

ABSTRACT
Macroautophagy/autophagy is an intracellular recycling system that delivers cytoplasmic organelles and materials to lysosomes for degradation. This process is operated by autophagy-related (ATG) genes and tightly controlled by stress-responsive signaling pathways. Our recent study revealed that autophagy programs are transcriptionally suppressed by the BET family protein BRD4. This repression is alleviated during nutrient deprivation through the AMPK-SIRT1 pathway. Our findings therefore provide new insights into the regulation of autophagy.
KEYWORDS: AMPK, autophagy, BRD4, BRD4-NUTM1/BRD4-NUT, EHMT2/G9a/KMT1C, KAT8/hMOF, lysosome, selective autophagy, SIRT1, transcriptional regulation of autophagy
Macroautophagy (hereafter referred to as autophagy) is a conserved catabolic process that maintains cellular homeostasis. While many autophagy-related (ATG) genes that constitute the core autophagy machinery have been identified, the regulatory mechanisms that modulate autophagic activity are less explored.
To obtain insights into the regulatory mechanisms of autophagy, we performed an RNAi screen in Drosophila melanogaster S2R+ cells expressing the autophagy marker GFP-LC3. We found that double-stranded RNA against fs(1)h (female sterile [1] homeotic) causes an increase in GFP-LC3 puncta, an indicator of autophagosome accumulation. Fs(1)h belongs to the Bromodomain and Extra-Terminal (BET) family, which consists of ubiquitously expressed BRD2 (bromodomain containing 2), BRD3, BRD4, and the testis-specific member BRDT (bromodomain testis associated) in mammals. To validate the screening results, BRD2, BRD3, and BRD4 were silenced and their effects on autophagy were determined. Knockdown of BRD4, but not BRD2 or BRD3, increases LC3 lipidation.
BRD4 functions as a scaffold protein that binds to acetylated histones via tandem bromodomains and recruits transcriptional regulators to promoter and enhancer regions. By using various assays, we showed that BRD4 knockdown activates a series of autophagy processes including phagophore/autophagosome formation, the fusion of autophagosomes with lysosomes, and the subsequent degradation step. Similarly, the BET inhibitor JQ1, a small molecule that binds to BET bromodomains and interferes with their binding to acetylated histones, also promotes autophagy, suggesting that BRD4 regulates autophagy in a manner dependent on its bromodomains. In addition, BRD4 knockdown and JQ1 administration also increase autophagosome formation in mice. Therefore, these results characterize BRD4 as an evolutionarily conserved autophagy repressor.
As BRD4 is a transcriptional regulator, we speculated that BRD4 represses autophagy by a transcriptional mechanism. Indeed, RNA-sequencing and subsequent qPCR validation experiments revealed that BRD4 silencing upregulates several autophagy genes whose products are involved in phagophore/autophagosome formation and the fusion of autophagosomes with lysosomes. Of note, we also observed a significant upregulation of lysosomal genes involved in proteolysis, glycan degradation, and lysosome biogenesis. Consistent with this, BRD4 knockdown increases lysosomal protein levels, LAMP1 (lysosomal associated membrane protein 1)- and LysoTracker-positive lysosomal compartments, and the activities of lysosomal enzymes such as CTSB (cathepsin B) and HEXB/β-hexosaminidase. These results therefore demonstrate that BRD4 negatively regulates lysosomal function and biogenesis as well as autophagic activity.
We next investigated the molecular mechanism by which BRD4 represses autophagy and lysosome gene expression. As TFEB (transcription factor EB) is recognized as a master transcriptional regulator of the autophagy-lysosome pathway, we first examined whether the mechanism involves TFEB and its family members TFE3 (transcription factor binding to IGHM enhancer 3) and MITF (melanogenesis-associated transcription factor). However, BRD4 knockdown still enhances autophagic flux in TFEB, TFE3 and MITF triple knockdown cells, indicating that BRD4 regulates autophagy in a manner independent of the TFEB family members. Instead, further analyses revealed that BRD4 connects histone modifications to the repression of autophagy gene expression. BRD4 binds to the promoter regions of autophagy and lysosome genes via histone H4K16 acetylation catalyzed by KAT8/hMOF (lysine acetyltransferase 8). It then recruits the histone methyltransferase EHMT2/G9a (euchromatic histone lysine methyltransferase 2) which dimethylates H3K9 and represses autophagy gene transcription (Fig. 1A). Upon nutrient deprivation, the histone deacetylase SIRT1 (sirtuin 1) dissociates from its inhibitory molecule CCAR2/DBC1 (cell cycle and apoptosis regulator 2) in an AMPK (AMP-activated protein kinase)-dependent manner. SIRT1 then becomes active and deacetylates H4K16, which in turn displaces BRD4 from the promoter regions and leads to transcriptional activation of autophagy genes (Fig. 1B).
Figure 1.
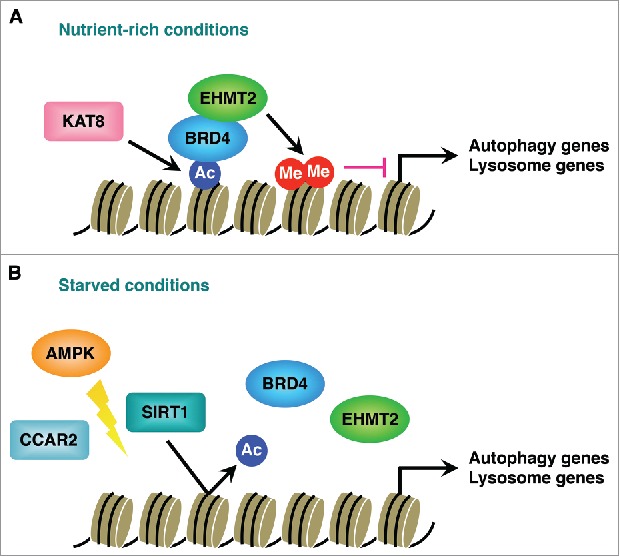
Transcriptional regulation of autophagy and lysosome genes by BRD4. (A) BRD4 recruitment to autophagy and lysosome gene promoters is mediated by the histone acetyltransferase KAT8/hMOF and H4K16 acetylation (Ac). BRD4 then interacts with the histone methyltransferase EHMT2/G9a, which represses autophagy gene transcription by dimethylating (MeMe) H3K9 under nutrient-rich conditions. (B) Nutrient starvation causes the dissociation of SIRT1 from its inhibitory protein CCAR2/DBC1 in a manner dependent on AMPK. SIRT1-dependent deacetylation of H4K16 leads to BRD4 dissociation from the promoters and transcriptional activation of autophagy genes.
Autophagy degrades various substrates in nonselective and selective manners, which are termed bulk autophagy and selective autophagy, respectively. Interestingly, we found that BRD4 knockdown promotes bulk autophagy and the autophagic degradation of mutant HTT (huntingtin) aggregates (in a process termed aggrephagy) but does not affect the clearance of damaged mitochondria or Salmonella enterica serovar Typhimurium (mitophagy and xenophagy). Bulk degradation of cytoplasmic constituents is typically activated during nutrient shortage and provides cells with nutrient sources to maintain cell survival. BRD4 knockdown further enhances autophagic activity in response to starvation and confers resistance to starvation-induced cell death in an autophagy-dependent manner. These results show that BRD4 knockdown influences certain types of autophagy.
Chromosomal translocation of the NUTM1/NUT (NUT midline carcinoma family member 1) gene with the BRD4 gene drives NUT midline carcinoma (NMC), a rare aggressive subtype of carcinoma. We found that the fusion gene product BRD4-NUTM1 functions as a suppressor of autophagy in NMC. BRD4-NUTM1 knockdown upregulates a subset of autophagy and lysosome genes and enhances autophagic flux and lysosomal biogenesis. Intriguingly, knockdown of wild-type BRD4, which is expressed from the unaffected allele, has only a comparatively slight effect on LC3 lipidation, implying that BRD4-NUTM1 is a dominant repressor of autophagy in NMC.
In summary, we demonstrate that BRD4 restricts autophagic and lysosomal activities by repressing the expression of autophagy and lysosome genes under nutrient-rich conditions, and derepression of this program contributes to autophagy activation during nutrient deprivation. Considering that transcriptional activation of the autophagy programs occurs over a longer time period compared with the rapid response of the core autophagy machinery to MTOR (mechanistic target of rapamycin) and AMPK signaling pathways, this derepression mechanism seems to function to sustain autophagic activity during prolonged starvation. In addition, although we showed that BRD4-NUTM1 strongly suppresses autophagy in NMC, whether autophagy repression contributes to NMC development and progression remains unknown. Similarly, the relevance of autophagy regulation by BRD4 to human diseases is still unexplored. Future studies should therefore uncover the significance of transcriptional repression of autophagy by BRD4 and BRD4-NUTM1 in different contexts.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Work in the Ryan laboratory is supported by Cancer Research UK (C596/A17196) and Worldwide Cancer Research (16–1194).