

ABSTRACT
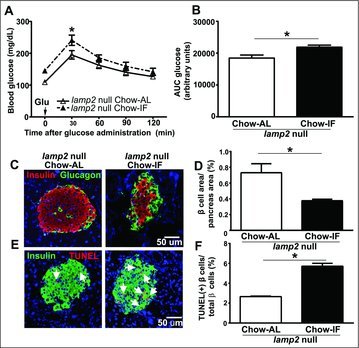
Obesity-induced diabetes is characterized by hyperglycemia, insulin resistance, and progressive beta cell failure. In islets of mice with obesity-induced diabetes, we observe increased beta cell death and impaired autophagic flux. We hypothesized that intermittent fasting, a clinically sustainable therapeutic strategy, stimulates autophagic flux to ameliorate obesity-induced diabetes. Our data show that despite continued high-fat intake, intermittent fasting restores autophagic flux in islets and improves glucose tolerance by enhancing glucose-stimulated insulin secretion, beta cell survival, and nuclear expression of NEUROG3, a marker of pancreatic regeneration. In contrast, intermittent fasting does not rescue beta-cell death or induce NEUROG3 expression in obese mice with lysosomal dysfunction secondary to deficiency of the lysosomal membrane protein, LAMP2 or haplo-insufficiency of BECN1/Beclin 1, a protein critical for autophagosome formation. Moreover, intermittent fasting is sufficient to provoke beta cell death in nonobese lamp2 null mice, attesting to a critical role for lysosome function in beta cell homeostasis under fasting conditions. Beta cells in intermittently-fasted LAMP2- or BECN1-deficient mice exhibit markers of autophagic failure with accumulation of damaged mitochondria and upregulation of oxidative stress. Thus, intermittent fasting preserves organelle quality via the autophagy-lysosome pathway to enhance beta cell survival and stimulates markers of regeneration in obesity-induced diabetes.
KEYWORDS: autophagy, beta cells, diabetes, intermittent fasting, lysosomes
Introduction
Diabetes is a major contributor to morbidity and mortality from multiple causes, including cardiovascular disease and chronic kidney disease. With an increasingly obese population, type 2 diabetes (T2D) is becoming a worldwide pandemic, with nearly one in 10 adults affected and more than 1.5 million deaths per year. By 2050, nearly one in 3 adults may be diagnosed with T2D.1 The pathogenesis of T2D involves a downward spiral of hyperglycemia, insulin resistance, impairments in insulin secretion and beta cell death culminating in overt insulin-dependence.2 Given the increasing burden of T2D, developing strategies to reverse or prevent these metabolic derangements and beta cell loss is of utmost importance.
The prevention of T2D by dietary modifications, including caloric restriction and intermittent fasting (IF), has been described in human clinical trials.3 Caloric restriction can extend life span in rodents, an effect that is associated with improvement in insulin sensitivity 4 and markers of metabolic health in humans;5 however, caloric restriction is often difficult to sustain. An alternative dietary regimen to caloric restriction is intermittent fasting (IF), which may have beneficial effects on longevity and metabolism in humans.6-8 IF prevents the development of diabetes and promotes fat loss and lean body mass retention in rodent models.9-12 However, the mechanisms by which IF leads to these benefits are unknown.
The autophagy-lysosome pathway is both induced by IF and implicated in the development of T2D.13-18 Our group has recently demonstrated that IF preconditions the myocardium towards ischemia-reperfusion injury, via stimulation of the autophagy-lysosome pathway.19 Here we have tested the hypothesis that IF improves glucose handling in obesity-induced diabetes via the autophagy-lysosome pathway. Our results demonstrate that IF improves glucose tolerance in high-fat diet (HFD) fed mice compared to ad libitum controls via preservation of beta cell mass and function. In LAMP2-deficient mice, a model of lysosomal insufficiency, the benefits of IF on glucose metabolism are lost, and IF paradoxically promotes increased beta cell apoptosis and exacerbates autophagic impairment. IF also provokes impaired glucose tolerance in Becn1 (Beclin 1, autophagy related) haplo-insufficient mice, indicating an important mechanistic role for autophagy and lysosomal function. Interestingly, IF also stimulated nuclear expression of the transcription factor NEUROG3, a marker of beta cell regeneration, in wild-type but not in LAMP2- or BECN1-deficient mice. These findings indicate that IF ameliorates HF diet-induced glucose intolerance by preserving beta cell mass and function via entrainment of the autophagy-lysosome pathway, and underscore the need for careful evaluation of IF as a clinically sustainable therapeutic strategy to enhance beta cell health in obesity and diabetes.
Results
Intermittent fasting preserves beta cell mass and function to improve glucose regulation in diet-induced diabetes
We fed wild-type mice a HF-diet (see Table S1 for composition) for 12 wk to induce weight gain and glucose intolerance relative to chow-fed controls (P < 0.001, Fig. 1A). Mice were then randomized to 6 wk of IF or continued ad-lib feeding, during which both HF and chow-fed male mice exhibited significant weight loss compared to ad-lib fed groups (P < 0.001, Fig. 1B). This correlated with a 25% reduction in caloric intake in the setting of IF (P < 0.001, Fig. 1C), and reductions in cholesterol (total, LDL and HDL cholesterol; see Table S2). IF caused a decrease in fasting glucose despite continued HF feeding in obese mice, and improved glucose tolerance in both chow and HF-diet fed mice (Fig. 1D and E). Further, IF-induced improvements in glucose tolerance were evident prior to weight loss in chow-fed mice (Fig. S1A–C). Intermittent fasting for 6 wk did not improve the glucose response to insulin in obese mice (Fig. S2A and B), although glucose returned to baseline values rapidly in IF mice (which could indicate worsening insulin sensitivity, Fig. S2A), as compared with ad-lib fed controls. Furthermore, evaluation of AKT (Ser473) phosphorylation 10 min after insulin administration in the heart, liver, and skeletal muscle revealed marked blunting of insulin action in these tissues in HF-fed mice vs. chow controls (Figs. S3A and B and S4) indicating insulin resistance in obese mice, and IF did not improve HF-diet induced impairment in AKT phosphorylation. Taken together, these data suggest that IF improves glucose tolerance in mice fed chow or HF diets, but does not improve peripheral insulin resistance in diet-induced obesity. Interestingly, HF-fed mice were hyperinsulinemic as compared to chow-fed mice, but were unable to further enhance circulating insulin levels in response to glucose injection (Fig. 1F). In contrast, IF further increased fasting insulin levels and restored the glucose-induced surge in HF-fed mice (Fig. 1F). The pattern of circulating insulin C-peptide levels prior to and 30 min after glucose injection mirrored circulating insulin levels (Fig. S2C), confirming that IF stimulates both basal and glucose-induced insulin release in mice with diet-induced obesity.
Figure 1.
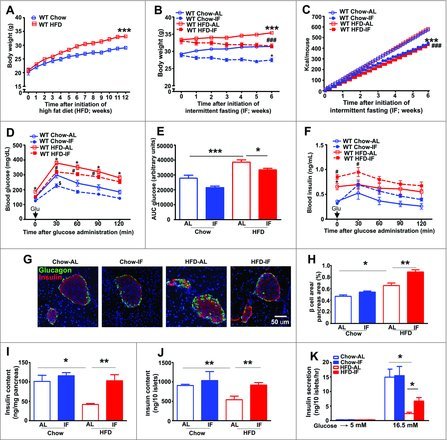
Intermittent fasting improves glucose regulation and preserves beta cell mass and function in mice with diet-induced obesity and diabetes. (A) Weight gain on high-fat diet (HFD, open red boxes, solid red line) as compared with chow feeding (open blue circles, solid blue line) in adult male C57BL/6 mice from 8 wk to 20 wk of age (n = 15 per group; ***P < 0.001 for HFD vs. chow). (B) Body weight after 6 wk of intermittent fasting (solid circles or boxes with dotted lines) in both chow (n = 15 per group) and high-fat feeding groups (n = 23 or 24/group, ***P < 0.001 for WT HFD-AL vs. WT chow-AL, ###P < 0.001 HFD-IF vs. HFD-AL). (C) Average cumulative caloric intake in mice treated as in (B) (***P < 0.001 for chow-IF vs. chow-AL, ###P < 0.001 HFD-IF vs. HFD-AL). (D, E) Glucose tolerance tests (GTT, D, *P < 0.05 for HFD-AL vs. chow-AL, #P < 0.05 HFD-IF vs. HFD-AL, $P < 0.05 for chow-IF vs. chow-AL), area under the curve for glucose measurement (E). (F) Insulin levels in response to glucose administration as in D (F, #P < 0.05 HFD-IF vs. HFD-AL, *P < 0.05 HFD-AL vs. chow-AL) in mice treated as in (B), n = 10 to 28/group. (G) Representative images of islets from mice treated as in (B); (n = 3 or 4/group) with immunostaining of beta cells (anti-insulin, red) and alpha cells (anti-glucagon, green); nuclei are blue (DAPI). (H) Quantification of beta cell area in pancreata from (G). (I) Insulin content per mg pancreas (n = 5 to 7/group; J) and in islets (n = 30 islets/mouse from n = 3 or 4 mice/group) from mice treated as in (B). (K) Glucose stimulated insulin release in isolated islets (n = 30 islets/mouse from n = 4 or 5 mice per group) as in (J). *P < 0.05 and **P < 0.001 for (H–K).
We therefore reasoned that effects of IF on glucose tolerance and insulin levels may be mediated by alteration in islet beta cell mass or glucose-stimulated insulin secretion. Thus, we evaluated beta cell area, beta/alpha cell ratio, total pancreatic insulin content, and islet insulin content. Beta cell area was significantly increased, but beta/alpha cell ratio was significantly decreased in ad-lib HF fed versus ad-lib chow-fed mice (Fig. 1G and H; Fig. S2D), consistent with a relative beta cell insufficiency in the face of insulin resistance and persistent hyperglycemia in diet-induced obesity.20 IF resulted in a significant increase in beta cell area as compared with ad-lib fed mice with diet-induced obesity (P = 0.0067 vs. HF ad-lib group; Fig. 1G and H) and restored the beta/alpha cell ratio after HF feeding to levels observed in wild-type mice on chow diet (P = 0.02 vs. HF ad-lib; Fig. 1G; Fig. S2D). Importantly, IF further increased circulating glucagon levels (that were increased by HF feeding versus chow), suggesting that the improvements in glucose tolerance with IF were independent of its effects on alpha cell mass or function, since glucagon itself does not improve glucose tolerance (Fig. S2E). Furthermore, HF feeding decreased pancreatic insulin content as compared with chow feeding (Fig. 1I), consistent with insulin deficiency observed with beta-cell failure in mice with obesity-induced diabetes.21 In contrast, the insulin content in HF fed mice subject to IF was similar to levels in chow-fed mice (102.8 ± 15.7 in HF-diet-IF vs. 41.8 ± 2.3 ng/mg of pancreas tissue in HF-diet-AL, P = 0.007 as compared with 101.2 ± 15.7 ng/mg in chow-AL; Fig. 1I). IF also restored the insulin content of individual islets, which was reduced by HF feeding as compared with ad-lib chow-fed controls (Fig. 1J). Furthermore, HF feeding impaired glucose-induced insulin secretion by 84.4% (2.41 ± 0.47 ng/10 islets/h in HF fed ad-lib vs. 14.94 ± 2.75 ng/10 islets/h in chow-fed ad-lib, P = 0.0146, Fig. 1K), while IF significantly increased glucose stimulated insulin secretion in obese mice (6.71 ± 1.19) as compared with HF ad-lib fed mice (P = 0.03, Fig. 1K). Thus, IF may improve glucose tolerance in intermittently fasted obese mice via dual mechanisms: increased insulin stores as well as increased insulin release.
Intermittent fasting rescues beta cell death in obesity-induced diabetes
We next hypothesized that IF increases beta cell mass by decreasing apoptotic beta cell death observed in islets from subjects with type 2 diabetes.22 Ad-lib HF feeding resulted in increased prevalence of TUNEL-positive beta cells as compared with chow control, and this was decreased by 49% after IF in HF fed obese mice as compared with the ad-lib HF diet group (Fig. 2A and B). To evaluate whether changes in beta cell proliferation were contributing to increased beta cell mass after IF, we examined expression of MKI67 (Ki-67), a marker for cellular proliferation in beta cells.23 HF-diet caused an increase in MKI67-positive ratio as compared with chow that was not significantly altered by IF (Fig. 2C and D). Thus, IF significantly decreases beta cell apoptosis without a major effect on cell proliferation.
Figure 2.
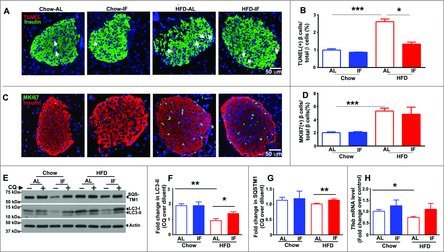
Intermittent fasting prevents beta cell loss in mice with diet-induced obesity. (A) Representative immunofluorescence images of pancreatic islets from mice fed HFD or chow diets for 12 wk and then subjected to intermittent fasting for 6 wk, for TUNEL (red, see arrows) and beta cells (green, anti-insulin) in (A). (B) Quantification of TUNEL-positive beta cell nuclei (n = 30 to 40 islets/group; 3 or 4 mice/group). (C) Representative images for MKI67 (MKI67, green), insulin (red) and nuclei (blue, DAPI) in mice treated as in A; (D) Quantification of MKI67(+) beta cells as a percentage of total beta cells. (E–G) Assessment of autophagix flux in islets by immunoblotting for LC3B and SQSTM1 in mice modeled as in (A) and injected with chloroquine (CQ, 60 mg/kg) or saline 4 h prior to sacrifice (E), with quantification of LC3B-II (F) and SQSTM1 (G); n = 3 mice/group. (H) Tfeb transcripts in islets from mice treated as in (A); n = 3 or 4 per group. *P < 0.05; **P < 0.001 and ***P < 0.0001.
Intermittent fasting restores autophagic flux in islets from mice with diet-induced obesity
Given prior observations that an intact autophagy machinery is required for beta cell survival 16, 17 and our studies indicating that intermittent fasting stimulates autophagic flux,19 we hypothesized that the beneficial effects of intermittent fasting on beta cell survival may involve stimulation of autophagy. Accordingly, we examined autophagic flux by comparing accumulation of autophagosome-bound LC3B-II (MAP1LC3B isoform II, as a marker of autophagosome abundance) and SQSTM1/p62 (an autophagy receptor and substrate) in the animals injected with chloroquine (to inhibit autophagosome-lysosome fusion), with animals treated with saline as control. Our data demonstrate that islets of mice fed a HF diet to provoke obesity-induced diabetes demonstrate impaired autophagic flux, as LC3B-II and SQSTM1 do not accumulate further in the setting of short-term chloroquine (CQ) treatment (Fig. 2E–G), as compared with intact flux (evidenced by increase in CQ vs. respective diluent treated samples) in control mice fed a chow diet. In contrast, intermittent fasting restored autophagic flux in islets from HF-fed mice, as evidenced by CQ-induced accumulation of LC3B-II and SQSTM1 versus their saline-injected counterparts (Fig. 2E–G). This impairment in autophagic flux in HF-fed mouse islets was accompanied by downregulation of Tfeb transcript abundance (by 27% vs. chow-fed mouse islets, Fig. 2H), the master regulator of autophagy-lysosome machinery24 that autoregulates its own transcription when activated,25 suggesting that TFEB inhibition plays a contributory role in the effects of high-fat diet on impairment of autophagic flux in islets. Indeed, intermittent fasting prevented Tfeb mRNA downregulation in islets from mice with diet-induced obesity (Fig. 2H), consistent with our prior observation that intermittent fasting activates TFEB in the myocardium.19
To examine if TFEB-induced autophagy-lysosome machinery activation plays a mechanistic role in beta cell death, we modeled glucolipotoxicity in INS-1 832/13 cells (a commonly studied in vitro model of beta cells)26 and examined the effect of loss-of-function and gain-of-function of TFEB, as well as inhibition of autophagy and lysosome function with knockdown of BECN1 and LAMP2, respectively, as we have previously described.19 Endogenous TFEB, BECN1 and LAMP1 were required to attenuate glucolipotoxicity (Fig. S5A and B), and exogenous TFEB was sufficient to attenuate glucolipotoxic cell death (Fig. S5C and D) supporting the notion that the TFEB-induced autophagy-lysosome pathway plays a critical functional role in protecting against beta cell death under diet-obesity stress, in vivo.
Intermittent fasting requires intact lysosome function to ameliorate glucose intolerance induced by high-fat feeding
Intact autophagic flux requires normal lysosome function, and we have previously found that the beneficial effect of IF on ischemic preconditioning requires intact autophagy-lysosome machinery.19 As a model of stress-induced lysosomal insufficiency, we studied female mice that were heterozygous null for the X-linked gene, Lamp2. 19 In published studies, we have found that while IF results in decreased myocardial ischemia-reperfusion injury in wild-type (WT) mice, Lamp2 heterozygous null mice do not benefit from IF.19 Accordingly, we reasoned that the metabolic benefits of IF on glucose homeostasis are dependent on an intact lysosomal machinery. Lamp2 heterozygous null female mice exhibited no differences in glucose or insulin tolerance on a chow diet as compared with C57/BL6 wild-type female controls (Fig. S6A–D), confirming that loss of one Lamp2 allele did not have discernable effects on glucose handling in the unstressed state, paralleling the lack of an observed phenotype in the unstressed heart19 and other organ systems.27 Subsequently, we fed Lamp2 heterozygous null mice and WT controls with HF-diet for 12 wk to generate a model of lysosomal insufficiency under metabolic stress. HF feeding resulted in similar levels of weight gain (Fig. S7A), glucose intolerance (Fig. S7B and C) and insulin resistance (Fig. S7D and E) in WT and mutant mice.
After 12 wk of HF feeding or chow diet as control, Lamp2 heterozygous null and WT female mice were randomized to 6 wk of IF or ad-lib with continued access to their original regimen of HF or chow diets. In contrast to male C57/BL6 mice (Fig. 1B), neither WT nor Lamp2 female mice exhibited weight loss after IF (Fig. 3A and B) despite a 15% reduction in caloric intake (Fig. 3C and D). Nevertheless, IF significantly improved glucose tolerance (Fig. 3E and G) in HF-fed WT female mice (mimicking the observations in male mice, see Fig. 1D and E) but not in HF-fed obese Lamp2 heterozygous null mice (Fig. 3F and H). Both basal and glucose-stimulated insulin levels were significantly increased after IF in obese WT but not in obese Lamp2 heterozygous null mice as compared with respective AL fed groups (Fig. 3I and J). Interestingly, IF did not improve insulin resistance in WT female or Lamp2 heterozygous null mice (Fig. S8A, B, D, and E). These findings indicate that the benefits of IF on glucose handling in obese WT female mice (Fig. 3E) and the absence of those benefits in obese Lamp2 heterozygous null mice (Fig. 3F) are primarily due to glucose-induced insulin availability (Fig. 3I and J).
Figure 3.
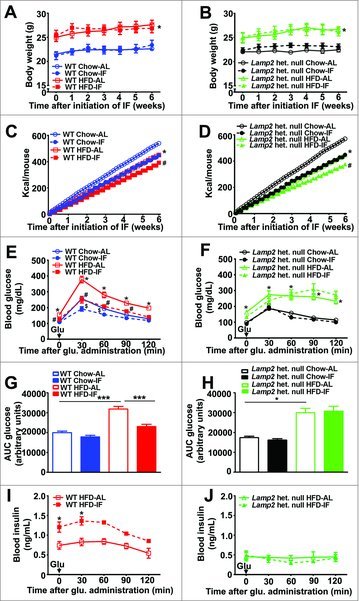
Intermittent fasting does not improve glucose tolerance in Lamp2 heterozygous null mice with obesity-induced diabetes. (A) Body weight in WT female mice fed HFD (red, squares) or chow (blue, circles) for 12 wk, during the subsequent 6 wk of intermittent fasting (closed squares or circles with dotted lines) or ad-lib feeding (open squares or circles with solid lines) while being continued on the respective diets (n = 5/ group for chow feeding, n = 10/group for HFD, *P < 0.05 HFD-AL vs. chow-AL). (B) Body weight in Lamp2 heterozygous null mice fed HFD (green, squares) or chow diets (black, circles) for 12 wk during the subsequent 6 wk of IF (closed squares or circles with dotted lines) or ad-lib feeding (open squares or circles with solid lines) while being continued on the respective diets (n = 6/group for HF diet and n = 3 or 4/group for chow diet; *P < 0.05 HFD-AL vs. chow-AL). (C and D) Caloric intake in mice treated as in (A and B), respectively (*P < 0.05 for HFD-AL vs. chow-AL, #P < 0.05 HFD-IF vs. HFD-AL). (E and F) GTTs in WT females (E, n = 10/ group, *P<0.05 for HFD-AL vs. chow-AL, #P < 0.05 HFD-IF vs. HFD-AL, $P < 0.05 for chow-IF vs. chow-AL) and Lamp2 heterozygous null mice (F, n = 3 to 6 per group, *P < 0.05 for HFD-AL vs. chow-AL) fed HFD or chow diets followed by IF or ad-lib access to respective diets. (G and H) AUC measurements from GTTs performed in (E and F). (I and J) Insulin levels upon glucose challenge in obese WT (I, n = 5/group) and Lamp2 heterozygous null mice (J, n = 3 or 4/group) after IF or ad-lib feeding on HFD for 6 wk. *P < 0.05; **P < 0.001 and ***P < 0.0001.
To examine the effects of complete loss of LAMP2, we studied lamp2 hemizygous null male mice and WT male mice. Interestingly, there were no differences in glucose tolerance or insulin resistance in WT male and lamp2 null male mice on chow diets (Fig. S9A–D), indicating that complete loss of LAMP2 is also well tolerated in the absence of metabolic stress, as previously described.28 Based on our prior observations that intermittent fasting provokes cardiomyopathy in the lamp2 null mice,19 we hypothesized that IF will be sufficient to induce metabolic derangements even in chow-fed mice. Indeed, IF provoked impaired glucose tolerance in male lamp2 null mice fed a chow diet (Fig. 4A and B) without a change in body weight (Fig. S10A) or an alteration in insulin tolerance (Fig. S10B and C). Taken together with the observations in wild-type male (Fig. 1D and E) and female (Fig. 3E–J)\ mice, these data demonstrate that IF significantly improves glucose tolerance in a manner that requires the presence of LAMP2, suggesting that intact lysosomal machinery is necessary for the beneficial effect of IF on glucose tolerance.
Figure 4.
Intermittent fasting worsens glucose tolerance in lamp2 null males fed a chow diet. (A) GTT performed in lamp2 male null mice after 6 wk IF (closed triangle, dotted line) or ad-lib feeding (open triangle, solid line) on a chow diet (n = 5/group); (B) AUC measurement from GTT in (A); *P < 0.05 for IF vs. AL for (A and B). (C) Representative images of islets from male lamp2 null mice showing beta cells (red, anti-insulin) and alpha-cells (green, anti-glucagon) for mice treated as in (A); nuclei are blue (DAPI). (D) Quantification of beta cell area from (C; n = 3 or 4 mice/group). (E) TUNEL staining (red) in islets costained with anti-insulin (green) and DAPI for mice treated as in (A; see arrows). (F) Quantification of TUNEL-positive beta cell nuclei in ad-lib versus IF treated mice fed a chow diet (n = 3 or 4 mice per group). *P < 0.05; **P < 0.001 and ***P < 0.0001.
Intermittent fasting does not preserve beta cell mass in a model of impaired lysosome function
Given that LAMP2 is necessary for the beneficial effects of IF on glucose tolerance, we hypothesized that IF-induced beta cell preservation would also be LAMP2 dependent. In male lamp2 null mice fed chow diet, where IF was sufficient to cause glucose intolerance, we observed a 48.6% decrease in beta cell area (Fig. 4C and D), a 33% decrease in the beta/alpha cell ratio (Fig. S10D) and a ∼2-fold increase in beta cell apoptosis in intermittently fasted as compared with ad-lib fed mice (Fig. 4E and F). These findings demonstrate that LAMP2 is essential to transduce the benefits of chronic intermittent fasting and restore metabolic homeostasis, and the absence of LAMP2 provokes fasting-induced beta cell death analogous to our prior observations of increased cardiomyocyte loss in intermittently fasted lamp2 null mice.19
To examine if IF-induced beta cell survival would also be LAMP2 dependent in the context of obesity-induced diabetes, we examined the islets of female Lamp2 heterozygous null mice and WT female mice. While IF increased insulin content in WT female mice in both the chow-fed and HF-fed groups (Fig. 5A), it induced a marked decrease in pancreatic insulin content in Lamp2 heterozygous null female mice as compared with the respective ad-lib fed groups (Fig. 5B). Whereas IF increased beta cell area and reversed the decline in beta/alpha cell ratio, and decreased beta cell apoptosis in obese WT female mice (as compared with ad-lib fed obese WT female mice; Fig. 5C–F; Fig. S8C); IF provoked a decline in beta cell area and beta/alpha cell ratio, and increased beta cell apoptosis in obese Lamp2 heterozygous null mice (as compared with ad-lib fed obese Lamp2 heterozygous null mice; Fig. 5G–J; Fig. S8F). These findings were similar to the effects of IF in chow-fed lamp2 null male mice (Fig. 4C–F). Therefore, the effects of IF on beta cell survival depend critically on intact lysosomal function, in so far as IF induces beta cell death in the absence of LAMP2.
Figure 5.
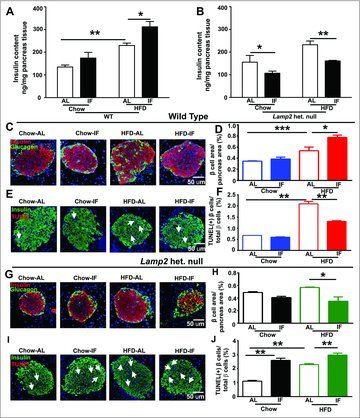
Intermittent fasting provokes beta cell loss in Lamp2 heterozygous null mice with obesity-induced diabetes. (A and B) Insulin content of whole pancreas in WT females (A) and Lamp2 heterozygous null female mice (B) after 12 wk HF diet or chow feeding followed by 6 wk of IF or ad-lib access to original dietary regimen (n = 4 to 6/ group). (C) Representative images of islets of WT female mice showing beta cells (red, anti-insulin) and alpha cells (green, anti-glucagon); (D) Quantification of beta cell area from (C; n = 3 or 4 mice/group); (E) TUNEL staining (red, arrows) in islets costained with anti-insulin (green) and DAPI. (F) Quantification of TUNEL-positive beta cell nuclei in WT females treated as in A (n = 3 or 4 mice/group); (G) Representative images of islets of Lamp2 heterozygous null female mice with staining for beta cells (red, anti-insulin) and alpha cells (green, anti-glucagon). (H) Quantification of beta cell area from (G; n = 3 or 4 mice/group); (I) TUNEL staining (red, arrows) in islets costained with anti-insulin (green) and DAPI in mice treated as in (B). and (J) Quantification of TUNEL-positive beta cell nuclei in in Lamp2 heterozygous null female mice in I (n = 3 or 4 mice/ group). *P < 0.05; **P < 0.001 and ***P < 0.0001.
Intermittent fasting provokes autophagosome accumulation with mitochondrial ultrastructural abnormalities in LAMP2-deficient mice
Autophagy plays an important role in homeostatic beta cell function and survival.14-16 We hypothesized that IF may exacerbate pancreatic beta cell death in LAMP2-deficient mice by impairing autophagy in the setting of impaired lysosome function provoked by loss of LAMP2.27 Since IF provoked a decline in pancreatic insulin content even in Lamp2 heterozygous null mice on a chow diet (Fig. 5B), we reasoned that impaired autophagic flux may be evident even in chow-fed mice after IF. In pancreatic beta cells, we observed punctate staining for LC3B and SQSTM1 (Fig. 6A), which are markers of autophagosomes and the autophagy-substrate, respectively.29,30 Quantitative morphometric analysis of LC3B and SQSTM1 revealed that LC3B and SQSTM1-positive puncta were significantly increased in beta cells from Lamp2 heterozygous null female mice, and IF further worsened the accumulation of LC3B and SQSTM1 puncta as compared with WT controls (Fig. 6B and C). A similar pattern was observed in lamp2 null male mice, whereby IF resulted in further increase in LC3B and SQSTM1 puncta, which were markedly increased in abundance as compared with WT mice even in the ad-lib fed state (Fig. 7A–C). These findings indicate impaired flux through macroautophagy with LAMP2 deficiency, which is worsened by repetitive fasting-induced autophagosome formation in the setting of impaired autophagosome-lysosome fusion.19, 27 To gain further insight into the pathological changes in LAMP2-deficient islets, the ultrastructure of beta cells was examined by transmission electron microscopy. Images from both Lamp2 heterozygous null female and lamp2 null male mice demonstrated accumulation of autophagic vacuoles and swollen mitochondria with rarified cristae, which were further worsened by IF (Fig. 6D and 7D); and accompanied by increased 4-HNE staining (a marker of oxidative stress), likely as a result of increased ROS levels in Lamp2-deficient beta cells (Fig. S11A–C), mimicking our previous observations in Lamp2-deficient myocardium subjected to intermittent fasting.19 This was accompanied by ultrastructural evidence for apoptotic beta cell nuclei in intermittently fasted lamp2 null male mice (Fig. 7D). These findings indicate that in the setting of defective autophagosome-lysosome fusion due to LAMP2 deficiency, IF-induced autophagic impairment results in the accumulation of damaged mitochondria and ROS upregulation with activation of cell death pathways.
Figure 6.
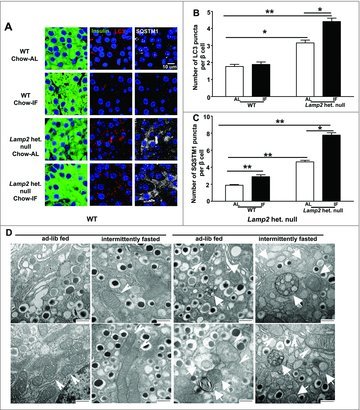
Intermittent fasting provokes autophagosome accumulation with mitochondrial ultrastructural abnormalities in Lamp2 heterozygous null beta cells with obesity-induced diabetes. (A) Representative images of WT or Lamp2 heterozygous null mice fed a chow diet ad-lib or subjected to 6 wk intermittent fasting with coimmunostaining for LC3B and SQSTM1 (anti-insulin, green; anti-LC3B, red; anti-SQSTM1, gray). (B) Quantification of LC3B puncta per beta cell nucleus (n = 3 or 4 mice per group). (C) Quantification of SQSTM1 puncta per beta cell nucleus (n = 3 or 4 mice per group). (D) Representative TEM images of beta cells from mice treated as in (A) with autophagic structures (white arrows) and mitochondria (white arrowheads). Scale bar: 500 nm. *P < 0.05; **P < 0.001 and ***P < 0.0001.
Figure 7.
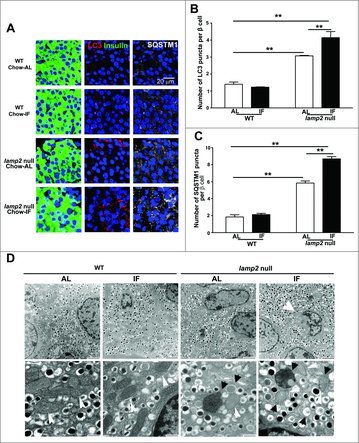
Intermittent fasting worsens autophagy impairment and provoke apoptosis in beta cells of lamp2 null male mice. (A) Representative images demonstrating expression of autophagy markers, LC3B and SQSTM1, in the islets from lamp2 null mice and wild-type controls fed a chow diet with ad-lib access to food or subjected to intermittent fasting for 6 wk (anti-insulin, green; anti-LC3B, red; anti-SQSTM1, gray). (B) Quantification of LC3B puncta from (A); n = 3 or 4 mice per group. (C) Quantification of SQSTM1 puncta from (A); n = 3 or 4 mice per group. (D) Representative electron micrographs of islets from mice treated as in (A). Top row demonstrates islet architecture. White arrow points to an apoptotic nucleus. Scale bar: 2 µm. Bottom row demonstrates organelle characteristics. Black arrows point to autophagic structures. White arrows point to mitochondria. Scale bar: 500 nm. **P < 0.001, ***P < 0.0001.
IF worsens glucose tolerance in obese Becn1 haplo-insufficient mice with defective autophagosome formation
Having determined that IF exacerbates autophagic impairment in the setting of lysosomal dysfunction, we tested whether mice with intact lysosomes but impaired autophagy would exhibit glucose intolerance after IF. Accordingly, we examined mice with partial deficiency of Becn1 (Becn1 heterozygous null, Becn1+/−), a protein essential for autophagosome formation.31 Becn1+/− mice were fed HF diet for 12 wk resulting in weight gain, glucose intolerance and insulin resistance relative to Becn1+/− chow-fed animals (Fig. S12A–E). Although body weights were similar in obese Becn1+/− mice subjected to 6 wk of IF or provided ad-lib access to HF diet (Fig. S13A), intermittently fasted obese Becn1+/− animals exhibited worsened glucose tolerance without altered insulin resistance as compared to ad-lib fed mice on HF diet (Fig. 8A and B; Fig. S13B and C). IF in HF-fed Becn1+/− mice did not increase beta cell area, but resulted in decreased beta/alpha cell ratio and increased TUNEL positivity in pancreatic beta cells, as compared with ad-lib fed Becn1+/− mice (Fig. 8C–F; Fig. S13D), indicating a lack of survival benefit of IF on beta cells in the setting of diet-induced obesity in Becn1+/− mice in contrast to the observations in wild-type mice (Fig. 2A and B; Fig. 5E and F). Consistent with an inability of Becn1+/− mice to form autophagosomes,31 we observed virtually no LC3B puncta (Fig. 8g and H; Fig. S14A and C). Examination of SQSTM1 revealed no differences between chow-fed Becn1+/− vs. Becn1+/+ controls (Fig. S14B and D), and increase in SQSTM1 puncta in HF-fed obese Becn1+/− vs. chow-fed Becn1+/− beta cells (SQSTM1 puncta/beta cell: 0.45 ± 0.06 in HF-fed vs. 0.10 ± 0.03 in chow-fed, P = 0.008, N = 3/group; also compare Fig. 8I and J with Fig. S14B and D) consistent with SQSTM1 accumulation due to impaired autophagic flux with diet-induced obesity as observed in male C57BL/6 mice (Fig. 2E–G). IF provoked a further increase in abundance of the autophagic substrate, SQSTM1, in obese Becn1+/− mice (Fig. 8G–J), attesting to worsening autophagic failure in the setting of IF. This was associated with accumulation of damaged mitochondria with rarified cristae in intermittently fasted Becn1+/− mice (Fig. 8K), pointing to impaired organelle quality control under stress. Moreover, in contrast to the effect of IF on improving beta cell function in wild-type mice (Fig. 1K), IF resulted in a further reduction in glucose-stimulated insulin release in islets from HF-fed Becn-1+/− mice with obesity-induced glucose intolerance (Fig. 8L). Therefore, mice with a specific defect in autophagosome formation exhibit glucose intolerance due to beta cell death and dysfunction in the setting of IF, attesting to a critical need for the macroautophagy function of lysosomes in transducing the metabolic effects of intermittent fasting in beta cells.
Figure 8.
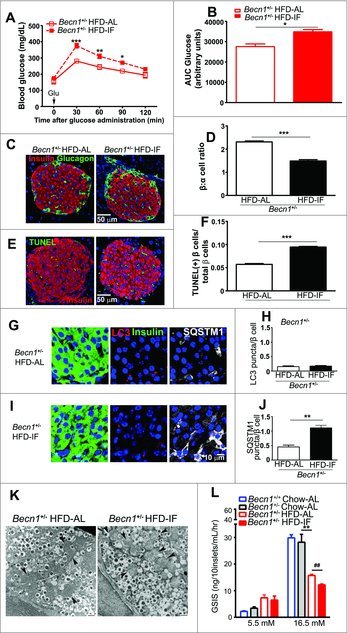
Intermittent fasting exacerbates glucose intolerance in Becn1+/− mice. (A) GTT in Becn1+/− mice fed a HFD for 12 wk followed by ad-lib feeding or intermittent fasting for 6 wk (n = 3/group). (B) AUC from GTT as in (A). (C) Representative images of islets from obese Becn1+/− mice after ad-lib feeding or intermittent fasting as in (A) with staining for beta cells (red, anti-insulin) and alpha cells (green, anti-glucagon). (D) Quantification of beta cell area from C (n = 3 mice/group). (E) TUNEL staining (red, arrows) in islets costained for insulin (green) with DAPI. (F) Quantification of TUNEL-positive beta cell nuclei in mice as in E (n = 3 mice/group). (G) Representative images of islets from Becn1+/− mice as in A with coimmunostaining for LC3B (red) with insulin (green); nuclei are blue (DAPI). (H) Quantification of LC3B puncta from (G; n = 3/group). (I) Representative images of islets from Becn1+/− mice as in (A) with coimmunostaining for SQSTM1 (gray) with insulin (green); nuclei are blue (DAPI). (J) Quantification of SQSTM1 puncta (n = 3/group). (K) Representative TEM images of beta cells from mice treated as in (A) with black arrowheads indicating mitochondria. Scale bar: 500 nm. (L) Glucose stimulated insulin release in isolated islets from Becn1+/− mice fed a HFD for 12 wk followed by ad-lib feeding or intermittent fasting for 6 wk, or Becn1+/− and littermate wild-type (Becn1+/+) mice fed chow ad-lib as controls (n = 30 islets/mouse from n = 3 or 4 mice per group) as in (J). Age matched Becn1+/− mice of both sexes was employed for the above analyses. *P < 0.05; **P < 0.001 and ***P < 0.0001.
Intermittent fasting induced markers of beta cell regeneration in an autophagy-lysosome dependent manner
Recent studies suggest beta cell regeneration as a potential therapy for T2D.23 We therefore tested whether IF may be expanding beta-cell mass via beta cell regeneration. As assayed by NEUROG3/Ngn3 nuclear localization, a marker of endocrine progenitor cells, we did not observe any evidence of pancreatic dedifferentiation in high-fat fed mice (Fig 9A and B), nor did we observe any effect of IF on beta cell regeneration in chow-fed male or female mice (Fig 9A). Interestingly, we observed that IF did induce an increase in NEUROG3 nuclear localization only in HF diet fed mice (in both sexes, Fig 9B). Neurog3 transcripts were downregulated in HFD-fed obese male mice as compared with chow-fed controls, and intermittent fasting was sufficient to increase Neurog3 transcripts in obese male mice (as compared with AL-fed obese controls; Fig. 9E). A transcriptional target of NEUROG3, namely NEUROD1,32 a basic helix-loop-helix transcription factor that transactivates insulin transcription and is critical for beta cell maturation33 was also transcriptionally upregulated by intermittent fasting in obese male mice (Fig. 9F). Taken together with the IF-induced increase in beta cell area in this setting (Fig. 1H and 5D), this finding suggests that IF may stimulate pancreatic regeneration in the setting of high-fat feeding. Surprisingly, we did not observe intermittent fasting-induced NEUROG3 nuclear localization in high-fat-fed Lamp2 heterozygous null mice and Becn1+/− mice (Fig. 9C and D) or chow-fed Lamp2 heterozygous null and lamp2 null mice (Fig. S15). Also, intermittent fasting did not result in transcriptional upregulation of Neurog3 in high-fat fed Lamp2 heterozygous null mice (0.96 ± 0.68-fold in IF vs. 1.02 ± 0.02-fold in AL, N = 3/group, P = NS) with a significant downregulation of Neurod1 (0.11 ± 0.08-fold in IF vs. 1.05 ± 0.19-fold in AL, N = 3/group, P = 0.01) Taken together with the lack of increase in beta cell area with intermittent fasting in these autophagy-lysosome deficient mice (see Fig. 4D, 5H, and 8D), these data indicate that IF may provoke pancreatic beta cell regeneration in a manner that requires an intact autophagy-lysosome pathway.
Figure 9.
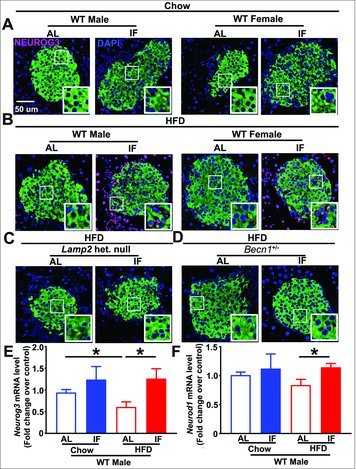
Intermittent fasting increases NEUROG3 expression in high-fat fed mice. (A) Representative images showing immunohistochemical staining for the transcription factor NEUROG3 (anti-NEUROG3, red), insulin (anti-insulin, green) in (A) chow-fed wild-type male and female mice subjected to ad-libitum feeding or intermittent fasting; (B) HFD-fed wild-type male and female mice subjected to ad-libitum feeding or intermittent fasting; and (C and D) HFD-fed Lamp2 heterozygous null female mice (C) or and Becn1+/− mice (D) subjected to ad-libitum feeding or intermittent fasting. Representative from n = 3 mice/group. (E and F) Neurog3 (E) and Neurod1 (F) transcripts in islets from chow and HFD-fed male mice subjected to ad-libitum feeding or intermittent fasting; N = 6 or 7/group. * indicates P < 0.05 by post-hoc test.
Discussion
A combination of genetic and environmental factors has contributed to a growing epidemic of obesity and T2D. Pharmacological therapies for T2D are insufficient to control adverse sequelae,34,35 highlighting the need for sustainable preventive strategies. Caloric restriction and exercise are the chief lifestyle recommendations for patients with T2D, although multiple studies have demonstrated that chronic caloric restriction is not sustainable for the overwhelming majority of patients.36,37 Intermittent fasting is a clinically sustainable strategy that has demonstrated metabolic benefits in humans8,38 and animals,9-11 but little is known about the mechanisms by which IF may result in improved metabolic health.
We have demonstrated that 6 wk of intermittent fasting improves glucose tolerance in high-fat fed WT mice with diet-induced obesity. Overall glucose tolerance is determined predominantly by insulin secretion, insulin action, as well as insulin-independent glucose effectiveness.39 Because IF does not improve insulin resistance as measured by insulin tolerance tests or biochemically determined insulin signaling, it is unlikely that IF improves glucose tolerance by modifying insulin action. Furthermore, the significantly increased levels of insulin measured during glucose tolerance tests are more consistent with a model whereby IF increases insulin secretion in HF fed mice. IF does not improve insulin resistance but increases pancreatic beta cell mass, insulin levels, and glucose stimulated insulin secretion. Additionally, IF decreases beta cell apoptosis as compared to ad-lib high-fat feeding, and stimulates markers of beta cell regeneration. These findings strongly suggest that IF improves glucose tolerance via directly affecting pancreatic islet beta cell health, in a manner that is independent of insulin resistance.
Interestingly, the beneficial effects of IF were consistently observed in male and female WT mice, although weight loss only occurred in male mice after fasting, thus indicating that weight loss is not required for the salutary benefits of IF on pancreatic beta cell survival and function (Table S3). In fact, it is surprising that intermittently fasted male mice fed a HF diet had no change in insulin tolerance, despite weight loss. Female mice did not lose weight during IF and exhibited worsening insulin resistance based on AUC measurements. One possible explanation for these findings is that IF per se had negative effects on insulin resistance in female mice, with neutral effects in male mice due to offsetting effects of weight loss on insulin action. Our data examining insulin-induced AKT phosphorylation in heart, skeletal muscle, and liver do not reveal further worsening with IF in male mice with diet-induced obesity (Fig. S3). Alternatively, IF may have resulted in upregulation of counter-regulatory mechanisms that increase glucose in the setting of hypoglycemia such as glucagon (Fig. S2D), and potentially others including growth hormone, cortisol, or catecholamines. Interestingly, growth hormone modulation of hepatic autophagy has recently been shown to protect mice against hypoglycemia.40 In this context, it is tempting to speculate that IF-induced increases in autophagic flux may protect mice against hypoglycemia.
While multiple studies have demonstrated a critical role for autophagy in preservation of beta cell homeostasis,16-18, 20, 41 recent findings indicate that flux through the macroautophagy machinery may be downregulated during acute starvation to prevent inappropriate release of insulin in settings of nutrient deprivation.42 In contrast, the observed accumulation of damaged organelles and autophagosomes in human islets from diabetic patients 15 and in mice with diet-induced obesity (Fig. 2E-G) suggests that impaired autophagic flux may be a key contributory factor to beta cell loss and dysfunction during conditions of nutrient excess. Our data demonstrate that impaired autophagic flux and decreased transcript levels of the autophagy master regulator TFEB accompany beta cell loss in islets from mice with diet-induced obesity (Fig. 2E and F). The observation that lysosomes are required for fasting-induced insulin granule degradation42 as well as maintenance of autophagic flux43 for beta cell homeostasis41, 44 indicates that stimulation of lysosome function is an integrative mechanism for achieving homeostasis during fasting and for facilitating organelle quality control under metabolic stress in beta cells. Indeed, our data demonstrate that the salutary effects of IF on beta cell health require intact lysosomal machinery. Consequently, while IF restores impaired autophagic flux in islets of wild-type obese mice, IF worsens autophagy-impairment in chow-fed LAMP2-deficient mice, as evident by the accumulation of autophagosome-bound LC3B and SQSTM1, an autophagy substrate. When the autophagy-lysosome pathway is genetically perturbed, such as in LAMP2-deficient beta cells, IF worsens organelle quality control. Since damaged mitochondria require an intact autophagy-lysosome pathway for their removal, the accumulation of these organelles triggers apoptotic signaling.43
In addition to a critical role for lysosomes with IF, our studies in the Becn1 haplo-insufficient mice indicate the need for intact autophagy initiation machinery for prevention of beta cell death under diabetic stress. Prior studies also support that autophagic machinery is necessary for beta cell health; studies on Atg7-deficient mice have suggested that autophagy is essential for maintaining normal beta cell architecture and insulin secretion.16, 18, 41, 44 However, the autophagy-lysosome pathway may play a tissue-specific role in diabetes, as an adipose-specific knockout of Atg7 results in increased insulin sensitivity and decreased white adipose mass,45 and lamp2 null male mice are reported to be resistant to diet-induced obesity.28 Furthermore, Atg7 heterozygosity exacerbates disease progression in ob/ob mice, an effect that is ameliorated by trehalose, a stimulator of autophagy-lysosome pathway.46 Thus, our finding that IF fails to ameliorate peripheral insulin resistance while improving beta cell function may be due to tissue-specific roles for the autophagy-lysosome pathway in diabetic disease progression.
A novel finding of our study is that after HF-feeding IF stimulates markers of beta cell regeneration including induction of NEUROG3 expression in the nucleus, a marker of endocrine progenitor cells,23, 47 and upregulation of its transcriptional target, NEUROD1, a known transactivator of insulin transcription that plays a critical role in beta cell maturation.32, 33 Our findings are in agreement with the recent description that a fasting-mimicking diet promotes NEUROG3 nuclear localization and beta cell regeneration in genetic and pharmacological models of diabetes,47 and extend those findings to a clinically relevant model of diet-induced obesity and diabetes. Our findings suggest that IF alone, rather than a fasting mimicking diet, may be sufficient to provoke beta cell differentiation in diet-induced obesity. Furthermore, our finding that beta cell regeneration was dependent on an intact autophagy-lysosome pathway is consistent with prior findings that regeneration is downstream of mammalian target of rapamycin.47 Future studies with lineage tracing and targeted ablation strategies will be required to confirm beta cell regeneration in this model.
In this study, we employed models with global ablation of LAMP2 and BECN1 that do not demonstrate basal defects in glucose handling (see Fig. S5, S8, and S11), as mice with beta cell-specific autophagy defects demonstrate metabolic derangements in the unstressed state.16, 17 Our findings corroborate the notion that intact autophagy is essential for beta cell survival not only under homeostatic conditions, but also proteotoxic and gluco-lipotoxic stress.18, 20, 41 Additionally, we identify IF as a therapeutic target that can be entrained to enhance beta cell survival during persistent diabetogenic stress. Perhaps the utility of IF or time restricted feeding48 may be due to the restoration of periods of nutrient deprivation, leading to oscillations in MTOR activity. Indeed, intermittent dosing of the MTOR inhibitor rapamycin has recently been shown to improve the metabolic side-effect profile of the drug.49 We demonstrate that repetitive fasting, a potent stimulus for pulsed activation of the autophagy-lysosome pathway,7, 10, 12, 19, 38 can be employed as a sustainable strategy towards diabetic prevention and treatment. These studies point to exciting therapeutic possibilities of intermittent activation of the autophagy-lysosome pathway, perhaps via small molecules such as trehalose or pharmacological approaches such as rapamycin, to address the cellular derangements that contribute to diabetes progression.
Materials and methods
Studies with mice
Eight-wk old C57BL/6J mice and Becn1+/− mice were obtained from Jackson Laboratories. lamp2 null mice have been previously described.27 Animal studies were approved by the Animal Studies Committee at the Washington University School of Medicine. Animals were maintained in a temperature-controlled room (22°C) on a 12 h light/dark cycle (lights on at 6:00 AM). All mice were divided into 2 groups and fed either a chow (Lab Diet, 5053) or high-fat diet (HF-diet, Harlan-Teklad, TD.96132) for 12 wk prior to initiation of IF for 6 wk. Diet compositions are presented in Table S1. Mice were then randomized into 2 groups, an intermittent fasting (IF) group or ad-lib fed control group.
Intermittent fasting
Adult mice were fed chow or HF-diet, while housed on cedar/pine chip bedding. IF was performed with total food deprivation and ad-lib access to water from 12:00 PM to 12:00 PM of the following day to implement alternate periods of 24-h fasting and feeding. Daily food intake per cage was weighed to calculate average daily food intake per mouse. Mice were weighed at weekly intervals on fed days. Terminal studies were initiated in the morning subsequent to an overnight period of feeding.
Glucose and insulin tolerance tests
Glucose and insulin tolerance tests were performed on mice fasted for 5 h in the morning. Glucose (1 g/kg body weight) or insulin (0.75 U/kg body weight) was administered via intraperitoneal injection. Blood glucose concentrations were measured with a Contour TS glucometer (Bayer, Parsipanny, NJ). Glucose measurements were performed at baseline and 30, 60, 90, and 120 min after glucose injection. To assess insulin action at the tissue level, mice fasted for 5 h were injected with insulin 0.75 U/kg intraperitoneally and tissues were harvested 10 min later.
Immunohistochemistry
Paraffin-embedded mouse islet sections were deparaffinized using xylene and rehydrated with graded alcohol. Antigen retrieval was carried out by microwaving the sections in 1 mM EDTA, pH 8.0, for 15 min at high power followed by blocking in 1% BSA (Santa Cruz Biotechnology, sc-2323), 4% normal donkey serum (Abcam, ab166643) for 1 h at room temperature. Indirect immunofluorescence was performed using primary antibodies, rabbit anti-insulin (1:100; Cell Signaling Technology, 3014S), guinea pig anti-insulin (1:100; abcam, ab7842), mouse anti-MKI67/Ki-67 (1:100; Dako, M7249), mouse anti-glucagon(1:500; Sigma, G2654); and secondary antibodies, namely, Alexa Fluor 594 donkey anti-rabbit (1:500), Alexa Fluor 680 donkey anti-mouse, Alexa Fluor 488 donkey anti-guinea and Alexa Fluor 488 donkey anti-mouse (1: 500; Invitrogen; A21207, A10038, A11073 and A21202 respectively). Nuclei were counterstained with 4′, 6-diamindino-2-phenylindole (DAPI; Vector Laboratories, H1200). Immunohistochemical detection of NEUROG3 was performed with anti-NEUROG3/Ngn3 (LifeSpan BioSciences, Inc., C97692/53897; 1:1,000), as previously described.23, 47, 50 Images were acquired using a Zeiss Confocal LSM 700 Laser Scanning confocal microscope (Need address) using 40X Zeiss Plan-Neofluar 40X/1.3 and 63X/1.4 oil immersion objectives. Beta cell area was assessed as previously described.51
Assessment of TUNEL positivity
TUNEL staining was performed on paraffin embedded mouse pancreas using ApopTag Red In-situ Apoptosis Detection Kit (Millipore, S7165) following the manufacturer's instructions.
Mouse islet isolation and immunoblotting
Islets were isolated from C57BL/6J mice (20 to 30 g) by collagenase (Sigma type XI, C-7657) digestion as described previously.52 Briefly, pancreatic tissues were perfused with 5 ml collagenase solution (HBSS without calcium and magnesium [Gibco, 14175–079], 10 mM HEPES, pH 7.4) and placed into 50-ml conical tubes with 2.5 ml of collagenase solution. After 15 min of incubation in a 37°C water bath and intermittent agitation, islets were rinsed and separated on a 70 µm nylon cell strainer (BD Falcon, 352350) and then placed in RPMI 1640 culture medium (Gibco, 11875–085) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 8.0 mM glucose, 100 units/ml penicillin, 100 µg/ml streptomycin and cultured overnight. For assessment of autophagic flux, mice were injected with chloroquine (60 mg/kg; Sigma, C6628) or equivalent volume of phosphate-buffered saline (Gibco, 14190–136), i.p., 4 h prior to sacrifice for islet isolation. 150 to 200 islets/mouse were subjected to crude extraction of proteins in RIPA buffer (Sigma, R0278) containing protease and phosphatase inhibitor cocktail (Thermo Scientific, 78442) and subjected to immunoblotting as previously described.19 Antibodies employed were as follows: LC3B (Novus Biologicals, NB100–2220), SQSTM1 (ab5416, Abcam); phospho-AKT (Ser473; Cell Signaling Technology, 9271), total AKT (Cell Signaling Technology, 9272), TFEB (Bethyl Labs, A303–673A), BECN1/Beclin-1 (Abcam, ab16998), LAMP2 (mouse monoclonal for rat tissue, kind gift of Michel Jadot (Universite de Namur, Belgium) as previously described for detecting LAMP2 of rat origin53), ACTA1/actin (Sigma, A2066) and GAPDH (Abcam, ab22555).
Insulin secretion and islet insulin content
Insulin secretion was measured by static incubation for 60 min with RPMI 1640 medium containing 5 mM or 16.5 mM glucose. Measurements were performed in triplicate in samples of 10 islets, each. For insulin content determination, 10 islets in triplicate from each group were washed with 0.05% BSA-containing phosphate-buffered saline solution and subject to acid/ethanol extraction. Insulin concentrations were determined via an insulin ELISA kit (Millipore, EZRMI-13K,). Serum glucagon was assayed with Glucagon ELISA kit (Mercodia, 10–1281-01); and serum C-peptide levels were determined with an ELISA kit (ALPCO, 80-CPTMS-E01) following the manufacturer's instructions.
Quantitative PCR analysis
Total RNA was extracted from islets (50 to 100) from each mouse using the Qiagen RNAeasy kit (74104) followed by reverse transcriptase reaction and quantitative PCR analysis was performed as described previously,19 on QuantStudio 3 Real-Time PCR system (Applied Biosystems, A28136). Following primer sets were employed: Tfeb: forward 5′- GTCTAGCAGCCACCTGAACGT-3′, reverse 5′-ACCATGGAGGCTGTGACC TG-3′; Gapdh: forward 5′ -ACTCCCACTCTTCCACCTTC-3′, reverse 5′-TCTTGCTCAGTGTCCTTGC-3′; 3′; Neurog3: forward 5′-TCCGAAGCAGAAGTGGGTGACT-3′, reverse 5′- CGGCTTCTTCGCTGTTTGCTGA-3′; Neurod1: forward 5′- CCTTGCTACTCCAAGACCCAGA-3′, reverse 5′- TTGCAGAGCGTCTGTACGAAGG-3′.
Detection of ROS in pancreas sections
We performed immunostaining for 4-HNE adducts on paraffin embedded tissue with anti-4HNE antibody (Abcam, ab48506; 1:25 dilution) in pancreatic sections from mice modeled for diet-induced obesity, per the manufacturer's instructions, as previously described.54
Whole pancreas insulin content
After euthanasia, the pancreas was isolated and wet weight obtained. The pancreas was minced and homogenized with a mechanical homogenizer in acid/ethanol extraction buffer (1ml per 100 mg tissue; 77% 100 proof ethanol, 1.5% HCL). The extract was incubated at 4°C overnight and centrifuged at 400 X g for 30 min at 4°C. Supernatants were stored at −20°C until analysis of insulin ELISAs were performed at 1:5000 dilution.
Electron microscopy
Mouse pancreas was fixed in a modified Karnovsky fixative of 3% glutaraldehyde, 1% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4; followed by postfixation in 2% osmium tetroxide in 0.1 M sodium cacodylate buffer for 1 h. The tissue layers were then stained with 2% aqueous uranyl acetate for 30 min, dehydrated in series of graded ethanol concentrations and embeded in PolyBedTM 185 (Polysciences, 08792–1). Blocks were sectioned at 90-nm thickness, post stained with Venable lead citrate and viewed with a JEOL model 1200 EX electron microscope (JEOL, Tokyo, Japan). Digital images were acquired using the AMT Advantage HR (Advanced Microscopy Techniques, Danvers, MA) high definition CCD 1.3 megapixel TEM camera. N = 2 mice were examined in each group.
Studies with the INS1 832/13 cell line
INS-1 832/13 rat insulinoma cells,26 a kind gift from Dr. Christopher Newgard (Duke University School of Medicine, U.S.A.) were transduced with adenoviral particles coding for shRNAs targeting rat Tfeb, Becn1 and Lamp2 or shLacZ as control; or adenoviruses for overexpression of human TFEB or LacZ as control, as we have previously described.19, 55 After 48 h, these cells were modeled for gluco-lipotoxicity by culturing in a high glucose medium (30 mM) followed by palmitate treatment (0.4 mM) for 18 h and terminal assessment of cell death the Live-Dead Cytotoxicity Viability kit for mammalian cells (Invitrogen, L3224), per the manufacturer's instructions as described previously.19
Statistical analysis
Results are expressed as mean ± SEM. Assumptions of normality and equality of variance were verified, and statistical differences were assessed with the unpaired 2-tailed Student t test for 2 experimental groups, one-way and two-way ANOVA with Bonferroni correction where appropriate (Prism, Version 5.2). Nonparametric tests were employed for data that was not normally distributed. A 2-tailed P value of less than 0.05 was considered statistically significant.
Supplementary Material
Funding Statement
This study was supported by grants from National Institutes of Health (HL107594; and DRC, Grant No. P30 DK020579) and Department of Veterans Affairs (I01BX000448, 1I01BX001969) to A.D, from the NIH (R01 DK098584) to MSR; and the Deutsche Forschungsgemeinschaft to P.S. AJ was supported by K08HL138262-01 and 5-T32-HL07081–40 from the NHLBI. We also acknowledge support from the NIH Shared Instrumentation Grant (S10 RR027552) for support through the Hope Center Neuroimaging Core; and support from the Electron Microscopy Core and Advanced Imaging and Tissue Analysis Core of the Digestive Disease Research Core Center (DDRCC) at Washington University School of Medicine.
Abbreviations
- AL
ad. libitum
- CQ
chloroquine
- GTT
glucose tolerance test
- GSIS
glucose stimulated insulin secretion
- HF
high-fat diet
- ITT
insulin tolerance test
- IF
intermittent fasting
- LAMP2
lysosomal-associated membrane protein 2
- MAP1LC3B (LC3B)
microtubule-associated proteins 1A/1B light chain 3B
- NEUROG3
Neurogenin 3
- SQSTM1
Sequestosome 1
- T2D
Type 2 diabetes
- TFEB
Transcription factor EB
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Clay F. Semenkovich, Washington University, for helpful scientific discussions.
References
- [1].Polonsky KS. The past 200 years in diabetes. N Engl J Med. 2012; 367:1332 40. https://doi.org/ 10.1056/NEJMra1110560 PMID:23034021 [DOI] [PubMed] [Google Scholar]
- [2].Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006; 444:840 6. https://doi.org/ 10.1038/nature05482. PMID:17167471 [DOI] [PubMed] [Google Scholar]
- [3].Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, Keinänen-Kiukaanniemi S, Laakso M, Louheranta A, Rastas M, et al.. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001; 344:1343 50. https://doi.org/ 10.1056/NEJM200105033441801. PMID:11333990 [DOI] [PubMed] [Google Scholar]
- [4].Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science 1996;273:59 63. https://doi.org/ 10.1126/science.273.5271.59. PMID:8658196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fontana L, Meyer TE, Klein S, Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci U S A. 2004; 101:6659 63. https://doi.org/ 10.1073/pnas.0308291101. PMID:15096581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fontana L, Partridge L, Longo VD. Extending healthy life span–from yeast to humans. Science. 2010; 328:321 6. https://doi.org/ 10.1126/science.1172539. PMID:20395504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014; 19:181 92. https://doi.org/ 10.1016/j.cmet.2013.12.008. PMID:24440038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, Dubeau L, Yap LP, Park R, Vinciguerra M, et al.. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. 2015; 22:86 99. https://doi.org/ 10.1016/j.cmet.2015.05.012. PMID:26094889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Baumeier C, Kaiser D, Heeren J, Scheja L, John C, Weise C, Eravci M, Lagerpusch M, Schulze G, Joost HG, et al.. Caloric restriction and intermittent fasting alter hepatic lipid droplet proteome and diacylglycerol species and prevent diabetes in NZO mice. Biochim Biophys Acta. 2015; 1851:566 76. https://doi.org/ 10.1016/j.bbalip.2015.01.013. PMID:25645620 [DOI] [PubMed] [Google Scholar]
- [10].Wan R, Camandola S, Mattson MP. Intermittent fasting and dietary supplementation with 2-deoxy-D-glucose improve functional and metabolic cardiovascular risk factors in rats. FASEB J. 2003; 17:1133 4. PMID:12709404 [DOI] [PubMed] [Google Scholar]
- [11].Pedersen CR, Hagemann I, Bock T, Buschard K. Intermittent feeding and fasting reduces diabetes incidence in BB rats. Autoimmunity. 1999; 30:243 50. https://doi.org/ 10.3109/08916939908993805. PMID:10524500 [DOI] [PubMed] [Google Scholar]
- [12].Anson RM, Guo Z, de Cabo R, Iyun T, Rios M, Hagepanos A, Ingram DK, Lane MA, Mattson MP. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc Natl Acad Sci U S A. 2003; 100:6216 20. https://doi.org/ 10.1073/pnas.1035720100. PMID:12724520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gonzalez CD, Lee MS, Marchetti P, Pietropaolo M, Towns R, Vaccaro MI, Watada H, Wiley JW. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011; 7:2 11. https://doi.org/ 10.4161/auto.7.1.13044. PMID:20935516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fujimoto K, Hanson PT, Tran H, Ford EL, Han Z, Johnson JD, Schmidt RE, Green KG, Wice BM, Polonsky KS. Autophagy regulates pancreatic beta cell death in response to Pdx1 deficiency and nutrient deprivation. J Biol Chem 2009; 284:27664 73. https://doi.org/ 10.1074/jbc.M109.041616. PMID:19654319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marchetti P, Masini M. Autophagy and the pancreatic beta-cell in human type 2 diabetes. Autophagy. 2009; 5:1055 6. https://doi.org/ 10.4161/auto.5.7.9511. PMID:19657235 [DOI] [PubMed] [Google Scholar]
- [16].Shigihara N, Fukunaka A, Hara A, Komiya K, Honda A, Uchida T, Abe H, Toyofuku Y, Tamaki M, Ogihara T, et al.. Human IAPP-induced pancreatic beta cell toxicity and its regulation by autophagy. J Clin Invest. 2014; 124:3634 44. https://doi.org/ 10.1172/JCI69866. PMID:25036706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim J, Cheon H, Jeong YT, Quan W, Kim KH, Cho JM, Lim YM, Oh SH, Jin SM, Kim JH, et al.. Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. J Clin Invest. 2014; 124:3311 24. https://doi.org/ 10.1172/JCI69625. PMID:25036705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rivera JF, Costes S, Gurlo T, Glabe CG, Butler PC. Autophagy defends pancreatic beta cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest. 2014; 124:3489 500. https://doi.org/ 10.1172/JCI71981. PMID:25036708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Godar RJ, Ma X, Liu H, Murphy JT, Weinheimer CJ, Kovacs A, Crosby SD, Saftig P, Diwan A. Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy. 2015; 11:1537 60. https://doi.org/ 10.1080/15548627.2015.1063768. PMID:26103523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, et al.. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008; 8:325 32. https://doi.org/ 10.1016/j.cmet.2008.08.009. PMID:18840363 [DOI] [PubMed] [Google Scholar]
- [21].Marchetti P, Masini M. Autophagy and the pancreatic beta-cell in human type 2 diabetes. Autophagy. 2009; 5:1055 6. https://doi.org/ 10.4161/auto.5.7.9511. PMID:19657235 [DOI] [PubMed] [Google Scholar]
- [22].Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003; 52:102 10. https://doi.org/ 10.2337/diabetes.52.1.102. PMID:12502499 [DOI] [PubMed] [Google Scholar]
- [23].Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014; 19:872 82. https://doi.org/ 10.1016/j.cmet.2014.03.010. PMID:24746806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Settembre C, Di MC, Polito VA, Garcia AM, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al.. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429 33. https://doi.org/ 10.1126/science.1204592. PMID:21617040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Settembre C, De CR, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, et al.. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. NatCell Biol. 2013; 15(6):647 58. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000; 49:424 30. https://doi.org/ 10.2337/diabetes.49.3.424. PMID:10868964 [DOI] [PubMed] [Google Scholar]
- [27].Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000; 406:902 6. https://doi.org/ 10.1038/35022595. PMID:10972293 [DOI] [PubMed] [Google Scholar]
- [28].Yasuda-Yamahara M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Haneda M, Ugi S, et al.. Lamp-2 deficiency prevents high-fat diet-induced obese diabetes via enhancing energy expenditure. Biochem Biophys Res Commun. 2015; 465:249 55. https://doi.org/ 10.1016/j.bbrc.2015.08.010. PMID:26271596 [DOI] [PubMed] [Google Scholar]
- [29].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3B, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000; 19:5720 8. https://doi.org/ 10.1093/emboj/19.21.5720. PMID:11060023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005; 171:603 14. https://doi.org/ 10.1083/jcb.200507002. PMID:16286508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al.. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003; 112:1809 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Huang HP, Liu M, El-Hodiri HM, Chu K, Jamrich M, Tsai MJ. Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol Cell Biol. 2000; 20:3292 307. https://doi.org/ 10.1128/MCB.20.9.3292-3307.2000. PMID:10757813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gu C, Stein GH, Pan N, Goebbels S, Hornberg H, Nave KA, Herrera P, White P, Kaestner KH, Sussel L, et al.. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 2010; 11:298 310. https://doi.org/ 10.1016/j.cmet.2010.03.006. PMID:20374962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hayward RA, Reaven PD, Emanuele NV, Investigators V. Follow-up of glycemic control and cardiovascular outcomes in type 2 Diabetes. N Engl J Med 2015; 373:978. PMID:26332555 [DOI] [PubMed] [Google Scholar]
- [35].Action to Control Cardiovascular Risk in Diabetes Study G, Gerstein HC, Miller ME, Byington RP, Goff DC Jr., Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, et al.. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358:2545 59. https://doi.org/ 10.1056/NEJMoa0802743. PMID:18539917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wing RR, Rosen RC, Fava JL, Bahnson J, Brancati F, Gendrano Iii IN, Kitabchi A, Schneider SH, Wadden TA. Effects of weight loss intervention on erectile function in older men with type 2 diabetes in the Look AHEAD trial. J Sex Med. 2010; 7:156 65. https://doi.org/ 10.1111/j.1743-6109.2009.01458.x. PMID:19694925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sumithran P, Prendergast LA, Delbridge E, Purcell K, Shulkes A, Kriketos A, Proietto J. Long-term persistence of hormonal adaptations to weight loss. N Engl J Med. 2011; 365:1597 604. https://doi.org/ 10.1056/NEJMoa1105816. PMID:22029981 [DOI] [PubMed] [Google Scholar]
- [38].Harvie MN, Pegington M, Mattson MP, Frystyk J, Dillon B, Evans G, Cuzick J, Jebb SA, Martin B, Cutler RG, et al.. The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: a randomized trial in young overweight women. Int J Obes. 2011; 35:714 27. https://doi.org/ 10.1038/ijo.2010.171. PMID:20921964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, McGuinness OP; NIH Mouse Metabolic Phenotyping Center Consortium . Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech. 2010; 3:525 34. https://doi.org/ 10.1242/dmm.006239. PMID: 20713647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang Y, Chen ML, Zhou Y, Yi L, Gao YX, Ran L, Chen SH, Zhang T, Zhou X, Zou D, et al.. Resveratrol improves hepatic steatosis by inducing autophagy through the cAMP signaling pathway. Mol Nutr Food Res. 2015; 59:1443 57. https://doi.org/ 10.1002/mnfr.201500016. PMID: 25943029 [DOI] [PubMed] [Google Scholar]
- [41].Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, et al.. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metabo. 2008; 8:318 24. https://doi.org/ 10.1016/j.cmet.2008.08.013. PMID:18840362 [DOI] [PubMed] [Google Scholar]
- [42].Goginashvili A, Zhang Z, Erbs E, Spiegelhalter C, Kessler P, Mihlan M, Pasquier A, Krupina K, Schieber N, Cinque L, et al.. Insulin granules. Insulin secretory granules control autophagy in pancreatic beta cells. Science. 2015; 347:878 82. https://doi.org/ 10.1126/science.aaa2628. PMID:25700520 [DOI] [PubMed] [Google Scholar]
- [43].Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Developmental cell. 2004; 6:463 77. https://doi.org/ 10.1016/S1534-5807(04)00099-1. PMID:15068787 [DOI] [PubMed] [Google Scholar]
- [44].Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, et al.. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008; 8:325 32. https://doi.org/ 10.1016/j.cmet.2008.08.009. PMID:18840363 [DOI] [PubMed] [Google Scholar]
- [45].Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009; 119:3329 39. PMID:19855132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lim YM, Lim H, Hur KY, Quan W, Lee HY, Cheon H, Ryu D, Koo SH, Kim HL, Kim J, et al.. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun. 2014; 5:4934. https://doi.org/ 10.1038/ncomms5934. PMID:25255859 [DOI] [PubMed] [Google Scholar]
- [47].Cheng CW, Villani V, Buono R, Wei M, Kumar S, Yilmaz OH, Cohen P, Sneddon JB, Perin L, Longo VD. Fasting-mimicking diet promotes Ngn3-driven beta-cell regeneration to reverse diabetes. Cell. 2017; 168:775–88 e12. https://doi.org/ 10.1016/j.cell.2017.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chaix A, Zarrinpar A, Miu P, Panda S. Time-restricted feeding is a preventative and therapeutic intervention against diverse nutritional challenges. Cell Metab. 2014; 20:991 1005. https://doi.org/ 10.1016/j.cmet.2014.11.001. PMID:25470547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, Pumper CP, Kimple ME, Lamming DW.. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell. 2016; 15:28 38. https://doi.org/ 10.1111/acel.12405. PMID:26463117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Remedi MS, Kurata HT, Scott A, Wunderlich FT, Rother E, Kleinridders A, Tong A, Brüning JC, Koster JC, Nichols CG. Secondary consequences of beta cell inexcitability: identification and prevention in a murine model of K(ATP)-induced neonatal diabetes mellitus. Cell Metab. 2009; 9:140 51. https://doi.org/ 10.1016/j.cmet.2008.12.005. PMID:19187772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Fujimoto K, Hanson PT, Tran H, Ford EL, Han Z, Johnson JD, Schmidt RE, Green KG, Wice BM, Polonsky KS. Autophagy regulates pancreatic beta cell death in response to Pdx1 deficiency and nutrient deprivation. J Biol Chem. 2009; 284:27664 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yang C, Diiorio P, Jurczyk A, O'Sullivan-Murphy B, Urano F, Bortell R. Pathological endoplasmic reticulum stress mediated by the IRE1 pathway contributes to pre-insulitic beta cell apoptosis in a virus-induced rat model of type 1 diabetes. Diabetologia. 2013; 56:2638 46. https://doi.org/ 10.1007/s00125-013-3044-4. PMID:24121653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012; 125:3170 81. https://doi.org/ 10.1161/CIRCULATIONAHA.111.041814. PMID:22592897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Liou GY, Storz P. Detecting reactive oxygen species by immunohistochemistry. Methods Mol Biol. 2015; 1292:97 104. https://doi.org/ 10.1007/978-1-4939-2522-3_7. PMID:25804750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy. 2012; 8:297 309. https://doi.org/ 10.4161/auto.18658. PMID:22302006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.