

ABSTRACT
The knowledge gap separating the molecular and cellular underpinnings of Parkinson disease (PD) and its pathology hinders treatment innovation. Adding to this difficulty is the lack of a reliable biomarker for PD. Our previous studies identify a link of 2 PD proteins, PINK1/PRKN Parkin to a mitochondrial motor adaptor RHOT1/Miro-1, which mediates mitochondrial motility and mitophagy. Here we review our recent paper showing that a third PD protein, LRRK2, also targets RHOT1 and regulates mitophagy, and pathogenic LRRK2 disrupts this function. Notably, we discover impairments in RHOT1 and mitophagy in sporadic PD patients with no known genetic backgrounds, pointing to RHOT1-mediated mitophagy as a convergent pathway in PD. This novelty opens new doors in PD research toward RHOT1-based therapy and biomarker development.
KEYWORDS: LRRK2, mitochondrial motility, mitophagy, Miro, PINK1, Parkin, Parkinson, sporadic
Prior to the initiation of mitophagy that clears the entire damaged mitochondria, RHOT1/Miro—an outer mitochondrial membrane (OMM) protein that anchors the microtubule motors to mitochondria—needs to be removed from the mitochondrial surface and is subsequently degraded in proteasomes (Fig. 1). Consequently, mobile mitochondria stop their movement and this facilitates the isolation and sequestration of damaged mitochondria. Our previous studies have demonstrated that 2 Parkinson disease (PD) proteins, PINK1 (PTEN-induced putative kinase 1) and PRKN/Parkin, target RHOT1 for removal from damaged mitochondria (Fig. 1), suggesting the intriguing possibility that a fraction of PD cases may be caused by the failure to remove RHOT1 and to clear dysfunctional mitochondria. To explore this possiblity, we set out to determine the relevance of RHOT1-mediated mitophagy in PD by using skin cells and neurons directly cultured from PD patients. Our research was guided by the following questions: Do damaged mitochondria stop movement before they undergo mitophagy in human neurons? Do defective mitochondria accumulate in cells from patients with pathogenic PINK1 or PRKN mutations and thereby cause neuronal cell death? Does neurodegeneration arise similarly in non-PINK1 or -Parkin-related PD?
Figure 1.
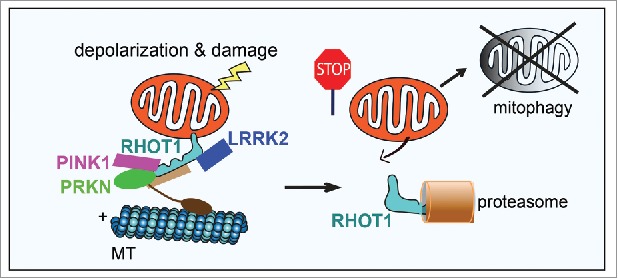
Schematic representation of the sequential events occurring after mitochondrial depolarization. (1) Depolarization triggers recruitments of PINK1, PRKN, and LRRK2 to RHOT1. (2) These proteins target RHOT1 for removal from the OMM of damaged mitochondria causing mitochondrial arrest. (3) RHOT1 is subsequently degraded by proteasomes. (4) Mitochondria are ultimately degraded in lysosomes via mitophagy. MT, microtubules.
Although dopaminergic neurons in the substantia nigra are found to be the most susceptible neuronal type in PD patients, PINK1 or PRKN are ubiquitously expressed and thus their mutations should affect cell types not limited to dopaminergic neurons. Because skin fibroblasts are notably easy to obtain and culture, we detected RHOT1 and mitophagy in fibroblasts from healthy controls and PD patients. We established a powerful readout in human fibroblasts: after mitochondrial depolarization in wild-type cells, RHOT1 is rapidly degraded, and multiple mitochondrial markers are subsequently degraded indicating the occurrence of mitophagy. Using this readout, we screened a total of 16 cell lines from PD patients, including sporadic patients with no known mutations and familial patients with mutations in LRRK2 (leucine rich repeat kinase 2), PINK1, or PRKN. Mutations in LRRK2 are the most common cause of familial PD as well as a risk factor for sporadic PD, and sporadic cases account for about 90% of total PD cases. To our surprise, we found that RHOT1 is resistant to depolarization-triggered degradation and mitophagy is impaired not only in mutant PINK1 and PRKN cells, but also in mutant LRRK2 cells, and even in those from sporadic patients. The significance of this finding is 2-fold: first it points to RHOT1 as a common molecular underpinning in a large population of PD; and second it raises the possibility that these fibroblast lines and this novel phenotypic readout can be used to screen for therapeutic interventions and diagnostic innovations.
To determine whether this impairment in RHOT1 and mitophagy we discovered in patients' skin cells also occurs in neurons, we converted fibroblast lines from 3 familial patients with the G2019S mutation in LRRK2 and 2 sporadic patients to induced pluripotent stem cells and then differentiated those cells to dopaminergic neurons. We live-imaged and monitored mitochondrial motility and mitophagy in neuronal axons. In wild-type neurons, we observed a sequence of mitochondrial events occurring after mitochondrial depolarization. Mitochondrial length is first shortened, then mitochondrial motility in both anterograde and retrograde directions is arrested, and finally mitophagy is triggered and mitochondrial clearance is induced. In all patients' neuronal axons, however, we discovered a significant delay in halting damaged mitochondrial motility and in starting mitophagy. Consequently, depolarized mitochondria are accumulated increasing neuronal vulnerability to oxidative stress. Thus, this cellular defect in mitophagy exists in both skin cells and neurons in culture. In human patients, this defect may lead to cell death only in the most vulnerable neurons, for example, aging dopaminergic neurons in the substantia nigra. These neurons have unusually elevated cellular stress caused by intense neuronal activities, dopamine metabolism, and elaborate axonal networks, and may contain more damaged mitochondria and be more susceptible to impaired mitophagy.
Our human neuron model demonstrates that removal of RHOT1 from damaged mitochondria is slowed by LRRK2G2019S, delaying mitochondrial arrest and mitophagy. Based on this model, we reasoned that mildly lowering the basal level of RHOT1 by RNAi in LRRK2G2019S neurons may correct this defect. Indeed, we revealed that RHOT1 RNAi rescues the delay in mitochondrial arrest and mitophagy in LRRK2G2019S neurons, and remarkably also protects LRRK2G2019S neurons from oxidative stress. We next tested the neuroprotective effect of lowering RHOT1 protein levels in vivo. We used a Drosophila model with overexpressed human LRRK2G2019S that exhibits locomotor defects and dopaminergic neurodegeneration. We knocked down Miro by RNAi in those flies, and found that the approach completely restored their larval crawling, adult climbing and jumping abilities, and dopaminergic neuronal numbers. These results in both cultured human neurons and in vivo indicate that the RHOT1 Miro-dependent pathway may constitute a major part of LRRK2-linked PD pathogenesis. Our findings warrant increased examination of RHOT1 as a potential target for PD, particularly considering that its partial inhibition could alleviate defects in both familial and sporadic PD patients.
To explore the mechanism by which LRRK2G2019S slows removal of RHOT1 from the OMM, we examined the possibility that wild-type LRRK2 directly promotes RHOT1 removal, and that LRRK2G2019S loses this function. This scenario may require LRRK2 to form a complex with RHOT1. We were interested to find that mitochondrial depolarization triggers recruitment of LRRK2 from the cytosol to the mitochondria and binding of LRRK2 to RHOT1 (Fig. 1), but not of LRRK2G2019S, in both induced pluripotent stem cell-derived neurons and fibroblasts. We further showed that LRRK2G2019S may act in a loss-of-function manner because cells lacking LRRK2 yielded the same impact on RHOT1 as cells bearing LRRK2G2019S. In addition, applying an LRRK2 kinase inhibitor, or using an LRRK2 kinase-dead mutation did not affect the action of wild-type LRRK2 on RHOT1, suggesting that the kinase activity of LRRK2 is not required for RHOT1 removal.
Taken together, our recent data reveal a shared molecular signature in clinically and genetically distinct patients. Future validation in a larger cohort of patients with broader genetic backgrounds will determine whether a common, RHOT1-dependent mechanism underlies all, or a subset of PD patients. Our studies continue to pursue the increasingly promising role RHOT1 and mitophagy play in transforming diagnosis, evaluation, and treatment of PD.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.