

Glutamyl(E)-prolyl(P) tRNA Synthetase (EPRS) is an exception to the “one amino acid–one aminoacyl-tRNA synthetase (AARS)” rule. Class I ERS and class II PRS are fused into a single polypeptide that aminoacylates Glu and Pro, respectively, to cognate tRNAs for protein synthesis.1 They were joined via a non-catalytic linker in a unicellular animal-like organism ∼800 Mya, and have remained fused with limited exceptions. EPRS is a constituent of the cytosolic multi-AARS complex (MSC) containing 9 synthetases and 3 non-synthetase proteins in mice and humans. The linker region is central to several EPRS noncanonical activities beyond aminoacylation. For example, interferon-γ (IFN-γ) induces phosphorylation of Ser999 in the linker, inducing translation-inhibition of inflammation-related mRNAs by the GAIT system in myeloid cells.1 Also, Ser990 phosphorylation in the linker directs antiviral defense upon infection.2 Finally, we have recently reported that insulin-stimulated Ser999 phosphorylation in adipocytes influences adiposity and aging.3 In all cases, phosphorylation induces EPRS release from the parental MSC for execution of its noncanonical activities, supporting the quarter century-old hypothesis that AARS phosphorylation might induce extra-aminoacylation activities.4
Insulin in adipocytes and IFN-γ in myeloid cells stimulate mammalian target of rapamycin complex 1 (mTORC1) to activate ribosomal protein S6 kinase 1 (S6K1) that in turn phosphorylates EPRS at Ser999 (Fig. 1, left).3 Utilizing knock-in mice bearing phospho-deficient (Ser999-to-Ala) and phospho-mimetic (Ser999-to-Asp) mutations in Eprs, we observed, unexpectedly, that EPRS links mTOR signaling and obesity-related metabolic processes. Homozygous EprsA/A mice on standard rodent diet displayed several loss-of-function phenotypes observed in S6K1–/– and adipocyte-specific raptor (mTORC1-constituent)-deficient mice including low body weight, reduced fat mass, and increased life-span (Fig. 1, right).5,6 The body weight phenotype of S6K1–/– was substantially rescued by introduction of the phosphomimetic EprsD/D allele, providing in vivo, epistatic evidence for pathway linearity. Investigation of the underlying molecular mechanism revealed that insulin induces EPRS phosphorylation and consequent release from the MSC in cultured adipocytes. Freed EPRS binds fatty acid transport protein 1 (FATP1) and conveys it to the plasma membrane where it facilitates long-chain fatty acid (LCFA) uptake for increased triglyceride synthesis (Fig. 1, left). This newly discovered pathway complements other adipogenic, mTOR-dependent pathways, e.g., SREBP-1-induced lipid synthesis.
Figure 1.
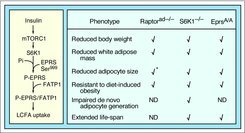
The insulin-stimulated pathway of EPRS Ser999 phosphorylation and long-chain fatty acid (LCFA) uptake in adipocytes (left). Phenotypic comparison of EprsA/A, S6K1–/–, and adipose-specific raptor knockout (Raptorad–/–) mice (right). Asterisk indicates phenotype observed in mice fed high-fat diet; ND, not determined.
In mice, the tissue-specific metabolic phenotype resulting from mutation of a single amino acid in an AARS was not anticipated. Our results extended the mTORC1-S6K1 signaling axis that influences adiposity and aging to include 2 new constituents – EPRS and FATP1. However, the relationship between the phenotypes of EprsA/A and FATP1–/– mice is equivocal due to conflicting reports regarding adiposity in the latter.3 Moreover, our studies do not illuminate the somewhat contentious mechanism of action of FATP1 – early studies suggested it facilitated inward LCFA transport, but later studies suggested intracellular metabolic trapping driven by a coenzyme A ligase activity inherent in FATP1, or in a bound enzyme. These results raise 2 important conundrums: What is the origin of the observed tissue specificity of EPRS-mediated LCFA influx as the insulin-activated TOR pathway is operative in non-adipose tissues? Equally surprising is the Ser999 phospho-site, which is completely unrelated to the consensus S6K1 recognition sequence, RXRXXS/T. A clue to both puzzles might be found by elucidating the role of cyclin-dependent kinase 5 (Cdk5), which is found primarily in neuronal cells, but also in adipocytes and monocytes, and is essential for EPRS Ser999 phosphorylation in monocytes.3
The EprsA/A mice do not precisely phenocopy S6K1–/– mice with respect to the earlier onset of body weight reduction in the latter. Moreover, breeding the S6K1–/– with mice bearing an EprsD/D mutation incompletely normalized body weight. These observations suggest as-yet unidentified targets of S6K1 might contribute to the metabolic phenotype. The mTORC1-S6K1 axis regulates a myriad of downstream pathways involved in functions that are most likely EPRS-independent. For example, the relatively early appearance of low body weight in S6K1–/– mice might be due to reduced H2B phosphorylation which impairs mesenchymal stem cell differentiation in the adipocyte lineage. Likewise, FATP1 is not likely to be the sole, or even principal, metabolic target of phosphorylated EPRS, and we reported that EPRSA/A mice exhibit elevated lipolysis and fatty acid oxidation.1 Interestingly, EprsD/D mice, like mice with constitutive activation of S6K1, do not exhibit increased adiposity or body weight compared with wild-type. These opposing effects upon activation or inactivation of a specific gene are consistent with a “switch regulation pathway” (www.its.caltech.edu/∼bi190/SG5.pdf), i.e., inactivation of one pathway arm is sufficient to inhibit weight gain, but activation of multiple pathway arms are required to drive weight gain.
Median life-span of mice in the laboratory and humans in the developed world is ∼750–800 d and ∼75–80 years, respectively. Thus, the 118-day extension observed in EPRSA/A mice is comparable to an increase of ∼12 y in humans. Increased longevity in EprsA/A mice is consistent with mice subjected to dietary restriction and defects in related pathways, e.g., rapamycin-treated and Irs1–/– mice.5 Inhibition of the mTORC1-S6K1 axis has been proposed to extend life by a mechanism independent of of global translation-inhibition, possibly involving translation of a subset of mRNAs or by translation-independent mechanisms. The latter is consistent with our results depending on EPRS phosphorylation in adipocytes. Adult EprsA/A mice are apparently healthy as indicated by improved glucose- and insulin-tolerance, and the absence of hepatosteatosis. However, detailed information about health-span, i.e., disease-free physiologic state, and cognitive capabilities has not been determined. Activation of EPRS by 2 classes of organismal stress, i.e., inflammatory and metabolic, suggests a central, pluripotent role in stress response. A function of LeuRS in regulating amino acid-responsiveness of mTORC1 suggests a general metabolic role for the AARS family.7
Noncanonical functions unrelated to protein synthesis have been reported for many AARS's. In most cases, appended domains not present in bacterial homologues drive the function. EPRS is not an exception as several of its noncanonical functions, including binding of FATP1 and the inflammation-repressive GAIT complex, are directed by the linker joining the catalytic synthetase domains. Indeed, the 3 RNA- and protein-binding WHEP domains, and the 3 stimulus-dependent phosphorylation sites, all reside within the linker. The anti-viral activity is the sole exception, in which function is driven by the N-terminus GST-like domain, but only after phosphorylation in the linker region.2 Because the linker domain in EPRS, and its phosphorylation, is not essential for protein synthesis, it represents an attractive therapeutic target for anti-obesity and -aging pharmaceuticals.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Arif A, Jia J, Mukhopadhyay R, Willard B, Kinter M, Fox PL. Two-site phosphorylation of EPRS coordinates multimodal regulation of noncanonical translational control activity. Mol Cell 2009; 35:164-80; PMID:19647514; https://doi.org/10.1016/j.molcel.2009.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lee EY, Lee HC, Kim HK, Jang SY, Park SJ, Kim YH, Kim JH, Hwang J, Kim JH, Kim TH, Arif A, Kim SY, Choi YK, Lee C, Lee CH, Jung JU, Fox PL, Kim S, Lee JS, Kim MH. Infection-specific phosphorylation of glutamyl-prolyl tRNA synthetase induces antiviral immunity. Nat Immunol 2016; 17:1252-62; PMID:27595231; https://doi.org/10.1038/ni.3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Arif A, Terenzi F, Potdar AA, Jia J, Sacks J, China A, Halawani D, Vasu K, Li X, Brown JM, Chen J, Kozma SC, Thomas G, Fox PL. EPRS is a critical mTORC1-S6K1 effector that influences adiposity in mice. Nature 2017; 542:357-61; PMID:28178239; https://doi.org/10.1038/nature21380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Clemens MJ. Does protein phosphorylation play a role in translational control by eukaryotic aminoacyl-tRNA synthetases? Trends Biochem Sci 1990; 15:172-5; PMID:2193433; https://doi.org/10.1016/0968-0004(90)90153-3 [DOI] [PubMed] [Google Scholar]
- [5].Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009; 326:140-4; PMID:19797661; https://doi.org/10.1126/science.1177221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004; 431:200-5; PMID:15306821; https://doi.org/10.1038/nature02866 [DOI] [PubMed] [Google Scholar]
- [7].Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, Ha SH, Ryu SH, Kim S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012; 149:410-24; PMID: 22424946; https://doi.org/10.1016/j.cell.2012.02.044 [DOI] [PubMed] [Google Scholar]