitophagy. Nat Protocols. 2017;12:183-194. doi: 10.1038/nprot.2016.159. PMID:28005069 [DOI] [PubMed] [Google Scholar]
- [48].Nadol JB., Jr Hearing loss. N Engl J Med. 1993;329:1092-102. doi: 10.1056/NEJM199310073291507. PMID:8371732 [DOI] [PubMed] [Google Scholar]
- [49].Waqas M, Zhang S, He Z, Tang M, Chai R. Role of Wnt and Notch signaling in regulating hair cell regeneration in the cochlea. Frontiers Med. 2016;10(3):237-49. doi: 10.1007/s11684-016-0464-9. PMID:27527363 [DOI] [PubMed] [Google Scholar]
- [50].Ni W, Zeng S, Li W, Chen Y, Zhang S, Tang M, Sun S, Chai R, Li H. Wnt activation followed by Notch inhibition promotes mitotic hair cell regeneration in the postnatal mouse cochlea. Oncotarget. 2016;7(41):66754-68. doi: 10.18632/oncotarget.11479. PMID:27564256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wu J, Li W, Chen L, Chen Y, Cheng C, Sun S, Tang M, Chai R, Li H. Co-regulation of the Notch and Wnt signaling pathways promotes supporting cell proliferation and hair cell regeneration in mouse utricles. Scientific Reports. 2016;6:29418. doi: 10.1038/srep29418. PMID:27435629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li W, You D, Chen Y, Chai R, Li H. Regeneration of hair cells in the mammalian vestibular system. Frontiers Med. 2016; 10:143. doi: 10.1007/s11684-016-0451-1. PMID:27189205 [DOI] [PubMed] [Google Scholar]
- [53].Waqas M, Guo L, Zhang S, Chen Y, Zhang X, Wang L, Tang M, Shi H, Bird PI, Li H, et al.. Characterization of Lgr5+ progenitor cell transcriptomes in the apical and basal turns of the mouse cochlea. Oncotarget. 2014;7:41123-41. doi: 10.18632/oncotarget.8636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lu X, Shan S, Qi J, Li W, Liu L, Zhang Y, et al.. Bmi1 regulates the Proliferation of Cochlear supporting cells via the canonical wnt signaling pathway. Mol Neurobiol. 2017;54:1326-1339. PMID:26843109 [DOI] [PubMed] [Google Scholar]
- [55].Liu L, Chen Y, Qi J, Zhang Y, He Y, Ni W, Li W, Zhang S, Sun S, Taketo MM, et al.. Wnt activation protects against neomycin-induced hair cell damage in the mouse cochlea. Cell Death Dis. 2016;7:e2136. doi: 10.1038/cddis.2016.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Guan M, Fang Q, He Z, Li Y, Qian F, Qian X, et al.. Inhibition of ARC decreases the survival of HEI-OC-1 cells after neomycin damage in vitro. Oncotarget. 2016;7:66647-66659. doi: 10.18632/oncotarget.11336. PMID:27556499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yu X, Liu W, Fan Z, Qian F, Zhang D, Han Y, Xu L, Sun G, Qi J, Zhang S, et al.. c-Myb knockdown increases the neomycin-induced damage to hair-cell-like HEI-OC1 cells in vitro. Scientific Reports. 2017;7:41094. doi: 10.1038/srep41094. PMID:28112219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Forge A, Schacht J. Aminoglycoside Antibiotics. Audiol Neurotol. 2000;5:3-22. doi: 10.1159/000013861. PMID:10686428 [DOI] [PubMed] [Google Scholar]
- [59].Huang T, Cheng AG, Stupak H, Liu W, Kim A, Staecker H, Lefebvre PP, Malgrange B, Kopke R, Moonen G, et al.. Oxidative stress-induced apoptosis of cochlear sensory cells: otoprotective strategies. Int J Dev Neurosci. 2000;18:259-70. doi: 10.1016/S0736-5748(99)00094-5. PMID:10715580 [DOI] [PubMed] [Google Scholar]
- [60].Wu WJ, Sha SH, Schacht J. Recent advances in understanding aminoglycoside ototoxicity and its prevention. Audiol Neurotol. 2002;7:171-4. doi: 10.1159/000058305. PMID:12053140 [DOI] [PubMed] [Google Scholar]
- [61].He Z, Sun S, Waqas M, Zhang X, Qian F, Cheng C, Zhang M, Zhang S, Wang Y, Tang M, et al.. Reduced TRMU expression increases the sensitivity of hair-cell-like HEI-OC-1 cells to neomycin damage in vitro. Scientific Reports. 2016;6:29621. doi: 10.1038/srep29621. PMID:27405449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wang X, Martindale JL, Liu Y, Holbrook NJ. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem J. 1998;333 (Pt 2):291-300. doi: 10.1042/bj3330291. PMID:9657968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kannan K, Jain SK. Oxidative stress and apoptosis. Pathophysiol Official J Int Society Pathophysiol. 2000;7:153-63. PMID:10996508 [DOI] [PubMed] [Google Scholar]
- [64].Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radical Biol Med. 2000;29:323-33. doi: 10.1016/S0891-5849(00)00302-6 [DOI] [PubMed] [Google Scholar]
- [65].Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463-77. doi: 10.1016/S1534-5807(04)00099-1. PMID:15068787 [DOI] [PubMed] [Google Scholar]
- [66].Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia–ischemia induced brain injury. Neurobiol Dis. 2008;32:329-39. doi: 10.1016/j.nbd.2008.07.022. PMID:18760364 [DOI] [PubMed] [Google Scholar]
- [67].Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, et al.. Inhibition of Autophagy Prevents Hippocampal Pyramidal Neuron Death after Hypoxic-Ischemic Injury. Am J Pathol. 2008;172:454-69. doi: 10.2353/ajpath.2008.070876. PMID:18187572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Moreau K, Luo S, Rubinsztein DC. Cytoprotective roles for autophagy. Curr Opin Cell Biol. 2010;22:206-11. doi: 10.1016/j.ceb.2009.12.002. PMID:20045304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Dunn WA, Cregg JM, Kiel JAKW, Klei IJVD, Oku M, Sakai Y, Sibirny AA, Stasyk OV, Veenhuis M. Pexophagy: the selective autophagy of peroxisomes. Autophagy 2005;1:75-83. doi: 10.4161/auto.1.2.1737. PMID:16874024 [DOI] [PubMed] [Google Scholar]
- [70].Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245-53. doi: 10.1016/j.abb.2007.03.034. PMID:17475204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Schrader B. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. Plos Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. PMID:17132049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kissová I, Plamondon LT, Brisson L, Priault M, Renouf V, Schaeffer J, Camougrand N, Manon S. Evaluation of the roles of apoptosis, autophagy, and mitophagy in the loss of plating efficiency induced by Bax expression in yeast. J Biol Chem. 2006;281:36187-97. doi: 10.1074/jbc.M607444200. PMID:16990272 [DOI] [PubMed] [Google Scholar]
- [73].Bellot G, Garciamedina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol Cell Biol. 2009;29:2570-81. doi: 10.1128/MCB.00166-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27:6229-42. doi: 10.1128/MCB.02246-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhang H, Boschmarce M, Shimoda LA, Tan YS, Jin HB, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892-903. doi: 10.1074/jbc.M800102200. PMID:18281291 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [76].Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829-40. doi: 10.1091/mbc.E04-06-0477. PMID:15331765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Yan X, Contento AL, Nguyen PQ, Bassham DC. Degradation of oxidized proteins by autophagy during oxidative stress in Arabidopsis. Plant Physiol. 2007;143:291-9. PMID:17098847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhang YL, Cao YJ, Zhang X, Liu HH, Tong T, Xiao GD, Yang YP, Liu CF. The autophagy-lysosome pathway: a novel mechanism involved in the processing of oxidized LDL in human vascular endothelial cells. Biochem Biophys Res Commun. 2010;394:377-82. doi: 10.1016/j.bbrc.2010.03.026 [DOI] [PubMed] [Google Scholar]
- [79].Tanemura M, Ohmura Y, Deguchi T, Machida T, Tsukamoto R, Wada H, Kobayashi S, Marubashi S, Eguchi H, Ito T, Nagano H, Mori M, Doki Y.. Rapamycin Causes Upregulation of Autophagy and Impairs Islets Function Both. In Vitro and In Vivo. Am J Transplantation Official J Am Society Transplantation Am Society Transplant Surgeons. 2012;12:102-14. doi: 10.1111/j.1600-6143.2011.03771.x [DOI] [PubMed] [Google Scholar]
- [80].Hartford CM, Ratain MJ. Rapamycin: something old, something new, sometimes borrowed and now renewed. Clinical Pharmacol Therapeutics. 2007;82:381-8. doi: 10.1038/sj.clpt.6100317 [DOI] [PubMed] [Google Scholar]
- [81].Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889-92. doi: 10.1073/pnas.79.6.1889. PMID:6952238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of Autophagosome Formation Using Apg5-Deficient Mouse Embryonic Stem Cells. J Cell Biol. 2001;152:657-68. doi: 10.1083/jcb.152.4.657. PMID:11266458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circulation Res. 2007;100:914-22. doi: 10.1161/01.RES.0000261924.76669.36. PMID:17332429 [DOI] [PubMed] [Google Scholar]
- [84].Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis – an effect rescued by Bcl-xL. Cell Death Differentiation. 2010;17:268-77. doi: 10.1038/cdd.2009.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Sinha S, Colbert CL, Becker N, Wei Y, Levine B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 2008;4:989-97. doi: 10.4161/auto.6803. PMID:18797192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Cell Dev Biol. 2011;27:107-32. doi: 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- [87].Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458-67. doi: 10.1038/nrm2708. PMID:19491929 [DOI] [PubMed] [Google Scholar]
- [88].Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015;25:354-63. doi: 10.1016/j.tcb.2015.02.002. PMID:25759175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mei H, Sun S, Bai Y, Chen Y, Chai R, Li H. Reduced mtDNA copy number increases the sensitivity of tumor cells to chemotherapeutic drugs. Cell Death Dis. 2015;6:e1710. doi: 10.1038/cddis.2015.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Pattingre S, Tassa A, Qu X, Garuti R, Xiao HL, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927-39. doi: 10.1016/j.cell.2005.07.002. PMID:16179260 [DOI] [PubMed] [Google Scholar]
- [91].Zhou L, Wang H, Ren H, Hu Q, Ying Z, Wang G. Bcl-2 Decreases the Affinity of SQSTM1/p62 to Poly-Ubiquitin Chains and suppresses the aggregation of misfolded protein in neurodegenerative disease. Mol Neurobiol. 2015;52:1-10. doi: 10.1007/s12035-014-8908-1. PMID:25092125 [DOI] [PubMed] [Google Scholar]
- [92].Ueno T, Komatsu M. Autophagy in the liver: functions in health and disease. Nat Rev Gastroenterol Hepatol. 2017;14(3):170-84. doi: 10.1038/nrgastro.2016.185 [DOI] [PubMed] [Google Scholar]
- [93].White Z, Terrill J, White RB, Mcmahon C, Sheard P, Grounds MD, Shavlakadze T. Voluntary resistance wheel exercise from mid-life prevents sarcopenia and increases markers of mitochondrial function and autophagy in muscles of old male and female C57BL/6J mice. Skeletal Muscle. 2016;6:45. doi: 10.1186/s13395-016-0117-3. PMID:27964759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Moon JH, Lee JH, Lee YJ, Park SY. Autophagy flux induced by ginsenoside-Rg3 attenuates human prion protein-mediated neurotoxicity and mitochondrial dysfunction. Oncotarget. 2016;7(52):85697-708. PMID:27911875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Li X, Fan Q, Xu W, Xue Z, Wang L. Inhibition of autophagy increased AGE|[sol]|ROS-mediated apoptosis in mesangial cells. Cell Death Dis. 2016;7:e2445. doi: 10.1038/cddis.2016.322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, Ren H, Chen D, Han J, Ying Z, et al.. TDP‐43 loss of function increases TFEBSS activity and blocks autophagosome–lysosome fusion. Embo J. 2015;35:121-42. doi: 10.15252/embj.201591998. PMID:26702100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Small DM, Coombes JS, Bennett N, Johnson DW, Gobe GC. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology. 2012;17:311-21. doi: 10.1111/j.1440-1797.2012.01572.x. PMID:22288610 [DOI] [PubMed] [Google Scholar]
- [98].Chen Y, Yu H, Zhang Y, Li W, Lu N, Ni W, He Y, Li J, Sun S, Wang Z, et al.. Cotransfection of Pax2 and Math1 promote in situ cochlear hair cell regeneration after neomycin insult. Scientific Reports. 2013;3:2996. doi: 10.1038/srep02996. PMID:24141260 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.