

Abstract
Autophagic and mitophagic defects are consistently observed in Alzheimer's disease-affected brains. However, the mechanistic defects underlying these anatomical lesions remained unexplained. We have delineated a molecular cascade by which PSEN1 and PSEN2 (presenilins 1 and 2) control PINK1 transcription and function by an AICD-mediated FOXO3a-dependent mechanism. Further, we establish that PARK2 (parkin) acts upstream to PINK1 and regulates its function by a PSEN-dependent mechanism. Our study thus demonstrates a functional interplay between PSEN and PINK1 and establishes a feedback process by which PARK2 and PINK1 could control mitochondrial dysfunction and autophagic processes in various neurodegenerative pathologies including Alzheimer's and Parkinson's diseases.
KEYWORDS: γ-secretase, AICD, Alzheimer disease, FOXO3/Foxo3a, in vivo, mitophagy, PARK2/parkin, Parkinson disease, PINK1, transgenic mice, transcription
Alzheimer disease (AD) is the largest age-related neurodegenerative syndrome, the anatomical lesions of which have been extensively documented. Besides primarily described canonical senile plaques and neurofibrillary tangles, additional stigmata include mitochondrial dysfunction and alterations of autophagy/mitophagy processes. The molecular defects accounting for altered macroautophagic/autophagic functions remained to be elucidated. However, the link between the accumulation of autophagic vacuoles and autophagosome maturation on the one hand and Aβ overload on the other hand in AD-affected brains, indicates that autophagic perturbation could well be at the center of gravity of the neurodegenerative process.
Both familial and sporadic AD is tightly linked to proteolytic dysfunctions resulting in either enhanced APP (amyloid β [A4] precursor protein) processing or altered Aβ degradation, respectively. One of the key enzymes involved in APP processing is γ-secretase, an acidic proteolytic multiprotein complex responsible for the ultimate enzymatic step yielding Aβ and its C-terminal counterpart AICD (APP intracellular domain). The catalytic core of the γ-secretase complex is mainly harbored by PSEN1 (presenilin 1) and to a lesser extent, by its parent protein PSEN2. Most of the familial cases of AD are due to autosomal dominant mutations located in PSEN1.
Recently, we delineated a molecular cascade where PSEN1 plays a central role in modulating the mitophagic process in cells as well as in AD-like transgenic mice models. The bedrock of our reasoning to explore the role of PSEN1 in mitochondrial defects taking place in AD stands on several key observations. First, PINK1 (PTEN induced putative kinase 1) controls mitochondrial physiology and mitophagy. Second, the expression of PTEN that controls PINK1 is drastically altered in selective areas of AD-affected brains. Third, PSEN1 controls the levels of PTEN in a γ-secretase-independent manner. Our study first demonstrated that PSEN1 affects Pink1 promoter transactivation, mRNA levels and protein expression. This regulation is clearly linked to PSEN1-dependent enzymatic function because PSEN1-induced modulation of PINK1 can be fully prevented both by γ-secretase inhibitors, in vitro and in vivo in AD mice models, and PSEN1 mutations that abolish its catalytic activity. Interestingly, PSEN1-mediated control of PINK1 is fully independent of PTEN, corroborating previous data showing that PSEN1-linked modulation of PTEN is γ-secretase-independent.
Whether PSEN1-mediated PINK1 modulation is indeed APP dependent remained a matter of question because PSEN1 had been shown to cleave several tens of natural substrates. We resolved this issue by showing that APP expression increases Pink1 mRNA and protein levels, whereas App gene knockout abolishes this phenotype. Accordingly, we showed that PSEN1 overexpression and γ-secretase inhibitors do not affect Pink1 mRNA levels in App knockout cells. Thus, endogenous APP mediates the PSEN1-induced γ-secretase-mediated effect on PINK1.
We and others previously documented the transcription factor function of AICD. Thus, this APP fragment was envisioned as the likely APP catabolite controlling Pink1 transcription. Indeed, we showed that AICD overexpression accounts for all APP-related effects on PINK1. This was further strengthened by our demonstration that an APP construct lacking the C-terminal moiety corresponding to the AICD domain fails to modulate PINK1. We established that AICD needs Foxo-3a (forkhead box O3) as a cofactor to trigger its effect on Pink1 transcription and expression.
To examine the functional link between AICD and PINK1, we assessed the influence of AICD overexpression on MAP1LC3/LC3 and SQSTM1/p62, 2 proteins that drive autophagosome formation and autophagic degradation in cells but also in vivo after adenoviral expression. We also examined the influence of AICD on TIMM23 and TOMM20, 2 markers allowing monitoring of mitophagy. AICD expression triggers increased LC3-II and reduced SQSTM1, TIMM23 and TOMM20 expression, suggesting the promotion of a pro-autophagic phenotype. Of utmost importance, AICD-mediated control of the autophagic process was fully abolished by PINK1 depletion.
We previously established that besides its well-characterized ubiquitin-ligase function, PARK2 could also behave as a transcription factor. Its first identified transcriptional target is the tumor suppressor Trp53/p53. Later, we established that PARK2 can upregulate Psen1 transcription while it lowers that of Psen2. This intriguing phenotype was reminiscent of our observation that endogenous PSEN1 and PSEN2 trigger opposite effects on Pink1 mRNA and protein expression. Thus, while PSEN1 enhances Pink1 promoter transactivation, PSEN2 induces its reduction. This led us to question whether PARK2 could control Pink1 in a PS-dependent manner. Three lines of independent evidence allowed us to conclude that it was indeed the case. First, PARK2 overexpression increases Pink1 mRNA levels and promoter transactivation, while depletion of endogenous PARK2 triggers the opposite phenotype. Second, PARK2-induced modulation of PINK1 is fully independent of PTEN. Third, the PARK2-associated effect on PINK1 is fully abolished by PS depletion.
The demonstration that PARK2 can act upstream of PINK1 and modulate its expression breaks the dogma that PINK1 occurs always and uniquely upstream to PARK2 to recruit it to damaged mitochondria. Overall, we have delineated a molecular cascade that links PARK2 and PINK1 by a PS/γ-secretase-associated and AICD-mediated transcriptional process (Fig. 1).
Figure 1.
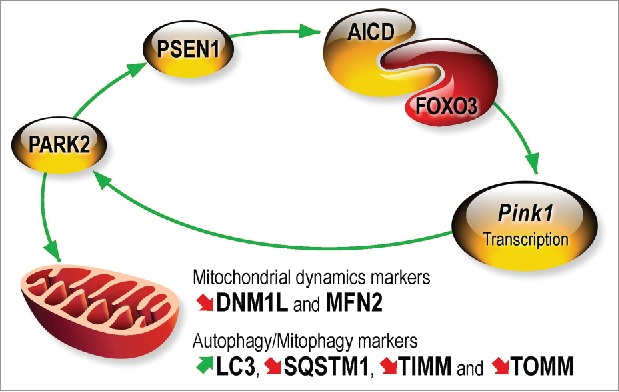
Molecular cascade linking PARK2 and PINK1. PARK2 upregulates Psen1 promoter transactivation and increases its protein and mRNA levels. PSEN1-associated γ-secretase activity targets APP thereby yielding AICD that interacts physically with FOXO3 to increase Pink1 gene transcription and protein expression. AICD lowers DNM1L/Drp1 and MFN2 (mitofusin 2), increases LC3-II and decreases SQSTM1/p62, TIMM23 (TIMM) and TOMM20 (TOMM) expression. AICD-mediated control of mitochondrial dynamics and mitophagy is totally abolished by Pink1 gene invalidation. PINK1 recruits PARK2 to damaged mitochondria. Thus, PARK2-PINK1 interplay drives the homeostasis of the 2 proteins and regulates mitochondrial dynamics and the mitophagic process that can be disrupted in pathological conditions.
Our work shows that a feedback loop could exist between PARK2 and PINK1 and that this interplay could drive a cellular homeostasis of these proteins. This interplay could well underlie a physiological regulatory process by which cells could promote an adaptive response aimed at regulating mitochondrial function and dynamics. Our study also shows that proteins involved in distinct neurodegenerative diseases could functionally interact in common cellular and molecular pathways that could be altered in pathologies. Thus, we previously showed that Parkinson-disease associated mutations of PARK2 impair its transcriptional function and could thus alter PS expression and associated control of PINK1. Conversely, in Alzheimer disease, mutations of PS or age-related alteration of the degradation process could lead to increased levels of AICD and, thereby, could directly condition PINK1 expression and associated functions. Thus, our work could help to consider in a more comprehensive manner, putative protein candidates, the alterations of which could explain certain common neuroanatomical and functional alterations intervening in apparently distinct pathologies.
We wish to acknowledge the contributions of all authors appearing on the original article. This work was supported by the Conseil départemental des Alpes Maritimes and the Foundation Claude Pompidou. This work has been developed and supported through the LABEX (Excellence Laboratory, program investment for the future) DISTALZ (Development of Innovative Strategies for a Transdisciplinary approach to Alzheimer's disease and the Hospital University Federation (FHU) OncoAge. Thomas Goiran has been funded by the Ligue Contre le Cancer.